Chapter 130 Capillary Hemangioblastoma of the Retina and von Hippel–Lindau Disease
Von hippel–lindau disease
Von Hippel–Lindau (VHL) (MIM193300) is an autosomal dominant neoplastic disorder in which multiple benign or malignant tumors and cysts with specific histopathologic features develop in the tissues of the central nervous system: the brain, spinal cord, inner ear, and retina and in visceral organs: the kidney, adrenal gland, pancreas, epididymis and broad ligament.1 VHL is a rare disorder (with an approximate incidence of 1 in 36 000 live births) and has a penetrance of over 90% by 65 years of age.
History
Over a century ago, von Hippel described retinal capillary hemangioblastomas transmitted through several generations of family members in a small number of pedigrees.2 In 1926, the Swedish pathologist, Lindau, observed the link between retinal and cerebellar hemangioblastomas as well as cysts in the kidney, pancreas, and epididymis as part of a familial syndrome.3 Melmon and Rosen established the first clinical diagnostic criteria for VHL in their landmark summary manuscript in 1965.4 Seizinger and colleagues discovered the linkage of the VHL gene to the short arm of chromosome 3 in 1988.5 Latif and colleagues identified the VHL tumor suppressor gene in 1993.6 The disease results from a germ line mutation in the VHL gene, which is located on the short arm of chromosome 3, 3p25–26.6
Genetic associations
In 1993, the discovery of the VHL gene,6 located on the short arm of chromosome 3 (3p25–26), was a seminal step in the molecular study of VHL disease. The isolation of the gene for VHL disease has led to significant advances in understanding how the VHL protein functions and how its inactivation may result in disease. The VHL gene is ubiquitously expressed and the protein acts to degrade particular transcription factors called hypoxia inducible factors (HIFs).7 HIFs are produced in response to low tissue oxygen levels, or hypoxia, and serve to upregulate proteins that can help reverse hypoxia, such as erythropoietin (EPO), vascular endothelial growth factor (VEGF), and platelet-derived growth factor (PDGF-β).8 Loss of VHL function by mutation of the VHL gene results in unregulated high levels of HIF, which in turn produces high levels of downstream gene products including VEGF, erythropoietin, PDGF-β, and transforming growth factor-α (TGF-α).9–11 The vascular nature of the VHL tumors may result from the HIF-mediated increased levels of VEGF and PDGF-β, which are known to support the proliferation of endothelial cells and pericytes, respectively.12 Moreover, increased vascular permeability of the tumor vessels resulting from increased levels of VEGF may also account for the edema seen with lesions of VHL.
The mutations in the VHL gene that result in VHL disease are highly varied, and may range from single base pair substitution in a single amino acid codon to the complete deletion of the gene.13,14 Although many of these mutations are thought to impair the ability of VHL to regulate HIF, the diversity in the manifestations of VHL disease suggests that different mutations may actually affect VHL protein function in different ways, and possibly even confer novel functions.15 Correlating the genotype to the phenotype might help elucidate how different types of VHL mutations result in disease. For example, the location of the missense mutation in the VHL gene correlates significantly with the prevalence and the phenotype of the eye disease, which would also in turn influence visual acuity loss in the affected patients.16 Such analyses might also help us understand the mechanisms by which the VHL mutations affect the eye.
Clinical presentation
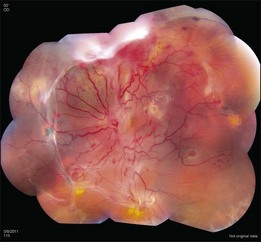
The retinal capillary hemangioblastomas, often the first manifestation of VHL, range from very small capillary abnormalities (Fig. 130.1) to large lesions that cause major visual impairment. The clinical appearance of the retinal capillary hemangioblastoma is very typical and diagnostic of VHL. The initial appearance of a retinal capillary hemangioblastoma is a subtle red or grayish dot no larger than a few hundred microns. As the proliferation of the vascular tumors (mostly composed of capillaries, with often a fibrotic component) progresses, secondary alterations occur to produce a distinctive clinical appearance that is often nodular (Fig. 130.2). The blood vessels leading to and away from the tumor become characteristically dilated and markedly enlarged (Fig. 130.3). This tumor, although often located in the retinal periphery, can lead to edema and hard exudates both around the tumor and in the center of the macula (Fig. 130.4). Without treatment, retinal capillary hemangioblastomas can grow and displace the normal structures of the retina and also cause an exudative retinal detachment. The fibrosis that accompanies these hemangioblastomas may result in tractional retinal detachment, with hemorrhaging and finally, neovascular glaucoma. Rarely, these tumors regress spontaneously.17 The tumors are located predominantly in the retinal periphery and less frequently on or around the optic disc (see below).
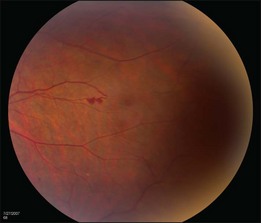
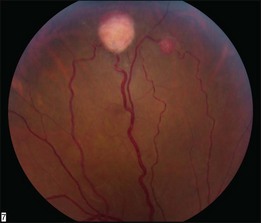
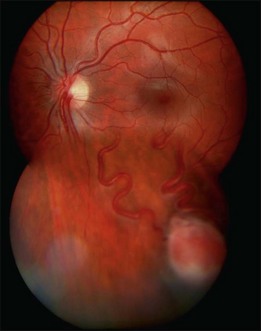
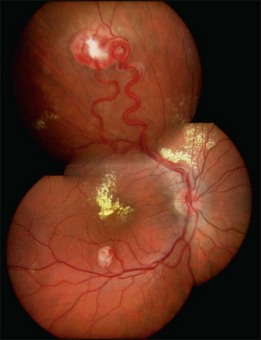
Optic nerve capillary hemangioblastomas or juxtapapillary capillary hemangioblastomas can start as small lesions at the optic disc or in the peripapillary area (Fig. 130.5). They can remain relatively slow growing for many years. However, with either increased growth (Figs. 130.6) or increased vasopermeability, exudative changes can occur (Fig. 130.7). Retinal edema, with retinal hard exudates, especially localized to the macula, can decrease vision. In addition, these exudative changes can lead to exudative retinal detachment (Fig. 130.8).
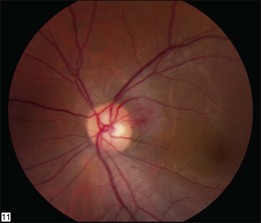
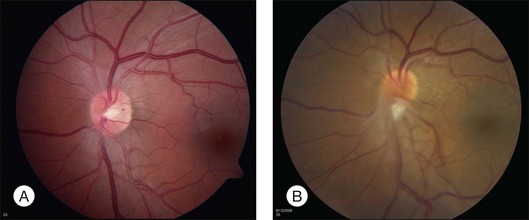
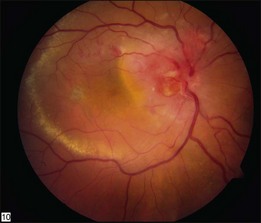
Fig. 130.7 Optic nerve tumor of this right eye has resulted in macular edema and marked retinal hard exudate.
Patients are often asymptomatic and VHL lesions tend to progress slowly. The retinal lesions can be detected in children to adults in the eighth decade of life on a routine exam.18,19 Symptoms such as decreased vision or strabismus may result in the detection of VHL in children. Decreased visual acuity can also cause adults to seek medical help with subsequent detection of the disease. The ophthalmologist plays an important role in initiating the medical work-up for VHL. Genetic testing has proven to be helpful in early diagnosis and clinical screening for VHL.
Clinical diagnosis
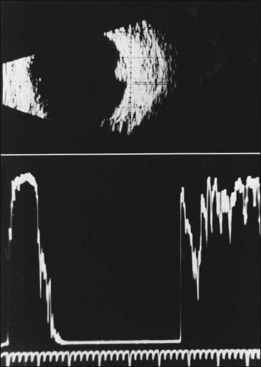
The diagnosis of the retinal capillary hemangioblastoma is primarily clinical, while other studies may help to confirm the diagnosis; there is no pathognomonic diagnostic tool. Fundus photography, especially with montage techniques or a wide-angle camera to capture the location and size of the peripheral lesions may be helpful in following the growth or regression of the lesions. Fluorescein angiography typically shows early leakage and marked hyperfluorescence and this may persist or decrease in the late phases of the study. Macular edema associated with these lesions may also be detected on fluorescein angiography. Optical coherence tomography may also be useful in such cases of macular edema but adds little to the diagnosis of retinal capillary hemangioblastoma (Fig. 130.9). Ultrasonography shows an acoustically solid retinal mass with a smooth anterior border with variable but mostly medium reflectivity (Fig. 130.9). Orbital shadowing and choroidal excavation are not seen.
Epidemiology of ocular lesions of von hippel–lindau disease
The true prevalence of the ocular manifestations of this rare systemic disease is difficult to ascertain. Most case series have reported higher prevalence of ocular disease as patients are referred to the ophthalmologists because of visual symptoms.20 The prevalence of ocular VHL was assessed in a study in which patients were referred because they were identified in the community as affected with VHL and not because of visual symptoms.21 The affected probands and their family members were invited to participate in a study at the National Institutes of Health for screening of VHL and its complications. This series of patients was diagnosed, examined and treated for various lesions of VHL by a team of experts including genetic counselors, medical geneticists, radiologists, pathologists, neurosurgeons, urologists, radiation oncologists, ophthalmologists, and otolaryngologists.
Over the period of 1988–2000, 335 (38%) affected individuals from a cohort of 890 patients of 220 unrelated pedigrees were found to have ocular involvement.21 Of those affected with ocular VHL, 42.1% were unilateral and 57.9% were bilateral. Approximately 85% were located in the peripheral retina, while 15% were juxtapapillary. Their ages ranged from 7 to 84 (mean 36 years with standard deviation ± 15 years), and 189 (47%) were male. In this population, 386 (95%) were white, seven (1.7%) were black, ten (2.5%) were Hispanic, and three (0.7%) were Asian.
Causes of vision loss
Visual loss from RCHs is generally caused by exudation from the tumor, causing retinal edema, or glial proliferation on the surface of the tumor resulting in retinal striae and distortion and eventually tractional and/or exudative retinal detachment.10 Interestingly, while tumor numbers did not increase significantly as a function of age, the risk of vision loss was found to increase with age, as was found in another cross-sectional series.22 The risk of severe vision loss also increased with the presence of juxtapapillary lesions and increasing peripheral tumor number and the extent of the retinal involvement.11 In the cohort studied at the National Eye Institute, approximately 77% had vision of 20/20 or better. The overall prevalence of legal blindness from ocular VHL was low at 5.7% with vision less than 20/160 in the better-seeing eye. Although this number appeared low, about 20% of all the patients with ocular VHL had some unilateral visual impairment, thus the burden of disease is still moderately high.
In about 8% of the NEI population, patients were found to have retinal neovascularization that mimicked diabetic retinal neovascularization.23 The retinal neovascularization most likely represents one end of the spectrum of hemangioblastoma formation. These patients tended to be younger and a majority of them had it as the sole finding in one eye, while the other eye had evidence of retinal capillary hemangioblastoma. The retinal neovascularization was also found often at the optic disc with an epiretinal membrane. The removal of the neovascularization along with the epiretinal membrane with vitrectomy and membrane peeling can result in visual acuity improvement. Other retinal lesions seen on the optic nerve include retinal vascular hamartomas.
Patients with systemic VHL also experienced vision loss from neurological lesions such as expanding cerebellar hemangioblastomas that may produce sustained elevated intracranial pressure, causing pressure on the optic nerves and resultant optic atrophy. Following surgical resection of the cerebellar lesions, the visual acuity can usually be expected to improve; but rarely, visual impairment may remain. Other extraocular causes of visual loss include hemangioblastomas that occur in the retrobulbar space, intracranial optic nerve, chiasm and/or the optic tract.24–29 Approximately 5.3% of cases of intracranial optic nerve/tract hemangioblastomas were documented in the NEI population.
Other manifestations of VHL that may come to an ophthalmologist’s attention include hypertensive retinopathy from pheochromocytomas, retinal vascular hamartomas,30 chiasmal syndromes,31 afferent pupillary defects from optic nerve hemangioblastomas,28 and papilledema (due to CNS lesions, optic nerve lesions, or pseudopapilledema with peripapillary RCH).32–34 Retinal function in general may be affected in VHL patients as demonstrated by electroretinography, although the mechanism for this dysfunction is obscure.35
Pathology of ocular lesions
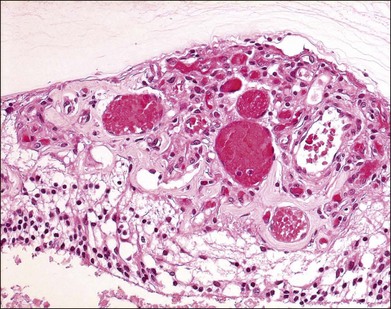
The pathology of retinal capillary hemangioblastoma has been proposed to be either a congenital, hamartomatous-type lesion or a benign vascular neoplastic process.36,37 Retinal capillary hemangioblastomas have mainly thin-walled, capillary-like or slightly larger, blood vessels that are separated by plump, vacuolated, foamy, “stromal” cells (Fig. 130.10).38 Such cells are strikingly similar to cerebellar hemangioblastoma and renal cell carcinoma cells found in VHL disease (Fig. 130.11). In the past, ultrastructural and immunohistochemical studies have suggested that these “stromal” cells may represent lipidized fibrous astrocytes or glial cells.39,40 Other investigators had previously suggested that these were not glial cells but more likely “vasoformative stem cells”.41 A study of cerebellar hemangioblastomas showed that these “stromal” cells may originate from developmentally arrested angioblasts.42 Expression of stem cell markers was evaluated in ocular lesions.43 The data suggested that capillary hemangioblastomas associated with VHL are comprised of developmentally arrested stem cells including hemangioblasts, endothelial and neuronal progenitor cells. There was coexpression of erythropoietin and its receptor that may not only mediate the development arrest of these progenitor cells, but may also induce proliferation.
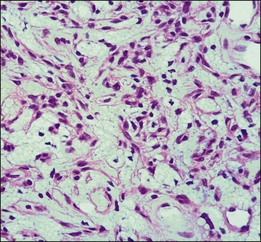
Using microdissection and molecular techniques, the “stromal” cells of the cerebellar hemangioblastomas and renal cell carcinomas were demonstrated to contain the VHL gene alterations.44,45 As stated by Knudson, it is believed that malignant neoplasms in VHL follow the “two-hit” theory, in which one allele is constitutionally inactivated (at the germline), while the other allele is subsequently inactivated (second hit) at the somatic target organ level.46 The VHL lesions, cerebellar and renal tumors showed the gene deletion in the “stromal” cells. The loss of heterozygosity was seen in these cells while the vascular component retained the heterozygosity.
Using microdissection and polymerase chain reaction amplification to study the ocular tissues from both surgical enucleation and autopsy eyes, the VHL gene alterations in six retinal lesions were evaluated.47 Similarly, these analyses showed the loss of heterozygosity in the VHL gene detected in the stromal cells of the lesion. This correlates with the inactivation of the gene, leading to the loss of the VHL tumor suppressor gene. The pVHL, the VHL suppressor gene product, is known to downregulate vascular endothelial growth factor (VEGF) expression. With the absence of pVHL, VEGF is upregulated, resulting in neovascularization on and around these retinal capillary hemangioblastomas. With this presumed mechanism of disease, there are implications for potential therapies for VHL lesions.
Histopathologic examination of peripheral lesions successfully treated by xenon photocoagulation,48 argon laser,48,49 and cryotherapy50 demonstrated occlusion of capillaries composing the angioma, gliosis, and fibrous metaplasia of retinal pigment epithelium.
Treatment
The treatment of the retinal capillary hemangioblastomas is based upon ablative therapy, which includes laser photocoagulation, cryotherapy, radiation, photodynamic therapy, or by surgical excision. The efficacy and the applicability of the therapy will depend on the location and size of the lesions. Small lesions are easy to treat successfully, while large lesions are difficult to treat. Photocoagulation with argon laser can fully treat small retinal capillary hemangioblastomas in most locations.49,51–53 However, for those tumors that are too large or located in the extreme periphery of the retina, cryotherapy may be indicated. Both photocoagulation and cryotherapy of peripheral tumors can often lead to massive retinal hard exudate accumulation and retinal edema in the macula, contributing to further decrease in vision following treatment. Fortunately, this so-called “ablatio fugax” may be transient. Although pre- and postoperative therapy with steroids, either locally or systemically, has not been assessed, it may be considered to decrease this side effect.
If the retinal capillary hemangioblastoma is located on the optic nerve, the treatment is much more difficult. Marked adverse side-effects including altitudinal visual field loss and central scotomas are associated with treatment of such tumors with laser photocoagulation.54 Photodynamic therapy has been used in these optic disc lesions with limited success and somewhat mixed results.55,56
Rarely, radiation may be useful in cases that do not respond to conventional laser or cryotherapy.57,58 There are anecdotal reports of such success, but the benefits are not uniform. The usefulness of radiation and its role in management of retinal capillary hemangioblastomas needs to be further defined.
Contraction of the fibrovascular lesion of the retinal capillary hemangioblastomas can lead to tractional retinal detachment. Vitrectomy can be performed on patients with either tractional or exudative retinal detachment.59 Vitreous hemorrhage from large tumors can also be treated by vitrectomy. However, the visual recovery following vitrectomy will only be maintained if the underlying tumors can be eradicated or their growth be interrupted.
Anti-angiogenic treatments
The understanding of the VHL protein function and tumorigenesis in VHL disease as noted previously, has led to treatments targeting the molecular biology of the disease, instead of the ablative approaches. The use of systemically administered antiangiogenic therapy has been reported for ocular VHL lesions. The use of SU5416, an intravenously administered inhibitor of VEGF receptor-2, has been reported in a few case reports. Aiello et al.,60 reported a single case of a juxtapapillary RCH treated with SU5416 that did not decrease in size with treatment but was associated with improvement in visual function, both in acuity and in visual field. Girmens et al.,61 reported a case of a treated peripheral tumor that did not shrink in size but decreased in exudation. Madhusudan et al.,62 found in a series of six similarly treated patients, only two achieved stability or improvement in their ocular lesions. SU5416 had significant adverse effects that precluded its further use. In a case report, bevacizumab, a humanized anti-VEGF antibody, was also used systemically, resulting in a transient reduction in exudation, but no improvement in visual outcome.63 Another case report using intravenous bevacizumab did not appear beneficial as there was subsequent development of new VHL ocular lesions.64
Local therapy of VHL-associated retinal capillary hemangioblastoma using a combination of an antiangiogenic agent (bevacizumab intravitreally) with one session of photodynamic therapy, has been reported.65 This resulted in a durable decrease in exudation, tumor regression, and improved visual acuity. However, in a prospective, pilot study of five patients treated with pegaptanib, an aptamer inhibitor of VEGF isoform 165, given every 6 weeks, there was no vision improvement and only minimal anatomical improvement.66 A similar prospective pilot study testing intravitreal ranibizumab also failed to show visual improvement or positive functional changes.67 Both of these studies showed decrease in the exudation but no change in the tumor size. In patients with persistent proliferative vitreoretinopathy who were treated with vitrectomy with one injection of bevacizumab, there was again no change in tumor size but there was no recurrence of the proliferative vitreoretinopathy.68
Screening and genetic testing for von hippel–lindau disease
The diagnosis of von Hippel–Lindau disease is often based on the following clinical criteria: (1) A positive family history and a CNS hemangioblastoma (including retinal hemangioblastomas), pheochromocytoma, or clear cell renal carcinoma is sufficient for diagnosis, and (2) if there is no family history, for diagnosis, the individual must have two or more CNS hemangioblastomas, or one CNS hemangioblastoma and a visceral tumor (with the exception of epididymal and renal cysts, which are frequent in the general population).4,69,70
When a patient presents with capillary hemangioblastomas in the absence of a family history or other systemic symptoms, genetic testing may be extremely useful.71 Advances in genetic testing have increased the detection of mutations from peripheral blood leucocytes from 75% to nearly 100%.72 Since the genetic testing detects mutation in almost 100% of documented affected families, serial clinical surveillance studies are recommended for family members who have the mutations.
The diagnosis of cases arising de novo (the first affected member of a family) may be more challenging and this may occur in up to 20% of the pedigrees.73 The initial mutation in these de novo cases might be so-called mosaicism in which some, but not all, tissues carry this new mutation. Consequently, such patients might have clinical signs of the disease, but the genetic tests from all peripheral leucocytes might be negative. The earlier the new mutation occurs in embryogenesis, the various types of cells that carry this mutation will be more numerous. Mosaicism can occur as a mutated gene in somatic tissue only, in germline tissue only, or in both. The risks to a carrier of a new mutation and to their offspring are very different in these three circumstances.
Since systemic VHL involves diverse organs with a high frequency of multiple neoplasms that are progressive, an integrated multispecialty team would provide the optimum assessment and therapy of these patients. This team should include neurosurgeons, urologic oncologists, ophthalmologists, neuroradiologists, otolaryngologists, pathologists, geneticists, genetic counselors, and rehabilitation specialists. Comprehensive serial screening and routine scheduled follow-up are essential for proper care (Table 130.1).74 These recommendations from one group of researchers are not the result of consensus building among the various VHL research groups. They do, however, provide general guidelines which may assist in the care of these patients.
Table 130.1 Recommended intervals for screening individuals at risk for von Hippel–Lindau disease
| Exam or procedure | Age of screening (frequency) |
|---|---|
| Ophthalmoscopy | Infancy (yearly) |
| Plasma or 24-hour urinary catecholamines and metanephrines | ~2 years (yearly and when blood pressure is elevated) |
| MRI of brain and spine | ~11 years of age (yearly) |
| CT and MRI of internal auditory canals | Onset of symptoms, hearing loss, tinnitus, vertigo |
| Ultrasound of abdomen | ~8 years (MRI as indicated) |
| CT of abdomen | ~18 years or earlier if clinically indicated (yearly) |
| Audiology testing | When clinically indicated |
(Adapted from Choyke PL, Glenn GM, Walther MM, et al. Von Hippel–Lindau disease: genetic, clinical, and imaging features. Radiology 1995;194:629–42.74)
Conclusion
Prior to the development of routine comprehensive screening surveys, the median survival of patients with VHL was less than 50 years of age, and the main causes of death were complications associated with renal cell carcinomas and central nervous system (CNS) hemangioblastomas.9,18,20–22,75–77 Improved surveillance, earlier diagnosis of lesions by modern imaging and laboratory studies, improvements in treatment, and increased knowledge of this disease have improved prognosis and reduced the complications related to these tumors.
1 Lonser RR, Glenn GM, Walther M, et al. Von Hippel–Lindau disease. Lancet. 2003;361:2059–2067.
2 von Hippel E. Uber eine sehr seltene erkrankung der netzhaut. Graefes Arch Ophthalmol. 1904;59:83–106.
3 Lindau A. Studien ber kleinbirncysten bau. Pathogenese und beziehungen zur angiomatosis retinae. Acta Pathol Microbiol Scand. 1926;3:S1–S28.
4 Melmon KL, Rosen SW. Lindau’s disease. Review of the literature and study of a large kindred. Am J Med. 1964;36:595–617.
5 Seizinger BR, Rouleau GA, Ozelius LJ, et al. Von Hippel–Lindau disease maps to the region of chromosome 3 associated with renal cell carcinoma. Nature. 1988;332:268–269.
6 Latif F, Tory K, Gnarra J, et al. Identification of the von Hippel–Lindau disease tumour suppressor gene. Science. 1993;260:1317–1320.
7 Maxwell PH, Wiesener MS, Chang GW, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–275.
8 Semenza GL. Regulation of mammalian O2 homeostasis by hypoxia-inducible factor 1. Annu Rev Cell Dev Biol. 1999;15:551–578.
9 Bohling T, Hatva E, Kujala M, et al. Expression of growth factors and growth factor receptors in capillary hemangioblastoma. J Neuropathol Exp Neurol. 1996;55:522–527.
10 Reifenberger G, Reifenberger J, Bilzer T, et al. Coexpression of transforming growth factor-alpha and epidermal growth factor receptor in capillary hemangioblastomas of the central nervous system. Am J Pathol. 1995;147:245–250.
11 Carmeliet P, Dor Y, Herbert JM, et al. Role of HIF-1alpha in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature. 1998;394:485–490.
12 Kaelin WG, Jr. Molecular basis of the VHL hereditary cancer syndrome. Nat Rev Cancer. 2002;2:673–682.
13 Zbar B, Kishida T, Chen F, et al. Germline mutations in the von Hippel–Lindau disease (VHL) gene in families from North America, Europe, and Japan. Hum Mutat. 1996;8:348–357.
14 Beroud C, Joly D, Gallou C, et al. Software and database for the analysis of mutations in the VHL gene. Nucleic Acids Res. 1998;26:256–258.
15 Czyzyk-Krzeska MF, Meller J. Von Hippel–Lindau tumor suppressor: not only HIF’s executioner. Trends Mol Med. 2004;10:146–149.
16 Mettu P, Agron E, Samtani S, et al. Genotype-phenotype correlation in ocular von Hippel–Lindau (VHL) disease: The effect of b missense mutation position on ocular VHL phenotype. Invest Ophthalmol Vis Sci. 2010;51:4464–4470.
17 Whitson JT, Welch RB, Green WR. Von Hippel–Lindau disease: case report of a patient with spontaneous regression of a retinal angioma. Retina. 1986;6:253–259.
18 Maher ER, Yates JRW, Harries R, et al. Clinical features and natural history of von Hippel–Lindau disease. Q J Med. 1990;77:1151–1163.
19 Jennings AM, Smith C, Cole DR, et al. Von Hippel–Lindau disease in a large British family: clinicopathological features and recommendations for screening and follow-up. Q J Med. 1988;66:233–249.
20 Singh AD, Shields CT, Shields JA. Von Hippel–Lindau disease. Surv Ophthalmol. 2001;46:117–142.
21 Wong WT, Agrón E, Coleman HR, et al. Clinical characterization of retinal capillary hemangioblastomas in a large population of patients with von Hippel–Lindau disease. Ophthalmology. 2008;115:181–188.
22 Webster AR, Maher ER, Moore AT. Clinical characteristics of ocular angiomatosis in von Hippel–Lindau disease and correlation with germline mutation. Arch Ophthalmol. 1999;117:371–378.
23 Wong WT, Yeh S, Chan CC, et al. Retinal vascular proliferation as an ocular manifestation of von Hippel–Lindau disease. Arch Ophthalmol. 2008;126:637–643.
24 Meyerle CB, Dahr SS, Wetjen NM, et al. Clinical course of retrobulbar hemangioblastoma in von Hippel–Lindau Disease. Ophthalmology. 2008;115:1382–1389.
25 Kupersmith MJ, Berenstein A. Visual disturbances in von Hippel–Lindau Disease. Ann Ophthalmology. 1981;2:195–197.
26 In S, Miyagi J, Kojho N, et al. Intraorbital optic nerve hemangioblastoma with von Hippel–Lindau disease. Case report. J Neurosurg. 1982;56:426–429.
27 O’Reilly GV, Rumbaugh CL, Bowens M, et al. Supratentorial haemangioblastoma: the diagnostic roles of computed tomography and angiography. Clin Radiol. 1980;32:389–392.
28 Kerr DJ, Scheithauer BW, Miller GM, et al. Hemangioblastoma of the optic nerve: Case report. Neurosurgery. 1995;36:573–581.
29 Baggentos M, Chew E, Butman JA, et al. Progressive peritumoral edema defining the optic fibers and resulting in reversible visual loss. J Neurosurg. 2008 Aug;109:313–317.
30 Schmidt D, Neumann HPH. Retinal vascular hamartoma in von Hippel–Lindau disease. Arch Ophthalmol. 1995;113:1163–1167.
31 Balcer LJ, Galetta SL, Curtis M, et al. Von Hippel–Lindau disease manifesting as a chiasmal syndrome. Surv Ophthalmol. 1995;39:302–306.
32 Gass JD, Braunstein R. Sessile and exophytic capillary angiomas of the juxtapapillary retina and optic nerve head. Arch Ophthalmol. 1980;98:1790–1797.
33 Yimoyines DJ, Topilow HW, Abedin S, et al. Bilateral peripapillary exophytic retinal hemangioblastomas. Ophthalmology. 1982;89:1388–1392.
34 Darr JL, Hughes RP, MaNair JN. Bilateral peripapillary retinal hemangiomas. Arch Ophthalmol. 1966;75:77–81.
35 Lubinski W, Krzystolik K, Cybulski C, et al. Retinal function in the von Hippel–Lindau disease. Doc Ophthalmol. 2003;106:271–280.
36 Welch RB. Von Hippel–Lindau disease: the recognition and treatment of early angiomatosis retinae and the use of cryosurgery as an adjunct to therapy. Trans Am Ophthalmol Soc. 1970;68:367–424.
37 Font RL, Ferry AP. The phakomatoses. Int Ophthalmol Clin. 1972;12:1–50.
38 Green WR. Retina: capillary hemangioma. In: Spencer WH, ed. Ophthalmic pathology: An atlas and textbook. Philadelphia: WB Saunders; 1996:709–718.
39 Jakobiec FA, Font RL, Johnson FB. Angiomatosis retinae: an ultrastructural study and lipid analyses. Cancer. 1976;38:2042–2056.
40 Grossniklaus HE, Thomas JW, Vigneswaran N, et al. Retinal hemangioblastoma: a histologic, immunohistochemical, and ultrastructural evaluation. Ophthalmology. 1992;99:140–145.
41 Mottow-Lippa L, Tso MO, Peyman GA, et al. Von Hippel angiomatosis. A light, electron microscopic, and immunoperoxidase characterization. Ophthalmology. 1983;90:848–855.
42 Vortmeyer AO, Frank S, Jeong SY, et al. Developmental arrest of angioblastic lineage initiates tumorigenesis in von Hippel–Lindau disease. Cancer Research. 2003;63:7051–7055.
43 Chan CC, Chew EY, Shen D, et al. Expression of stem cells markers in ocular hemangioblastomas associated with von Hippel–Lindau (VHL) disease. Mol Vis. 2005;11:697–704.
44 Lubensky IA, Gnarra JR, Bertheau P, et al. Allelic deletions of the VHL gene detected in multiple microscopic clear cell renal lesions in von Hippel–Lindau disease patients. Am J Pathol. 1996;149:2089–2094.
45 Vortmeyer AO, Gnarra JR, Emmert-Buck MR, et al. Von Hippel–Lindau gene deletion detected in stromal cell component of a cerebellar hemangioblastoma associated with von Hippel–Lindau disease. Hum Pathol. 1997;28:540–543.
46 Knudson AG, Jr. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci USA. 1971;68:820–823.
47 Chan CC, Vortmeyer AO, Chew, et al. VHL gene deletion and enhanced VEGF gene expression detected in the stromal cells of retinal angioma. Arch Ophthalmol. 1999;117:625–630.
48 Goldberg MF. Clinicopathologic correlation of von Hippel angiomas after xenon arc and argon laser photocoagulation. In: Peyman GA, Apple DJ, Sanders DR. Intraocular tumors. New York: Appleton-Century-Crofts; 1977:219–234.
49 Annesley WH, Leonard BC, Shields JA, et al. Fifteen year review of treated cases of retinal angiomatosis. Trans Sect Ophthalmol Am Acad Ophthalmol Otolaryngol. 1977;83:446–453.
50 Watzke RC. Cryotherapy for retinal angiomatosis: a clinico-pathologic report. Doc Ophthalmol. 1973;34:405–411.
51 Singh AD, Nouri M, Shields CL, et al. Treatment of retinal capillary hemangioma. Ophthalmology. 2002;109:1799–1806.
52 Rosa RH, Goldberg MF, Green WR. Clinicopathologic correlation of argon laser photocoagulation of retinal angiomas in a patient with von Hippel–Lindau disease followed for more than 20 years. Retina. 1996;16:145–156.
53 Schmidt D, Natt E, Neumann HP. Long-term results of laser treatment for retinal angiomatosis in von Hippel–Lindau disease. Eur J Med Res. 2000;5:47–58.
54 Garcia-Arumí J, Sararols LH, Cavero L, et al. Therapeutic options for capillary papillary angiomas. Ophthalmology. 2000;107:48–54.
55 Schmidt-Erfurth UM, Kusserow C, Barbazetto IA, et al. Benefits and complications of photodynamic therapy of papillary capillary angiomas. Ophthalmology. 2002;109:1256–1266.
56 Sachdeva R, Dadgostar H, Kaiser PK, et al. Verteporfin photodynamic therapy of six eyes with.
57 Palmer JD, Gragoudas ES. Advances in treatment of retinal angiomas. Int Ophthalmol Clin. 1997;37:150–170.
58 Matsuo T, Himei K, Ichimura K, et al. Long-term effect of external beam radiotherapy of optic disc hemangioma in a patient with von Hippel–Lindau disease. Acta Med Okayama. 2011;65:135–141.
59 Gaudric A, Krivosic V, Duguid G, et al. Vitreoretinal surgery for severe retinal capillary hemangiomas in von Hippel–Lindau disease. Ophthalmology. 2011;118:142–149.
60 Aiello LP, George DJ, Cahill MT, et al. Rapid and durable recovery of visual function in a patient with von Hippel–Lindau syndrome after systemic therapy with vascular endothelial growth factor receptor inhibitor su5416. Ophthalmology. 2002;109:1745–1751.
61 Girmens JF, Erginay A, Massin P, et al. Treatment of von Hippel–Lindau retinal hemangioblastoma by the vascular endothelial growth factor receptor inhibitor SU5416 is more effective for associated macular edema than for hemangioblastomas. Am J Ophthalmol. 2003;136:194–196.
62 Madhusudan S, Deplanque G, Braybrooke JP, et al. Antiangiogenic therapy for von Hippel–Lindau disease. JAMA. 2004;291:943–944.
63 von Buelow M, Pape S, Hoerauf H. Systemic bevacizumab treatment of a juxtapapillary retinal haemangioma. Acta Ophthalmol Scand. 2007;85:114–116.
64 Wackernagel W, Lackner EM, Pilz S, et al. Von Hippel–Lindau disease: treatment of retinal haemangioblastomas by targeted therapy with systemic bevacizumab. Acta Ophthalmol. 2010;88:e271–e272.
65 Ziemssen F, Voelker M, Inhoffen W, et al. Combined treatment of a juxtapapillary retinal capillary haemangioma with intravitreal bevacizumab and photodynamic therapy. Eye. 2007;21:1125–1126.
66 Dahr SS, Cusick M, Rodriguez-Coleman H, et al. Intravitreal anti-vascular endothelial growth factor therapy with pegaptanib for advanced von Hippel–Lindau disease of the retina. Retina. 2007;27:150–158.
67 Wong WT, Liang K, Hammel K, et al. Intravitreal ranibizumab therapy for retinal capillary hemangioblastoma related to von Hippel–Lindau Disease. Ophthalmol. 2008;115:1957–1964.
68 Tano R, Kakurai K, Sakurai T, et al. Intravitreal bevacizumab (avastin) combined with vitrectomy for recurrences of proliferative vitreoretinopathy in von Hippel–Lindau disease. Acta Ophthalmol. 2011. [Epub ahead of print]
69 Lamiell JM, Salazar FG, Hsia YE. Von Hippel–Lindau disease affecting 43 members of a single kindred. Medicine. 1989;68:1–29.
70 Escourolle R, Poirer J. Manual of neuropathology, 2nd edn. Philadelphia: WB Saunders; 1978. p. 49–51
71 Patel RJ, Appukuttan B, Ott S, et al. DNA-based diagnosis of the von Hippel–Lindau Syndrome. Am J Ophthalmol. 2000;129:258–260.
72 Stolle C, Glenn G, Zbar B, et al. Improved detection of germline mutations in the von Hippel–Lindau disease tumour suppressor gene. Hum Mutat. 1998;12:417–423.
73 Sgambati MT, Stolle C, Choyke PL, et al. Mosaicism in von Hippel–Lindau disease: lessons from kindreds with germline mutations identified in offspring with mosaic parents. Am J Hum Genet. 2000;66:84–91.
74 Choyke PL, Glenn GM, Walther MM, et al. Von Hippel–Lindau disease: genetic, clinical, and imaging features. Radiology. 1995;194:629–642.
75 Richard S, Campello C, Taillandier L, et al. Haemangioblastoma of the central nervous system in von Hippel–Lindau disease. French VHL Study Group. J Intern Med. 1998;243:547–553.
76 Neumann HP, Eggert HR, Scheremet R, et al. Central nervous system lesions in von Hippel–Lindau syndrome. J Neurol Neurosurg Psychiatry. 1992;55:898–901.
77 Lamiell JM, Salazar FG, Hsia YE. Von Hippel–Lindau disease affecting 43 members of a single kindred. Medicine. 1989;68:1–29.