Table 46-1
Chemotherapeutic Drug Overview
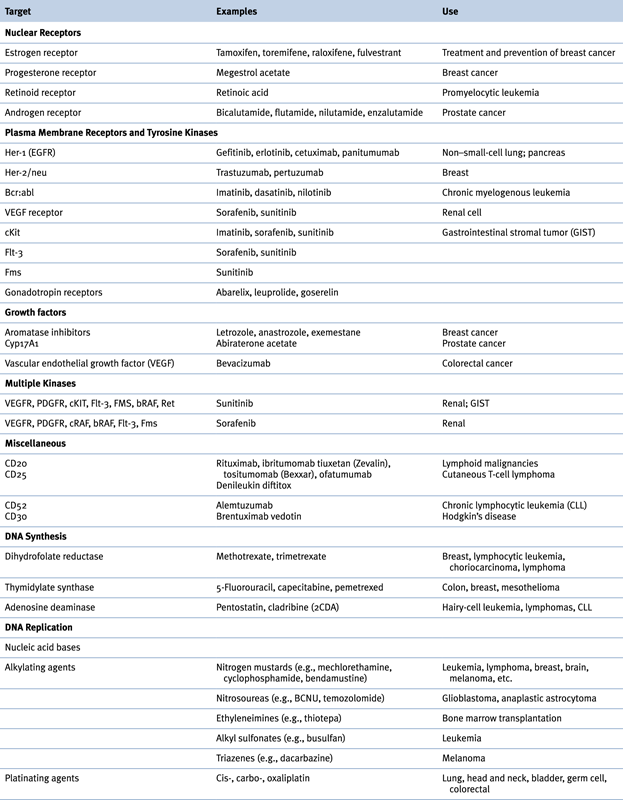
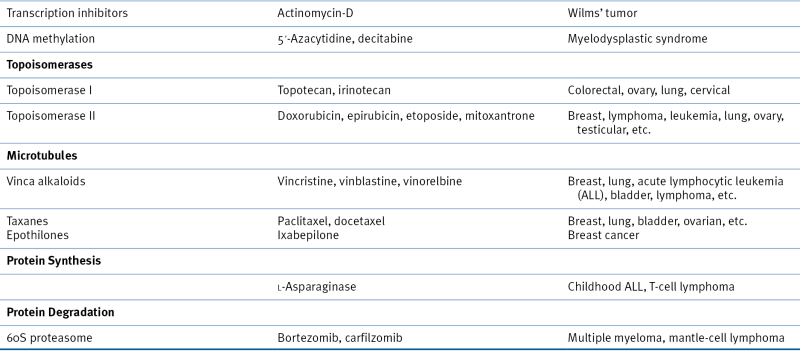
Table 46-2
Drugs That Alter Nucleic Acid Synthesis and Function
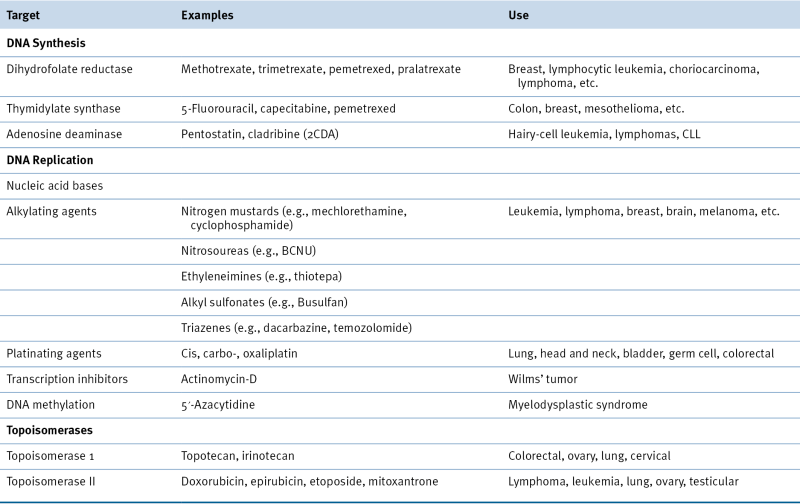
Table 46-3
Drugs with Other Mechanisms

Drugs Affecting Growth Factors and Growth Factor Receptors
Drugs Affecting Growth Factors
Sex Hormones
Gonadotropin-Releasing Hormones
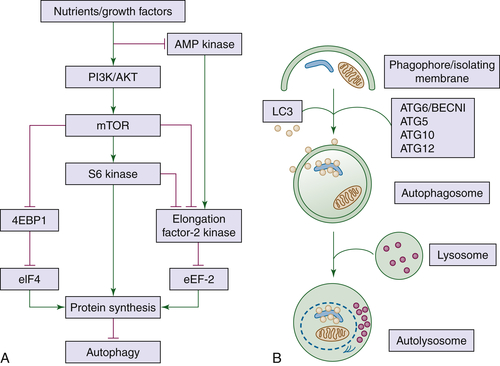
Vascular Endothelial Growth Factor
Drugs Affecting Growth Factor Receptors
Steroid Hormone Receptors
Selective Estrogen Receptor Modulators
Drugs that Affect Oncogenic Growth Factor Pathways
Epidermal Growth Factor Receptor Family
Drugs That Target Cancer Stem Cells
Drugs That Target the Immune System
Drugs That Alter Nucleic Acid Synthesis and Function
Inhibitors of Nucleic Acid Synthesis
Inhibitors of DNA Topoisomerase
Alkylating Agents
Platinating Agents
Epigenetic Modulators
Drugs that Affect the Mitotic Apparatus
Vinca Alkaloids, Taxanes, and Epothilones
Drugs Affecting Microtubule Polymerization
Drugs Affecting Microtubule Depolymerization
Drugs That Affect Protein Synthesis and Degradation
1. Human myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell . Nat Med . 1997 ; 3 : 730 – 737 .
2. Cancer stem cells, micro RNAs, and therapeutic strategies including natural products . Cancer Metastasis Rev . 2012 ; 31 733-571 .
3. Successful medical adrenalectomy with amino-glutethimide. Role of altered drug metabolism . JAMA . 1974 ; 230 : 1661 – 1665 .
4. Abiraterone and increased survival in metastatic prostate cancer . N Engl J Med . 2011 ; 364 : 1995 – 2005 .
5. Angiogenesis . Annu Rev Med . 2006 ; 57 : 1 – 18 .
6. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo . Nature . 1993 ; 362 : 841 – 844 .
7. Humanization of an anti-vascular endothelial growth factor monoclonal antibody for the therapy of solid tumors and other disorders . Cancer Res . 1997 ; 57 : 4593 – 4599 .
8. Role of angiogenesis inhibitors in colorectal cancer: sensitive and insensitive tumors . Curr Cancer Drug Targets . 2012 ; 12 : 303 – 315 .
9. Effects of tamoxifen vs raloxifene on the risk of developing invasive breast cancer and other disease outcomes: the NSABP Study of Tamoxifen and Raloxifene (STAR) P-2 trial . JAMA . 2006 ; 295 : 2727 – 2741 .
10. Increased survival with enzalutamide in prostate cancer after chemotherapy . N Engl J Med . 2012 ; 367 : 1187 – 1197 .
11. The neu oncogene encodes an epidermal growth factor receptor-related protein . Nature . 1986 ; 319 : 226 – 230 .
12. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene . Science . 1987 ; 235 : 177 – 182 .
13. Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer . N Engl J Med . 2005 ; 353 : 1673 – 1684 .
14. Status of epidermal growth factor receptor antagonists in the biology and treatment of cancer . J Clin Oncol . 2003 ; 21 : 2787 – 2799 .
15. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib . N Engl J Med . 2004 ; 350 : 2129 – 2139 .
16. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy . Science . 2004 ; 304 : 1497 – 1500 .
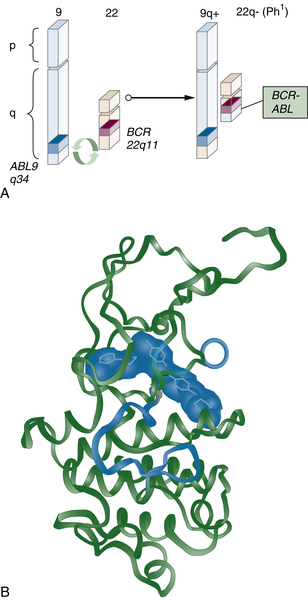
17. Hematologic and cytogenetic responses to imatinib mesylate in chronic myelogenous leukemia . N Engl J Med . 2002 ; 346 : 645 – 652 .
18. Denileukin diftitox: a concise clinical review . Expert Rev Anticancer Ther . 2005 ; 5 : 33 – 38 .
19. Inhibition of mutated, activated BRAF in metastatic melanoma . N Engl J Med . 2010 ; 363 : 809 – 819 .
20. Sorafenib: scientific rationales for single-agent and combination therapy in clear-cell renal cell carcinoma . Clin Genitourin Cancer . 2005 ; 4 : 167 – 174 .
21. Sunitinib maleate . Nat Rev Drug Discov . 2006 ; 5 : 279 – 280 .
22. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer . N Engl J Med . 2010 ; 363 : 1693 – 1703 .
23. Efficacy and safety of vismodegib in advanced basal-cell carcinoma . N Engl J Med . 2012 ; 366 : 2171 – 2179 .
24. Adoptive immunotherapy for cancer: harnessing the T cell response . Nat Rev Immunol . 2012 ; 12 : 269 – 281 .
25. Improved survival with ipilimumab in patients with metastatic melanoma . N Engl J Med . 2010 ; 363 : 711 – 723 .
26. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer . N Engl J Med . 2012 ; 366 : 2455 – 2465 .
27. The antifolates: evolution, new agents in the clinic, and how targeting delivery via specific membrane transporters is driving the development of the next generation of folate analogs . Current Opin Invest Drugs . 2010 ; 11 : 1409 – 1423 .
28. Pemetrexed (ALIMTA), a novel multitargeted antineoplastic agent . Clin Cancer Res . 2004 ; 10 : 4276 – 4280 .
29. Capecitabine: effective oral fluoropyrimidine chemotherapy . Expert Opin Pharmacother . 2005 ; 6 : 1231 – 1239 .
30. Evaluation of the antitumor activity of gemcitabine (2′,2′-difluoro-2′-deoxycytidine) . Cancer Res . 1990 ; 50 : 4417 – 4422 .
31. Clofaribine in pediatric acute leukemia: current findings and issues . Pediatr Blood Cancer . 2012 ; 59 : 417 – 422 .
32. DNA topoisomerase poisons as antitumor drugs . Annu Rev Biochem . 1989 ; 58 : 351 – 375 .
33. Camptothecins: from bench research to hospital wards . Cancer Res . 1994 ; 54 : 1431 – 1439 .
34. From bedside to bench to bedside to clinical practice: an odyssey with irinotecan . Clin Cancer Res . 2006 ; 12 : 1658 – 1660 .
35. Landmark article Sept. 21, 1946: Nitrogen mustard therapy. Use of methyl-bis(beta-chloroethyl)amine hydrochloride and tris(beta-chloroethyl)amine hydrochloride for Hodgkin’s disease, lymphosarcoma, leukemia and certain allied and miscellaneous disorders . By Louis S. Goodman, Maxwell M. Wintrobe, William Dameshek, Morton J. Goodman, Alfred Gilman and Margaret T. McLennan . JAMA . 1984 ; 251 : 2255 – 2261 .
36. Temozolomide and treatment of malignant glioma . Clin Cancer Res . 2000 ; 6 : 2585 – 2597 .
37. Bendamustine: rebirth of an old drug . J Clin Oncol . 2009 ; 27 : 1492 – 1501 .
38. Inhibition of cell division in Escherichia coli by electrolysis products from a platinum electrode . Nature . 1965 ; 205 : 698 – 699 .
39. Cellular and molecular pharmacology of oxaliplatin . Mol Cancer Ther . 2002 ; 1 : 227 – 235 .
40. FR901228, a novel antitumor bicyclic depsipeptide produced by Chromobacterium violaceum No. 968. I. Taxonomy, fermentation, isolation, physico-chemical and biological properties, and antitumor activity . J Antibiot (Tokyo) . 1994 ; 47 : 301 – 310 .
41. The epigenomics of cancer . Cell . 2007 ; 128 : 683 – 692 .
42. Results from a pivotal, open label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy . J Clin Oncol . 2012 ; 30 : 631 – 636 .
43. Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anti cancer drug . Nat Biotechnol . 2007 ; 1 : 84 – 90 .
44. Camptothecin and taxol: discovery to clinic. Thirteenth F. Bruce Cain Memorial Award Lecture . Cancer Res . 1995 ; 55 : 753 – 760 .
45. Promotion of microtubule assembly in vitro by taxol . Nature . 1979 ; 277 : 665 – 667 .
46. SPARC expression correlates with tumor response to albumin-bound paclitaxel in head and neck cancer patients . Transl Oncol . 2009 ; 2 : 59 – 64 .
47. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer . N Engl J Med . 2004 ; 351 : 1513 – 1520 .
48. The role of MAP4 expression in the sensitivity to paclitaxel and resistance to vinca alkaloids in p53 mutant cells . Oncogene . 1998 ; 16 : 1617 – 1624 .
49. Epothilones, a new class of microtubule-stabilizing agents with a taxol-like mechanism of action . Cancer Res . 1995 ; 55 : 2325 – 2333 .
50. Paclitaxel at ultra low concentrations inhibits angiogenesis without affecting cellular microtubule assembly . Anticancer Drugs . 2003 ; 14 : 13 – 19 .
51. The discovery and development of brentuximab vedotin for use in relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma . Nat Biotechnol . 2012 ; 30 : 631 – 637 .
53. Bortezomib: proteasome inhibition as an effective anticancer therapy . Annu Rev Med . 2006 ; 57 : 33 – 47 .
54. Analysis of expression of nuclear factor kappa B (NF-kappa B) in multiple myeloma: downregulation of NF-kappa B induces apoptosis . Br J Haematol . 2001 ; 115 : 279 – 286 .
55. Subcutaneous versus intravenous administration of bortezomib in patients with relapsed multiple myeloma: a randomised, phase 3, non-inferiority study . Lancet Oncol . 2011 ; 12 : 431 – 440 .
56. A matter of life or death (or both): understanding autophagy in cancer . Clin Cancer Res . 2006 ; 12 : 1961 – 1965 .
∗ We focus primarily on drugs that have achieved approval from the U.S. Food and Drug Administration (FDA); numerous drugs against new targets are in development, but a comprehensive description is beyond the scope of this chapter.






