Cardiac Stretch–Activated Channels and Mechano-Electric Coupling
The processes that underlie excitation-contraction coupling (ECC; see chapter 16), as well as contractile activity of cardiac muscle, form major foci of basic and applied research. In contrast, the relevance of the mechanical environment for cardiac electrical activity—mechano-electric coupling (MEC)—is often overlooked or ignored. Thus, mechano-electric dissociation is often introduced in experimental research on purpose, by applying pharmacologic uncouplers, to reduce or abolish motion artifacts that interfere with the fidelity of electrical signals, even though this uncoupling alters observed electrical behavior.1 In conceptual terms, cardiac MEC complements ECC, integrating cardiac electrical and mechanical activity into an intracardiac regulatory loop (Figure 14-1).
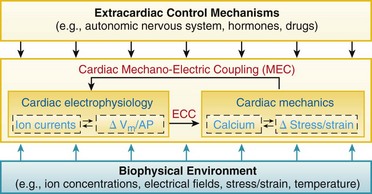
Figure 14-1 Schematic view of cardiac electromechanic integration. Within the heart (center box), electrical behavior steers mechanical activity via excitation-contraction coupling (ECC) but, in turn, it is affected by mechano-electric coupling (MEC). This involves a multitude of interdependent feed-forward and feed-back pathways that alter transsarcolemmal and intracellular ion transport, membrane and action potential configuration (ΔVm/AP), calcium handling, and stress–strain dynamics. Intracardiac electromechanic interactions are modulated by the biophysical environment of the heart (bottom box), and form a target of extracardiac control (top box).
In fact, manifestations of cardiac MEC are present at all levels of structural and functional integration, from in situ and ex vivo whole heart, over in vitro tissue and cells, to subcellular domains such as membrane patches. Indeed, patch-clamp identification of cardiac stretch-activated ion channels (SACs) by Guharay and Sachs2 was pivotal not only for the development of quantitative insight into mechanisms underlying MEC, but also for the advancement of the topic beyond the perception of a scientifically ill-founded clinical curiosity. This chapter will discuss acute electrophysiologic responses of the heart to mechanical stimulation and the involvement of SAC.
Functional Relevance of Cardiac Mechano-Electric Coupling
Effects of cardiac mechanical stimulation on heart rate and rhythm have been reported in the medical literature for more than a century. To name a few key contributions: pioneering work by Felice Meola2a and Ferdinand Riedinger2b in the late nineteenth century identified Commotio cordis (or Commotio thoracica) as an independent pathologic entity where cardiac rhythm disturbances of varying severity are initiated by nonpenetrating mechanical stimulation of the precordium in the absence of visible structural damage to the heart. In the early twentieth century, Eduard Schott29 reported that precordial fist thumps can be used to pace otherwise asystolic ventricles, such as in Adams-Stokes syndrome. At the same time, Francis Bainbridge2c famously identified the positive chronotropic response of the heart to increased venous return.
Mechanical Induction of Nonphysiologic Rhythms
The fact that mechanical stimuli of sufficient amplitude can be used to pace otherwise quiescent hearts has been illustrated by Franz et al., using Langendorff-perfused rabbit heart preparations where the ventricles (rendered asystolic by ablation of the atrioventricular node) were stimulated by periodic inflation of an intraventricular balloon (Figure 14-2, A).
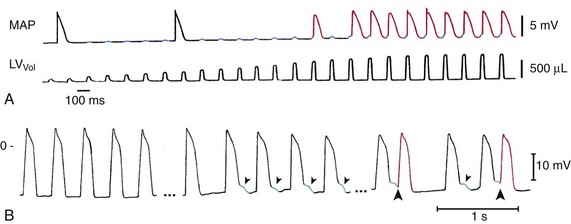
Figure 14-2 Mechanically induced premature ventricular beats (PVB). A, Mechanical pacing of the Langendorff-perfused asystolic rabbit heart. Pulsatile left-ventricular (LV) distention (bottom trace, showing increasing LV balloon inflation) causes diastolic membrane depolarization (top trace, blue highlights). The amplitude of depolarization increases with the mechanical stimulus and, upon reaching threshold levels, gives rise to the generation of action potentials (APs; top trace, red) with each volume pulse. Note that the first two APs are spontaneous escape beats, unrelated to the mechanical stimuli. B, Cardiac contraction–induced tissue depolarization (blue) and PVB induction (red) in patients. MAP recording before (left) and during (middle and right) inflation of a balloon in the right-ventricular outflow tract, which gives rise to early afterdepolarization-like events (blue) and PVB-induction (red). LVVol, volume pulses; MAP, monophasic AP. (A, From Franz MR, Cima R, Wang D, et al: Electrophysiological effects of myocardial stretch and mechanical determinants of stretch-activated arrhythmias. Circulation 86:968–978, 1992. B, From Levine JH, Guarnieri T, Kadish AH, et al: Changes in myocardial repolarization in patients undergoing balloon valvuloplasty for congenital pulmonary stenosis: evidence for contraction-excitation feedback in humans. Circulation 77:70–77, 1988.)
Similar behavior is believed to underlie “fist pacing” in asystolic patients. Energy levels required for mechanical PVB induction by precordial impact have been established by defibrillation pioneer Zoll et al.3 in human volunteers as 0.04 to 1.5 J. For comparison, the lower end of this energy range is equivalent to dropping a golf ball (46 g) from a height of 9 cm (3.5 in).
The study by Zoll et al.3 found that, in anaesthetized dogs, impacts with energies tenfold greater than the PVB-induction threshold do not induce repetitive responses, ventricular tachycardia (VT), or VF, even if applied in the relative refractory period.3 Overall, this is in keeping with the notion that arrhythmogenesis requires the combination of trigger and sustaining mechanisms, so that consequences of isolated ectopic beats, whether mechanically induced or not, are usually benign.
Although these examples illustrate the effects of the external mechanical environment on cardiac MEC (see Figure 14-1, bottom box), mechanical PVB induction can also arise because of the heart’s own contractile activity (see Figure 14-1, middle box). This is illustrated in Figure 14-2, B, which shows MAP recordings from a patient undergoing pulmonary balloon valvuloplasty. In this procedure, the stenosed RV outflow valve is widened by insertion and inflation of a balloon. During balloon inflation, RV contractions are isovolumic (no ejection) and give rise to significantly increased RV peak-pressures. This increase is generally associated with AP-shortening, diastolic depolarization, and occurrence of early afterdepolarization (EAD)-like events. If supra-threshold, these depolarization can give trigger PVB.4 As before, against an otherwise inconspicuous myocardial background, self-sustained repetitive activity is not normally induced.
In contrast, in the presence of preexisting pathologies, PVB can give rise to sustained arrhythmias, as shown in whole-animal studies of pathologically prolonged QT intervals, conducted by Volders et al. In their model, QT-prolongation was induced acutely by pharmacologic block of the slowly activating delayed rectifier potassium current. On this background, additional β-adrenergic stimulation by bolus injection of isoproterenol gives rise to ventricular after contractions of increasing amplitude (up to 25 mm Hg). Originating from near-endocardial locations, their onset precedes (by tens of milliseconds) EAD-like epicardial potentials and alterations in descending T-wave morphology. With increasing amplitude, these apparently contraction-induced afterdepolarizations eventually reach threshold for PVB-induction, followed by torsades de pointes arrhythmic activity.5
Under certain conditions, acute mechanical stimulation can be sufficient to give rise to both trigger and sustaining mechanisms for maintained arrhythmias, even in otherwise healthy myocardium. A prominent example of this is Commotio cordis. A number of risk factors for the mechanical induction of such rhythm disturbances have been identified, based on experimental observations from the pioneering work of Schlomka6 to modern studies by Link.7 Key risk factors include (1) type of impact (impulse-like stimulation, whose arrhythmogenic risk is inversely related to projectile compliance and contact area); (2) impact energy (large subcontusional forces, reaching more than 100 J in competitive sports); (3) impact location (precordial, or in human also spinal, areas that offer efficient energy transmission from body surface to myocardium); and (4) impact timing. Factors 1 to 3 can be regarded as permissive: only if they are all present does timing become decisive.8 This presumably explains why the vast majority of precordial (or spinal) impacts result in relatively benign heart rhythm changes, if any.
The critical time-window for the worst possible outcome of Commotio cordis, induction of VF, overlaps with the T wave. The T wave, during which myocardial electrophysiologic heterogeneity is maximal, has long been associated with a period of increased susceptibility to arrhythmogenesis by electrical stimulation, the so-called vulnerable window. Compared with electrical stimulation, the vulnerable window for mechanical VF-induction is surprisingly narrow (∼15 ms, just before the peak of the T wave in anaesthetized pig).9
Quantitative computational modeling suggests that mechanically induced VF is favored during a short period of time only, when the mechanically stimulated tissue volume overlaps with the trailing wave of excitation.10,11 In this setting, sustained arrhythmogenesis occurs because, in addition to mechanical PVB induction in tissue that has regained excitability (trigger), the intersection of mechanically affected myocardium and the trailing repolarization wave gives rise to a functional block zone around which reentry develops (sustaining mechanism; Figure 14-3). In three-dimensional tissue, the ectopic activation front forms around a transmurally directed conelike volume of tissue in which mechanical activation reaches the AP threshold.12 The more forceful the impact, the closer to fully transmural is the affected tissue cone. In addition, the more focal the stimulus, the closer to perpendicular is this ectopic wavefront, relative to the cardiac surface and, by implication, to the trailing wave-end. This relationship enhances the arrhythmogenic potential, as seen in classic S1/S2 cross-stimulus experiments, and could hold a key to understanding risk factors 1 and 2.
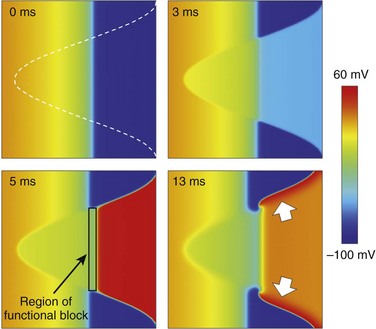
Figure 14-3 Effect of local mechanical stimulation during the critical window of the T wave in a two-dimensional model of ventricular free wall consisting of 256 × 256 electrotonically coupled guinea pig ventricular cell models. A local impact to the epicardium (right-hand side of the grid), timed to occur at 40% tissue repolarization (see trailing wave-end of the previous activation, which travels from right to left; for color-coding, see scale bar), stimulates cation nonselective stretch-activated channels in the mechanically affected tissue (white outline, top left panel). This gives rise to depolarization in tissue that has regained excitability and early repolarization of tissue with positive membrane potentials (top right panel). If suprathreshold, stretch-induced depolarization causes ectopic excitation (bottom left panel). The intersection of trailing wave and mechanically induced new excitation forms a functional block zone. This causes the formation of two wave fronts that progress across the tissue near perpendicularly to the normal direction of electrical wave propagation (bottom-right, see white arrows) and give rise to the formation of two stable reentrant rotors of excitation. (Data from Garny A, Kohl P: Mechanical induction of arrhythmias during ventricular repolarisation: modelling cellular mechanisms and their interaction in 2D. Ann N Y Acad Sci 1015:133–143, 2004.)
In contrast to acute effects, MEC-contributions to arrhythmogenesis in chronic cardiac overload are more difficult to uncover. Usually, pathologies that involve cardiac pressure or volume overload develop slowly, and they are associated with pronounced structural and functional remodeling. In addition, the causes of overload and tissue remodeling could be proarrhythmogenic in their own right. Nonetheless, mechanical factors have been implicated in the domestication of atrial fibrillation (AF),13 whose dominant frequency in patients correlates with atrial pressure.14 Similarly, ventricular arrhythmogenesis has been linked to pressure and volume overload.15,16
A conceptually interesting approach to probing the relevance of mechanical factors for arrhythmogenesis in the chronically overloaded heart is the temporary removal of tissue strain. This can be achieved with the Valsalva maneuver, an attempt to forcefully exhale against the closed glottis. Intrathoracic pressure increases during the strain phase of the maneuver, reducing venous return and favoring arterial drainage from the chest. This condition leads to measurable reductions in cardiac dimensions. On this background, VT has been found to convert to sinus rhythm.17 In this study, 7 of 15 patients showed sustained and 2 showed transient cardioversion. Relief of ventricular wall stress, rather than autonomic nervous system–mediated responses, appears to be a causal contributor to this antiarrhythmic effect, because successful cardioversion can also be observed in the presence of pharmacologic17 or surgical denervation of the heart, such as in transplant recipients.18
Thus, acute stretch can cause diastolic depolarization, which can trigger ectopic excitation. Systolic or sustained stretch can contribute to arrhythmia sustenance by enhancing heterogeneities in excitability, refractoriness, and electrical load. These MEC effects have implications for preventive measures (e.g., chest protector design for sports involving fast-moving projectiles) and for interventions such as hemodynamic unloading, active and passive cardiac assist, or biventricular pacing. In addition, defibrillation threshold increases with ventricular preload,19 apparently not only because of geometric factors,20 but also because of regionally differing MEC-mediated strain effects that raise background electrophysiologic heterogeneity.21 Cardiac mechanosensitivity therefore adds an interesting dimension to the present discussion about whether a period of chest compressions should precede defibrillation attempts in cardiac arrest victims with prolonged delays to intervention.
Mechanical Termination of Nonphysiologic Rhythms
Acute mechanical stimulation, usually by precordial thump (PT), can be used as a means of cardiopulmonary resuscitation. PT has been reported to terminate arrhythmias, including asystole, VT, and rarely VF (Figure 14-4).
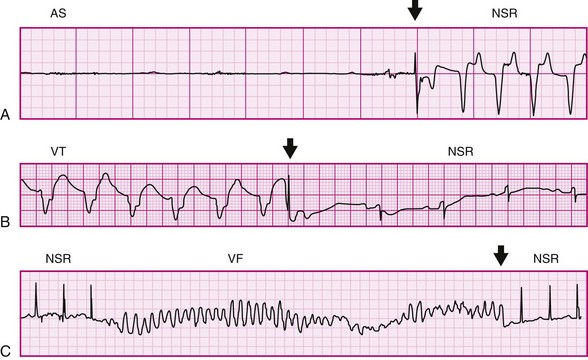
Figure 14-4 Electrocardiograph recordings of precordial thump (PT)-induced cardioversion in patients. A, Conversion of ventricular asystole (AS) to normal sinus rhythm (NSR) by PT. B, PT-conversion of ventricular tachycardia (VT). C, PT-induced termination of early ventricular fibrillation (VF). Arrows indicate PT application. (A, From Pellis T, Kette F, Lovisa D, et al: Utility of precordial thump for treatment of out of hospital cardiac arrest: a prospective study. Resuscitation 80:17–23, 2009. B, From Pennington JE, Taylor J, Lown B: Chest thump for reverting ventricular tachycardia. N Engl J Med 283:1192–1195, 1970. C, From Barrett JS: Chest thumps and the heartbeat. N Engl J Med 284:393, 1971.)
In the emergency resuscitation setting, PT is often the fastest resuscitative procedure available. PT is delivered with the ulnar side of the clinched fist, from a height of approximately 20 cm (8 in), followed by active retraction to emphasize the impulse like nature of the stimulus. The energy levels required for PT-based termination of VT and VF are one to two orders of magnitude higher than those involved in mechanical pacing of the acutely asystolic ventricle (4 to 10 J vs. 0.04 to 1.5 J in adults).22 The more powerful thumps are applied preferentially to the lower sternum, rather than the left sternal edge, which is targeted for fist-pacing or precordial percussion.23
Efficacy of PT in real-life settings is difficult to calibrate and compare, because patient backgrounds vary, mechanical impact properties are not normally monitored, controlled study designs are not usually possible, and single case reports in particular suffer from positive data publication bias. The available information from case series involving tens24 to 100 patients25 suggest that fist-pacing of the bradycardic heart has higher success rates (up to 90%)25 than PT-version of tachyarrhythmias (1% to 60%).24,26 Only three small prospective studies have investigated the clinical utility of PT.26–28 They showed that PT is ineffective in most tachyarrhythmias, that acutely-asystolic arrest (for which PT was initially described in 1920)29 may be the most amenable target for mechanical cardioversion, and that negative side effects are rare (none in >700 cases).26–28
In cases where PT causes successful termination of tachyarrhythmias, it is believed to act primarily via depolarizing excitable tissue in the excitable gaps.30 However, as these gaps may be in myocardium at sites distant from the tissue underneath the precordium, high PT energy levels are necessary and, if there is a multitude of them (e.g., in VF), positive outcomes are rare.
In the asystolic heart, fist-pacing can trigger cardiac excitation and active contraction. The hemodynamic efficacy of such mechanically induced cardiac contractions is not different from electrically stimulated beats,31 and both are about twice as productive (in terms of volume output) as chest compressions, even if performed optimally.32 This efficacy confers resuscitatory value to fist-pacing even when normal sinus rhythm is not immediately reinstated; of course, PT should not delay implementation of other established resuscitation measures.
The most common manifestation of PT effects in ECG recordings, in particular in the asystolic heart, is an impact-induced electrical artifact with an electrical axis that tends to resemble the direction of normal QRS-complexes. This manifestation suggests that mechanically induced excitation in the quiescent heart can proceed along a pathway that has an overall trajectory similar to that of normal activation. It is possible, therefore, that earliest excitation is triggered preferentially either in cells of the secondary/tertiary pacemaker/conduction tissue of the heart (e.g., Purkinje fibers that have long been known to be mechanosensitive),33 or in subendocardial locations, which appear to be more mechanosensitive than subepicardial tissue.5 This possibility is supported by transmural differences in expression levels of potassium-selective SAC (SACK) such as TREK-134 and cation nonselective SAC (SACNS) such as TRPC1.35
Overall, the range of cardiac electrophysiologic responses to mechanical stimulation is not principally different from those caused by electrical energy delivery. Both can be used to pace, cardiovert, or arrest the heart. Concepts such as the vulnerable window for VF-induction apply to both electrical and mechanical stimuli. Even the effective energy ranges required for pacing, cardioversion and defibrillation are not entirely dissimilar. For example, external electrical defibrillation is usually achieved by extracorporeal energy application of 150 to 250 J; of this, only 4% traverses the heart,36 yielding a cardiac energy delivery of 6 to 10 J, similar to that for the PT-version of tachyarrhythmia. These similarities should not be surprising, because mechanical energy can be converted into a transmembrane current by ion fluxes through SAC at the intervention’s target site: the cardiac cell.
Mechanical Modulation of Pacemaking
Diastolic stretch of cardiac tissue causes membrane depolarization, if strong enough to induce any change. In working myocardium, such depolarization may trigger PVB (see Figure 14-2), while in conduction33 and pacemaker tissue37 of many species an increase in beating rate (BR) is observed (Figure 14-5). This positive chronotropic response to stretch is seen predominantly in mammals with low resting heart rates, such as guinea pigs, rabbits, cats, dogs, and humans. In fast-beating murine hearts, in contrast, sustained stretch reduces BR, although apparently via the same underlying mechanism: SACNS activation.38
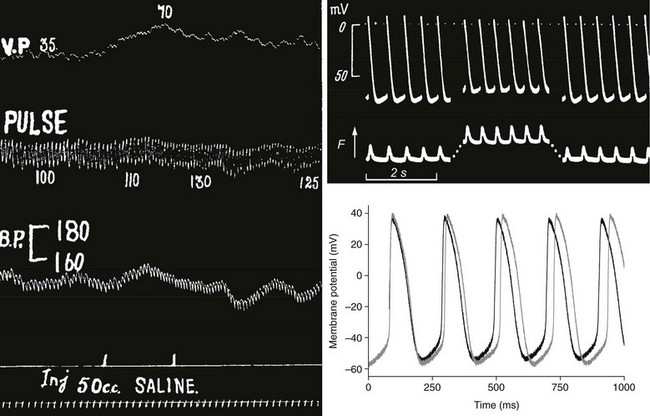
Figure 14-5 Positive chronotropic response to stretch in whole animal, isolated tissue, and single pacemaker cell. Left, Classic observation by Francis Bainbridge, showing that intravenous injection of saline (bottom) raises venous pressure (top) and pulse rate (second from top) without coinciding change in arterial blood pressure (BP) in the anesthetized dog. Top right, Sharp electrode recording of sinoatrial node (SAN) pacemaker cell potential (top) and tissue tension (bottom, contraction pointing upward) during application of a 30% area increase to isolated right-atrial tissue containing the SAN (stretch indicated by the increase in resting tension). Telltale signs of MEC effects are reduction of maximal diastolic and maximal systolic potentials, and increased beating rate. Bottom right, Axial stretch by 7% (black curve) of rabbit single SAN cell gives rise to electrophysiologic changes that match those seen at tissue level. (A, From Bainbridge FA: The influence of venous filling upon the rate of the heart. J Physiol 50:65–84, 1915. B, From Deck KA: Dehnungseffekte am spontanschlagenden, isolierten Sinusknoten. Pflug Arch 280:120–130,1964. C, From Cooper PJ, Lei M, Cheng LX, Kohl P. Axial stretch increases spontaneous pacemaker activity in rabbit isolated sinoatrial node cells. J Appl Physiol 89:2099–2104, 2000.)
From a systematic point of view, early induction of the next heartbeat in response to increased venous return is advantageous, unless volume throughput is limited by inflow, such as in species with already very high heart rates. In species with an upright body posture, however, a chief and evidently overriding requirement for survival is the control of cardiac output pressure to ensure brain perfusion. Therefore, heart rate responses to hemodynamic stimuli that affect both venous return and arterial pressure, such as most changes in body posture (including standard tilt-table experiments), will be determined primarily by arterial pressure control patterns. This obscured the identification of the positive chronotropic response to stretch in human until Donald and Shepherd39 dissociated the increase in venous return from arterial pressure changes, by passively elevating the legs of healthy volunteers in supine position, confirming the positive chronotropic response in humans.39
Similar mechanosensitive behavior is believed to underlie the nonnervous component of respiratory sinus arrhythmia (RSA). Dynamic changes in thoracoabdominal pressure gradients favor venous blood return to the heart during inspiration, causing a relative increase in right atrial filling and an associated rise in heart rate. Although this mechanical component contributes little to RSA at rest (when modulation of vagal innervation is the dominant driver), it is a major cause of RSA during peak exercise in healthy subjects (when vagal tone is reduced, while respiratory effort and associated pressure gradients are enhanced),40,41 and explains the presence of RSA in heart transplant recipients.41
Cardiac Stretch–Activated Ion Channels
Transsarcolemmal Channels
The mammalian heart contains both SACs and VACs. SACs respond instantaneously to mechanical stimuli, whereas VACs tend to show a significant lag-time (up to minutes) between the onset of cell volume changes and ion channel response. VACs, possibly encoded by CIC-3,42 are understood to be important contributors to cardiac electrophysiology in chronic settings, such as ischemic or postischemic cell swelling and hypertrophy (where VAC are constitutionally active).43 In the context of electrophysiologic responses to beat-by-beat changes in the mechanical environment, however, SACs are implied as a key mechanism underlying MEC.
SACs were discovered in the 1980s, initially in cultured avian skeletal muscle by Guhara and Sachs,2 and confirmed in mammalian cardiomyocytes by Craelius et al.44 SACs show either little selectivity for (predominantly monovalent) cations (SACNS, for nonselective), or they preferentially conduct potassium ions (SACK). These selectivity profiles determine their transmembrane current reversal potentials, which are half-way between AP-plateau and resting potentials for SACNS (usually between 0 and –25 mV), and close to the potassium equilibrium-potential for SACK (approximately –95 mV in cardiomyocytes).
Like other phenomenological classifications, mechanosensitive ion channel categories are not absolute, and there is overlap with other types of ion channels. Several mechanosensitive channels are also voltage or ligand sensitive, and vice versa, voltage or ligand activated channels can be modulated by their mechanical environment. Thus, the hyperpolarization-activated cyclic nucleotide gated (HCN) channel is mechanically modulated,45 as is the adenosine triphosphate (ATP)-inactivated potassium channel (KATP), whose open probability is increased by stretch in atrial46 and ventricular myocytes.47 Mechanosensitivity could explain why these two ion channel populations appear to be less active when studied in vitro, compared with predictions based on in situ observations. The contribution of the funny current, if (HCN equivalent in cardiac pacemaker cells) to pacemaking, for example, could be underestimated under conditions of reduced external load, such as in isolated cell and tissue preparations. Likewise, in vitro activation of KATP channels can occur only after reaching abnormally low ATP levels in mechanically unloaded cells. This would be in keeping with the in situ observation that prevention of systolic stretch (or paradoxical segment lengthening) of ischemic myocardium, achieved using a tripodlike mechanical clamp, reduces or delays extracellular potassium accumulation in the anaesthetized pig.16
SACs typically respond to a range of mechanical stimuli, including local membrane deformation, changes in cell curvature, lateral compression, and axial stretch. Whether transfer of mechanical energy to the ion channel protein occurs mainly via the cytoskeleton, or via the lipid bilayer, is a matter of debate. It is likely that both are involved to individually varying degrees. Some SACs, such as the transient receptor potential channel (TRPC1), can be activated in pure lipid bilayers.48 Other SACs are sensitive to cytoskeletal integrity, which could either promote or prevent their opening.49
The actual mechanisms of cardiac SAC activation are not well established. Open and closed state data from large conductance prokaryotic mechanosensitive channels (MscL) have identified one mode of action. This action involves an irislike increase in pore dimensions during channel opening, involving an increase in the outer circumference of the protein in the plane of the sarcolemma, combined with a reduction in transsarcolemmal dimensions.50 These configurational changes would be favored by increased lipid bilayer tension and by membrane thinning, both of which could underlie the mechanosensitive gating of the channel. In this context, any channel whose area projection in the plane of the cell membrane increases during opening should be sensitive to lipid bilayer tension. The question “why do SAC channels exist?” could be replaced, therefore, by asking “should not more channels be at least modulated by their mechanical environment?” Recent reports indicate that the range of mechanically modulated ion channels is indeed significantly larger than generally assumed.45,51
At the same time, no sequence or structure homologues of MscL have been identified in mammalian myocardium. However, recent evidence suggests that TRPC channels (such as TRPC148 or TRPC652) underlie cardiac sarcolemmal SACNS, whereas cardiac SACK appear to include two pore-domain channels (e.g., TREK-1),53 inwardly rectifying channels (e.g., Kir),54 and ligand-activated channels (e.g., KATP, BKCa).46,55 Another promising candidate is formed by piezo proteins that have been shown recently to assemble into large (protein-tetramers containing more than 100 transmembrane domains) mechanosensitive channels in a range of species, from flies to mammals.56
Detailed functional description of SAC has been complicated by the fact that some of them (in particular SACNS) appear to be located in membrane areas that are not easily accessible to patch-clamp investigations in adult mammalian ventricular cells, such as T-tubules57 and caveolae.58 Single-ion channel data have therefore been obtained mainly on neonatal or atrial cells.
Nontranssarcolemmal Channels
Calcium release from and reuptake into the SR have been shown to be modulated in cardiac cells by the mechanical environment, chiefly invoking length-dependent changes in calcium buffer capacity of contractile filaments and direct or secondary effects of transsarcolemmal ion fluxes (e.g., Ca2+-fluxes, or Na+-fluxes that affect the Ca2+-balance via sodium-calcium exchange). However, SR calcium release events (“sparks”) become more frequent during acute axial stretch, even in resting cardiomyocytes and in the absence of extracellular sodium and calcium (Figure 14-6).59 Similarly, stimulation of cultured atrial and ventricular cardiomyocytes by the application of small fluid jets can increase calcium spark rate in a way that appears to draw on mitochondrial calcium.60
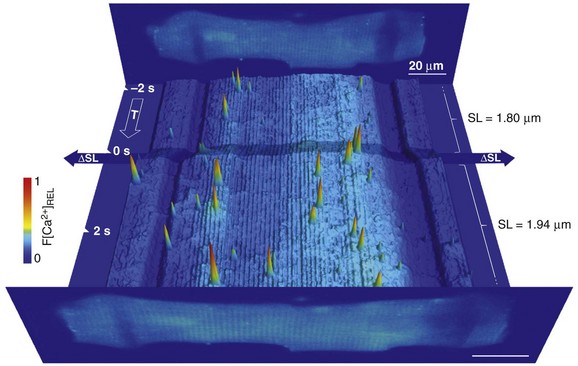
Figure 14-6 Time course of relative Fluo-4 signal intensity in a rat ventricular resting cardiomyocyte, illustrating dynamic changes in spatially resolved Ca2+ concentration before and during axial cell stretch. Upright panels show cell images, averaged from 10 confocal XY scans, before (back) and after (front) stretch application. Low signal-intensity areas, overlapping the cell, reveal carbon fiber (CF) positions (scale bars, 20 µm). In between, fluorescence intensity in each confocal XY scan was added along the y-axis and plotted as a pseudo-3D XT-sequence of relative Ca2+ fluorescence. Note the corrugated appearance of background intensity, which is indicative of sarcomere length (SL). Axial stretch was applied by lateral movement of both CF (shading across XT plot indicates period of CF movement), increasing SL in the affected area by approximately 8%. This is associated with an increase in spark rate. (Data from Iribe G, Ward CW, Camelliti P et al: Axial stretch of rat single ventricular cardiomyocytes causes an acute and transient increase in Ca2+ spark rate. Circ Res 104:787–795, 2009. Figure design courtesy Dr. Alan Garny, University of Oxford.)
In other cell types, the nuclear envelope has been found to contain SACs that contribute to calcium signaling in this domain,61 and it would not be surprising if the same applied to the heart.
Finally, connexin channels can be mechanosensitive. Connexins are best known for linking the cytosols of two contacting cells; although “sarcolemmal,” they do not connect a cell’s inside to the outside in this configuration. Noncardiac connexin46 has been shown to be mechanosensitive.62 It will be interesting to see whether the same applies to cardiac isoforms, such as connexin43, whether located in the sarcolemmal or in the internal membranes of cardiac mitochondria.63 In fact, mitochondria could be more important players in cardiac MEC than customarily assumed, because they are able to generate significant intracellular forces during mitochondrial swelling,64 and hence possibly link metabolic disturbances to mechanisms involved in MEC.
Pharmacologic Probes
All these substances have drawbacks. Gadolinium suffers from a lack in specificity (overlapping concentration range for block of L-type calcium, sodium, and rapid delayed rectifier potassium channels, as well as of the sodium-calcium exchanger) and from precipitation in bicarbonate/phosphate-buffered solutions (although both Gd3+ and gadolinium salts may affect SACs). Aminoglycosidic antibiotics are neither strictly selective (e.g., L-type calcium channel block with a half-maximal inhibition at 1 to 2 mM), nor are they necessarily reliable tools for acute SAC block in situ (if they were, prescribing them as antibiotics would be unlikely). Finally, high-purity GsMTx-4,65 although selective and effective in both D- and L-configurations, still suffers from limited availability and high cost.
Given the above limitations, streptomycin has emerged as a popular pharmacologic probe to study SACs in vitro, where it efficiently and reasonably specifically (at micromolar concentrations) blocks whole-cell currents activated by axial stretch (Figure 14-7). In this context, caution is advised when using cultured cells to study mechanosensitive behavior, because many culture media contain streptomycin. In addition, efficacy in vitro cannot simply be extrapolated to the in situ setting, where it would appear that cardiac SACs, accessible to GsMTx-4, can be protected from acute block by streptomycin.66
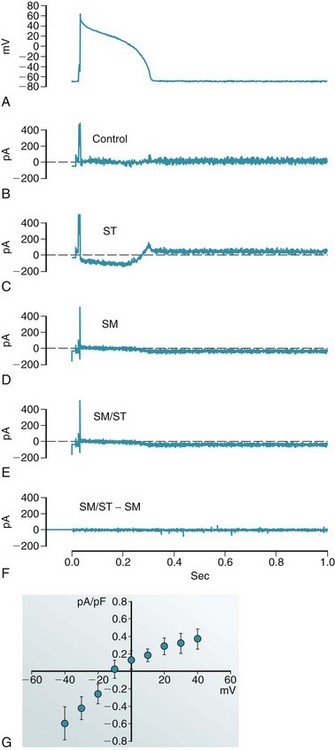
Figure 14-7 Action potential (AP) clamp recording of whole-cell current, induced by axial stretch of guinea pig ventricular myocyte, in the absence and presence of 40 µM of streptomycin. A, AP recorded in control conditions and reapplied to the same cell as a voltage-command (AP clamp). Compensation currents (B-E) illustrate the cell’s response to interventions. Note that compensation currents have the opposite polarity to native transmembrane currents. B, Control. C, After application of 5% stretch (ST). D, After return to control length and application of streptomycin (SM). E, During 5% stretch in the presence of streptomycin (SM/ST). F, Difference current (E minus D), illustrating that any SM-resistant stretch-induced currents are negligible. G, Current-voltage relation of the stretch-induced whole-cell current, measured during AP-clamp repolarization from +40 to –40 mV. This current appears to be commensurate with SACNS and is completely abolished with 40 µM of streptomycin. (Data from Lei M, Cooper PJ, Kohl P. Unpublished.)
The mechanisms of action of SAC block have not been resolved, but they might include screening of negative charges (popular blockers are cations with net charges ranging from 2+ for streptomycin to 5+ for GsMTx-4), interactions with the lipid bilayer, and open channel block (for detail on SACs as pharmacologic targets, see the review by White67).
Manifestations of Cardiac Stretch–Activated Ion Channel Activation
Cell-Level Responses
As a rule, diastolic stretch gives rise to depolarization if it is of sufficient amplitude to cause any changes in membrane potential (Vm; Figure 14-8). This can best be explained by the activation of SACs whose reversal potential is positive to the resting cell membrane (i.e., SACNS).
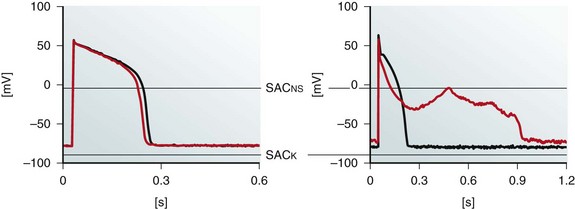
Figure 14-8 Effects of sustained moderate (≤5%) and severe (≥10%) axial stretch on guinea pig isolated ventricular myocyte action potential (AP) morphology. Perforated patch-clamp recordings show AP shortening in the absence of diastolic membrane potential changes during moderate stretch, applied using a pair of carbon fibers (left), whereas more severe distension gives rise to diastolic depolarization, early AP shortening, and crossover of AP repolarization, yielding early afterdepolarization-like behavior (right). Black, Control; red, during stretch; SACNS and SACK indicate differences in reversal potential of SAC populations with different ion selectivity. Note different time scales. (Data from Kohl P, Nesbitt AD, Cooper PJ et al: Sudden cardiac death by Commotio cordis: role of mechano-electric feedback. Cardiovasc Res 50:280-289, 2001.)
The effects of stretch during the AP are less clear, because AP shortening, crossover of repolarization, and delayed repolarization have all been reported. Some of this discrepancy could be explained by differences in recording techniques, which can affect stretch targets. For instance, sharp electrodes and a perforated or ruptured patch can give rise to opposite AP duration changes, hinging on differential effects of these techniques on the interaction between SACs and calcium handling.68 Stretch configuration matters as well, because moderate stretch initially affects only AP duration (the AP plateau is electrophysiologically more labile than the resting potential), whereas more severe distension alters diastolic behavior, potentially causing crossover of late AP repolarization and EAD-like behavior (see Figure 14-8). Another confounding aspect is related to the repolarization level at which AP duration is measured. Because early AP shortening may coincide with late AP-prolongation, this can give rise to apparently contradictory changes in reported AP duration.
Whereas stretch of normal myocardium appears to preferentially target SACNS (whose block fully eradicates whole-cell current responses in isolated cells; see Figure 14-7), this could be different under conditions of metabolic impairment. Thus, KATP channels that are normally quiescent in atrial and ventricular cardiomyocytes respond to pipette suction46 and axial stretch47 when preactivated by a reduction in ATP concentration. Coactivation of a potassium-selective channel by ATP reduction and stretch will give rise to a less-positive reversal potential of the “net whole-cell current” induced by stretch in the ATP-starved heart. This might explain the reduced efficacy of PT (less efficient eradication of excitable gaps by reduced net inward current) in myocardium with severe metabolic impairment.47
Stretch effects may vary not only with disease, but also with species, age, cell type, and location. For example, subendocardial ventricular cardiomyocytes appear to be more likely than subepicardial cells to respond to mechanical stimulation with EAD-like behavior.5 Differences in mechanosensitivity can be caused by (1) regionally or transmurally varying levels of relevant mechanical stimuli; (2) different mechanical properties of cell and tissue components involved in the transmission of external mechanical stimuli to the actual mechanotransducers; (3) variable expression or responsiveness of mechanotransducers; and (4) distinct electrophysiologic background properties of affected cells and tissues. The latter appears to underlie species differences in SAN pacemaker responses to mechanical stimulation.
The response of the SAN pacemaker cells to stretch seems to chiefly involve SACNS activation, as shown in rabbit SAN isolated cells during axial stretch.69 The principal effects of stretch on SAN cell AP-morphology are qualitatively equivalent to those observed in SAN tissue (see Figure 14-5). They include (in addition to truncation of diastolic and systolic Vm-maxima) acceleration of spontaneous diastolic depolarization and of early AP repolarization, which can both act to increase pacemaker rate, combined with a slowing of late AP repolarization when Vm is below the SACNS reversal potential, which would prolong the pacemaker cycle and reduce beating rate if it dominated the response. In other words, SAN pacemaker potential changes that move toward the reversal potential of SACNS (–11 mV in rabbit SAN cells)69 are accelerated by stretch, whereas those that move away from the reversal potential are slowed.
If one compares the SAN pacemaker potential waveforms of slow- and fast-beating mammalian heart (e.g., guinea pig or rabbit versus mouse or rat), it is apparent that murine SAN pacemaker potentials have a very different AP morphology: both upstroke and initial repolarization are extremely fast, followed by an extended late repolarization phase. The percentage of the pacemaker cycle during which Vm moves away from the SACNS reversal potential dominates murine SAN pacemaking (71% in mouse, compared to 46% in rabbit)66; this might underlie the negative chronotropic response to sustained stretch, observed in murine hearts. Importantly, both negative and positive chronotropic responses to sustained stretch can be abolished by the application of GsMTx-4,66 confirming the pivotal contribution of SACNS. Thus, identical mechanisms can give rise to opposite responses, depending on the electrophysiologic background of affected cells. This reemphasizes the need for caution when extrapolating observations between species, such as from mouse to human.
Of course, SAN tissue in situ will not normally be stretched throughout the entire cycle, and more refined experimental study designs are needed to apply mechanical stimulation in a cycle-dependent manner. In addition, it will be interesting to explore whether individual components of the “voltage- and calcium-clocks” that drive pacemaking are directly mechanosensitive in native SAN cells (e.g., HCN or SR-release channels) and whether effects secondary to other MEC-actions, such as changes in intracellular calcium concentration, determine pacemaker responses to stretch.70
Tissue- and Organ-Level Effects
On the “output side,” many of the standard techniques used to record electrophysiologic consequences of mechanical stimulation in tissue and organ preparations report ensemble properties (including ECG, MAP, and optical mapping). In this context, it is important to recall that the heart contains a large number of different cell types, the majority of which are not cardiomyocytes. These types include endothelial cells, fibroblasts, smooth muscle, and intracardiac neurons, all of which are mechanosensitive and can affect cardiac electrophysiologic responses to mechanical stimulation. In addition, stretch can influence conduction velocity (reports in the literature are divided between increase, reduction, and no change, whereas reported effects depend on stretch amplitudes and could differ in conduction system versus working muscle),70 which would be important for the interpretation of electrophysiologic ensemble data, and for their pathophysiologic relevance.
Given this complexity, the identification of SAC contributions to cardiac MEC in multicellular preparations has relied largely on pharmacologic probes. For most SAC types, specific and efficient openers are not available, whereas available blockers suffer from a lack of cardiomyocyte specificity, as well as the limitations discussed earlier. In addition, it is not sufficient for a blocker to be SAC specific at the single cell level, but it must also be effective upon acute application to native tissue. This is not necessarily the case for streptomycin,66 which calls for careful interpretation, in particular of apparently negative observations at the tissue level.
Despite these notes of caution, the available evidence overwhelmingly demonstrates that block of SACNS abolishes stretch-induced changes in SAN pacemaker rate. It can also prevent the mechanical induction of arrhythmogenic triggers, such as single PVB,71 and the mechanical promotion of sustained arrhythmias, such as the preload-dependent amplification of burst-pacing–induced AF.72 In contrast, block of SACK can increase PVB inducibility.55 Interestingly, however, SACK block may still prevent mechanically induced development of VF.73 Whether this is caused by removal of an arrhythmia-sustaining effect (ectopic excitation is still observed), a shift in the space-time sensitive narrow vulnerable window for mechanical VF-induction, or another mechanism, remains to be confirmed.
Summary and Outlook
The currently available data suggest that mechanical stimulation, acting via sarcolemmal SACs, gives rise to changes in cardiomyocyte Vm and AP morphology. SACNS-mediated effects in particular appear to be sufficient to explain quantitatively the majority of acute functional consequences of cardiac MEC quantitatively in physiologic conditions (Figure 14-9). Sufficiency should not, however, be confused with validity, necessity, or exclusivity. There are complex interactions of SAC effects with other mediators of mechano-electric integration, in particular those affecting calcium.74 SACs can contribute to altered calcium handling either directly (e.g., via changes in SR calcium release or via transsarcolemmal Ca2+-flux through SACNS), or indirectly (via changes in Vm/AP morphology that alter voltage-sensitive Ca2+ fluxes, or through SACNS Na+-influx with knock-on effects on the balance of transsarcolemmal ion exchange).
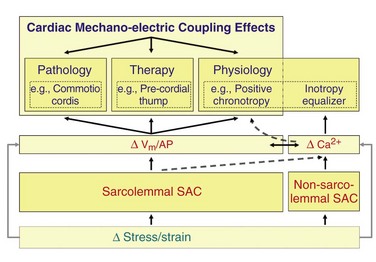
Figure 14-9 Schematic representation of functional relevance (blue) and key mechanisms (red) of acute cardiac MEC. Effects of MEC are particularly striking in the context of induction and termination of arrhythmias (top left and middle), and can be explained by effects of stretch-activated ion channels (SACs) on cardiomyocyte electrophysiology. The physiologic relevance of cardiac SAC (top right) is not documented as well, which in addition to preload-dependent modulation of pacemaking rate is likely to include an additional role: the matching of individual cell contractility to external demand. The latter is linked to mechanical modulation of cellular calcium handling (ΔCa2+). Dashed lines indicate some of the possible links between classic MEC components and cell calcium, such as from SAC-effects on calcium handling, to modulation of SAN pacemaking (see text for detail). Vm, Membrane potential; AP, action potential.
Such matching of local contractility to dynamically varying external loads has been shown experimentally in mechanically linked cardiac muscle preparations,75 and it could act as an equalizer of the inotropic state of individual cardiomyocytes (see Figure 14-9). This mechanism could underlie efficient function of cells across regionally and temporally varying myocardial stress–strain gradients in the healthy heart—in particular as the activity of individual cells is not controlled by neuromuscular junctions, in contrast to skeletal muscle. Disturbances of this balance can be arrhythmogenic if they are sustained, because even small wall-motion abnormalities in patients are associated with increased dispersion of repolarization.76
So—What Next?
• Development of improved tools to assess strain and stress in cells and tissue, such as those heralded by the advent of fluorescent force-sensors for live-cell studies,77 and by stretchable electronics for whole-heart research78
• Identification of the mechanisms underlying SAC activation, including open and closed state SAC structures, differential contributions of stress and strain, and characterization of links between externally applied and internally sensed mechanical clues, including transmission to nonsarcolemmal ion channels59
• Integration of MEC research with mechano-mechanic and acute mechanochemical transduction studies, such as involving reactive oxygen species79,80
• Exploration of cross-talk between MEC and pharmacologic interventions, including the identification of MEC components as drug targets and modulators81
References
1. Brines, L, Such-Miquel, L, Gallego, D, et al. Modifications of mechanoelectric feedback induce1d by 2,3-butanedione monoxime and blebbistatin in Langendorff-perfused rabbit hearts. Acta Physiol. 2012; 206:29–41.
2. Guharay, F, Sachs, F. Stretch-activated single ion channel currents in tissue-cultured embryonic chick skeletal muscle. J Physiol. 1984; 352:685–701.
2a. Meola, F. La commozione toracica. G Int Sci Med. 1879; 1:923–937.
2b. Riedinger, F. Über Brusterschütterung. In: Festchrift zur dritten Saecularfeier der Alma Julia Maximiliana Leipzig. Leipzig: Verlag von F. C. W. Vogel; 1882:221–234.
2c. Bainbridge, FA. The influence of venous filling upon the rate of the heart. J Physiol. 1915; 50:65–84.
3. Zoll, PM, Belgard, AH, Weintraub, MJ, et al. External mechanical cardiac stimulation. N Engl J Med. 1976; 294:1274–1275.
4. Levine, JH, Guarnieri, T, Kadish, AH, et al. Changes in myocardial repolarization in patients undergoing balloon valvuloplasty for congenital pulmonary stenosis: evidence for contraction-excitation feedback in humans. Circulation. 1988; 77:70–77.
5. Gallacher, DJ, Van de Water, A, van der Linde, H, et al. In vivo mechanisms precipitating torsades de pointes in a canine model of drug-induced long-QT1 syndrome. Cardiovasc Res. 2007; 76:247–256.
6. Schlomka, G. Commotio cordis und ihre Folgen. Die Einwirkung stumpfer Brustwandtraumen auf das Herz. Ergeb inn Med Kinderhkd. 1934; 47:1–91.
7. Link, MS. Commotio cordis: sudden death from blows to the chest wall. In: Kohl P, Sachs F, Franz MR, eds. Cardiac Mechano-Electric Coupling and Arrhythmias. Oxford: Oxford University Press, 2011. [325–239].
8. Kohl, P, Nesbitt, AD, Cooper, PJ, et al. Sudden cardiac death by Commotio cordis: role of mechano-electric feedback. Cardiovasc Res. 2001; 50:280–289.
9. Link, MS, Wang, PJ, Pandian, NG, et al. An experimental model of sudden cardiac death due to low-energy chest-wall impact (Commotio cordis). NEJM. 1998; 338:1805–1811.
10. Garny, A, Kohl, P. Mechanical induction of arrhythmias during ventricular repolarisation: modelling cellular mechanisms and their interaction in 2D. Ann NY Acad Sci. 2004; 1015:133–143.
11. Li, W, Kohl, P, Trayanova, N. Induction of ventricular arrhythmias following a mechanical impact: a simulation study in 3D. J Mol Histol. 2004; 35:679–686.
12. Garny, A, Noble, D, Kohl, P. Dimensionality in cardiac modelling. Prog Biophys Mol Biol. 2005; 87:47–66.
13. Allessie, MA, Ausma, J, Schotten, U. Electrical, contractile and structural remodeling during atrial fibrillation. Cardiovasc Res. 2002; 54:230–246.
14. Yoshida, K, Ulfarsson, M, Oral, H, et al. Left atrial pressure and dominant frequency of atrial fibrillation in humans. Heart Rhythm. 2011; 8:181–187.
15. Moreno, J, Zaitsev, AV, Warren, M, et al. Effect of remodelling, stretch and ischaemia on ventricular fibrillation frequency and dynamics in a heart failure model. Cardiovasc Res. 2005; 65:158.
16. Bollensdorff, C, Lab, MJ. Stretch effects on potassium accumulation and alternans in pathological myocardium. In: Kohl P, Sachs F, Franz MR, eds. Cardiac Mechano-Electric Coupling and Arrhythmias. Oxford: Oxford University Press; 2011:173–179.
17. Waxman, MB, Wald, RW, Finley, JP, et al. Valsalva termination of ventricular tachycardia. Circulation. 1980; 62:843–851.
18. Ambrosi, P, Habib, G, Kreitmann, B, et al. Valsalva manoeuvre for supraventricular tachycardia in transplanted heart recipient. Lancet. 1995; 346:713.
19. Dosdall, DJ, Doppalapudi, H, Ideker, RE. Mechanical modulation of defibrillation and resuscitation efficacy. In: Kohl P, Sachs F, Franz MR, eds. Cardiac Mechano-Electric Coupling and Arrhythmias. Oxford: Oxford University Press; 2011:374–380.
20. Trayanova, N, Li, WH, Eason, J, et al. Effect of stretch-activated channels on defibrillation efficacy. Heart Rhythm. 2004; 1:67–77.
21. Li, W, Gurev, V, McCulloch, AD, et al. The role of mechanoelectric feedback in vulnerability to electric shock. Prog Biophys Mol Biol. 2008; 97:461–478.
22. Pellis, T, Kohl, P. Anti-arrhythmic effects of acute mechanical stimulation. In: Kohl P, Sachs F, Franz M, eds. Cardiac Mechano-Electric Coupling and Arrhythmias. Oxford: Oxford University Press; 2011:361–368.
23. Eich, C, Bleckmann, A, Schwarz, SKW. Percussion pacing – an almost forgotten procedure for haemodynamically unstable bradycardias? A report of three case studies and review of the literature. Brit J Anaesth. 2007; 98:429–433.
24. Befeler, B. Mechanical stimulation of the heart: its therapeutic value in tachyarrhythmias. Chest. 1978; 73:832–838.
25. Klumbies, A, Paliege, R, Volkmann, H. Mechanische Notfallstimulation bei Asystolie und extremer Bradykardie. Z Ges. 1988; 43:348–352.
26. Amir, O, Schliamser, JE, Nemer, S, et al. Ineffectiveness of precordial thump for cardioversion of malignant ventricular tachyarrhythmias. Pacing Clin Electro Physiol. 2007; 30:153–156.
27. Haman, L, Parizek, P, Vojacek, J. Precordial thump efficacy in termination of induced ventricular arrhythmias. Resuscitation. 2009; 80:14–16.
28. Pellis, T, Kette, F, Lovisa, D, et al. Utility of pre-cordial thump for treatment of out of hospital cardiac arrest: a prospective study. Resuscitation. 2009; 80:17–23.
29. Schott, E. Über Ventrikelstillstand (Adam-Stokes’sche Anfälle) nebst Bemerkungen über andersartige Arrhythmien passagerer Natur. (“On Ventricular Standstill (Adam-Stokes Attacks) together with other Arrhythmias of Temporary Nature. ”). Dtsch Arch Klin Med. 1920; 131:211–229.
30. Pennington, JE, Taylor, J, Lown, B. Chest thump for reverting ventricular tachycardia. N Eng J Med. 1970; 283:1192–1195.
31. Chan, L, Reid, C, Taylor, B. Effect of three emergency pacing modalities on cardiac output in cardiac arrest due to ventricular asystole. Resuscitation. 2002; 52:117–119.
32. Iseri, LT, Allen, BJ, Baron, K, et al. Fist pacing, a forgotten procedure in bradysystolic cardiac arrest. Am Heart J. 1987; 113:1545–1550.
33. Kaufmann, R, Theophile, U. Automatie-fördernde Dehnungseffekte an Purkinje-Fäden, Papillarmuskeln und Vorhoftrabekeln von Rhesus-Affen. Pflüg Arch. 1967; 297:174–189.
34. Tan, JH, Liu, W, Saint, DA. Differential expression of the mechanosensitive potassium channel TREK-1 in epicardial and endocardial myocytes in rat ventricle. Exp Physiol. 2004; 89:237–242.
35. Stones, R, Calaghan, SC, Billeter, R, et al. Transmural variations in gene expression of stretch-modulated proteins in the rat left ventricle. Pflug Arch. 2007; 454:545–549.
36. Lerman, BB, Deale, OC. Relation between transcardiac and transthoracic current during defibrillation in humans. Circ Res. 1990; 67:1420–1426.
37. Deck, KA. Dehnungseffekte am spontanschlagenden, isolierten. Sinusknoten. Pflüg Arch. 1964; 280:120–130.
38. Cooper, PJ, Ravens, U. Mechanical modulation of pacemaker electrophysiology. In: Kohl P, Sachs F, Franz MR, eds. Cardiac Mechano-Electric Coupling and Arrhythmias. Oxford: Oxford University Press; 2011:95–102.
39. Donald, DE, Shepherd, JT. Reflexes from the heart and lungs: physiological curiosities or important regulatory mechanisms. Cardiovasc Res. 1978; 12:449–469.
40. Casadei, B, Moon, J, Johnston, J, et al. Is respiratory sinus arrhythmia a good index of cardiac vagal tone in exercise? J Appl Physiol. 1996; 81:556–564.
41. Bernardi, L, Salvucci, F, Suardi, R, et al. Evidence for an intrinsic mechanism regulating heart rate variability in the transplanted and the intact heart during submaximal dynamic exercise? Cardiovasc Res. 1990; 24:969–981.
42. Duan, DD. The ClC-3 chloride channels in cardiovascular disease. Acta Pharmacol Sin. 2011; 32:675–684.
43. Baumgarten, CM, Clemo, HF. Swelling-activated chloride channels in cardiac physiology and pathophysiology. Prog Biophys Mol Biol. 2003; 82:25–42.
44. Craelius, W, Chen, V, El-Sherif, N. Stretch activated ion channels in ventricular myocytes. Bosci Rep. 1988; 8:407–414.
45. Lin, W, Laitko, U, Juranka, PF, et al. Dual stretch responses of mHCN2 pacemaker channels: accelerated activation, accelerated deactivation. Biophys J. 2007; 92:1559–1572.
46. Van Wagoner, DR, Lamorgese, M. Ischemia potentiates the mechanosensitive modulation of atrial ATP-sensitive potassium channels. Ann N Y Acad Sci. 1994; 723:392–395.
47. Kohl, P, Bollensdorff, C, Garny, A. Effects of mechano-sensitive ion channels on ventricular electrophysiology: experimental and theoretical models. Exp Physiol. 2006; 91:307–321.
48. Maroto, R, Raso, A, Wood, TG, et al. TRPC1 forms the stretch-activated cation channel in vertebrate cells. Nature Cell Biol. 2005; 7:179–185.
49. Janmey, PA. The cytoskeleton and cell signalling: component localization and mechanical coupling. Physiol Rev. 1998; 78:763–781.
50. Perozo, E, Kloda, A, Cortes, DM, et al. Physical principles underlying the transduction of bilayer deformation forces during mechanosensitive channel gating. Nat Struct Biol. 2002; 9:696–703.
51. Morris, CE, Juranka, PF. Nav channel mechanosensitivity: activation and inactivation accelerate reversibly with stretch. Biophys J. 2007; 93:822–833.
52. Dyachenko, V, Christ, A, Gubanov, R, et al. Misalignment of sarcomeres by mechanical stimuli: an input signal for integrin dependent modulation of ion channels? Prog Biophys Mol Biol. 2008; 97:196–216.
53. Terrenoire, C, Lauritzen, I, Lesage, F, et al. TREK-1-like potassium channel in atrial cells inhibited by β-adrenergic stimulation and activated by volatile anesthetics. Circ Res. 2001; 89:336–342.
54. Tamargo, J, Caballero, R, Gomez, R, et al. Pharmacology of cardiac potassium channels. Cardiovasc Res. 2004; 62:9–33.
55. Iribe, G, Jin, H, Naruse, K. Role of sarcolemmal BKCa channels in stretch-induced extrasystoles in isolated chick hearts. Circ J. 2011; 75:2552–2558.
56. Coste, B, Xiao, B, Santos, JS, et al. Piezo proteins are pore-forming subunits of mechanically activated channels. Nature. 2012; 483:176–181.
57. Zeng, T, Bett, GCL, Sachs, F. Stretch-activated whole cell currents in adult rat cardiac myocytes. Am J Physiol. 2000; 278:H548–H557.
58. Kohl, P, Cooper, PJ, Holloway, H. Effects of acute ventricular volume manipulation on in situ cardiomyocyte cell membrane configuration. Prog Biophys Mol Biol. 2003; 82:221–227.
59. Iribe, G, Ward, CW, Camelliti, P, et al. Axial stretch of rat single ventricular cardiomyocytes causes an acute and transient increase in Ca2+ spark rate. Circ Res. 2009; 104:787–795.
60. Belmonte, S, Morad, M. ‘Pressure-flow’-triggered intracellular Ca2+ transients in rat cardiac myocytes: possible mechanisms and role of mitochondria. J Physiol. 2008; 586:1379–1397.
61. Itano, N, Okamoto, S, Zhang, D, et al. Cell spreading controls endoplasmic and nuclear calcium: a physical gene regulation pathway from the cell surface to the nucleus. Proc Natl Acad Sci U S A. 2003; 100:5181–5186.
62. Bao, L, Sachs, F, Dahl, G. Connexins are mechanosensitive. Am J Physiol. 2004; 278:C1389–C1395.
63. Miro-Casas, E, Ruiz-Meana, M, Agullo, E, et al. Connexin43 in cardiomyocyte mitochondria contributes to mitochondrial potassium uptake. Cardiovasc Res. 2009; 83:747–756.
64. Kaasik, A, Kuum, M, Joubert, F, et al. Mitochondria as a source of mechanical signals in cardiomyocytes. Cardiovas Res. 2010; 87:83–91.
65. Bowman, CL, Gottlieb, PA, Suchyna, TM, et al. Mechanosensitive ion channels and the peptide inhibitor GsMTx-4: history, properties, mechanisms and pharmacology. Toxicon. 2007; 49:249–270.
66. Cooper, PJ, Kohl, P. Species- and preparation-dependence of stretch effects on sino-atrial node pacemaking. Ann N Y Acad Sci. 2005; 1047:324–335.
67. White, E. Mechanosensitive channels: therapeutic targets in the myocardium? Current Pharmaceutical Design. 2006; 12:3645–3663.
68. Calaghan, SC, Belus, A, White, E. Do stretch-induced changes in intracellular calcium modify the electrical activity of cardiac muscle? Prog Biophys Mol Biol. 2003; 82:81–95.
69. Cooper, PJ, Lei, M, Cheng, L-X, et al. Axial stretch increases spontaneous pacemaker activity in rabbit isolated sinoatrial node cells. J Appl Physiol. 2000; 89:2099–2104.
70. Quinn, TA, Kohl, P. Mechano-sensitivity of cardiac pacemaker function: pathophysiological relevance, experimental implications, and conceptual integration with other mechanisms of rhythmicity. Prog Biophys Mol Biol. 2012; 110:257–268.
71. Hansen, DE, Borganelli, M, Stacy, GPJ, et al. Dose-dependent inhibition of stretch-induced arrhythmias by gadolinium in isolated canine ventricles. Evidence for a unique mode of antiarrhythmic action. Circ Res. 1991; 69:820–831.
72. Bode, F, Sachs, F, Franz, MR. Tarantula peptide inhibits atrial fibrillation. Nature. 2001; 409:35–36.
73. Link, MS, Wang, PJ, VanderBrink, BA, et al. Selective activation of the K+ATP channel is a mechanism by which sudden death is produced by low-energy chest-wall impact (commotio cordis). Circulation. 1999; 100:413–418.
74. ter Keurs, HE, Wakayama, Y, Sugai, Y, et al. Role of sarcomere mechanics and Ca2+ overload in Ca2+ waves and arrhythmias in rat cardiac muscle. Ann NY Acad Sci. 2006; 1080:248–267.
75. Solovyova, O, Katsnelson, L, Konovalov, P, et al. Activation sequence as a key factor in spatio-temporal optimization of myocardial function. Phil Trans Roy Soc. 2006; 364:1367–1383.
76. Opthof, T, Sutton, P, Coronel, R, et al. The association of abnormal ventricular wall motion and increased dispersion of repolarization in humans is independent of the presence of myocardial infarction. Front Physiol. 2012; 3:235.
77. Meng, F, Suchyna, TM, Sachs, F. A fluorescence energy transfer-based mechanical stress sensor for specific proteins in situ. FEBS J. 2008; 275:3072–3087.
78. Kim, DH, Ghaffari, R, Lu, N, et al. Flexible and stretchable electronics for biointegrated devices. Annu Rev Biomed Eng. 2012; 14:113–128.
79. Dyachenko, V, Rueckschloss, U, Isenberg, G. Modulation of cardiac mechanosensitive ion channels involves superoxide, nitric oxide and peroxynitrite. Cell Calcium. 2009; 45:55–64.
80. Prosser, BL, Ward, CW, Lederer, WJ. X-ROS signaling: rapid mechano-chemo transduction in heart. Science. 2011; 333:1440–1445.
81. Beyder, A, Strege, PR, Reyes, S, et al. Ranolazine decreases mechanosensitivity of the voltage-gated sodium ion channel NaV1. 5: A novel mechanism of drug action. Circulation. 2012; 125:2698–2706.