[level-membership-for-anesthesiology-category]
Cardiac cycle: Control and synchronicity
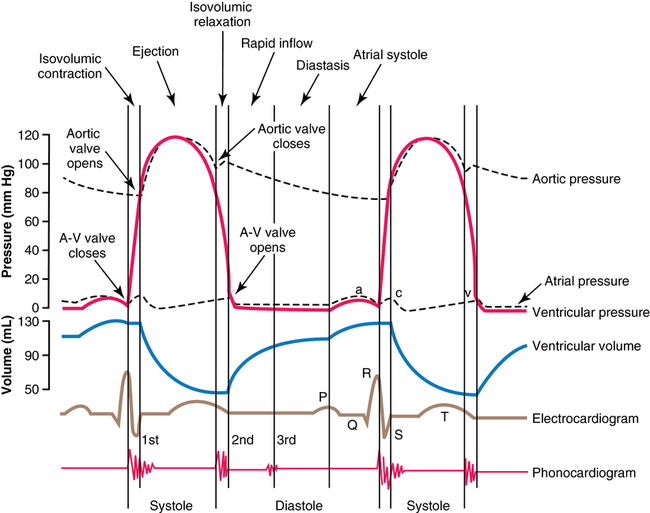
At the end of systole, the second period, diastole, or relaxation of the ventricle—takes place during four phases: isovolumic relaxation, rapid inflow, diastasis, and atrial systole (the final three phases constitute ventricular filling) (Figures 33-1 and 33-2). During isovolumic relaxation, the ventricular pressure rapidly drops below that of the arterial pressure, and the semilunar valves snap shut. For approximately 0.06 sec, the ventricular pressure continues to decrease without any change in ventricular volume. Once the ventricular pressure drops below the atrial pressure, the AV valves open and blood rapidly fills the ventricles. This period of rapid blood inflow is augmented by a negative intracavitary pressure (diastolic suction) created by the rapid myocardial relaxation. Once relaxation is complete and elastic distention of the ventricle begins, the slowing of blood return, termed diastasis, immediately precedes atrial systole. Effective atrial systole contributes up to about 20% of ventricular filling and completes the period of diastole.
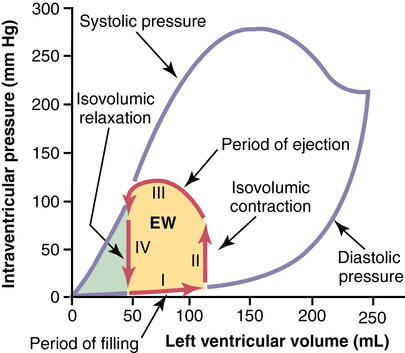
The maximum systolic pressure for the left ventricle is between 250 and 300 mm Hg but varies widely between individuals. The heavy red lines show a volume-pressure curve during a normal cardiac cycle, with EW representing the net external work of the heart. (From Guyton AC, Hall JE. Textbook of Medical Physiology. 11th ed. Philadelphia: Elsevier Saunders; 2005.)
Control
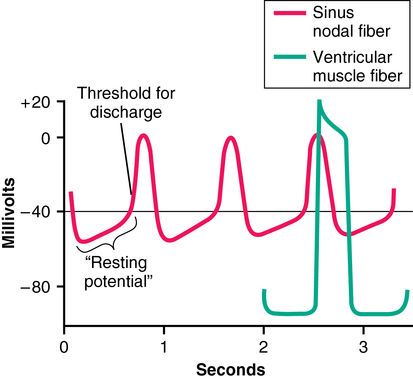
Rhythmicity of the cycle resides within the cells of the SA node because these cell membranes are inherently more “leaky” to Na+ and Ca2+ ions than are cell membranes of the ventricular conduction system. The influx of Na+ and Ca2+ creates a less negative resting membrane potential (−55 mV), a voltage at which many of the fast sodium channels have become inactivated. Therefore, depolarization is a result of the activation of the slow sodium-calcium channels, which results in a slower upslope of depolarization and a slower repolarization period in comparison with APs of ventricular muscle (Figure 33-3). The passive diffusion of Na+ due to its high concentration in the extracellular fluid outside the nodal fibers continues to depolarize the membrane. Once the threshold of −40 mV is reached, the sodium-calcium channels are activated, and an AP is generated. The sodium-calcium channels are quickly inactivated, and potassium channels are opened and allow the positively charged K+ ions to diffuse out of the cell until the resting membrane potential is hyperpolarized again at −55 mV. Finally, potassium channels close, with the inward-leaking Na+ and Ca2+ ions counterbalancing the outward flux of K+ ions. The process repeats itself, eliciting another cycle. The cells in the SA node control the heart rate because they depolarize more rapidly than does the rest of the conducting system (SA node rate, 70 to 80/min; AV node rate, 40 to 60/min; Purkinje fiber rate, 15 to 40/min).
[/level-membership-for-anesthesiology-category][not-level-membership-for-anesthesiology-category]
Cardiac cycle: Control and synchronicity
At the end of systole, the second period, diastole, or relaxation of the ventricle—takes place during four phases: isovolumic relaxation, rapid inflow, diastasis, and atrial systole (the final three phases constitute ventricular filling) (Figures 33-1 and 33-2). During isovolumic relaxation, the ventricular pressure rapidly drops below that of the arterial pressure, and the semilunar valves snap shut. For approximately 0.06 sec, the ventricular pressure continues to decrease without any change in ventricular volume. Once the ventricular pressure drops below the atrial pressure, the AV valves open and blood rapidly fills the ventricles. This period of rapid blood inflow is augmented by a negative intracavitary pressure (diastolic suction) created by the rapid myocardial relaxation. Once relaxation is complete and elastic distention of the ventricle begins, the slowing of blood return, termed diastasis, immediately precedes atrial systole. Effective atrial systole contributes up to about 20% of ventricular filling and completes the period of diastole.
The maximum systolic pressure for the left ventricle is between 250 and 300 mm Hg but varies widely between individuals. The heavy red lines show a volume-pressure curve during a normal cardiac cycle, with EW representing the net external work of the heart. (From Guyton AC, Hall JE. Textbook of Medical Physiology. 11th ed. Philadelphia: Elsevier Saunders; 2005.)
