Cardiac Disorders
Perspective
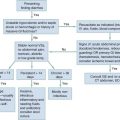
The second scenario represents more of a challenge to the emergency physician: the child with an undiagnosed congenital or acquired cardiac disorder who presents to the emergency department with nonspecific or concerning signs and symptoms (Box 171-1). This chapter focuses on some of the more common and life-threatening cardiac disorders in infants and children who present to the emergency department, with an emphasis on rapid evaluation, stabilization, and management of these disorders.
Principles of Disease
Fetal and Neonatal Circulation
Some of the key features of the fetal circulation that differ from the circulation of a child are the ductus venosus, the ductus arteriosus, and a patent foramen ovale. During fetal development, blood is oxygenated by the placenta, flows to the fetus through the umbilical vein, bypasses the fetal liver through the ductus venosus, and returns to the fetal heart through the inferior vena cava. Blood returning from the inferior vena cava then enters the right atrium and is preferentially shunted across to the left atrium through the patent foramen ovale (Fig. 171-1). The blood in the left atrium is then pumped from the left ventricle to the aorta. The oxygenated blood ejected through the ascending aorta is preferentially directed to the fetal coronary and cerebral circulations.
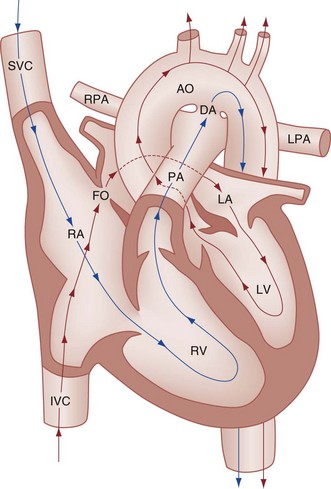
Deoxygenated blood that returns to the right atrium through the superior vena cava crosses the tricuspid valve and is pumped into the fetal pulmonary artery through the right ventricle. Because the fetal pulmonary vascular resistance (PVR) is higher than the fetal systemic vascular resistance (SVR), this deoxygenated blood bypasses the nonoxygenated fetal lungs through the patent ductus arteriosus (see Fig. 171-1). This poorly oxygenated blood enters the aorta through the patent ductus arteriosus and then mixes with the well-oxygenated blood in the descending aorta. The mixed blood in the descending aorta then returns to the placenta for oxygenation through the two umbilical arteries.
Pathophysiology of Cardiovascular Compensatory Responses
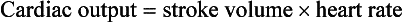
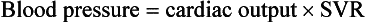
The young myocardium is inefficient and unable to increase its contractility in response to demand.1 When more cardiac output is needed, infants and children respond with an increase in heart rate. Therefore, bradycardia in infants and young children is an ominous sign that connotes a severely compromised cardiac output. Children develop the adult capacity to increase their stroke volume to improve overall cardiac output by 8 to 10 years of age.2
On the basis of the first physiologic formula, as stroke volume decreases, a compensatory increase in the heart rate will be necessary to preserve a normal cardiac output. A decrease in stroke volume can be produced by a weak “pump,” decreased volume in the circulation, or both. The most common cause of decreased stroke volume in children is hypovolemia due to dehydration. Other causes of decreased stroke volume in children may be implicated (Box 171-2). Thus tachycardia is the first compensatory cardiovascular response when the stroke volume is decreased. If tachycardia alone is not enough to maintain a normal cardiac output, the next compensatory physiologic mechanism to preserve perfusion is an increase in the SVR. This increase in SVR is exhibited as an increase in the diastolic blood pressure, which in turn accounts for a narrowed pulse pressure. The clinical examination findings of the extremities of a child with an increased SVR include pallor, mottling, cool skin, delayed capillary refill time (>2 seconds), and weak or thready distal pulses.
Pathophysiology of Cyanosis
Cyanosis is a clinical sign caused by the presence of deoxygenated blood in the capillary beds, most readily observed in the mucous membranes, conjunctiva, nail beds, and skin. The presence of cyanosis usually means that there are at least 4 to 5 g/dL of deoxyhemoglobin in the blood, which correlates with an oxygen saturation of about 80 to 85%.3 Central cyanosis results from a decrease in pulmonary ventilation and oxygenation, a decrease in pulmonary perfusion, the shunting of deoxygenated blood directly into the systemic circulation, or the presence of abnormal hemoglobin. Cyanosis in the neonate can be due to a variety of cardiac, pulmonary, or hematologic causes. Cardiac causes of cyanosis include congenital lesions with right-to-left shunts and cardiac lesions with decreased or increased pulmonary blood flow. Common pulmonary causes of cyanosis include bronchiolitis, pneumonia, and pulmonary edema. Methemoglobinemia can be one of the hematologic causes of cyanosis.
Clinical Features of Cyanosis
The region of the body that is cyanotic can provide important clinical clues to the cause of the cyanosis. Central cyanosis involves the lips, tongue, and mucous membranes, whereas peripheral cyanosis (acrocyanosis) involves the hands and feet. Acrocyanosis is a common phenomenon in neonates caused by cold stress and peripheral vasoconstriction. Central cyanosis reflects a pathologic origin and is an ominous sign. Infants with cyanosis secondary to a congenital heart defect may not exhibit as much respiratory distress compared with the infant with cyanosis due to a pulmonary cause. Thus a cardiac cause of central cyanosis may be more likely than a purely pulmonary cause in the child who appears “comfortably blue.” Another important clinical clue to the cause of central cyanosis is that cyanosis of cardiac origin usually worsens with crying, whereas cyanosis due to a pulmonary cause may improve when the infant cries.4 Cyanotic congenital heart defects with right-to-left shunting will demonstrate a minimal improvement with supplemental oxygen, whereas cyanosis of a purely pulmonary origin typically exhibits a significant improvement with supplemental oxygen (Table 171-1).
Table 171-1
Clinical Clues to Help Distinguish between Cardiac and Pulmonary Causes of Central Cyanosis
| CARDIAC ETIOLOGY | PULMONARY ETIOLOGY | |
| Respiratory status | May be “comfortably blue” | Respiratory distress |
| Response to crying | Worsening cyanosis | Improved cyanosis |
| Response to oxygen | Minimal or no improvement | Improvement with oxygen |
Clinical Features and Diagnostic Strategies: the Cardiac Evaluation
The key elements that should be elicited in the history of a child with a known underlying cardiac disorder are listed in Box 171-3. Early consultation with the child’s cardiologist or cardiac surgeon is extremely useful.
History
Infants with an underlying congenital heart disorder may exhibit diaphoresis during feeds and poor weight gain secondary to congestive heart failure (CHF). The cause of the infant’s hypoxia—cardiac or pulmonary—may be ascertained by the age at onset and the events surrounding a change in color. For example, an infant who sweats during feeding may exhibit a splanchnic steal from anomalous coronary arteries, causing transient ischemia, pain, color change, and diaphoresis that resolve after eating.1 A child with an undiagnosed congenital heart defect may take longer to feed, frequently pausing to catch his or her breath, with subsequent poor weight gain and gradually increased work of breathing due to having CHF and pulmonary edema. Respiratory tract infections are common during childhood and may cause an acute deterioration in a child with an underlying cardiac disorder. In turn, children with congenital heart disease (CHD) with large left-to-right shunts and increased pulmonary blood flow tend to have a higher incidence of lower respiratory tract infections. Acute respiratory distress in these patients may be from a combination of pulmonary and cardiac factors (e.g., CHF).
Chest Pain
The majority of pediatric chest pain cases are noncardiac in origin and benign in nature. Common causes include musculoskeletal or chest wall pain, costochondritis, asthma exacerbations, pneumonia, pleurisy, gastritis, and gastroesophageal reflux. An often underdiagnosed cause of acute chest pain in the child or adolescent is referred to as the precordial catch syndrome (also known as Texidor’s twinge). The pain is sharp, nonradiating, and located in the left periapical area of the chest wall; it occurs suddenly and is often exacerbated during inspiration but is not associated with dyspnea.5 Patients may report that the pain took their breath away or that they were afraid to move; it typically resolves within a few minutes and is not associated with dysrhythmias or other sequelae. The pathophysiologic mechanism of precordial catch syndrome is unknown; the pain may originate from the parietal pleura or costal cartilage.5 If an ECG and a chest radiograph are performed, findings are normal in these patients. The pain is fleeting but may recur at any time and at any age; giving the patient’s symptoms a name emphasizes certainty in and familiarity with the diagnosis and reassures family members concerned about a cardiac etiology of their child’s chest pain.6
Physical Examination
Vital Signs and Blood Pressures
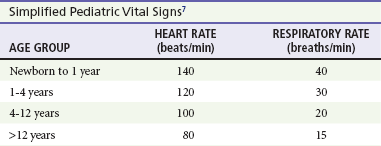
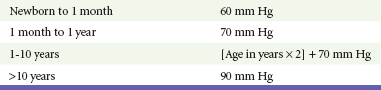
A mild resting tachypnea or tachycardia may be the only clinical clue to an underlying cardiovascular disorder. Age-related variables in heart rates, respiratory rates, and blood pressures can be a source of frustration and confusion to those clinicians who do not manage pediatric patients on a routine basis. Although there are numerous tables of pediatric vital signs with variations based on sleep or awake states, one can easily recall a rough estimate of the normal pediatric heart rates and respiratory rates by a simplified table of pediatric vital signs (Table 171-2).7 The methods to calculate the normal expected blood pressures and hypotensive blood pressures are also listed in Table 171-2.8
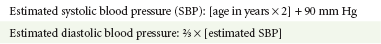
An accurate blood pressure reading is accomplished by use of a cuff that covers two thirds of the upper arm or thigh. A cuff that is too narrow will overestimate the patient’s true blood pressure, and a cuff that is too large will underestimate the true blood pressure. Any child with a suspected cardiac disorder should have blood pressures measured in both arms. If the blood pressure in the left arm is significantly lower than the blood pressure in the right arm, a coarctation of the aorta proximal to the origin of the left subclavian artery should be suspected. Blood pressures should be measured in the thighs in any child with a suspected aortic coarctation or documented hypertensive blood pressures in the upper extremities. The mere presence of femoral pulses does not rule out clinically the possibility of a coarctation of the aorta. Even with an appropriately sized cuff, the blood pressures in the thighs can be 10 to 20 mm Hg higher than the blood pressures in the upper extremities because of the lack of well-designed blood pressure cuffs for the legs. Therefore, if the measured blood pressure in the lower extremities is lower than the blood pressure in the upper extremities, coarctation of the aorta should be suspected. Pulse oximetry readings that are lower in the legs than in the upper extremities are also suggestive of either a coarctation of the aorta or a right-to-left-shunt across a patent ductus arteriosus.9
Cardiac Auscultation
Cardiac murmurs are produced by turbulent blood flow through the heart. The presence of a cardiac murmur may not be associated with an underlying cardiac defect, however. The location, intensity, quality, timing, and radiation of the murmur determine whether the murmur is suggestive of an underlying cardiac pathologic condition. Although systolic murmurs can be present without any underlying anatomic abnormalities, diastolic murmurs are always considered pathologic in nature. Some of the other criteria that would suggest an underlying anatomic cardiac abnormality are listed in Box 171-4. Murmurs may be difficult to appreciate in the noisy emergency department setting and given the degree of tachycardia that is often present even in normal infants. However, the location of the murmur may be a valuable clinical tool in determining the underlying anatomic origin of the murmur (Box 171-5).
Hyperoxia Test
The hyperoxia test is an important bedside diagnostic tool to help differentiate between cardiac and pulmonary causes of central cyanosis. This test consists of assessment of the rise in arterial oxygenation with the administration of 100% oxygen. An arterial blood gas is measured on room air (if tolerated) and repeated after several minutes on high-flow oxygen (100% oxygen); the two blood gas analyses are then compared. When the child is breathing high-flow oxygen, an arterial oxygen partial pressure (PaO2) of more than 250 mm Hg virtually excludes hypoxia due to CHD—a “passed” hyperoxia test.10 An arterial oxygen reading of less than 100 mm Hg (in a child without obvious pulmonary disease) is consistent with a right-to-left shunt and is highly predictive of CHD—a “failed” hyperoxia test.10 Values between 100 and 250 mm Hg may indicate lesions with intracardiac mixing. Pulse oximetry is not an appropriate substitute for an arterial blood gas analysis; it is not sensitive enough to determine “pass or fail” of the test because a child breathing high-flow oxygen and registering 100% on pulse oximetry may actually have a PaO2 anywhere between 80 and 680 mm Hg.1 Prolonged administration of 100% oxygen may cause some theoretic problems, such as closure of the ductus arteriosus in those infants with critical left-sided heart obstructions or pulmonary vasodilation (which would potentially worsen pulmonary vascular congestion).3 However, oxygen should not be withheld initially in critically ill infants; continuous reassessment is crucial in the evaluation and management of children with suspected CHD.1
Chest Radiography
Three features of the chest radiograph (Fig. 171-2) that deserve special attention are the cardiac size (cardiothoracic ratio), the cardiac shape (silhouette), and the degree of pulmonary vascular markings. The easiest method to determine the heart size in children is to determine the cardiothoracic ratio, which is obtained by comparing the largest transverse diameter of the cardiac shadow on the posteroanterior view of the chest radiograph with the widest internal diameter (measured from the inside rib margin to the widest point above the costophrenic angles) of the chest. The films should be obtained during maximal inspiration whenever possible.
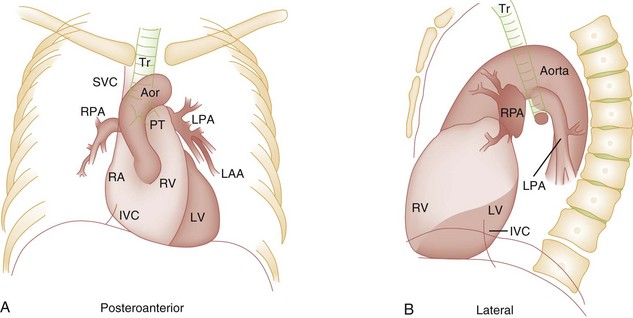
The normal cardiothoracic ratio in children is approximately 50%. The cardiothoracic ratio is not very accurate in newborns and small infants, in whom a good inspiratory view is rarely obtained.11 A cardiac silhouette that is larger than normal may be due to a shunt lesion, cardiomegaly, or pericardial effusion.11 An enlarged heart shadow on a chest radiograph more reliably reflects a problem with volume overload rather than pressure overload. Problems with pressure overload are better represented on the ECG.
The cardiac size can be falsely increased in infants by the presence of the thymus, which can be seen in the mediastinum on the chest radiograph from birth until about 5 years of age. The thymic borders are typically wavy in appearance and sometimes can be seen as the classic “sail sign” along the superior right border of the heart (Fig. 171-3). The thymic shadow may not be visible radiographically in infants during times of physiologic stress but should reappear when the infant recovers.
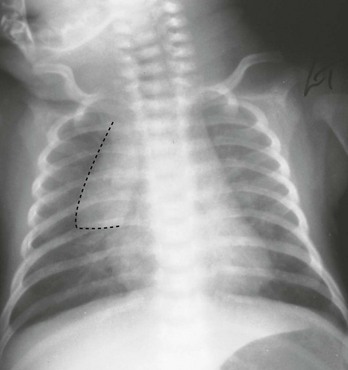
Figure 171-3 Thymic shadow demonstrating the “sail sign” along the right cardiac border (dotted line).
The three classic cardiac silhouettes seen in patients with congenital heart defects are the boot-shaped heart of tetralogy of Fallot (Fig. 171-4), the egg-on-a-string silhouette of transposition of the great vessels, and the snowman-shaped or figure-of-eight heart of total anomalous pulmonary venous return.
In a normal left-sided aortic arch, the aorta descends to the left of the midline and displaces the tracheal air shadow slightly toward the right of midline above the level of the carina. In contrast to this, the tracheal air shadow may be midline or deviated toward the left in the presence of a right-sided aortic arch.3 It is important to note this finding because a right-sided aortic arch is found in up to 25% of the children with tetralogy of Fallot.9 Rib notching secondary to increased collateral blood flow along the intercostal vessels can sometimes be appreciated between the fourth and eighth ribs in older children with undiagnosed coarctation of the aorta but is rarely visualized in children with coarctation of the aorta who are younger than 5 years.11
Electrocardiography
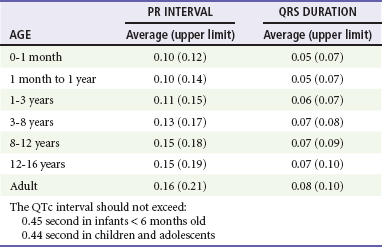
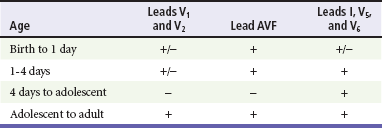
The electrocardiographic findings in infants and children can sometimes be problematic because various components of the ECGs change according to the child’s age (Table 171-3).8 At birth, the muscle mass of the right ventricle is greater than that of the left ventricle; this is demonstrated by right axis deviation on the neonatal ECG. By the end of the first month of life, the left ventricle assumes dominance. By 6 months of age, the left ventricular to right ventricular mass ratio is 2:1, which then reaches the adult ratio of 2.5:1 by adolescence. The durations of the PR interval, QRS complex, and QT intervals increase with age.

Biochemical Markers
As in adults, cardiac troponin T (cTnT) and cardiac troponin I (cTnI) are highly sensitive and specific in children for myocardial damage.11 Reference values are slightly higher for neonates younger than 3 months; normal and indeterminate values will depend on the bioassay used.11 The indications for troponin testing in children include suspected cardiac ischemia (of any etiology), myocarditis, and myocardial dysfunction in sepsis syndrome.11,12 Several studies have evaluated the use of plasma B-type natriuretic peptide (BNP) levels in the assessment and management of CHF in adults.13 Studies of BNP levels in children have demonstrated a similar correlation of elevated levels in children with CHF, and these also correlated with the clinical symptoms of heart failure and the ejection fraction as measured by echocardiography.14 The clinician is urged to refer to the particular range of age-specific values from the laboratory kit used at his or her institution.
Specific Disorders
Perspective
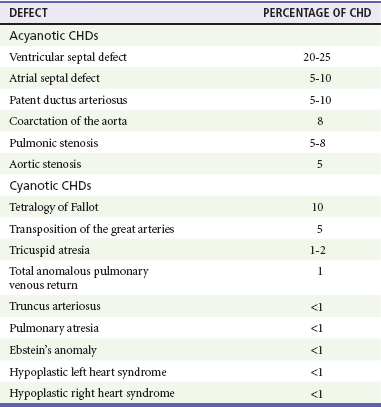
The incidence of CHD in the United States has remained fairly constant at approximately 1%, or 8 to 10 cases per 1000 live births. This equates to approximately 32,000 infants born each year with some form of CHD15 (Table 171-4). Although a large percentage of CHD is now detected with prenatal ultrasonograms, one study also recommended routine pulse oximeter readings in all newborns before discharge from the nursery as an additional inexpensive screening tool for CHD.16
Clinical Features
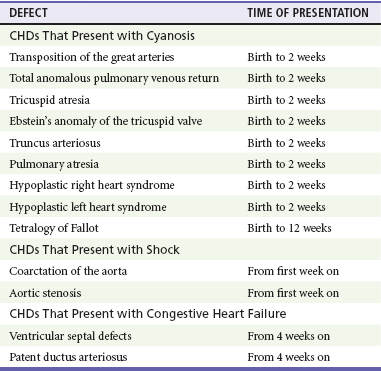
The age, severity of symptoms, and time of presentation of a child with CHD vary by the specific defect, complexity and severity of the defect, and timing of the normal physiologic changes that occur as the fetal circulation transitions to that of a neonate (Table 171-5). The more severe or complex CHD lesions may not be clinically apparent immediately after birth. However, as the ductus arteriosus begins to close in the first several weeks of life, cardiac defects with obstructive lesions of the pulmonary or systemic circulations will be unmasked, and these infants will present clinically with acute cyanosis, shock, or both. Even the harsh systolic murmur of a large, isolated ventricular septal defect may not be heard until about the fourth to sixth week of life when the left-to-right shunt across the ventricular septal defect increases because of the decrease in the PVR. In general, the more severe the anatomic defect is (i.e., lack of pulmonary blood flow or lack of systemic blood flow), the earlier in life these conditions will be manifested with cyanosis and shock.
Diagnostic Strategies
The emergency physician must rely on several key elements of the clinical examination in addition to findings on the chest radiograph and ECG to narrow the diagnostic possibilities. For example, with use of the data in Box 171-6, the presence of cyanosis, a grade 3/6 systolic ejection murmur best heard at the midleft sternal border, a boot-shaped heart, and a decreased pulmonary blood flow on the chest radiograph with evidence of right ventricular hypertrophy on the ECG suggest tetralogy of Fallot. Only a brief discussion of some of the more common CHDs is presented in this chapter.
Management
One unique pharmacologic intervention that can be lifesaving in infants involves the use of prostaglandin E1 (PGE1) to maintain the patency of the ductus arteriosus. CHD that is manifested within the first 2 to 3 weeks of life with a sudden onset of cyanosis or cardiovascular collapse is typically due to duct-dependent cardiac lesions (Box 171-7).4
Closure of the ductus arteriosus in patients with these specific cardiac lesions causes life-threatening situations by either an interruption of blood flow to the lungs, producing cyanosis (i.e., tricuspid atresia), or a disruption of blood flow to the systemic circulation, producing shock (i.e., hypoplastic left heart syndrome).4 Box 171-8 details one suggested method to prepare this potentially lifesaving PGE1 infusion. The PGE1 infusion is typically started at 0.05 to 0.1 µg/kg/min (the method of preparation described in Box 171-8). A known adverse reaction to a PGE1 infusion is apnea; one should perform endotracheal intubation on these infants before the initiation of the PGE1 infusion. Not only will intubation provide a secure airway, but controlled ventilation will also help decrease the infant’s work of breathing, shunting much needed cardiac output and metabolic demands from the overtaxed respiratory apparatus. Other adverse reactions to a PGE1 infusion include fever, seizures, bradycardia, hypotension, flushing, and decreased platelet aggregation.
Acyanotic Congenital Heart Defect
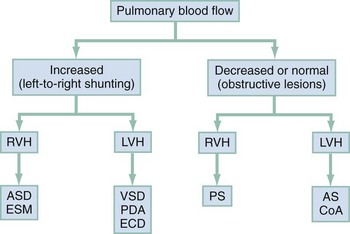
Acyanotic CHD can be further subdivided (Fig. 171-5) into obstructive lesions (i.e., pulmonic stenosis, aortic stenosis, and coarctation of the aorta) and lesions characterized by left-to-right shunts with an associated increase in pulmonary blood flow (i.e., ventricular septal defects, atrial septal defects, patent ductus arteriosus, and endocardial cushion defects). These acyanotic lesions usually are manifested within the first 6 months of life with symptoms of CHF; however, atrial septal defects can remain asymptomatic until adulthood.
Perspective.: Ventricular septal defects are the most common congenital cardiac defects and account for 20 to 25% of all cases of CHD. Spontaneous closure occurs in 30 to 40% of all ventricular septal defects overall and in 50 to 70% of smaller ventricular septal defects.17
Clinical Features.: The degree of symptoms is dependent on the size of the ventricular septal defect and the degree of PVR. Most ventricular septal defects are clinically asymptomatic (minimal or no left-to-right shunting) immediately after birth because of the high PVR. When the PVR decreases to the normal levels at 6 to 8 weeks after birth, left-to-right shunting can then occur, and the typical systolic murmur of a ventricular septal defect will then be appreciated. Small ventricular septal defects may remain completely asymptomatic throughout childhood. Approximately 10% of the infants with large ventricular septal defects will eventually have signs and symptoms of CHF (e.g., poor feeding and poor growth) by 2 to 3 months of age because of the increased pulmonary blood flow. Older children with ventricular septal defects may exhibit signs of decreased exercise tolerance and recurrent pulmonary infections. If moderate to large ventricular septal defects are not surgically corrected, irreversible changes in the pulmonary vasculature may begin to occur as early as 6 to 12 months of age, which will result in an elevation of the PVR and pulmonary hypertension. This in turn can lead to a reversal of the shunt direction across the ventricular septal defect to now become a right-to-left shunt, Eisenmenger’s syndrome, with resultant cyanosis.
Diagnostic Strategies.: The chest radiograph in children with small ventricular septal defects may be entirely normal, but cardiomegaly with increased pulmonary vascular markings is usually present with untreated moderate to large ventricular septal defects. The ECG of moderate-sized ventricular septal defects typically reveals left ventricular hypertrophy, but biventricular hypertrophy may be present in ventricular septal defects with large left-to-right shunting.
Management.: All ventricular septal defects, regardless of the size of the defect, are at risk for bacterial endocarditis because of the high velocity of turbulent blood flow through them.
Traditional closure of ventricular septal defects required open heart surgery. Today, however, a transcatheter closure technique that avoids the inherent risks and complications of open heart surgery and cardiopulmonary bypass has supplanted traditional methods.18–20
Perspective.: Atrial septal defects account for 5 to 10% of all cases of CHD. The majority of infants and children with atrial septal defects remain clinically asymptomatic until adulthood. Spontaneous closure has been reported in up to 40% of the cases within the first 5 years of life.17
Clinical Features.: Large atrial septal defects or those associated with comorbid conditions, such as bronchopulmonary dysplasia, can be manifested with symptoms of CHF and pulmonary overcirculation (e.g., dyspnea with feedings, poor weight gain, and frequent lower respiratory tract infections).3 The majority of atrial septal defects are discovered when a suspicious murmur is detected on a routine physical examination. A widely split and fixed S2 is a characteristic finding of atrial septal defects.
Diagnostic Strategies.: The chest radiograph of children with atrial septal defects will reveal varying degrees of cardiomegaly, right atrial and right ventricular enlargement, and a prominent main pulmonary artery segment and increased pulmonary vascular markings. The ECG will reveal varying degrees of right axis deviation and right ventricular hypertrophy. All patients with unrepaired atrial septal defects will have symptoms if pulmonary hypertension ensues. Patients with large atrial septal defects that are not detected and repaired are at risk for development of Eisenmenger’s syndrome. Unlike ventricular septal defects, uncomplicated atrial septal defects are not associated with high risk of bacterial endocarditis because of the lower turbulence and velocity of blood flow through the atrial septal defects.
Management.: The traditional closure of atrial septal defects, like that of ventricular septal defects, required open heart surgery to place a patch over the septal defect. Newer therapies involving closures with septal occluder devices placed by the transcatheter approach have been described.21,22 Antiplatelet therapy during the 6-month period after placement of the device is typically given and is safe and effective in preventing thrombus formation on the surface of the septal occluder device.
Eisenmenger’s Syndrome.: Eisenmenger’s syndrome can occur in any large left-to-right shunt defect that is not surgically corrected. When large ventricular septal defects and atrial septal defects are not surgically corrected, irreversible changes can occur in the pulmonary arterioles, leading to pulmonary vascular obstruction and pulmonary hypertension. As the degree of pulmonary hypertension increases, the PVR may then begin to exceed the SVR. This causes right-sided pressures to exceed those on the left, causing right-to-left shunting. The reversal in the direction of shunt flow produces cyanosis. Other clinical features of patients who have Eisenmenger’s syndrome include chest pain, dyspnea on exertion, and hemoptysis.23
Perspective.: Coarctation of the aorta accounts for approximately 8% of all CHD, and up to 50% of patients with coarctation of the aorta also have an associated bicuspid aortic valve.24 The area of coarctation can occur proximal to the insertion of the ductus arteriosus (preductal type) or distal to the insertion of the ductus arteriosus (postductal type). The majority of cases (89%) are of the postductal type.24
Clinical Features.: The severity of the symptoms and the age at time of presentation are dependent on the location of the coarctation, the degree of narrowing, and the presence of any other associated cardiac defects. Infants with the rarer, preductal type of coarctation of the aorta may also exhibit differential cyanosis if the ductus arteriosus remains open.25 The upper half of the body is perfused with well-oxygenated blood supplied by the left ventricle and the ascending aorta. However, the lower half of the body will appear cyanotic, as it is largely perfused by right-to-left shunting of deoxygenated blood from the patent ductus arteriosus into the descending aorta. Infants with the preductal type of coarctation of the aorta will present with signs of circulatory failure and shock when the ductus arteriosus begins to close. The astute clinician may find a “brachial-femoral delay” by palpating both pulses simultaneously.26
Most of the asymptomatic cases of the more common postductal coarctation of the aorta are diagnosed as a result of a cardiology referral for a systolic murmur or a hypertension workup, but infants with severe postductal coarctation of the aorta can also present during the first few weeks of life with signs of circulatory failure and shock. If a child is discovered to have hypertension on a routine physical examination, it is necessary to obtain blood pressure measurements in the lower extremities to assess the possibility of coarctation of the aorta. A systolic blood pressure in the right arm that is 15 to 20 mm Hg higher than that in the legs is sufficient evidence to suspect coarctation of the aorta because the systolic blood pressure in the legs is normally higher than that in the arms.25 If the systolic pressure in the right arm is higher than that in the left arm, the area of coarctation is probably preductal and located proximal to the origin of the left subclavian artery. In general, diastolic blood pressures are similar in the upper and lower extremities.
Diagnostic Strategies.: The chest radiograph will most often reveal a normal-sized cardiac silhouette and normal pulmonary vascular markings, but notching along the lower borders of the posterior fourth to eighth ribs due to the pressure of the dilated collateral vessels may be exhibited in children older than 5 years. The absence of rib notching, however, does not rule out the possibility of coarctation of the aorta. The ECG typically reveals a left axis and left ventricular hypertrophy. Suspected cases of coarctation of the aorta should be imaged with transthoracic echocardiography or cardiac magnetic resonance imaging to confirm and to define the coarctation26; in stable patients, this can be done on an outpatient basis.
Management.: Definitive surgical repair of coarctation of the aorta involves resection of the narrowed section of the aorta with an end-to-end anastomosis. Complications of undiagnosed cases are related to the resultant hypertension and can include heart failure, hypertensive encephalopathy, and intracranial hemorrhages.
Cyanotic Congenital Heart Diseases
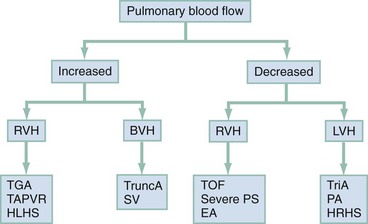
Cyanotic CHDs are a result of either decreased pulmonary blood flow to the lungs or right-to-left shunting of desaturated blood directly into the systemic circulation. They can be further subdivided into those conditions with an increased pulmonary blood flow and those lesions with decreased pulmonary blood flow (Fig. 171-6). The classic cyanotic CHD can be remembered by the five t‘s: truncus arteriosus, transposition of the great vessels, tricuspid atresia, tetralogy of Fallot, and total anomalous pulmonary venous return. Other forms of cyanotic CHD include Ebstein’s anomaly, pulmonary atresia, severe pulmonary stenosis, hypoplastic left heart syndrome, and hypoplastic right heart syndrome. Many of these cyanotic heart lesions are routinely detected either on prenatal ultrasonographic examinations or in the nursery; only tetralogy of Fallot is covered in this section.
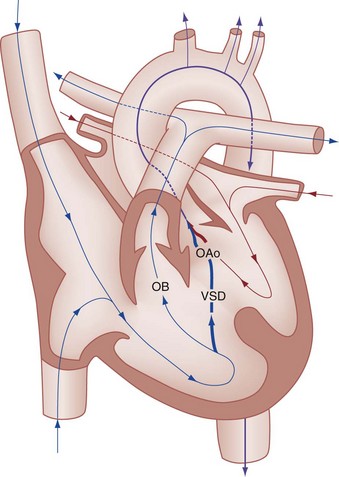
Perspective.: Tetralogy of Fallot accounts for approximately 10% of all cases of CHD and is the most common cause of cyanotic CHD beyond infancy. Tetralogy of Fallot is often associated with other cardiac defects, such as right-sided aortic arch (25% of patients), atrial septal defect (10% of patients), and anomalous origin of the left coronary artery.27 Tetralogy of Fallot arises from a single embryologic defect in which the subpulmonic conus fails to expand, resulting in the four abnormalities (Fig. 171-7): (1) right ventricular outflow tract obstruction; (2) large, unrestrictive, malaligned ventricular septal defect; (3) over-riding aorta that receives blood flow from both ventricles; and (4) right ventricular hypertrophy secondary to the high pressure load placed on the right ventricle by the right ventricular outflow tract obstruction. These anatomic defects collectively result in decreased pulmonary blood flow and varying degrees of right-to-left shunting of deoxygenated blood across the ventricular septal defect.
Clinical Features.: The degree of cyanosis and the age at presentation are directly dependent on the degree of right ventricular outflow tract obstruction. Infants with tetralogy of Fallot typically have worsening of their cyanosis during crying and feeding. Older children with tetralogy of Fallot may have cyanotic exacerbations during periods of physical exertion. Infants who have milder forms of right ventricular outflow tract obstruction may be acyanotic and are sometimes referred to as having a “pink” tetralogy of Fallot. However, the majority of the cases of tetralogy of Fallot exhibit some degree of cyanosis. Infants with severe right ventricular outflow tract obstruction exhibit profound cyanosis within the first few days of life and may even require a PGE1 infusion to preserve pulmonary blood flow by left-to-right shunting from the aorta into the main pulmonary artery through the patent ductus arteriosus.
Diagnostic Strategies.: The chest radiograph of a patient with cyanotic tetralogy of Fallot (see Fig. 171-4) reveals decreased pulmonary vascular markings and a boot-shaped heart (secondary to a concave main pulmonary artery segment along the superior aspect of the left border of the heart). The heart size in tetralogy of Fallot is normal, and a right-sided aortic arch may be seen in 25% of the cases. The ECG of cyanotic tetralogy of Fallot reveals right ventricular hypertrophy and a right axis deviation. Children with pink tetralogy of Fallot may not initially exhibit any degree of right ventricular hypertrophy, but these acyanotic forms of tetralogy of Fallot gradually develop the cyanotic form by 1 to 3 years of age.
A potentially life-threatening complication of tetralogy of Fallot that can be seen in a patient who presents to the emergency department is the so-called tet spell, which has also been referred to as the hypercyanotic spell or the hypoxic spell. Although these hypoxic spells can occur in children with other forms of CHD, they are most commonly seen in infants and children with tetralogy of Fallot and hence the term tet spells. These episodes occur most commonly in infants, with a peak incidence between 2 and 4 months of age.27
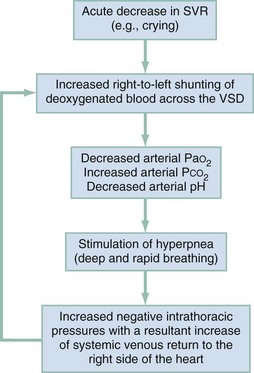
Any event that suddenly lowers the SVR, such as crying or defecation, will produce a large right-to-left shunt across the ventricular septal defect, beginning the vicious circle of a hypoxic spell. Acute hypovolemia and tachycardia can also precipitate tet spells. The large right-to-left shunt through the ventricular septal defect bypasses the lungs, which then causes a decrease in the PaO2, an increase in the PCO2, and a fall in the arterial pH. These metabolic changes then stimulate the respiratory centers in the brain to produce hyperpnea (deep and rapid respirations), which increases the negative intrathoracic pressure during inspiration, causing an increase in the systemic venous blood return to the right side of the heart. This increased volume of blood in the right ventricle is then shunted through the ventricular septal defect by the combination of the existing right ventricular tract outflow obstruction and the acute decrease in the SVR. This in turn further decreases the arterial oxygen saturation, perpetuating the hypoxic spell (Fig. 171-8).
Management.: The overall treatment goals for tet spells (Box 171-9) are to increase the SVR, to abolish the hyperpnea, and to correct the metabolic acidosis. Although supplemental oxygen should be provided, this alone will not reverse a tet spell because there is a decrease in the amount of pulmonary blood flow and an increase in the amount of right-to-left shunting across the ventricular septal defect. The infant should be picked up and placed in a knee-to-chest position. Older children can be placed in the squatting position. Both maneuvers are believed to increase the SVR and to decrease the amount of systemic venous blood return to the right side of the heart. Morphine (0.1-0.2 mg/kg) has been traditionally given intramuscularly to calm the child, to decrease catecholamine surge, and to decrease the respiratory rate. A theoretic adverse effect of morphine, however, is that it can cause systemic vasodilation (further decreasing the SVR) by endogenous histamine release.28 Although there are no current studies evaluating the use of other medications that may also suppress the respiratory centers, fentanyl and midazolam may be used for the same effect without the potential risk of endogenous histamine release. Ketamine (1-2 mg/kg intravenously or 3-5 mg/kg intramuscularly) has also been suggested for its sedative effect as well as for its effect on increasing the SVR.25 Sodium bicarbonate (1 mEq/kg intravenously) can be given to correct clinically suspected or documented (pH < 7.4) metabolic acidosis and to reduce the respiratory center—stimulating effects of acidosis. Most infants respond to these measures and exhibit an improvement in their oxygenation and a decrease in their degree of cyanosis. Infants whose condition does not improve with these measures may require a vasopressor such as phenylephrine to increase the SVR and thereby to decrease the degree of right-to-left shunting across the ventricular septal defect. An intravenous fluid bolus may also be considered to increase the volume of blood flow through the pulmonary artery. Propranolol has also been used as an adjunct to break the cycle of a tet spell. Although the exact pharmacophysiologic mechanisms by which propranolol accomplishes this is uncertain, it is thought to increase the SVR and perhaps to promote an increase in the pulmonary blood flow by reducing spasms of the right ventricular outflow tract obstruction.
Postoperative Complications of Congenital Heart Defects
A variety of postoperative complications can be seen in patients who present to the emergency department weeks to months after cardiac surgery. The types of complications that could occur in each case depend on the original underlying cardiac defect and the surgical procedure that was used to correct that defect. Some of the complications that may be seen in the emergency department include thrombosis of a shunt-conduit with decreased flow, increased shunt-conduit flow with resultant CHF, atrial and ventricular dysrhythmias, heart blocks, myocardial ischemia, and endocarditis. The size of the cardiac silhouette and the degree of pulmonary blood flow visualized on the chest radiograph may provide valuable clues as to whether there is an increased or decreased blood flow through a surgical conduit that was created to provide an improvement in blood flow to the pulmonary system.29 Comparison of the child’s other postoperative chest radiographs can help determine whether there has been a change in the heart size and pulmonary vascularity.
Respiratory Syncytial Virus Infections in Infants and Children with Congenital Heart Defects
Respiratory syncytial virus (RSV) is the most common cause of lower respiratory tract infections in infants and children worldwide, with the majority of children being infected at least once by 2 years of age. Reinfection occurs commonly throughout life.29 RSV lower respiratory tract infections account for more than 125,000 pediatric admissions annually in the United States, with a fatality rate of 6.3 deaths per 100,000 patients up to 4 years of age (Box 171-10).30,31 Children with CHD who have RSV infections tend to have a higher rate of intensive care unit admissions and require mechanical ventilation more frequently than those children who do not have CHD. Children with CHD who require hospitalization for RSV infection have a fatality rate that is two to six times greater than that of children without CHD.30,31 RSV is responsible for a mortality rate of 40% in infants with CHD and up to 70% in infants with CHD and associated pulmonary hypertension.
Two products can be used to prevent RSV infections. Both of these preparations require monthly administration before the onset of the peak RSV season, which typically runs from November through March in most of the United States. The two currently available preparations are palivizumab (Synagis, a humanized monoclonal antibody) and RSV immune globulin (RespiGam).32 The use of palivizumab in the prevention of RSV infections in high-risk infants and children has largely replaced RSV immune globulin because palivizumab can be given intramuscularly, requires a 100-fold lesser volume, and has a 50-fold smaller protein load than RSV immune globulin.33
In 1998 the Food and Drug Administration gave approval for the use of palivizumab in prevention of RSV lower respiratory tract infections in selected infants and children who were deemed to have a higher risk of severe infections. During the 4-year period from 1998 to 2002, several multicenter studies involving more than 24,000 subjects (including data from the Palivizumab Outcomes Registry) have demonstrated the efficacy of palivizumab in preventing hospitalizations of high-risk infants and children.34 During this same 4-year period, another multicenter study involving six countries demonstrated a 45% relative reduction in hospitalization of 1287 high-risk children younger than 2 years with CHD who were given monthly intramuscular injections of palivizumab during a 5-month period.33 This study also demonstrated a significant reduction in the total number of hospital days, supplemental oxygen requirement, total number of days in the intensive care unit, and total number of days requiring intubation in the group that received the monthly palivizumab intramuscular injections. On the basis of these results, the Food and Drug Administration in September 2003 gave approval for the use of palivizumab in infants and children with hemodynamically significant CHD (Box 171-11). The American Academy of Pediatrics emphasizes that prophylaxis with palivizumab should be initiated before the onset of the RSV season. RSV can persist on environmental surfaces for several hours; the best method to prevent the spread of RSV-contaminated secretions within the emergency department setting is meticulous handwashing and wearing of a mask in caring for children with a documented or suspected RSV infection.
Congestive Heart Failure
Diagnostic Strategies
The chest radiograph typically reveals an enlargement of the cardiac silhouette and varying degrees of pulmonary congestion. An echocardiogram will be able to assess the ejection fraction as well as to identify underlying anatomic defects. Plasma BNP has been reported to be helpful in differentiating cardiac from pulmonary causes of dyspnea in children.35 Other diagnostic studies to consider are case specific and depend on the suspected cause of the child’s CHF.
Management
Acute stabilization of any child who presents with CHF includes administration of supplemental oxygen and agents to augment cardiac contractility and to improve cardiac output. Children who present in severe respiratory distress secondary to pulmonary edema may require intubation to support oxygenation and ventilation. Children with respiratory distress due to pulmonary congestion may also benefit from elevation of the head and upper torso. Continuous positive airway pressure or biphasic positive airway pressure ventilation may be useful initially to avert the need for endotracheal intubation. Plasma BNP levels have been used also to monitor the response to treatment regimens in patients with CHF.14
Diuretics and inotropic agents are the mainstay for treatment of the majority of children with CHF. Furosemide (Lasix) in a dose of 1 mg/kg is the most common loop diuretic used to increase renal perfusion and to improve urine output. With the proper use of furosemide and digoxin (Table 171-6), most children with CHF will demonstrate a favorable response. The narrow therapeutic index of digoxin requires that levels be monitored closely to prevent iatrogenic digoxin toxicity, which could cause a worsening of the preexisting CHF.
Table 171-6
Digoxin Dosing for Infants and Children with Congestive Heart Failure8
| AGE | TOTAL DIGITALIZING DOSE (ORAL)* | DAILY MAINTENANCE DOSE (ORAL) |
| Premature | 20 µg/kg/24 hr | 5 µg/kg/day |
| Full term | 30 µg/kg/24 hr | 8-10 µg/kg/day |
| <2 years | 40-50 µg/kg/24 hr | 10-12 µg/kg/day |
| 2-10 years | 30-40 µg/kg/24 hr | 8-10 g/kg/day |
| >10 years | 0.75-1.25 mg/24 hr | 0.125-0.25 mg/day |
Intravenous digoxin is supplied as 100 µg/mL and 250 µg/mL solutions.
Oral digoxin suspension is supplied as a 50 µg/mL elixir.
Therapeutic range: 0.8-2.0 µg/L.
*The intravenous dose of digoxin is equal to 75% of the oral dose (except in children older than 10 years, in whom the intravenous dose is the same as the oral dose).
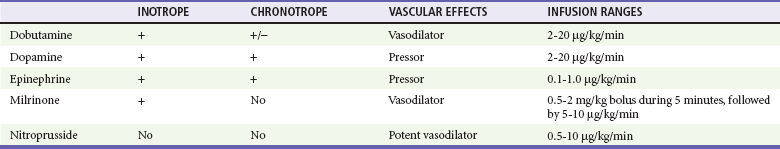
Other inotropic agents that are used for the treatment of CHF in infants and children are dopamine, dobutamine, and epinephrine (Table 171-7). Standardized drips have been established and have replaced the dated “rule of six.”36 Dopamine is an endogenous catecholamine with complex cardiovascular effects. Dopamine can increase renal blood flow in low doses (2-5 µg/kg/min) and will increase the cardiac contractility and heart rate in moderate doses (5-10 µg/kg/min). Dopamine stimulates cardiac beta1-adrenergic receptors both directly and indirectly through the release of endogenous norepinephrine stored in the cardiac sympathetic nerves. Because of this, some of the inotropic effects of dopamine may be reduced in those patients with decreased endogenous myocardial norepinephrine stores (i.e., patients with chronic CHF and neonates). At higher doses (10-20 µg/kg/min), dopamine will also increase the SVR, but excessive vasoconstriction may compromise end-organ perfusion at infusion rates of more than 20 µg/kg/min. If additional inotropic effects are required, the addition of either dobutamine or epinephrine infusions may be preferable to an increase in the dopamine infusion to more than 20 µg/kg/min. The major toxicities of dopamine are tachycardia, vasoconstriction, and ventricular ectopy.
Pediatric Dysrhythmias
Dysrhythmias are not as common in children as they are in adults. The most common cause of cardiopulmonary arrest in infants and children is the untreated progression of respiratory failure or shock rather than a primary cardiac dysrhythmia.37 Accordingly, the most common arrest rhythm that will confront the emergency physician will be asystole or bradycardia rather than ventricular fibrillation or ventricular tachycardia. When confronted with a child with a primary cardiac dysrhythmia, the physician must identify and treat the underlying cause quickly and systematically. The conditions in children who are at risk for development of dysrhythmias are listed in Box 171-12. Various medications, drugs, and toxins can also precipitate dysrhythmias in children. Even those medications that are used to treat underlying cardiac problems, such as digoxin, amiodarone, and procainamide, can themselves precipitate dysrhythmias. Drugs of abuse (e.g., cocaine and crystal methamphetamine, among others) and overdose of prescription medications (e.g., cyclic antidepressants) should be considered in the evaluation of any previously healthy adolescent patient who presents with an acute dysrhythmia.
Pediatric dysrhythmias can be divided into three broad categories of rhythms by their effect on the child’s pulse: slow (sinus bradycardia and heart blocks), fast (supraventricular tachycardia or ventricular tachycardia with a pulse), or absent (ventricular tachycardia without a pulse, ventricular fibrillation, pulseless electrical activity, or asystole).38 The most common dysrhythmia in children is supraventricular tachycardia, which occurs most commonly in infants and young children. Although supraventricular tachycardia can spontaneously occur in infants without any underlying structural cardiac defects, ventricular tachycardias typically are due to an underlying myocardial abnormality.
Management
A summary of the treatment algorithms for pediatric dysrhythmias and the most commonly used medications in treatment of these dysrhythmias are listed in Box 171-13. Although some medications can be used to treat only atrial tachycardias (e.g., the use of adenosine for supraventricular tachycardia) or ventricular tachycardias (e.g., the use of lidocaine for ventricular tachycardia), amiodarone and procainamide can be used for a wide array of both atrial and ventricular dysrhythmias, including supraventricular tachycardia and ventricular tachycardia.38–40
Bradydysrhythmias
Sinus Bradycardia.: Bradycardia is defined as a heart rate that is slower than the lower limit of normal for a child’s age. According to the current definition by the American Heart Association (AHA) guidelines in Pediatric Advanced Life Support (PALS), clinically significant bradycardia in children is a heart rate slower than 60 beats/minute that is associated with poor systemic perfusion.38 Bradycardia is poorly tolerated in infants and children because they are not physiologically capable of increasing their stroke volume to maintain an adequate cardiac output in the face of significant bradycardia.
Tachydysrhythmias
Perspective.: Supraventricular tachycardia is the most common symptomatic dysrhythmia in infants and children.37 In children younger than 12 years, the most common cause of supraventricular tachycardia is a reentry mechanism due to an accessory atrioventricular pathway.41 No cardiac abnormalities are found in approximately half of the cases, and the Wolff-Parkinson-White syndrome is present in only 10 to 20%.42 The QRS complex is narrow (<0.08 second) in up to 90% of the cases of pediatric supraventricular tachycardia.43 The type of supraventricular tachycardia that occurs most commonly in infants and children involves a reentrant mechanism that uses an accessory pathway and the atrioventricular node. The orthodromic reentry phenomenon involves the normal antegrade conduction from the atria to the ventricles down the atrioventricular node with retrograde conduction back from the ventricles to the atria by the accessory pathway. Orthodromic conduction will produce a narrow–QRS complex supraventricular tachycardia. The less common reentry mechanism is the antidromic form in which conduction from the atria to the ventricles first goes antegrade down the accessory pathway then retrograde back to the atria by the atrioventricular node. Antidromic conduction will produce a wide–QRS complex supraventricular tachycardia. Supraventricular tachycardia in a child with a preexisting bundle branch block can also result in a wide-complex supraventricular tachycardia. The ECG in Figure 171-9 reveals a case of a wide-complex supraventricular tachycardia in a child with Ebstein’s anomaly of the tricuspid valve who also presented with CHF (Fig. 171-10).
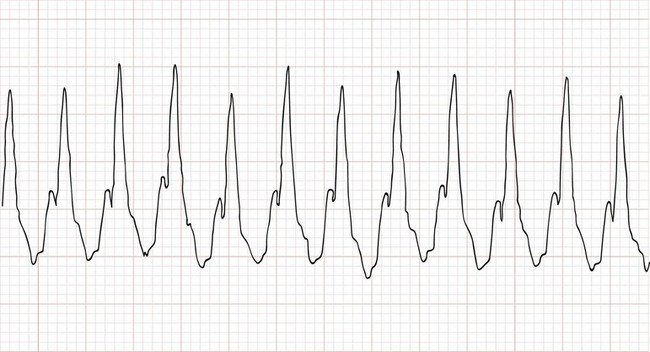
Figure 171-9 An example of an electrocardiogram showing a wide-complex supraventricular tachycardia at a rate of approximately 270 beats/minute in an infant with Ebstein’s anomaly of the tricuspid valve. This infant was in supraventricular tachycardia for approximately 2 days and presented with an acute exacerbation of her congestive heart failure, as evidenced by the cardiomegaly on the chest radiograph (see Fig. 171-10). Note that the cardiothoracic ratio in this infant is approximately 70%.
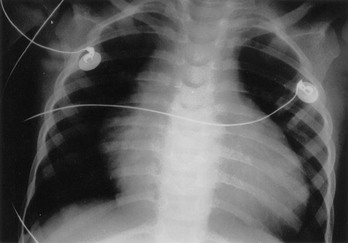
Figure 171-10 Chest radiograph of same infant as in Figure 171-9.
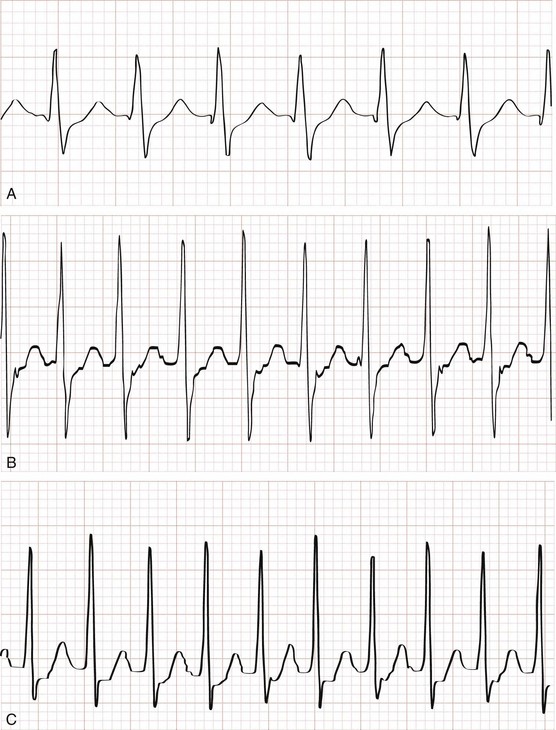
Clinical Features and Diagnostic Strategies.: The width of the QRS interval in patients with pediatric supraventricular tachycardia is most commonly narrow complex, with heart rates in infants usually higher than 220 beats/minute (Fig. 171-11). It is sometimes difficult to distinguish between sinus tachycardia and supraventricular tachycardia (Table 171-8).
Table 171-8
Clinical and Electrocardiographic Features to Differentiate Sinus Tachycardia from Supraventricular Tachycardia in Children38
| SINUS TACHYCARDIA | SUPRAVENTRICULAR TACHYCARDIA | |
| Precipitating events | Dehydration, fever, pain | No precipitating event |
| P waves on electrocardiogram | Present | Absent |
| Heart rate varies with activity | Yes | No |
| Beat-to-beat variability | Yes | Constant R-R intervals |
| Heart rate in infants (beats/minute) | Usually <220 | Usually >220 |
| Heart rate in children (beats/minute) | Usually <180 | Usually >180 |
Management.: Management of supraventricular tachycardia depends on the hemodynamic stability of the child. If the child with supraventricular tachycardia is hemodynamically unstable and intravenous access in not available, immediate cardioversion starting at 0.5 to 1 joule/kg is indicated. If the child does not convert with this initial cardioversion attempt, the energy dose can be doubled up to 2 joules/kg on subsequent attempts. If the child is hemodynamically stable, vagal maneuvers or adenosine, or both, can be attempted initially before cardioversion, depending on the individual case scenario. Regardless of the method selected to convert the supraventricular tachycardia, a continuous rhythm strip should be run to document the response to each conversion attempt. Vagal maneuvers (e.g., a bag containing a slurry of crushed ice and water to the face, blowing on an occluded straw, or blowing on the tip of a syringe) can be attempted before adenosine administration only in the child with hemodynamically stable supraventricular tachycardia. Application of ice to the face has been demonstrated to be a fairly effective method of converting supraventricular tachycardia in infants and children.44,45 One method to perform this maneuver is to fill a plastic bag or surgical glove with a slurry of crushed ice and water, which is then placed over the infant’s forehead, eyes, and bridge of the nose for 10 to 15 seconds. Care must be taken not to occlude the nose or mouth with the bag of ice water. External ocular pressure should be avoided; it can be dangerous in children because excessive pressure can lead to a ruptured globe. Carotid massage is less effective and is not recommended as a vagal maneuver in infants or children.38
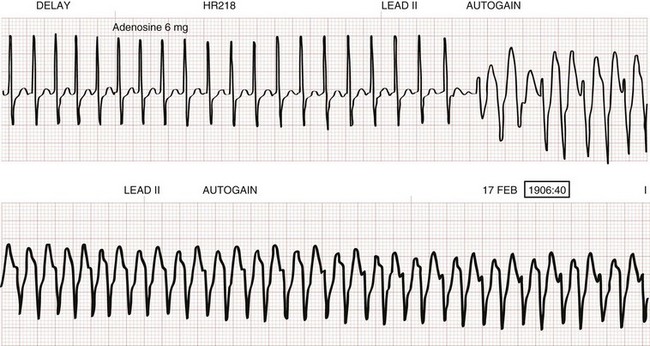
The initial dose of adenosine in children is 0.1 mg/kg with a maximum initial dose of 6 mg. If this initial dose of adenosine fails to convert the supraventricular tachycardia, the dose is then doubled to 0.2 mg/kg with a maximum of 12 mg/dose. This 0.2 mg/kg dose of adenosine can be attempted once more during the third adenosine dose. Elective cardioversion or esophageal overdrive pacing under conscious sedation may be required in children who fail to convert with adenosine. Asystole and various dysrhythmias, including adenosine-induced wide-complex tachycardia (secondary to an occult accessory conduction pathway), are complications of adenosine administration (Fig. 171-12). The use of verapamil to convert supraventricular tachycardia in infants and younger children should be avoided because of the potential of profound hypotension and cardiovascular collapse when this medication is administered in this age group.38
Atrial Flutter and Atrial Fibrillation.: Both atrial flutter and atrial fibrillation are rare in children and are usually associated with underlying heart conditions (i.e., CHD, status post–open heart surgical procedures that involved the atria, myocarditis, and digoxin toxicity). Hemodynamic stability of these two dysrhythmias depends on the rate of the ventricular response. Cardioversion is the treatment of choice for children who present with hemodynamically unstable atrial flutter or atrial fibrillation. The initial treatment priority in patients with hemodynamically stable atrial flutter and atrial fibrillation is first to slow down the rate of the ventricular response with medications such as digoxin, beta-blockers, or diltiazem. Once the ventricular rate is controlled, the rhythm can then be converted and suppressed with amiodarone, procainamide, or elective cardioversion. If the patient who presents with atrial flutter or atrial fibrillation is known to have an underlying Wolff-Parkinson-White syndrome, the four medications that should be avoided are the A-B-C-D medications (adenosine, beta-blockers, calcium channel blockers, and digoxin) because all of these medications preferentially block conduction down the atrioventricular node, leaving the accessory pathway open to conduct the atrial tachycardia to the ventricles at a potentially lethal rate.45,46 Under these circumstances, amiodarone, procainamide, or cardioversion would be the safer alternative. Consultation with the cardiologist and initiation of anticoagulation should also be considered to prevent a thromboembolic complication before conversion of either of these atrial dysrhythmias in the hemodynamically stable patient believed to have the rhythm disorder 48 to 72 hours before presentation.
Ventricular Tachycardia.: Ventricular tachycardia is not a common dysrhythmia in children. The majority of children with ventricular tachycardia have an underlying condition, such as post–cardiac surgery status, myocarditis, prolonged QT syndrome, drug or toxin exposures (e.g., cyclic antidepressants), or electrolyte abnormalities. The treatment of ventricular tachycardia will depend on whether a pulse is present and on the hemodynamic status of the patient (Box 171-14). Torsades de pointes is a unique type of polymorphic ventricular tachycardia characterized by QRS complexes that change in polarity and amplitude. Prolonged QT syndrome, underlying congenital cardiac defects, hypomagnesemia, and various medications (e.g., cyclic antidepressants) have been identified as known causes of torsades de pointes. The treatment of choice is intravenous magnesium. Class IA (i.e., procainamide) and class III (i.e., amiodarone) antidysrhythmic agents are contraindicated in the treatment of torsades de pointes because these two antidysrhythmic agents are capable of prolonging the QT interval, which could then precipitate the degeneration of the torsades de pointes into a more lethal rhythm.
Pulseless Rhythms
Ventricular Fibrillation and Pulseless Ventricular Tachycardia.: Ventricular fibrillation and pulseless ventricular tachycardia account for approximately 9 to 10% of out-of-hospital cardiac arrest cases in which a terminal rhythm was recorded.47,48 The survival rate for out-of-hospital ventricular fibrillation and pulseless ventricular tachycardia can be as high as 30%, whereas the survival rate from asystolic cardiac arrest is less than 1%.35 In a study of in-hospital cardiac arrests, a shockable rhythm was present during some point of the resuscitation in 25% of the children.49 The survival rate of children who initially exhibited shockable rhythms was higher than that of those children who presented with nonshockable rhythms. However, the survival rates in those children who later had a shockable rhythm at some time during their resuscitation were not as favorable.49–51 Ventricular fibrillation should also be suspected as the arrest rhythm in cases of commotio cordis or in cases of sudden cardiac arrest. Current arrhythmia detection algorithms for automated external defibrillators (AEDs) appear to have a high sensitivity and specificity for detection of shockable rhythms in children. Cardiopulmonary resuscitation (CPR) and the early use of AEDs by first responders in the field are encouraged; AEDs may be used in the hospital setting if more advanced equipment and staff are not readily available.52 The use of AEDs is now acceptable in all age ranges, including infants and children. Pediatric pads (attenuating pads) should be used whenever possible for children 1 year to 8 years old (or less than 25 kg).53–56 In infants younger than 1 year, manual defibrillation is preferred.52 If a manual defibrillator is unavailable or its use is delayed, an AED with a pediatric attenuator may be used; if neither is available, an AED without a dose attenuator may be used.52
The treatment algorithm and medication dosages for ventricular fibrillation and pulseless ventricular tachycardia are listed in Box 171-13. On the basis of the 2010 PALS guidelines, ventricular fibrillation and pulseless ventricular tachycardia are treated with a single defibrillation, followed immediately by 2 minutes of uninterrupted CPR.44 Although a single shock with a biphasic defibrillator has a high likelihood of terminating ventricular fibrillation, the resulting rhythm is typically a nonperfusing rhythm, which therefore requires CPR to maintain perfusion to the heart and brain until normal cardiac contractility can resume.44,56 Epinephrine is given with the second defibrillation, and antiarrhythmic agents are added to the treatment algorithm with the third defibrillation. Although biphasic defibrillators can convert ventricular fibrillation at lower dosages than with the previous monophasic defibrillators in adults, until more data are gathered on biphasic defibrillation dosages in children, the current AHA recommendation for pediatric defibrillation with biphasic defibrillators remains the same as that with monophasic defibrillators (i.e., starting at 2 joules/kg, followed by 4 joules/kg). The 2010 PALS guidelines recommend escalating doses for subsequent shocks, up to 10 joules/kg or the maximum adult dose delivered from the defibrillator.
Asystole and Pulseless Electrical Activity.: Asystole is the most common rhythm found in cases of out-of-hospital cardiac arrest in children and is associated with a less than 1% chance of survival.57–62 The treatment algorithm and medication dosages for asystole and pulseless electrical activity are listed in Box 171-13. Pulseless electrical activity can be a slow or fast rhythm and have a narrow or wide QRS complex. The use of high-dose epinephrine is no longer recommended.44,52
The key to survival from any pulseless electrical activity rhythm is to rapidly identify and to correct the underlying cause. The causes of pulseless electrical activity can be remembered by the six h‘s and five t‘s: hypovolemia, hypoxia, hydrogen ion (acidosis), hypokalemia/hyperkalemia, hypoglycemia, hypothermia, toxins, tamponade, tension pneumothorax, thrombosis, and trauma.38,44 The most common cause of pulseless electrical activity in children is profound hypovolemia. Therefore, an intravenous fluid bolus should always be considered as a therapeutic option during the treatment of pulseless electrical activity.
Special Resuscitation Situations in Children
Children with single-ventricle physiology (e.g., hypoplastic left heart syndrome, double-outlet right ventricle physiology, among others) after a palliative shunting procedure should be given standard resuscitation care.52 Heparin may be used in the pre-arrest or arrest of infants with a systemic–to–pulmonary artery shunt or right ventricle–to–pulmonary artery shunt to halt thrombus propagation in this low circulatory flow state.52 A target oxyhemoglobin saturation (SpO2) of approximately 80% is preferred in these children. End-tidal carbon dioxide readings after resuscitation may lag behind because of varying pulmonary blood flow changes that do not necessarily reflect cardiac output. The goal is to provide adequate preload with judicious fluids to balance systemic and pulmonary blood flow.52 If it is available, extracorporeal membrane oxygenation may be considered in these patients.52,53
Children with a history of pulmonary hypertension should also receive standard PALS care. Preload should be optimized with isotonic saline boluses. Inhaled nitric oxide in the intensive care unit may be given to reduce PVR.52,54 Intravenous prostacyclin is also used to decrease PVR and may be an option in the emergency department. Early contact with the child’s cardiologist and cardiothoracic surgeon is instrumental in the postresuscitative care of children with CHD.
Bacterial Endocarditis
Bacterial endocarditis involves an infection of the endothelial surfaces of the heart with a propensity for the valves. The incidence of bacterial endocarditis in children may be increasing slightly because of the many advances in surgical technology that are now allowing many children with very complex congenital heart lesions to survive. Children with indwelling intravenous lines with or without underlying CHD are also at risk for development of bacterial endocarditis. Although bacterial endocarditis most commonly occurs in children with an underlying CHD or an acquired cardiac lesion (e.g., acute rheumatic valvular heart disease), it can also occur in patients with no underlying anatomic defects of the valves or endocardium.63
Clinical Features and Diagnostic Strategies
The early clinical manifestations of bacterial endocarditis may be nonspecific. The child may simply present with only fever and tachycardia. However, bacterial endocarditis should be suspected in any child with an anatomic cardiac defect who presents with an unexplained fever. This diagnosis should always be considered in any child with a known CHD or an acquired cardiac lesion who presents with any of the conditions listed in Box 171-14. A new heart murmur is present in less than 50% of the bacterial endocarditis cases.64 The largest study of pediatric infective endocarditis to date65 found common presenting signs to be fever (99%), petechiae (21%), changing murmur (21%), dental caries (14%), and hepatosplenomegaly (14%); less common signs were CHF (9%), splinter hemorrhages (5%), Roth’s spots (5%), and Osler’s nodes (4%).
In addition to vigilance for the diagnosis of infective endocarditis, especially in children with CHD, the emergency physician should be aware of the indications for prophylaxis. In 2007 the AHA in conjunction with the American Academy of Pediatrics and the Infectious Diseases Society of America published revised guidelines for the prevention of infective endocarditis.66 The changes simplify and greatly narrow the recommendations to provide prophylaxis for only the higher risk patients and procedures (Box 171-15). For those children for whom prophylaxis is recommended, the indications are (1) all dental procedures and (2) any manipulation or perforation of the gingival or oral mucosa.66 Antibiotic prophylaxis for infective endocarditis is no longer recommended for gastrointestinal and genitourinary procedures (Box 171-16). The committee found that it is still reasonable to give prophylaxis for procedures on the respiratory tract, infected skin, or musculoskeletal tissue only for high-risk patients (Table 171-9).66
Table 171-9
Regimens for Prophylaxis of Infective Endocarditis
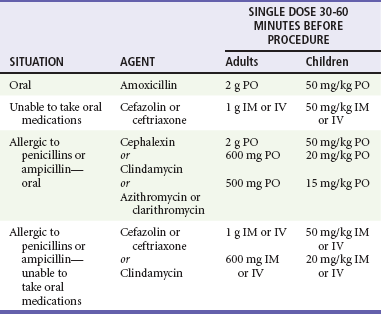
Modified from American Heart Association: Prevention of infective endocarditis. Guidelines from the American Heart Association. Circulation 116:1736, 2007.
Diagnostic studies for a child with suspected bacterial endocarditis include a complete blood cell count, C-reactive protein (CRP) assessment, measurement of erythrocyte sedimentation rate (ESR), three blood cultures,67 chest radiography, and electrocardiography. Cultures from scrapings of cutaneous emboli can also aid in the diagnosis. Although the definitive diagnostic study is the echocardiogram, it is only 80% sensitive in detecting the nidus of infection on the endocardium or valves.68 Streptococcus viridans and Staphylococcus aureus are the two most common offending organisms recovered from the blood cultures of children with bacterial endocarditis. Studies have shown that in children with CHD, 60% of the cases caused by staphylococcal species are methicillin resistant and associated with increased risk of mortality.69
Management
Antibiotics should be started immediately after blood culture samples have been obtained. Although the choice of intravenous antibiotics depends on the suspected source of seeding and the child’s immune status, a common recommended regimen includes an aminoglycoside plus a penicillinase-resistant penicillin such as oxacillin. If methicillin-resistant staphylococci are suspected, vancomycin should also be included in the initial empirical antibiotic regimen.70
A surge of antibiotic resistance to the three major causes of bacterial endocarditis—streptococci, staphylococci, and enterococci—has increased the mortality rate of bacterial endocarditis, now at 25 to 40%.71 Complications of bacterial endocarditis include systemic septic emboli, pulmonary emboli, central nervous system emboli with resultant neurologic deficits, dysrhythmias, CHF, myocarditis, myocardial abscesses, and valvular obstruction and destruction. Despite appropriate antibiotic treatment, surgical intervention to remove septic vegetations or valve replacement is sometimes necessary.
Myocarditis
Clinical Features
Myocarditis usually has a gradual onset with preceding upper respiratory tract infection symptoms. The presenting signs and symptoms of a child with myocarditis depend on the cause of the myocarditis, the age of the patient, and the degree of myocardial inflammation. The key to diagnosis of myocarditis in infants and children is to include it in the differential diagnosis of a child with symptoms disparate from the presentation. In mild cases, the only sign of myocarditis may be tachycardia. Tachycardia that is disproportionate to the degree of fever should alert the clinician to the possibility of myocarditis.72 Other presenting signs and symptoms include fever, myalgias, fatigue, tachypnea, wheezing, abdominal pain, and chest pain. More severe cases of myocarditis can even have signs and symptoms of acute CHF and various dysrhythmias. The physical examination may reveal a new murmur, a gallop rhythm, or a pericardial friction rub with muffled heart tones (if the myocarditis is also accompanied by pericarditis and subsequent pericardial effusion). In general, these are sick children with vague symptoms.
Diagnostic Strategies and Management
The echocardiogram is an essential component of the workup of any child with suspected myocarditis. The echocardiogram will not only be useful to assess the degree of left ventricular function, but it will also be able to detect associated pericardial effusion that may accompany the myocarditis. Although an endomyocardial biopsy is rarely required, it is the definitive method to confirm the diagnosis and origin of myocarditis. If the endomyocardial biopsy is performed, it will typically demonstrate a myocardial inflammation with lymphocytic and monocytic infiltrates. The goal of treatment is to maintain adequate cardiac output and to control any associated dysrhythmias. Children who present in CHF may require inotropic support and diuretics. Digoxin and the various pressors must be used very cautiously in children with myocarditis because the inflamed myocardium is sensitive to the dysrhythmogenesis of these medications. The use of beta-blockers is contraindicated,72 and the routine use of immunosuppressive agents remains controversial.73 Although the majority of children with acute viral myocarditis make a full recovery, a few patients will progress to dilated cardiomyopathy, which is characterized by dilated ventricles and impaired systolic contractility.
Pericarditis
Although the most common causes of pericarditis include bacterial and viral infections, other causes are ARF, systemic lupus erythematosus, uremia, postpericardiotomy syndrome, leukemia, lymphoma, and tuberculosis. Approximately 30% of pericarditis cases are due to bacteria such as pneumococcus, S. aureus, meningococcus, and Haemophilus influenzae.2 Approximately 30% of the purulent bacterial pericarditis cases occur in children younger than 6 years.28 Viral causes are common, but a specific viral pathogen is recovered in only 20 to 30% of the cases.28 Common viral causes include Coxsackieviruses, echoviruses, adenovirus, Epstein-Barr virus, and influenza viruses.
Clinical Features
The chest radiograph in a child with pericarditis may not reveal an enlarged cardiac silhouette, depending on the amount of fluid that has accumulated within the pericardial sac. If there is a large collection of fluid within the pericardial sac, the heart shadow on the chest radiograph will resemble a “water bottle” silhouette. Approximately 50% of pericarditis cases also have an associated pleural effusion.2
The classic electrocardiographic findings of pericarditis include diffuse ST segment elevation and diffuse T wave inversions in all leads. The classic electrocardiographic changes associated with pericarditis evolve through four phases (Fig. 171-13). During the initial phase, there is diffuse ST segment elevation in all leads secondary to subepicardial inflammation; PR segment depression may also be seen. During the second phase, the previously elevated ST segments begin to return to isoelectric baseline, and the T wave amplitudes begin to decrease with flattening of the T waves. During the third phase, although the ST segments are now back to isoelectric baseline, the T waves are now inverted. The fourth and final phase demonstrates complete resolution of the ST segment and T wave abnormalities. Diminished electrocardiographic voltages in all leads can also occur if there is a significant amount of fluid accumulated within the pericardial sac.
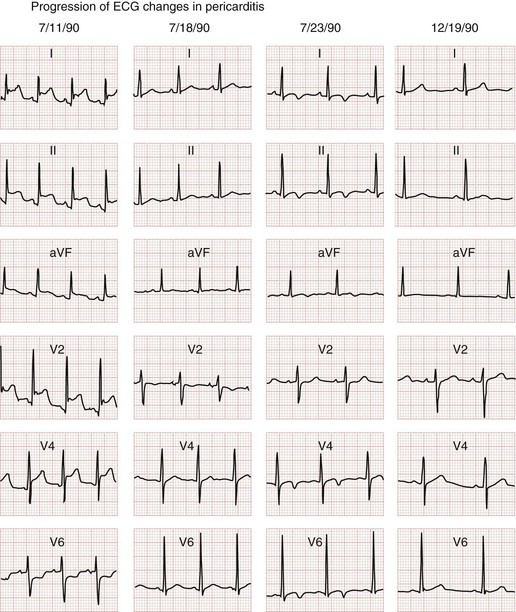
Management
The management of a child with pericarditis depends on both the suspected cause and the amount of fluid that has accumulated within the pericardial space. These children require admission for observation and analgesia with nonsteroidal anti-inflammatory drugs. Certainly those patients with fever, respiratory distress, or signs of CHF should be admitted and an echocardiogram performed emergently. An emergency pericardiocentesis will be required in those patients who have signs of acute cardiac tamponade. Any fluid that is aspirated from the pericardial space should be sent for routine cell counts, Gram’s stain, and cultures. Anti-inflammatory agents and appropriate antibiotics should also be initiated on the basis of the suspected cause. Steroids are reserved for refractory cases that are not responsive to these agents and would be considered only after an infectious etiology is ruled out.74
Kawasaki Disease
Kawasaki disease, originally described as mucocutaneous lymph node syndrome by Dr. Tomisaku Kawasaki in 1967, has emerged as a significant cause of acquired cardiac disease in children in the United States. An estimated 3000 to 5000 cases of Kawasaki disease are diagnosed annually in the United States. Up to 20% of untreated children have some degree of coronary artery abnormalities.75 This febrile, exanthematous, multisystem vasculitis is seen most commonly in children younger than 5 years with a male-to-female ratio of 1.5:1. The incidence is higher in Asian children and in certain geographic locations, such as Hawaii. Although the exact cause of this vasculitis of small and medium-sized vessels remains unknown, early clinical recognition and initiation of high-dose aspirin and intravenous immune globulin (IVIG) therapies have improved the morbidity and mortality rates of Kawasaki disease in children.
Clinical Features
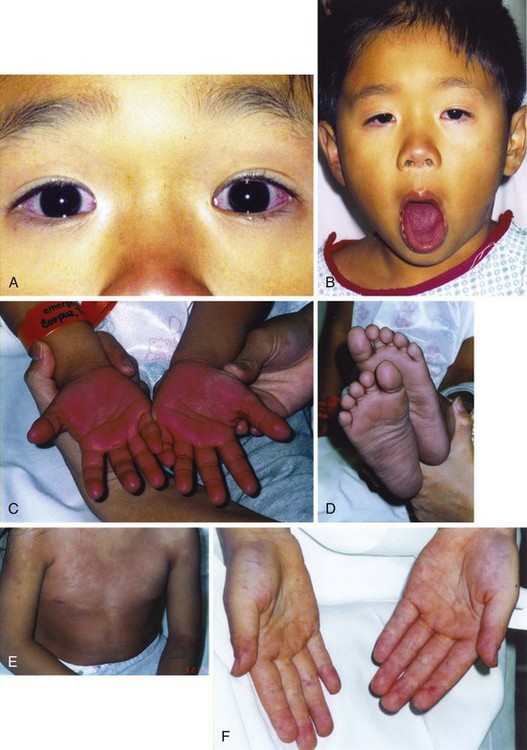
The key to prevention of the coronary artery complications of Kawasaki disease is first to recognize the clinical signs and symptoms of this disease. In addition to fever, the physical examination of a child with Kawasaki disease may reveal the typical findings as listed in Box 171-17 and illustrated in Figure 171-14. The classic features of Kawasaki disease may be manifested simultaneously or in series of days; a careful history and physical examination may elucidate the need for further testing. In addition, very young children may not have a classic presentation and require further investigation. All children with suspected Kawasaki disease, with either classic or incomplete features, should undergo echocardiography for detection of the presence and degree of coronary aneurysm.76
Incomplete Kawasaki Disease.
The classic presentation of Kawasaki disease is a clinical diagnosis of four or more of the five criteria in a child who is febrile 5 days or more. However, these strict criteria may miss a substantial number of children who present with incomplete Kawasaki disease. Any child may have an incomplete presentation, but this is mostly seen in infants younger than 6 months.76
The AHA’s Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease has published consensus guidelines on the approach to incomplete Kawasaki disease.71 The more inclusive criteria recommend that in a child who is febrile 5 days or more, the presence of two or three criteria should prompt further testing. A CRP of 3 mg/dL or more or an ESR of 40 mm/hr or more or both necessitate further laboratory investigations, such as those listed in Box 171-18. All of these children will also need echocardiography to assess for coronary aneurysm, and the recommendation is to treat empirically those whose supplementary laboratory values (see Box 171-18) are positive for systemic inflammation while echocardiographic confirmation is awaited.76
Differential Considerations
The palmar lesions of measles are discrete macular lesions (Fig. 171-14F), whereas the palmar finding in children with Kawasaki disease is diffuse erythema, which may later lead to desquamation (Fig. 171-14C).
Streptococcal disease, including pharyngitis and scarlet fever, can be confused with Kawasaki disease, but conjunctivitis and swelling of the hands and feet are unusual for streptococcal disease. Other infectious or autoimmune causes that may mimic Kawasaki disease include Rocky Mountain spotted fever, leptospirosis, Stevens-Johnson syndrome, and juvenile rheumatoid arthritis.76
Clinical Course
Kawasaki disease is postulated to be caused by an infectious agent that enters the respiratory tract and initiates an oligoclonal immunoglobulin A response, which activates lymphocytes, cytokines, and proteinases that weaken vessel walls and predispose the entire circulation to aneurysms.77 The main reason to recognize this disease entity in children is to initiate prompt therapies to prevent the cardiac complications of Kawasaki disease, which occur in two phases. Approximately 25% of patients have mild diffuse myocardial inflammation. This occurs during the acute febrile period and is characterized by tachycardia, a gallop rhythm, and nonspecific ST-T wave changes. Up to 5% of the children also exhibit some degree of CHF during this acute phase of their illness. This carditis usually resolves when the fever resolves. Pericardial effusions also occur in up to 20 to 40% of cases. Mild mitral and aortic regurgitation is also seen in 1 to 2% of untreated cases on echocardiographic examinations. This phase of the disease is mild and self-resolving. Therapy during this phase of the illness is primarily supportive.
The second phase of the disease involves coronary artery dilation, which usually peaks 2 to 4 weeks from the onset of the illness and is seen in 15 to 25% of the untreated patients with Kawasaki disease.78 Without the appropriate treatment, 15 to 20% of children with Kawasaki disease go on to have coronary aneurysms within 1 to 3 weeks from the onset of their illness.2 These coronary aneurysms can then lead to myocardial infarction, thrombosis, rupture, and a variety of ischemia-induced dysrhythmias. Significant risk factors for coronary aneurysmal formation include male gender, age younger than 1 year or older than 8 years, prolonged febrile period longer than 10 to 14 days, early myocarditis, anemia (hemoglobin <10 g/dL), white blood cell count greater than 30,000, increased band count, elevated ESR, elevated CRP level, low serum albumin levels, aneurysms involving other arteries (renal, axillary, or iliac), and giant coronary aneurysms (>8 mm in diameter). Death from Kawasaki disease is primarily due to myocardial infarction secondary to coronary artery occlusion. Giant coronary artery aneurysmal rupture is rare. Although most of the fatalities of Kawasaki disease occur within 6 weeks from the onset of the illness, sudden death can also occur many years after the illness. Prompt recognition and treatment have decreased this mortality rate from 2% to less than 0.01%.75
Management
The main goal of treatment during the acute febrile phase of Kawasaki disease is to provide supportive care and to decrease the inflammation of the myocardium and coronary arteries. The two major components of treatment include IVIG infusion and high-dose aspirin therapy, which together have an additive effect. This combination of IVIG and high-dose aspirin, when it is initiated within 10 days from the onset of the illness, can substantially decrease the progression to coronary artery dilation and aneurysm formation compared with aspirin therapy alone and results in a more rapid resolution of fever and the other indicators of acute inflammation.76 However, despite prompt treatment with IVIG and high-dose aspirin, 2 to 4% of the children still have coronary artery abnormalities.76
Aspirin is initiated at 80 to 100 mg/kg/day orally divided into an every-6-hour dosing regimen until the child is afebrile for 48 to 72 hours (or longer). This dose is then decreased to 3 to 5 mg/kg orally each day until the laboratory study results return to normal, which typically occurs 6 to 8 weeks after the onset of the disease. Aspirin therapy is continued beyond this period only in those children in whom coronary artery abnormalities are present. Ibuprofen can antagonize the antiplatelet effects of aspirin and should be avoided during treatment.76
The use of corticosteroids as primary treatment has not been well established. A multicenter, randomized, double-blind study to determine the efficacy of the addition of methylprednisolone to initial conventional therapy showed no difference in clinical outcome, including dimensions of coronary artery aneurysms, length of hospital stay, and rate of re-treatment with IVIG.79 The current recommendations include corticosteroids for refractory cases of Kawasaki disease, in which high-dose aspirin is continued, a second dose of IVIG is administered, and corticosteroids are added to decrease the risk of coronary artery aneurysm.76,80 Other complications, such as coronary aneurysmal thrombosis, likewise have limited data to guide management. Therapeutic options include streptokinase, tissue-type plasminogen activator, and interventional cardiac catheterization.76
The follow-up of children with Kawasaki disease depends on the degree and presence of carditis and coronary artery abnormalities detected on the initial echocardiogram. Other imaging modalities used to follow aneurysmal parameters include electron-beam computed tomography, coronary magnetic resonance angiography, and multislice spiral computed tomography.76 Those children with more severe cardiac abnormalities require close follow-up by a cardiologist who is experienced in managing the cardiac complications of Kawasaki disease. Overall, prompt diagnosis and appropriate therapies can prevent coronary aneurysm formation in up to 95% of the cases as well as result in rapid symptomatic improvement in up to 90% of the cases.81
Acute Rheumatic Fever
Clinical Features and Diagnostic Considerations
The diagnosis of ARF is based on the Jones criteria (Box 171-19). In addition to the Jones criteria, there must also be evidence of an antecedent streptococcal infection, which can be documented by a positive throat culture, a positive rapid streptococcal antigen test finding, or an elevated antistreptolysin O (ASO) titer. The ASO titer begins to rise 1 to 3 weeks after streptococcal infection, peaks at 3 to 5 weeks, and reliably falls to baseline after 6 months.82 The streptozyme test is not as reliable and therefore should not be used as a definitive test for evidence of an antecedent group A streptococcal infection.83 The diagnosis of ARF is made in a patient with a documented antecedent streptococcal infection who exhibits either two major criteria or one major plus two minor criteria. The most common presenting major criterion is the migratory polyarthritis, which commonly involves the larger joints of the extremities as well as the smaller tarsal joints in the foot and the smaller carpal joints in the hand. The carditis of ARF most commonly involves valvulitis of the mitral and aortic valves, which clinically is manifested as occult mitral or aortic insufficiency. The murmur of mitral insufficiency is characterized as a holosystolic murmur best heard over the apex with radiation to the axilla. The murmur of aortic insufficiency is characterized as a diastolic murmur that is best heard over the base of the heart. Innocent murmurs that are normally exacerbated with fever can be mistaken for the murmurs of mitral or aortic insufficiency. The other cardiac manifestations of ARF include CHF, pericarditis, and various degrees of heart block. The two dermatologic major criteria (erythema marginatum and subcutaneous nodules) and chorea occur less commonly than the migratory polyarthritis and carditis. Chorea may occur as the only manifestation of ARF. If arthritis is used as a major component, arthralgia cannot be used as a minor component to make the diagnosis. Likewise, if carditis is used as a major component, a prolonged PR interval cannot be used as a minor component.83
Cardiac Causes of Sudden Death in Young Athletes
In a large registry study of 1866 sudden deaths in young athletes, 1048 (56%) were found to be due to cardiovascular disease, and 416 (22%) were due to direct blunt trauma.84 The most common cardiovascular cause of sudden death in the athlete is hypertrophic cardiomyopathy, which accounts for up to 36% of the cardiovascular-related cases (Box 171-20).70
Specific Disorders
Congenital Coronary Artery Anomalies.: An additional 24% of the cases of sudden death are due to various anomalies of the coronary arteries.85 Congenital coronary artery anomalies are difficult to detect from a clinical standpoint, but 37% of those individuals who died of congenital coronary artery anomalies exhibited previous symptoms of exercise-induced syncope or chest pain.86 The exact pathophysiologic mechanism of sudden death in individuals with congenital coronary anomalies is unknown. Although there are a variety of congenital coronary artery anomalies, the most common potentially lethal lesion is the anomalous left coronary artery, in which the left main and right coronary arteries both arise from the right sinus of Valsalva. Individuals with this particular anomaly have a 46% incidence of sudden death, with more than 85% of the known cases of sudden death occurring during exercise.84 Congenital coronary artery hypoplasia is another uncommon cause of exercise-induced sudden death. Any athlete with exertional syncope or chest pain should be evaluated by a cardiologist for the possibility of a congenital coronary artery anomaly. If an anomaly is detected and surgically corrected, the athlete may resume full activity and participation in competitive sports.
Marfan Syndrome.: Individuals with Marfan syndrome should be evaluated for potential cardiac abnormalities before being allowed to participate in competitive sports. Clinical manifestations of the disease include tall and slender habitus, striae atrophicae of the skin (multiple stretch marks on skin), disproportionately long extremities compared with the trunk, scoliosis, pectus excavatum or carinatum, and dislocated lenses of the eye. Approximately 50% of patients with Marfan syndrome have cardiac manifestations, such as mitral valve prolapse or aortic dilation. The most serious cardiac complication of Marfan syndrome is the progressive dilation of the aorta with the potential risk of aortic rupture, which most commonly involves the descending portion of the aorta. Therefore, patients with Marfan syndrome should be prohibited from participation in contact sports. Those individuals who are known to have aortic dilation should also be prohibited from participation in any competitive sports regardless of the degree of contact involved.85 All patients with Marfan syndrome with or without cardiac involvement on their initial evaluation should be observed by a cardiologist with serial imaging studies of the aorta by echocardiography, magnetic resonance imaging, or computed tomography.
Perspective.: Although the nonobstructive form of hypertrophic cardiomyopathy is an uncommon cardiac malformation that occurs in only 0.2% of the general population, it is the single most common cardiac cause of sudden death in the young athlete.87–89 Hypertrophic cardiomyopathy is a familial disease that is inherited in an autosomal dominant fashion with variable penetrance. The hypertrophy of the left ventricle in this condition is idiopathic in nature and not due to chronic pressure overload conditions, such as systemic hypertension or aortic stenosis. The systolic left ventricular contractile function is vigorous but the thickened muscle of the left ventricle is stiff, resulting in impaired ventricular relaxation and high diastolic filling pressures.90
Clinical Features.: Some individuals with hypertrophic cardiomyopathy have experienced previous “warning” episodes of chest pain, dyspnea, syncope, or palpitations during vigorous activities. A family history of sudden unexplained death in young adults should also alert the clinician to the possibility of hypertrophic cardiomyopathy. The majority of young athletes who die of this condition have the nonobstructive form of hypertrophic cardiomyopathy, and the classic loud systolic ejection murmur that is present with the obstructive form may not be heard during the routine pre-sports physical examination.91
Current recommendations for pre-sports screening include a detailed family and personal history of known or suspected heart disease, physical examination, and 12-lead ECG. If any suspicion remains, further workup (e.g., echocardiography, Holter monitoring, other imaging studies) and referral to a cardiologist are indicated.91
Diagnostic Studies.: The electrocardiographic findings in individuals with hypertrophic cardiomyopathy typically reveal various degrees of left ventricular hypertrophy and left atrial enlargement. Other electrocardiographic findings include prominent Q waves in the inferolateral leads and diffuse T wave inversions. The most accurate study for the diagnosis of hypertrophic cardiomyopathy is the echocardiogram, which will demonstrate various degrees of left ventricular hypertrophy most commonly involving the ventricular septum in up to 90% of the cases.90 Those patients who have echocardiographic evidence of hypertrophic cardiomyopathy should be observed with serial echocardiographic examinations to monitor progression of the condition.
Management.: Although beta-blockers have been used in patients with hypertrophic cardiomyopathy, they have not been shown to prevent sudden death in these patients. The use of digoxin is contraindicated in patients with hypertrophic cardiomyopathy because its positive inotropic effect may worsen the left ventricular outflow obstruction. Sudden death in these patients with hypertrophic cardiomyopathy is thought to be due to exertion-induced ventricular fibrillation or pulseless ventricular tachycardia. Therefore, the current recommendation is that all individuals who are diagnosed with hypertrophic cardiomyopathy as well as those individuals with an equivocal diagnosis of hypertrophic cardiomyopathy should not participate in vigorous activities and competitive sports.
Perspective.: In 1957, Jervell and Lange-Nielsen first described the association of recurrent syncope, sudden death, and long QT interval in a series of deaf patients. Later, in 1963, Romano reported a similar association of symptoms with long QT intervals in patients with normal hearing. Both the Jervell–Lange-Nielsen and the Romano-Ward syndromes are inherited disorders with variable penetrance, characterized by a prolonged QT interval that has been associated with sudden death. The corrected QT interval (QTc) in normal individuals should not exceed 0.44 second in children or 0.42 second in adolescents. Individuals with QTc intervals longer than 0.55 second have a higher risk of sudden death. Prolongation of the QT interval predisposes the individual to ventricular tachycardia, torsades de pointes, and ventricular fibrillation, which is often initiated by a premature ventricular contraction occurring during the prolonged repolarization phase. In addition to the inherited syndromes of prolonged QT intervals, other causes of prolonged QT intervals include hypocalcemia, hypokalemia, hypomagnesemia, myocarditis, and medications (e.g., procainamide, erythromycin, cyclic antidepressants, phenothiazines, quinidine, organophosphates).
Clinical Features.: Symptoms in the young athlete that are suggestive of QT prolongation include exercise-induced palpitations, chest pains, syncope, dizziness, and atypical seizures. The young athlete who has any of these symptoms should be evaluated by a cardiologist, especially if the family history is positive for sudden unexplained death, cardiac problems, syncope, or deafness. Any young athlete who has been diagnosed with a prolonged QT syndrome should be prohibited from participation in competitive sports and vigorous activities. The growing popularity and presence of AEDs in public places and at sporting events can potentially save the lives of those athletes who suddenly collapse because of an underlying prolonged QT syndrome–induced nonperfusing ventricular dysrhythmia.
Management.: Treatment of a prolonged QT interval depends on the cause. Correction of any underlying metabolic disorder and discontinuation of a medication that induced the prolongation of the QT interval are the easiest therapies to implement. Magnesium sulfate is the drug of choice in the treatment of torsades de pointes. Antidysrhythmic agents that also can prolong the QT interval, such as procainamide and amiodarone, should be avoided. Therefore, the safest medication to use in a patient with prolonged QT interval–induced ventricular tachycardia or fibrillation is lidocaine. Beta-blockers have been used to prevent sudden ventricular dysrhythmias in those patients with the familial forms of QT prolongation. Adjunctive treatment in these selected patients also includes the insertion of pacemakers or internal defibrillators.
Commotio Cordis.: The phenomenon of commotio cordis occurs when an object such as a baseball strikes the chest and produces sudden death. This phenomenon most commonly occurs in children between 5 and 15 years of age with no known predisposing cardiac conditions.85,92 Although commotio cordis most commonly occurs in baseball, it has also been reported to occur in ice hockey, lacrosse, softball, and fist fights.78 In a few cases in which the cardiac rhythm was documented after the blunt trauma to the chest, the most common rhythm documented was ventricular fibrillation.93 The majority of patients who sustain commotio cordis do not survive, especially if they are not rapidly treated with defibrillation. If an AED is not immediately available and the patient is completely unresponsive with no pulse after sustaining a direct blow to the chest, some practitioners have proposed that performance of chest thumps during CPR may be of some benefit under this particular circumstance.










References
1. Horeczko, T, Young, KD. Congenital heart disease. In Strange GR, Ahrens WR, Schaftermeyer RW, Wiebe RA, eds.: Pediatric Emergency Medicine: A Comprehensive Study Guide, 3rd ed, New York: McGraw-Hill, 2009.
2. Fitzmaurice, L, Gerardi, MJ. Cardiovascular system. In: Gausche-Hill M, Fuch S, Yamamoto L, eds. APLS: The Pediatric Emergency Medicine Resource. 4th ed. Sudbury, Mass: Jones & Bartlett; 2004:106.
3. Woolridge, DP, Love, JC. Congenital heart disease in the pediatric emergency department. Part I: Pathophysiology and clinical characteristics. Pediatr Emerg Med Rep. 2002;7:69.
4. Sharieff, GQ, McCollough, M. The nightmare neonate: Life-threatening events in the first month of life. Emerg Med Pract. 2003;5:1.
5. Miller, AJ, Texidor, TA. Precordial catch, a neglected syndrome of precordial pain. JAMA. 1955;159:1364–1365.
6. Gumbiner, CH. Precordial catch syndrome. South Med J. 2003;96:38–41.
7. Inaba, AS. A simple way to remember pediatric vital signs. Contemp Pediatr. 2002;19:16.
8. Siegfried, BH, Henderson, TO. Cardiology. In: Gunn VL, Nechyba C, eds. The Harriet Lane Handbook: A Manual for Pediatric House Officers. 16th ed. Philadelphia: Mosby; 2002:123.
9. Park, MK. Physical examination. In: Park MK, ed. Pediatric Cardiology for Practitioners. 4th ed. St. Louis: Mosby; 2002:10.
10. Cloherty, JP, et al. Manual of Neonatal Care, 5th ed. Philadelphia: Lippincott Williams & Wilkins; 2004.
11. Adamcova, M. Troponins in children and neonates. Acta Pædiatr. 2003;92:1373–1375.
12. Fenton, KE, Sable, CA, Bell, MJ, Patel, KM, Berger, JT. Increases in serum levels of troponin I are associated with cardiac dysfunction and disease severity in pediatric patients with septic shock. Pediatr Crit Care Med. 2004;5:533–538.
13. Maisel, A. B-type natriuretic peptide in the diagnosis and management of congestive heart failure. Cardiol Clin. 2001;19:557.
14. Mir, TS, et al. Plasma concentrations of N-terminal pro–brain natriuretic peptide in control children from the neonatal to adolescent period and in children with congestive heart failure. Pediatrics. 2002;110:e76.
15. Hoffman, JI. The incidence of congenital heart disease. J Am Coll Cardiol. 2002;39:12.
16. Koppel, RI, et al. Effectiveness of pulse oxymetry screening for congenital heart disease in asymptomatic newborns. Pediatrics. 2003;111:451.
17. Park, MK. Left-to-right shunt lesions. In: Park MK, ed. Pediatric Cardiology for Practitioners. 4th ed. St. Louis: Mosby; 2002:129.
18. Thanopoulos, BD, et al. Transcatheter closure of perimembranous ventricular septal defects with the Amplatzer asymmetric ventricular septal defect occluder: Preliminary experience in children. Heart. 2003;89:918.
19. Chessa, M, et al. Transcatheter closure of congenital and acquired muscular ventricular septal defects using the Amplatzer device. J Invasive Cardiol. 2002;14:322.
20. Arora, R, et al. Transcatheter closure of congenital ventricular septal defects: Experience with various devices. J Interv Cardiol. 2003;16:83.
21. Brandt, RR. Transcatheter closure of atrial septal defect and patent foramen ovale in adult patients using the Amplatzer occlusion device: No evidence for thrombus deposition with antiplatelet agents. J Am Soc Echocardiogr. 2002;15:1094.
22. Wilkinson, JL. Interventional pediatric cardiology: Device closures. Indian J Pediatr. 2000;67(Suppl):30.
23. Niwa, K, et al. Eisenmenger syndrome in adults: Ventricular septal defects, truncus arteriosus, univentricular heart. J Am Coll Cardiol. 1999;34:223.
24. Park, MK. Obstructive lesions. In: Park MK, ed. Pediatric Cardiology for Practitioners. 4th ed. St. Louis: Mosby; 2002:155.
25. Chen, YB, Liberthson, RR, Freed, MD. Congenital heart disease. In: Lilly L, ed. Pathophysiology of Heart Disease: A Collaborative Project of Medical Students and Faculty. 3rd ed. Baltimore: Lippincott Williams & Wilkins; 2003:347.
26. Warnes, CA, et al. ACC/AHA 2008 guidelines for the management of adults with congenital heart disease: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Develop Guidelines for the Management of Adults With Congenital Heart Disease). Circulation. 2008;118:2395–2451.
27. Park, MK. Cyanotic congenital heart defects. In: Park MK, ed. Pediatric Cardiology for Practitioners. 4th ed. St. Louis: Mosby; 2002:174.
28. Gewitz, MH, Vetter, V. Cardiac emergencies. In: Fleisher GR, Ludwig S, eds. Textbook of Pediatric Emergency Medicine. 4th ed. Baltimore: Lippincott Williams & Wilkins; 2000:659.
29. Woods, WA, Schutte, DA, McCulloch, MA. Care of children who have had surgery for congenital heart disease. Am J Emerg Med. 2003;21:318.
30. Shay, DK, et al. Bronchiolitis-associated hospitalizations among US children. JAMA. 1999;282:1140.
31. Shay, DK, et al. Bronchiolitis-associated mortality and estimates of respiratory syncytial virus–associated deaths among US children. J Infect Dis. 2001;183:16.
32. American Academy of Pediatrics. Respiratory syncytial virus. In: Pickering LK, et al, eds. Red Book: 2003 Report of the Committee on Infectious Diseases. 26th ed. Elk Grove Village, Ill: American Academy of Pediatrics; 2003:523.
33. Feltes, TF, et al. Palivizumab prophylaxis reduces hospitalization due to respiratory syncytial virus in young children with hemodynamically significant congenital heart disease. J Pediatr. 2003;143:523.
34. Romero, JR. Palivizumab prophylaxis of respiratory syncytial virus disease from 1998 to 2002: Results from four years of palivizumab usage. Pediatr Infect Dis J. 2003;22(Suppl):46.
35. Koulouri, S, et al. Utility of B-type natriuretic peptide in differentiating congestive heart failure from lung disease in pediatric patients with respiratory distress. Pediatr Cardiol. 2004;25:341.
36. Vaidya, VU, et al. A computerized program for changing the rule of six to standardized drips. Pediatr Crit Care Med. 2005;6:109.
37. Young, KD, Seidel, JS. Pediatric cardiopulmonary resuscitation: A collective review. Ann Emerg Med. 1999;33:195.
38. Hazinski, MF, et al. Rhythm disturbances. In: Ralston M, ed. PALS Provider Manual. Dallas, Texas: American Heart Association; 2002:185.
39. Naccarelli, GV, et al. Amiodarone: Clinical trials. Curr Opin Cardiol. 2000;15:64.
40. Saul, JP, et al. Intravenous Amiodarone Pediatric Investigators: Intravenous amiodarone for incessant tachyarrhythmias in children: A randomized, double-blind, antiarrhythmic drug trial. Circulation. 2005;112:3470–3477.
41. O’Connor, M, McDaniel, N, Brady, WJ. The pediatric electrocardiogram, part II: Dysrhythmias. Am J Emerg Med. 2008;26:348–358.
42. Park, MK. Cardiac arrhythmias. In: Park MK, ed. Pediatric Cardiology for Practitioners. 4th ed. St. Louis: Mosby; 2002:333.
43. Manole, MD, Saladino, RA. Emergency department management of the pediatric patient with supraventricular tachycardia. Pediatr Emerg Care. 2007;23:176–185.
44. Ralston, M, et al. Recognition and management of cardiac arrest. In: Ralston M, ed. PALS Provider Manual. Dallas, Texas: American Heart Association; 2006:153.
45. Schlechte, EA, Boramanand, N, Funk, M. Supraventricular tachycardia in the pediatric primary care setting: Age-related presentation, diagnosis, and management. J Pediatr Health Care. 2008;22:289–299.
46. Cummins, RO. Stable tachycardias. In: Cummins RO, ed. ACLS Provider Manual. Dallas, Texas: American Heart Association; 2001:167.
47. Donoghue, AJ, et al. Out-of-hospital pediatric cardiac arrest: An epidemiologic review and assessment of current knowledge. Ann Emerg Med. 2005;46:512–522.
48. Young, KY, Gausche-Hill, M, McClung, CD, Lewis, RJ. A prospective, population-based study of the epidemiology and outcome of out-of-hospital pediatric cardiopulmonary arrest. Pediatrics. 2004;114:157.
49. Nadkami, VM, et al. First documented rhythm and clinical outcome from in-hospital cardiac arrest among children and adults. JAMA. 2006;295:50.
50. Smith, BT, et al. Ventricular fibrillation in pediatric cardiac arrest. Acad Emerg Med. 2006;13:525.
51. Samson, RA, et al. Outcomes of in-hospital ventricular fibrillation in children. N Engl J Med. 2006;354:2328.
52. Kleinman, ME, et al. Part 14: Pediatric advanced life support: 2010 American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Circulation. 2010;122(Suppl 3):S876–S908.
53. Alsoufi, B, et al. Survival outcomes after rescue extracorporeal cardiopulmonary resuscitation in pediatric patients with refractory cardiac arrest. J Thorac Cardiovasc Surg. 2007;134:952–959.
54. Morris, K, Beghetti, M, Petros, A, Adatia, I, Bohn, D. Comparison of hyperventilation and inhaled nitric oxide for pulmonary hypertension after repair of congenital heart disease. Crit Care Med. 2000;28:2974–2978.
55. Samson, RA, Berg, RA, Bingham, R. Use of automated external defibrillators for children: An update (an advisory statement from the Pediatric Advanced Life Support Task Force, International Liaison Committee on Resuscitation). Circulation. 2003;107:3250.
56. Jun, K. AEDs and children: Evidence for safe use now sufficient. Currents. 2003;14:11.
57. American Academy of Pediatrics Committee on Pediatric Emergency Medicine; American Academy of Pediatrics Section on Cardiology and Cardiac SurgeryMarkenson, D. Ventricular fibrillation and the use of automated external defibrillators in children. Pediatrics. 2007;120:1159.
58. Markenson, D, et al. Ventricular fibrillation and the use of automated external defibrillators in children. Pediatrics. 2006;120:e1368.
59. Berg, M, et al. Post-shock chest compression delays with automatic external defibrillator usage. Resuscitation. 2005;64:287.
60. Hickey, RW, Zuckerbraun, NS. Pediatric cardiopulmonary arrest: Current concept and future directions. Pediatr Emerg Med Rep. 2003;8:1.
61. Kochanek, PM, et al. Cerebral resuscitation after traumatic brain injury and cardiopulmonary arrest in infants and children in the new millennium. Pediatr Clin North Am. 2001;48:661.
62. Sirbaugh, PE, et al. A prospective, population-based study of the demographics, epidemiology, management, and outcome of out-of-hospital pediatric cardiopulmonary arrest. Ann Emerg Med. 1999;33:174.
63. Ferrieri, P, et al. From the Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease of the American Heart Association Council on Cardiovascular Disease in the Young: Unique features of infective endocarditis in childhood. Circulation. 2002;105:2115–2126.
64. Brook, MM. Pediatric bacterial endocarditis treatment and prophylaxis. Pediatr Clin North Am. 1999;46:275.
65. Martin, JM, Neches, WH, Wald, ER. Infective endocarditis: 35 years of experience at a children’s hospital. Clin Infect Dis. 1997;24:669–675.
66. American Heart Association. Prevention of infective endocarditis. Guidelines from the American Heart Association. Circulation. 2007;116:1736.
67. Lee, A, Mirrett, S, Reller, LB, Weinstein, MP. Detection of blood stream infections in adults: How many blood cultures are needed? J Clin Microbiol. 2007;45:3546.
68. Ferrieri, P, et al. Unique features of infective carditis in childhood. Pediatrics. 2002;109:931.
69. Yoshinaga, M, et al. Risk factors for in-hospital mortality during infective endocarditis in patients with congenital heart disease. Am J Cardiol. 2008;101:114.
70. American Academy of Pediatrics. Antimicrobial prophylaxis. In: Pickering LK, et al, eds. Red Book: 2003 Report of the Committee on Infectious Diseases. 26th ed. Elk Grove Village, Ill: American Academy of Pediatrics; 2003:782.
71. Baddour, LM, et al. Infective endocarditis: Diagnosis, antimicrobial therapy, and management of complications: A statement for healthcare professionals from the Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease, Council on Cardiovascular Disease in the Young, and the Councils on Clinical Cardiology, Stroke, and Cardiovascular Surgery and Anesthesia, American Heart Association: Endorsed by the Infectious Diseases Society of America. Circulation. 2005;111:e394–e434.
72. Freedman, SB, et al. Pediatric myocarditis: emergency department clinical findings and diagnostic evaluation. Pediatrics. 2007;120:1278.
73. Hia, CP, Yip, WC, Tai, BC, Quek, SC. Immunosuppressive therapy in acute myocarditis: An 18 year systematic review. Arch Dis Child. 2004;89:580–584.
74. Imazio, M, et al. Treatment of refractory recurrent pericarditis. J Cardiovasc Med. 2007;8:748.
75. American Academy of Pediatrics. Kawasaki syndrome. In: Pickering LK, et al, eds. Red Book: 2003 Report of the Committee on Infectious Diseases. 26th ed. Elk Grove Village, Ill: American Academy of Pediatrics; 2003:392.
76. Newburger, JW, et al. Diagnosis, treatment, and long-term management of Kawasaki disease: A statement for health professionals from the Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease, Council on Cardiovascular Disease in the Young, American Heart Association. Circulation. 2004;110:2747.
77. Falcini, F. Kawasaki disease. Curr Opin Rheumatol. 2006;18:33.
78. Hsieh, KS, et al. Treatment of acute Kawasaki disease: Aspirin’s role in the febrile stage revisited. Pediatrics. 2004;114:e689–e693.
79. Newburger, JW, et al. Randomized trial of pulsed corticosteroid therapy for primary treatment of Kawasaki disease. N Engl J Med. 2007;356:663.
80. Wooditch, A, Aronoff, S. Effect of initial corticosteroid therapy on coronary artery aneurysm formation in Kawasaki disease: A meta-analysis of 862 children. Pediatrics. 2005;116:989.
81. Sundel, RP. Rheumatologic emergencies. In: Fleisher GR, Ludwig S, eds. Textbook of Pediatric Emergency Medicine. 4th ed. Baltimore: Lippincott Williams & Wilkins; 2000:1191.
82. Rantz, LA, Randall, E, Rantz, HH. Antistreptolysin O; a study of this antibody in health and in hemolytic streptococcal respiratory disease in man. Am J Med. 1948;5:3.
83. Park, MK. Rheumatic fever. In: Park MK, ed. Pediatric Cardiology for Practitioners. 4th ed. St. Louis: Mosby; 2002:304.
84. Maron, BJ, Doerer, JJ, Haas, TS, Tierney, DM, Mueller, FO. Sudden deaths in young competitive athletes: Analysis of 1866 deaths in the United States, 1980-2006. Circulation. 2009;119:1085–1092.
85. Washington, RL. Most sudden deaths in adolescent athletes caused by cardiac conditions. Pediatr Ann. 2003;32:751.
86. Basso, C, et al. Clinical profile of congenital coronary artery anomalies with origin from the wrong aortic sinus leading to sudden death in young competitive athletes. J Am Coll Cardiol. 2000;35:1493.
87. Eckart, RE, Jones, SO, Shry, EA, Garrett, PD, Scoville, SL. Sudden death associated with anomalous coronary origin and obstructive coronary disease in the young. Cardiol Rev. 2006;14:161–163.
88. Basavarajaiah, S, et al. Prevalence of hypertrophic cardiomyopathy in highly trained athletes: Relevance to pre-participation screening. J Am Coll Cardiol. 2008;51:1033–1039.
89. Bar-Cohen, Y, Silka, MJ. Sudden cardiac death in pediatrics. Curr Opin Pediatr. 2008;20:517–521.
90. Chen, YB, Dec, GW, Lilly, LS. The cardiomyopathies. In: Lilly L, ed. Pathophysiology of Heart Disease: A Collaborative Project of Medical Students and Faculty. 3rd ed. Baltimore: Lippincott Williams & Wilkins; 2003:237.
91. Corrado, D, et al. Cardiovascular pre-participation screening of young competitive athletes for prevention of sudden death: Proposal for a common European protocol. Consensus Statement of the Study Group of Sport Cardiology of the Working Group of Cardiac Rehabilitation and Exercise Physiology and the Working Group of Myocardial and Pericardial Diseases of the European Society of Cardiology. Eur Heart J. 2005;26:516–524.
92. Maron, BJ, Gohman, TE, Kyle, SB, Estes, NA, Link, MS. Clinical profile and spectrum of commotio cordis. JAMA. 2002;287:1142–1146.
93. Geddes, LA, Roeder, RA. Evolution of our knowledge of sudden death due to commotio cordis. Am J Emerg Med. 2005;23:67–75.