CHAPTER 14 Cancer Genetics
Cell biology and molecular genetics have revolutionized our understanding of cancer in recent years; all cancer is a genetic disease of somatic cells because of aberrant cell division or loss of normal programmed cell death, but a small proportion is strongly predisposed by inherited germline mutations behaving as mendelian traits. However, this does not contradict our traditional understanding that, for many cancers, environmental factors are etiologically important, whereas heredity plays a lesser role. The latter is certainly true of the ‘industrial cancers’, which result from prolonged exposure to carcinogenic chemicals. Examples include cancer of the skin in tar workers, cancer of the bladder in aniline dye workers, angiosarcoma of the liver in process workers making polyvinyl chloride, and cancer of the lung (mesothelioma) in asbestos workers. Even so, for those who have been exposed to these substances and are unfortunate enough to suffer, it is possible that a significant proportion may have a genetic predisposition to the activity of the carcinogen. The link between cigarette smoking and lung cancer (as well as some other cancers) has been recognized for nearly half a century, but not all smokers develop a tobacco-related malignancy. Studies have shown that smokers with short chromosome telomeres (p. 31) appear to be at substantially greater risk for tobacco-related cancers than people with short telomeres who have never smoked, or smokers who have long telomeres, and another gene variant has been found to be more frequent in non-smokers who developed lung cancer.
Differentiation between Genetic and Environmental Factors in Cancer
Epidemiological Studies
It has long been recognized that people from lower socioeconomic groups have an increased risk of developing gastric cancer. Specific dietary irritants, such as salts and preservatives, or potential environmental agents, such as nitrates, have been suggested as possible carcinogens. Gastric cancer also shows variations in incidence in different populations, being up to eight times more common in Japanese and Chinese populations than in those of western European origin. Migration studies have shown that the risk of gastric cancer for immigrants from high-risk populations does not fall to that of the native low-risk population until two to three generations later. It has been suggested previously that this could be due to exposure to environmental factors at an early critical age. This may include early infection with Helicobacter pylori, which causes chronic gastric inflammation, and is associated with a five- to sixfold increased gastric cancer risk.
Family Studies
The frequency with which other family members develop the same cancer can provide evidence supporting a genetic contribution. The lifetime risk of developing breast cancer for a woman who lives until her mid-70s in Western Europe is at least 1 in 10. Family studies have shown that, for a woman who has a first-degree relative with breast cancer, the risk that she will also develop breast cancer is between 1.5 and 3 times the risk for the general population. The risk varies according to the age of onset in the affected family member: the earlier the age at diagnosis, the greater the risk to close relatives (p. 224).
Biochemical Factors
Biochemical factors can determine the susceptibility to environmental carcinogens. Examples include the association between slow-acetylator status and debrisoquine metabolizer status (p. 187) and a predisposition to bladder cancer, as well as glutathione S-transferase activity, which influences the risk of developing lung cancer in smokers.
Viral Factors
Subsequent studies have shown that certain viruses are tumor-forming or oncogenic in humans. A limited number of DNA viruses are associated with certain types of human tumors (Table 14.1), whereas a variety of RNA viruses, or retroviruses, cause neoplasia in animals. The study of the genetics and replicative processes of oncogenic retroviruses has revealed some of the cellular biological processes involved in carcinogenesis.
| Virus Family | Type | Tumor |
|---|---|---|
| Papova | Papilloma (HPV) | Warts (plantar and genital), urogenital cancers (cervical, vulval, and penile), skin cancer |
| Herpes | Epstein-Barr (EBV) | Burkitt lymphoma,* nasopharyngeal carcinoma, lymphomas in immunocompromised hosts |
| Hepadna | Hepatitis B (HBV) | Hepatocellular carcinomaa |
* For full oncogenicity, ‘co-carcinogens’ are necessary (e.g., aflatoxin B1 in hepatitis B–associated hepatocellular carcinoma).
Retroviruses
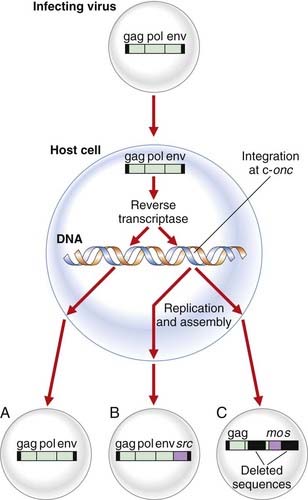
Retroviruses have their genetic information encoded in RNA and replicate through DNA by coding for an enzyme known as reverse transcriptase (p. 17), which makes a double-stranded DNA copy of the viral RNA. This DNA intermediate integrates into the host cell genome, allowing the appropriate proteins to be manufactured, resulting in repackaging of new progeny virions.
Naturally occurring retroviruses have only the three genes necessary to ensure replication: gag, encoding the structural proteins for the core antigens; pol, coding for reverse transcriptase; and env, the gene for the glycoprotein envelope proteins (Figure 14.1). Study of the virus responsible for the transmissible tumor in chickens, the so-called Rous sarcoma virus, identified a fourth gene that results in transformation of cells in culture, a model for malignancy in vivo. This viral gene, which transforms the host cell, is known as an oncogene.
Oncogenes
Identification of Oncogenes
Oncogenes have been identified by two types of cytogenetic finding in association with certain types of leukemia and tumor in humans. These include the location of oncogenes at chromosomal translocation breakpoints, or their amplification in double-minute chromosomes or homogeneously staining regions of chromosomes (p. 212). In addition, a number of oncogenes have also been identified by the ability of tumor DNA to induce tumors in vitro by DNA transfection.
Identification of Oncogenes at Chromosomal Translocation Breakpoints
Chromosome aberrations are common in malignant cells, which often show marked variation in chromosome number and structure. Certain chromosomes seemed to be more commonly involved and it was initially thought that these changes were secondary to the transformed state rather than causal. This attitude changed when evidence suggested that chromosomal structural changes, often translocations (p. 44), resulted in rearrangements within or adjacent to proto-oncogenes. It has been found that chromosomal translocations can lead to novel chimeric genes with altered biochemical function or level of proto-oncogene activity. There are numerous examples of both types, of which chronic myeloid leukemia is an example of the former and Burkitt lymphoma an example of the latter.
Chronic myeloid leukemia
In 1960, investigators in Philadelphia were the first to describe an abnormal chromosome in white blood cells from patients with chronic myeloid leukemia (CML). The abnormal chromosome, referred to as the Philadelphia, or Ph1, chromosome, is an acquired abnormality found in blood or bone marrow cells but not in other tissues from these patients. The Ph1 is a tiny chromosome that is now known to be a chromosome 22 from which long arm material has been reciprocally translocated to and from the long arm of chromosome 9 (Figure 14.2), i.e., t(9;22)(q34;q11). This chromosomal rearrangement is seen in 90% of those with CML. This translocation has been found to transfer the cellular ABL (Abelson) oncogene from chromosome 9 into a region of chromosome 22 known as the breakpoint cluster, or BCR, region, resulting in a chimeric transcript derived from both the c-ABL (70%) and the BCR genes. This results in a chimeric gene expressing a fusion protein consisting of the BCR protein at the amino end and ABL protein at the carboxy end, which is associated with transforming activity.
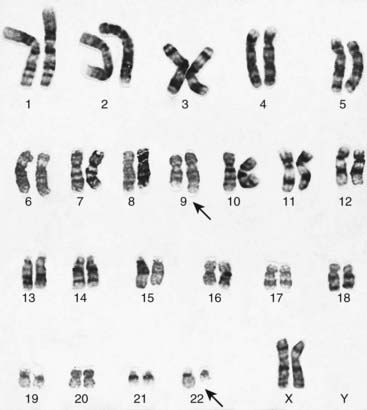
Burkitt lymphoma
An unusual form of neoplasia seen in children in Africa is a lymphoma that involves the jaw, known as Burkitt lymphoma, named after Dennis Burkitt, a medical missionary who first described the condition in the late 1950s. Chromosomal analysis has revealed the majority (90%) of affected children to have a translocation of the c-MYC oncogene from the long arm of chromosome 8 on to heavy (H) chain immunoglobulin locus on chromosome 14. Less commonly the MYC oncogene is translocated to regions of chromosome 2 or 22, which encode genes for the kappa (κ) and lambda (λ) light chains, respectively (pp. 196–197). As a consequence of these translocations, MYC comes under the influence of the regulatory sequences of the respective immunoglobulin gene and is overexpressed 10-fold or more.
Detection of Oncogenes by DNA Transfection Studies
The ability of DNA from a human bladder carcinoma cell line to transform a well established mouse fibroblast cell line called NIH3T3, as demonstrated by the loss of contact inhibition of the cells in culture, or what is known as DNA transfection, led to the discovery of the human sequence homologous to the ras gene of the Harvey murine sarcoma virus. The human RAS gene family consists of three closely related members, H-RAS, K-RAS, and N-RAS. The RAS proteins are closely homologous to their viral counterparts and differ from one another only near the carboxy termini. Oncogenicity of the ras proto-oncogenes has been shown to arise by acquisition of point mutations in the nucleotide sequence. In approximately 50% of colorectal cancers and 95% of pancreatic cancers, as well as in a proportion of thyroid and lung cancers, a mutation in a ras gene can be demonstrated. The RAS gene family has been shown to be the key pathway (RAS-MAPK) in neurofibromatosis type 1 (p. 298) and the Noonan/cardio-facio-cutaneous/Costello syndromes, all of which demonstrate some increased risk of tumor formation.
DNA transfection studies have also led to identification of other oncogenes that have not been demonstrated through retroviral studies. These include MET (hereditary papillary renal cell carcinoma), TRK, MAS, and RET (multiple endocrine neoplasia type 2, see Tables 14.5, 14.9).
Function of Oncogenes
Cancers have characteristics that indicate, at the cellular level, loss of the normal function of oncogene products consistent with a role in the control of cellular proliferation and differentiation in the process known as signal transduction. Signal transduction is a complex multistep pathway from the cell membrane, through the cytoplasm to the nucleus, involving a variety of types of proto-oncogene product involved in positive and negative feedback loops necessary for accurate cell proliferation and differentiation (Figure 14.3).
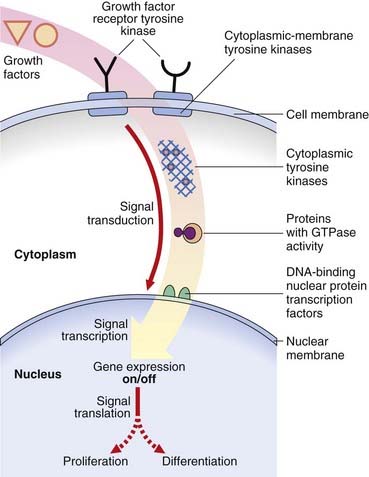
Types of Oncogene
Growth Factors
The transition of a cell from G0 to the start of the cell cycle (p. 39) is governed by substances called growth factors. Growth factors stimulate cells to grow by binding to growth factor receptors. The best known oncogene that acts as a growth factor is the v-SIS oncogene, which encodes part of the biologically active platelet-derived growth factor B subunit. When v-SIS oncoprotein is added to the NIH 3T3 cultures, the cells are transformed, behaving like neoplastic cells; that is, their growth rate increases and they lose contact inhibition. In vivo they form tumors when injected into nude mice. Oncogene products showing homology to fibroblast growth factors include HST and INT-2, which are amplified in stomach cancers and in malignant melanomas, respectively.
Growth Factor Receptors
Many oncogenes encode proteins that form growth factor receptors, with tyrosine kinase activity possessing tyrosine kinase domains that allow cells to bypass the normal control mechanisms. More than 40 different tyrosine kinases have been identified and can be divided into two main types: those that span the cell membrane (growth factor receptor tyrosine kinases) and those located in the cytoplasm (non-receptor tyrosine kinases). Examples of tyrosine kinases include ERB-B, which encodes the epidermal growth factor receptor, and the related ERB-B2 oncogene. Mutations, rearrangements, and amplification of the ERB-B2 oncogene result in ligand-independent activation, which has been associated with cancer of the stomach, pancreas, and ovary. Mutations in KIT occur in the hereditary gastrointestinal stromal tumor syndrome. These oncogenes are not activated by translocation (as in Burkitt lymphoma) but rather by point mutations. When germline or inherited, the mutations are not lethal, nor are they sufficient by themselves to cause carcinogenesis. In the case of MET (located on chromosome 7), the papillary renal cell carcinoma tumors are trisomic for chromosome 7 and two of the three copies of MET are mutant. A ratio of one mutant to one wild-type copy of MET is not sufficient for carcinogenesis, but a 2:1 ratio is.
Intracellular Signal Transduction Factors
DNA-Binding Nuclear Proteins
The FOS, JUN, and ERB-A oncogenes encode proteins that are specific transcription factors that regulate gene expression by activating or suppressing nearby DNA sequences. The function of MYC and related genes remains uncertain but appears to be related to alterations in control of the cell cycle. The MYC and MYB oncoproteins stimulate cells to progress from the G1 into the S phase of the cell cycle (p. 39). Their overproduction prevents cells from entering a prolonged resting phase, resulting in persistent cellular proliferation.
Cell-Cycle Factors
Cancer cells can increase in number by increased growth and division, or accumulate through decreased cell death. In vivo, most cells are in a non-dividing state. Progress through the cell cycle (p. 39) is regulated at two points: one in G1 when a cell becomes committed to DNA synthesis in the S phase, and another in G2 for cell division in the M (mitosis) phase, through factors known as cyclin-dependent kinases. Abnormalities in regulation of the cell cycle through growth factors, growth factor receptors, GTPases or nuclear proteins, or loss of inhibitory factors lead to activation of the cyclin-dependent kinases, such as cyclin D1, resulting in cellular transformation with uncontrolled cell division. Alternatively, loss of the factors that lead to normal programmed cell death, a process known as apoptosis (p. 85), can result in the accumulation of cells through prolonged cell survival as a mechanism of development of some tumors. Activation of the bcl-2 oncogene through chromosomal rearrangements is associated with inhibition of apoptosis, leading to certain types of lymphoma.
Tumor Suppressor Genes
Studies carried out by Harris and colleagues in the late 1960s, which involved fusion of malignant cells with non-malignant cells in culture, resulted in the suppression of the malignant phenotype in the hybrid cells. The recurrence of the malignant phenotype with loss of certain chromosomes from the hybrid cells suggested that normal cells contain a gene(s) with tumor suppressor activity that, if lost or inactive, can lead to malignancy and was acting like a recessive trait. Such genes were initially referred to as anti-oncogenes. This term was considered inappropriate because anti-oncogenes do not oppose the action of the oncogenes and are more correctly known as tumor suppressor genes. The paradigm for our understanding of the biology of tumor suppressor genes is the eye tumor retinoblastoma. It is important to appreciate, however, that a germline mutation in a tumor suppressor gene (as with an oncogene) does not by itself provoke carcinogenesis: further somatic mutation at one or more loci is necessary and environmental factors, such as ionizing radiation, may be significant in the process. At least 20 tumor suppressor genes have been identified.
Retinoblastoma
Retinoblastoma (Rb) is a relatively rare, highly malignant, childhood cancer of the developing retinal cells of the eye that usually occurs before the age of 5 years (Figure 14.4). If diagnosed and treated at an early stage, it is associated with a good long-term outcome.
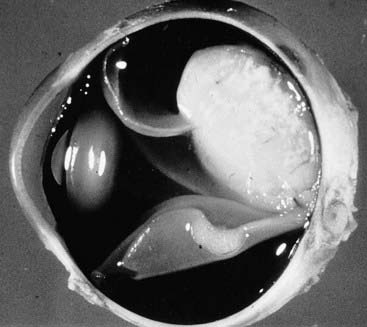
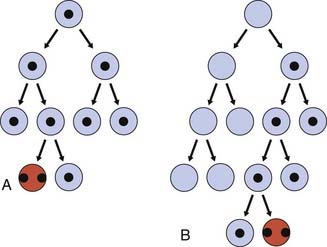
‘Two-Hit’ Hypothesis
In 1971, Knudson carried out an epidemiological study of a large number of cases of both types of Rb and advanced a ‘two-hit’ hypothesis to explain the occurrence of this rare tumor in patients with and without a positive family history. He proposed that affected individuals with a positive family history had inherited one non-functional gene that was present in all cells of the individual, known as a germline mutation, with the second gene at the same locus becoming inactivated somatically in a developing retinal cell (Figure 14.5). The occurrence of a second mutation was likely given the large number of retinal cells, explaining the autosomal dominant pattern of inheritance. This would also explain the observation that in hereditary Rb the tumors were often bilateral and multifocal. In contrast, in the non-heritable or sporadic form, two inactivating somatic mutations would need to occur independently in the same retinoblast cell (see Figure 14.5), which was much less likely to occur, explaining the fact that tumors in these patients were often unilateral and unifocal, and usually occurred at a later age than in the hereditary form. Hence, although the hereditary form of Rb follows an autosomal dominant pattern of inheritance, at the molecular level it is recessive because a tumor occurs only after the loss of both alleles.
It was also recognized, however, that approximately 5% of children presenting with Rb had other physical abnormalities along with developmental concerns. Detailed cytogenetic analysis of blood samples from these children revealed some of them to have an interstitial deletion involving the long arm of one of their number 13 chromosome pair. Comparison of the regions deleted revealed a common ‘smallest region of overlap’ involving the sub-band 13q14 (Figure 14.6). The detection of a specific chromosomal region involved in the etiology of these cases of Rb suggested that it could also be the locus involved in the autosomal dominant familial form of Rb. Family studies using a polymorphic enzyme, esterase D, which had previously been mapped to that region, rapidly confirmed linkage of the hereditary form of Rb to that locus.
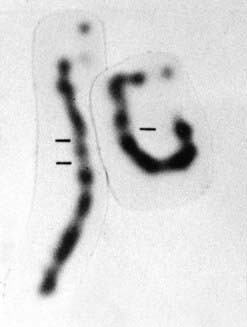
Loss of Heterozygosity
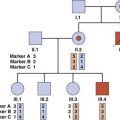
Analyses of the DNA sequences in this region of chromosome 13 in the peripheral blood and in Rb tumor material from children who had inherited the gene for Rb showed them to have loss of an allele at the Rb locus in the tumor material, known as loss of heterozygosity (LOH), or sometimes as loss of constitutional heterozygosity. An example of this is shown in Figure 14.7, A, in which the mother transmits the Rb gene along with allele 2 at a closely linked marker locus. The father is homozygous for allele 1 at this same locus, with the result that the child is constitutionally an obligate heterozygote at this locus. Analysis of the tumor tissue reveals apparent homozygosity for allele 2. In fact, there has been loss of the paternally derived allele 1 (i.e., LOH in the tumor material). This LOH is consistent with the ‘two-hit’ hypothesis leading to development of the malignancy as proposed by Knudson.
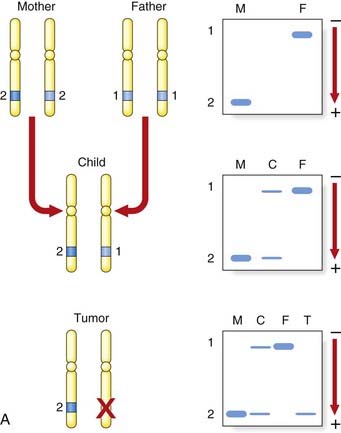
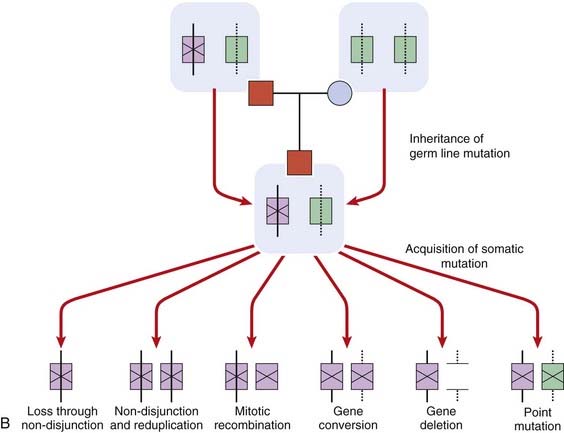
FIGURE 14.7 A, Diagrammatic representation of the loss of heterozygosity (LOH) in the development of a tumor. The mother (M) and father (F) are both homozygous for different alleles at the same locus, 2–2 and 1–1, respectively. The child (C) will therefore be constitutionally heterozygous, 1–2. If an analysis of DNA from a tumor at that locus reveals only a single allele, 2, this is consistent with LOH. B, Diagrammatic representations of the mechanisms by causing the ‘second hit’ leading to the development of retinoblastoma.
LOH can occur through several mechanisms, which include loss of a chromosome through mitotic non-disjunction (p. 43), a deletion on the chromosome carrying the corresponding allele, or a crossover between the two homologous genes leading to homozygosity for the mutant allele (Figure 14.7, B). Observation of consistent cytogenetic rearrangements in other malignancies has led to demonstration of LOH in a number of other cancers (Table 14.2). Subsequent to the observation of LOH, linkage studies of familial cases can be carried out to determine whether the familial cases of a specific type of malignancy are due to mutations at the same locus and thus lead to the identification of the gene responsible, as occurred with the isolation of the RB1 gene.
Table 14.2 Syndromes and Cancers that Show Loss of Heterozygosity and Their Chromosomal Location
| Syndrome or Cancer | Chromosomal Location |
|---|---|
| Retinoblastoma | 13q14 |
| Osteosarcoma | 13q, 17p |
| Wilms tumor | 11p13, 11p15, 16q |
| Renal carcinoma | 3p25, 17p13 |
| von Hippel-Lindau disease | 3p25 |
| Bladder carcinoma | 9q21, 11p15, 17p13 |
| Lung carcinoma | 3p, 13q14, 17p |
| Breast carcinoma | 11p15, 11q, 13q12, 13q14, 17p13, 17q21 |
| Rhabdomyosarcoma | 11p15, 17p13 |
| Hepatoblastoma | 5q, 11p15 |
| Gastric cancer | 1p, 5q, 7q, 11p, 13q, 17p, 18p |
| Familial adenomatous polyposis | 5q21 |
| Colorectal carcinoma | 1p, 5q21, 8p, 17p13, 18q21 |
| Neurofibromatosis I (NF1, von Recklinghausen disease) | 17q |
| Neurofibromatosis II (NF2) | 22q |
| Meningioma | 22q |
| Multiple endocrine neoplasia type I (MEN1) | 11q |
| Melanoma | 9p21, 17q |
| Ovarian | 11q25, 16q, 17q |
| Pancreatic | 9p21, 13q14, 17p13 |
| Prostate cancer | 1p36, 7q, 8p, 10q, 13q, 16q |
Function of Tumor Suppressor Genes
Although familial Rb was classically considered to be an autosomal dominant trait, demonstration of the action of the Rb gene as a tumor suppressor gene is consistent with it being a recessive trait, as originally suggested in the somatic cell hybridization studies carried out by Harris and colleagues. In other words, absence of the gene product in the homozygous state leads to the development of this particular tumor. In contrast to oncogenes, tumor suppressor genes are a class of cellular genes whose normal function is to suppress inappropriate cell proliferation (i.e., the development of a malignancy is due to a loss of function mutation, see p. 26).
The RB1 Gene/p110RB Protein
The RB1 gene specifies a 4.7-kilobase (kb) transcript that encodes a nuclear protein called p110RB, which associates with DNA and is involved in the regulation of the cell cycle. Fortuitously, research on the mechanism of action of the E1A oncogene of human adenovirus demonstrated that p110RB forms a complex with E2F-1, which is an E1A oncogene-regulated inhibitor of the transcription factor E2F. The complex so formed interferes with the ability of E2F to activate transcription of some key proteins required for DNA synthesis. When p110RB is in a hyperphosphorylated state, it does not interact readily with E2F-1, so permitting the cell cycle to proceed into the S phase (p. 39). Retinoblasts fail to differentiate normally in the presence of mutant p110RB.
p110RB interacts with several viral oncoproteins, such as the transforming proteins of simian virus (SV) 40 (large T antigen) and papilloma virus (E7 protein), and is inactivated, thereby liberating cells from normal growth constraints. These findings yield insight into the mechanisms of interaction between oncogenes, tumor suppressor genes, and the cell cycle.
TP53
Li-Fraumeni Syndrome
Because mutations in TP53 appear to be a common event in the genesis of many cancers, an inherited or germline mutation of TP53 would be expected to have serious consequences. This hypothesis was substantiated with the discovery of such a defect in persons with Li-Fraumeni syndrome. Members of families with this rare syndrome (p. 225), which is inherited as an autosomal dominant trait, are highly susceptible to developing a variety of malignancies at an early age, including sarcomas, adrenal carcinomas, and breast cancer. Point mutations in highly conserved regions of the TP53 gene (codons 245 to 258) have been identified in the germline of family members, with analysis of the tumor revealing loss of the normal allele.
Epigenetics and Cancer
Much of this chapter discusses familial cancer syndromes that follow mendelian inheritance, characterized by mutations in disease-specific genes. However, no discussion about cancer genetics is complete without considering epigenetic mechanisms. As discussed in Chapter 6 (p. 103), epigenetics refers to heritable changes to gene expression that are not due to differences in the genetic code. Such gene expression can be transmitted stably through cell divisions, both mitosis and meiosis. In cancer, much is now known about alterations to methylation status of the genome, both hypomethylation and hypermethylation, and in this section we also discuss telomere length and cancer.
DNA Methylation and Genomic Imprinting
The methylation of DNA is an epigenetic phenomenon (p. 103), and is the mechanism responsible for X-inactivation (p. 103) and genomic imprinting (p. 121). Methylation of DNA has the effect of silencing gene expression and maintaining stability of the genome, especially in areas where there is a vast quantity of repetitive DNA (heterochromatin), which might otherwise become erroneously involved in recombination events leading to altered regulation of adjacent genes. The relevance of this for cancer emerged in 1983 when studies showed that the genomes of cancer cells were hypomethylated compared with those of normal cells, primarily within repetitive DNA. This loss of imprinting (LOI) may lead to activation of an allele that is normally silent, and hence the high expression of a product that confers advantageous cellular growth. This appears to be an early event in many cancers and may correlate with disease severity. Chromosomal instability is strongly associated with increased tumor frequency, which has been clearly observed in mouse models, and all the ‘chromosome breakage’ syndromes (p. 288), which in humans are associated with a significant increased risk of cancer, particularly leukemia and lymphoma. LOI and removal of normal gene silencing may lead to oncogene activation, and hence cancer risk. LOI has been studied extensively at the IGF2/H19 locus on chromosome 11p15.5, previously discussed in Chapter 7 (p. 124). Insulin-like growth factor 2 (IGF2) and H19 are normally expressed from the paternal and maternal alleles, respectively (see Figure 7.27), but relaxed silencing of the maternal allele (i.e., hypomethylation, results in increased IGF2 expression). This has been shown to be the most common LOI event across a wide range of common tumor types (e.g., lung, liver, colon, ovary) as well as Wilms tumor, in which it was first identified.
Just as hypomethylation may lead to activation of oncogenes, the opposite effect of hypermethylation may also give rise to an increased cancer risk, in this case through silencing of tumor suppressor genes whose normal functions include inhibition of cell growth. The aberrant hypermethylation usually affects CpG nucleotide islands (C and G adjacent to each, p-phosphodiester bond), which are mostly unmethylated in somatic cells. This results in changes in chromatin structure (hypoacetylation of histone) that effectively silence transcription. When the genes involved in all sorts of cell regulatory activity are silenced, cells have a growth advantage. Early hypermethylation has been detected in colonic cancer. The effects of altered methylation leading to cancer are summarized in Figure 14.8, although the mechanism(s) that initiate the processes are poorly understood.
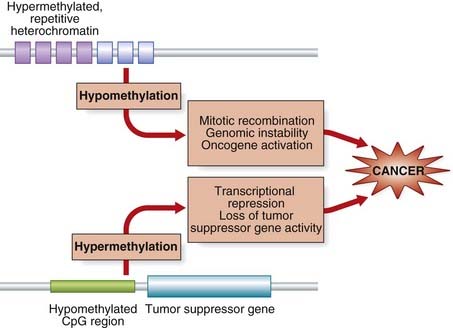
FIGURE 14.8 Methylation of DNA and cancer. The top schema shows a region of hypermethylated repetitive DNA sequence (heterochromatin). When this loses its methylation imprint, chromosome instability may result, which may lead to activation of oncogene(s). In the lower panel, hypomethylated stretches of CpG sequence (p. 219) become methylated, resulting in transcriptional suppression of tumor suppressor and cell regulatory genes.
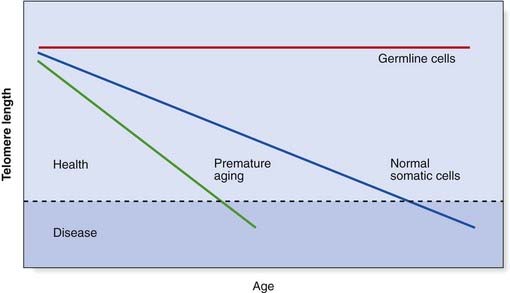
Telomere Length and Cancer
The ends of the chromosomes are known as telomeres (p. 31) and they are specialized chromatin structures that have a protective function. The sequence of DNA is specific and consists of multiple double-stranded tandem repeats as follows: TTAGGG. This sequence is typically about 10 to 15 kb long in human cells and is bound by specific proteins. It is also the substrate for telomerase, an enzyme that can lengthen the telomeres in those cells in which it is expressed. The final length of DNA at the very tip of the telomere is a single-stranded overhang of 150 to 200 nucleotides. Telomerase recognizes the 3′ end of the overhang, allowing lengthening to proceed.
Every cell division appears to result in the loss of TTAGGG repeats because conventional DNA polymerases cannot replicate a linear chromosome in its entirety, known as the ‘end-replication problem’. This progressive loss of telomere length is a form of cellular clock believed to be linked to both aging and human disease. When telomeres reach a critically short length, there is loss of protection and a consequence is chromosomal, and therefore genomic, instability, which means the cell is no longer viable. Short telomeres are now known to be a feature of the premature aging syndromes, such as ataxia telangiectasia, and other chromosome breakage disorders (p. 288), all of which are associated with premature onset of various cancers. It appears that the rate of telomere shortening is markedly increased in these conditions, so that cells and tissues literally ‘age’ more quickly. It is of great interest that some cancer cells express high levels of telomerase, so that cell viability is maintained. Most metastases have been shown to contain telomerase-positive cells, suggesting that telomerase is required to sustain such growth. However, cancer cells generally have shorter telomeres than the normal cells surrounding them, so telomerase activation in cancer rescues short telomeres and perpetuates genomically unstable cells.
Telomere length is therefore almost certainly a key concept in many cancers, as well as aging processes, even though the exact mechanisms remain to be elucidated. The relationship of telomere length to age and disease is displayed graphically in Figure 14.9.
Genetics of Common Cancers
It is estimated that about 5% of colorectal and breast cancers arise as a result of an inherited cancer susceptibility gene. A similar proportion of many other cancers are due to inherited predisposing genetic factors, but there are some notable exceptions in which only very low incidences of dominantly inherited carcinomas are recorded. These include the lung and cervix, as well as leukemias, lymphomas, and sarcomas. Here external agents or stimuli, and/or stochastic genetic events, are presumed to be the main factors. Nevertheless, studies of the common cancers—bowel or colorectal and breast cancer—have provided further insights into the genetics of cancer.
Colorectal Cancer
Multistage Process of Carcinogenesis
Similarly, allele loss of chromosome 5 markers occurs in approximately 40% of adenomatous polyps and 70% of carcinomas. Deletions on chromosome 17p in the region containing the TP53 gene occur in more than 75% of carcinomas, but this is an uncommon finding in small or intermediate-sized polyps. A region on 18q is deleted in about 10% of small adenomas, rising to almost 50% when the adenoma shows foci of invasive carcinoma, and in more than 70% of carcinomas (Figure 14.10). Genes at this locus include DCC (deleted in colorectal cancer), SMAD2, and SMAD4, the latter being part of the transforming growth factor-β (TGF-β) pathway. In some colorectal cancers mutations in the TGF-β receptor gene have been identified.
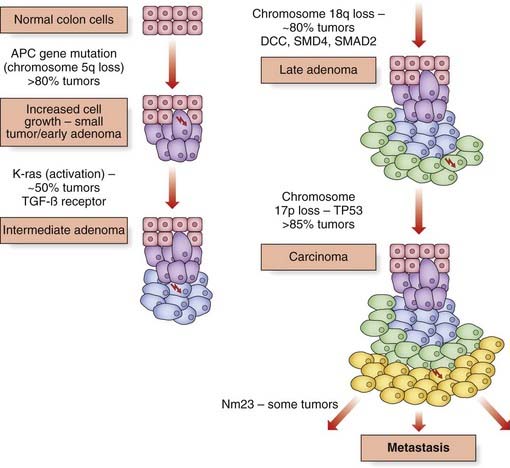
The multistage process of the development of cancer is likely, of course, to be an oversimplification. The distinction between oncogenes and tumor suppressor genes (Table 14.3) has not always been clear-cut—e.g., the RET oncogene and MEN2 (p. 93, Table 14.5). In addition, the same mutation in some of the inherited cancer syndromes (p. 225) can result in cancers at various sites in different individuals, perhaps as a consequence of the effect of interactions with inherited polymorphic variation in a number of other genes or a variety of environmental agents.
Table 14.3 Some Familial Cancers or Cancer Syndromes due to Tumor Suppressor Mutations
| Disorder | Gene | Locus |
|---|---|---|
| Retinoblastoma | RB1 | 13q14 |
| Familial adenomatous polyposis | APC | 5q31 |
| Li-Fraumeni syndrome | Tp53 | 17p13 |
| von Hippel–Lindau syndrome | VHL | 3p25-26 |
| Multiple endocrine neoplasia type II | RET | 10q11.2 |
| Breast–ovarian cancer | BRCA1 | 17q21 |
| Breast cancer | BRCA2 | 13q12-13 |
| Gastric cancer | CDH1 | 16q22.1 |
| Wilms tumor | WT1 | 11p13 |
| Neurofibromatosis I | NF1 | 17q12-22 |
Further insight into the processes involved in the development of colorectal cancer came from a rare cause of familial colonic cancer known as familial adenomatous polyposis.
Familial Adenomatous Polyposis
Approximately 1% of persons who develop colorectal cancer do so through inheritance of an autosomal dominant disorder known as familial adenomatous polyposis (FAP). Affected persons develop numerous polyps of the large bowel, which can involve its entirety (Figure 14.11). There is a high risk of carcinomatous change taking place in these polyps, with more than 90% of persons with FAP eventually developing bowel cancer.

FIGURE 14.11 Large bowel from a person with polyposis coli opened up to show multiple polyps throughout the colon.
(Courtesy Mr. P. Finan, Department of Surgery, General Infirmary, Leeds.)
The identification of an individual with FAP and an interstitial deletion of a particular region of the long arm of chromosome 5 (5q21) led to the demonstration of linkage of FAP to DNA markers in that region. Subsequent studies led to the isolation of the adenomatous polyposis coli (APC) gene. Analyses of the markers linked to the APC gene in cancers from persons who have inherited the gene for this disorder have shown LOH, suggesting a similar mechanism of gene action in the development of this type of bowel cancer.
Hereditary Non-Polyposis Colorectal Cancer—Lynch Syndrome
A proportion of individuals with familial colonic cancer may have a small number of polyps, and the cancers occur more frequently in the proximal, or right side, of the colon, which is sometimes called ‘site-specific’ colonic cancer. The average age of onset for colonic cancer in this condition is the mid-forties. This familial cancer-predisposing syndrome is inherited as an autosomal dominant disorder and has been known as hereditary non-polyposis colorectal cancer (HNPCC)—even though polyps may be present (the name helps to distinguish the condition from FAP). There is now a preference to return to the original eponymous designation of Lynch syndrome, specifically Lynch syndrome type I (site-specific, e.g., colorectal). There is also a risk of small intestinal cancers, including stomach, endometrial cancer, and a variety of others (see Table 14.5).
DNA mismatch repair genes
When looking for LOH, comparison of polymorphic microsatellite markers in tumor tissue and constitutional cells in persons with HNPCC somewhat surprisingly revealed the presence of new, rather than fewer, alleles in the DNA from tumor tissue. In contrast to the site-specific chromosome rearrangements seen with certain malignancies (pp. 211–212), this phenomenon, known as microsatellite instability (MSI), is generalized, occurring with all microsatellite markers analyzed, irrespective of their chromosomal location.
This phenomenon was recognized to be similar to that seen in association with mutations in genes known as mutator genes, such as the MutHLS genes in yeast and Escherichia coli. In addition, the human homolog of the mutator genes were located in regions of the human chromosomes to which HNPCC had previously been mapped, leading to rapid cloning of the genes responsible for HNPCC in humans (Table 14.4). The mutator genes code for a system of ‘proof-reading’ enzymes and are known as mismatch repair genes, which detect mismatched base pairs arising through errors in DNA replication or acquired causes (e.g., mutagens). The place of the TACSTD1 gene is unusual. It lies directly upstream of MSH2 and, when the last exons of the gene are deleted, transcription of TACSTD1 extends into MSH2, causing epigenetic inactivation of the MSH2 allele. However, deletions in this gene appear to be a rare cause of HNPCC.
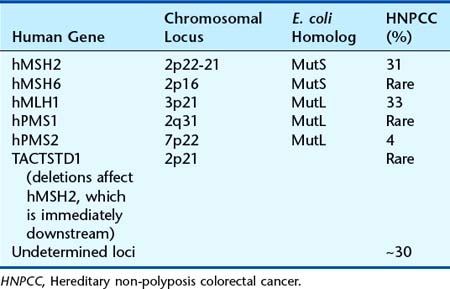
Individuals who inherit a mutation in one of the mismatch repair genes responsible for HNPCC are constitutionally heterozygous for a loss-of-function mutation (p. 26). Loss of function of the second copy through any of the mechanisms discussed in relation to LOH (p. 215) results in defective mismatch repair leading to an increased mutation rate associated with an increased risk of developing malignancy. Certain germline mutations, however, seem to have dominant-negative effects (p. 26). Although HNPCC accounts for a small proportion of colonic cancers, estimated as 2% to 4% overall, approximately 15% of all colorectal cancers exhibit MSI, the proportion being greater in tumors from persons who developed colorectal cancer at a younger age. Some of these individuals will have inherited constitutional mutations in one of the mismatch repair genes in the absence of a family history of colonic cancer. In addition, for women with a constitutional mismatch repair gene mutation, the lifetime risk of endometrial cancer is up to 50%.
Other Polyposis Syndromes
Juvenile Polyposis Syndrome
Autosomal dominant transmission is well described for a rare form of juvenile polyposis that may present in variety of ways, including bleeding with anemia, pain, intussusception, and failure to thrive. The polyps carry an approximate 13-fold increased cancer risk and, once diagnosed, regular surveillance and polypectomy should be undertaken. The average age at diagnosis of cancer is in the third decade, so that colectomy in adult life may be advisable. Two genes have been identified as causative: SMAD4 (18q) and BMPR1A (10q22). Both are components of the TGF-β signaling pathway (p. 85) and SMAD4 mutations, which account for about 60% of cases, appear to carry a higher malignancy potential and the possibility of large numbers of gastric polyps.
Peutz-Jegher Syndrome
Also autosomal dominant, this condition is characterized by the presence of dark melanin spots on the lips, around the mouth (Figure 14.12), on the palms and plantar areas, and other extremities. These are usually present in childhood and can fade in adult life. Patients often present with colicky abdominal pain from childhood from the development of multiple polyps that occur throughout the gastrointestinal tract, although they are most common in the small intestine. These are hamartomas but there is a significant risk of malignant transformation. There is an increased risk of cancers at other sites, particularly breast, uterus, ovary, and testis, and these tend to occur in early adult life. Regular screening for these cancers throughout life, from early adulthood, is warranted. Mutations in a serine threonine kinase gene, STK11, located on chromosome 19p, cause Peutz-Jegher syndrome.

Prostate Cancer
Analysis of prostate cancer tumor material has revealed LOH at several chromosomal locations. Segregation analysis of family studies of prostate cancer suggested that a single dominant susceptibility locus could be responsible, accounting for 9% of all prostate cancers and up to 40% of early-onset prostate cancers (diagnosed before age 55 years). Linkage analysis studies identified two major susceptibility loci, hereditary prostate cancer-1 and -2 (HPC1 and HPC2), and genome wide association studies have highlighted a number of other susceptibility loci of variable significance. It is possible in due course that testing of multiple susceptibility loci will enable identification of high risk individuals who can be offered surveillance. Mutations in the ribonuclease L gene (RNASEL) were identified in two families showing linkage to the HPC1 locus at 1q25. Mutations have been found in the ELAC2 gene at 17p11, the HPC2 locus, and, rarely, mutations in three genes—PTEN, MXI1, and KAI1—have been identified in a minority of families with familial prostate cancer. A small proportion of familial prostate cancer is associated with BRCA1 or BRCA2. Men who carry mutations in either BRCA1 or BRCA2 have an increased risk, and in one study, conducted in Ashkenazi Jews, men with such mutations had a 16% risk of prostate cancer by age 70 years, compared with 3.8% for the general population.
Genetic Counseling in Familial Cancer
Recognition of individuals with an inherited susceptibility to cancer usually relies on taking a careful family history to document the presence or absence of other family members with similar or related cancers. The malignancies that develop in susceptible individuals are often the same as those that occur in the population in general. There are a number of other features that can suggest an inherited cancer susceptibility syndrome in a family (Box 14.1).
Inherited Cancer-Predisposing Syndromes
Although most cancers from an inherited cancer syndrome occur at a specific site, families have been described in which cancers occur at more than one site in an individual or at different sites in various members of the family more commonly than would be expected. These families are referred to as having a familial cancer-predisposing syndrome. The majority of the rare inherited familial cancer-predisposing syndromes currently recognized are dominantly inherited, with offspring of affected individuals having a 50% chance of inheriting the gene and therefore of being at increased risk of developing cancer (Table 14.5). For the clinician, it is important to be aware of the physical signs that may point to as diagnosis, for example melanin spots around the mouth and lips (Peutz-Jegher syndrome), macrocephaly (Cowden disease), and dome-shaped skin papules (trichodiscomas; Figure 14.13) over the face and neck (Birt-Hogg-Dubé syndrome). In the latter condition, pneumothorax may be a presenting feature. There are also a number of syndromes, usually inherited as autosomal recessive disorders, with an increased risk of developing cancer associated with an increased number of abnormalities in the chromosomes when cultured, or what are known as the chromosomal breakage syndromes (p. 288).
Table 14.5 Inherited Family Cancer Syndromes, Mode of Inheritance, Gene Responsible and Chromosomal Site
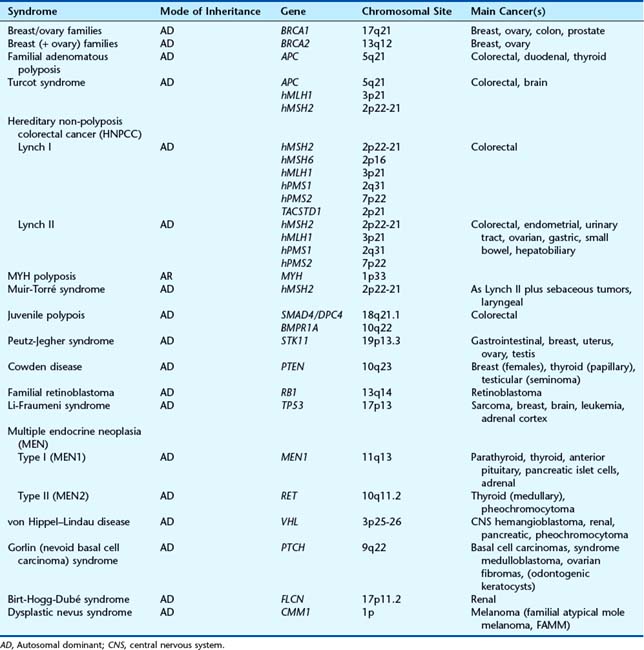

A number of different familial cancer-predisposing syndromes have been described, depending on the patterns of cancer occurring in a family. For example, persons with the Li-Fraumeni syndrome (p. 218) are at risk of developing adrenocortical tumors, soft-tissue sarcomas, breast cancer, brain tumors, and leukemia—sometimes at a strikingly young age. The cancer-predisposing syndrome HNPCC, also known as Lynch type I (site-specific colorectal cancer) and Lynch type II (confusingly, once known as the cancer family syndrome), in which family members are also at risk for a number of other cancers, including stomach, endometrial, breast, and renal transitional cell carcinomas. Progress at the molecular level (see Table 14.4) has highlighted the difficulty of classifying two types of Lynch syndrome—but despite this there is now a preference to return to this name. Further confusion can arise in consideration of Turcot syndrome, which is due to mutations in the APC gene and two of the mismatch repair genes, whereas Muir-Torré syndrome results from mutations in the hMSH2 mismatch repair gene. Individuals at risk in such families should, however, be screened for the appropriate cancers.
Inherited Susceptibility for the Common Cancers
The majority of persons at an increased risk of developing cancer because of their family history do not have one of the cancer-predisposing syndromes. The level of risk for persons with a family history of one of the common cancers such as bowel or breast cancer depends on several factors. These include the number of persons with cancer in the family, how closely related the person at risk is to the affected individuals, and the age at which the affected family member(s) developed cancer. A few families with a large number of members affected with one of the common cancers are consistent with a dominantly inherited cancer susceptibility gene. In most instances, there are only a few individuals with cancer in a family, and there is doubt about whether a cancer susceptibility gene is responsible or not. In such an instance, one relies on empirical data gained from epidemiological studies to provide risk estimates (Tables 14.6 and 14.7). With respect to mainly breast and ovarian cancers, in recent years the Manchester Scoring System (Table 14.8) has gained acceptance as a method of determining the likelihood of identifying a BRCA1 or BRCA2 mutation based on family history information. The derived score discriminates the likelihood of finding a mutation in one of these genes, and this provides a very useful clinical guide to genetic testing, which in many centers is set at a threshold of approximately 20%.
Table 14.6 Lifetime Risk of Colorectal Cancer for an Individual According to the Family History of Colorectal Cancer
| Population risk | 1 in 50 |
| One first-degree relative affected | 1 in 17 |
| One first-degree relative and one second-degree relative affected | 1 in 12 |
| One relative younger than age 45 years affected | 1 in 10 |
| Two first-degree relatives affected | 1 in 6 |
| Three or more first-degree relatives affected | 1 in 2 |
Data from Houlston RS, Murday V, Harocopos C, Williams CB, Slack J 1990 Screening and genetic counselling for relatives of patients with colorectal cancer in a family screening clinic. Br Med J 301:366–368.
Table 14.7 Lifetime Risk of Breast Cancer in Females According to the Family History of Breast Cancer
| Population risk | 1 in 10 |
| Sister diagnosed at 65–70 years of age | 1 in 8 |
| Sister diagnosed younger than age 40 years | 1 in 4 |
| Two first-degree relatives affected younger than age 40 years | 1 in 3 |
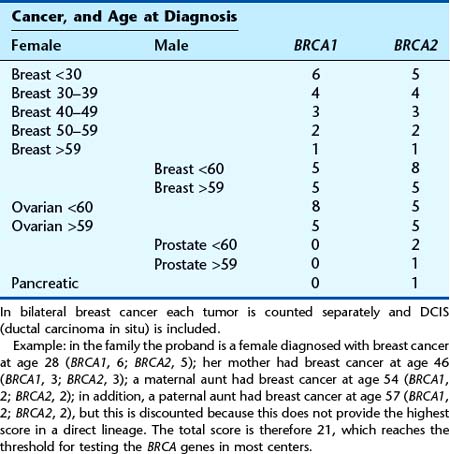
Screening for Familial Cancer
Screening of those at risk of familial cancer is usually directed at detecting the phenotypic expression of the genotype (i.e., surveillance for a particular cancer or its precursor). Screening can also include diagnostic tests that indirectly reveal the genotype, looking for other clinical features that are evidence of the presence or absence of the gene. For example, individuals at risk for FAP can be screened for evidence of the APC gene by retinal examination looking for areas of congenital hypertrophy of the retinal pigment epithelium, or what is known as CHRPEs. The finding of CHRPEs increases the likelihood of an individual at risk being heterozygous for the APC gene and therefore developing polyposis and malignancy. We now know that CHRPEs are seen in persons with FAP when mutations occur in the first part of the APC gene, an example of a genotype–phenotype correlation (p. 26).
More recently, identification of the gene responsible for a number of the cancer-predisposing syndromes, and determination of the genotypic status (i.e., presymptomatic testing, see p. 316), of an individual at risk allows more efficient delivery of surveillance screening for the phenotypic expression—e.g., renal cancer, central nervous system tumors and pheochromocytomas in von Hippel–Lindau disease (Table 14.9). For those who test negative for the family mutation, expensive and time-consuming screening is unnecessary. As more genes for cancer susceptibility are discovered, there will be an increasing number of conditions for which DNA testing will enable presymptomatic determination of genotypic status.
Table 14.9 Suggested Screening Guidelines for Persons at Significant Risk of Cancer: Familial Cancer-Predisposing Syndromes and Common Cancers
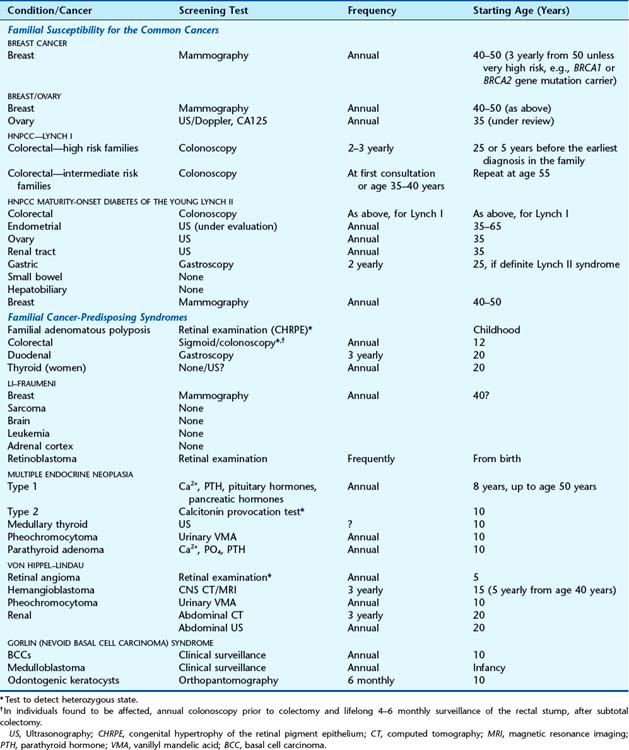
Familial Cancer-Predisposing Syndromes
Many familial cancer-predisposing syndromes are inherited as autosomal dominant traits that are fully penetrant, with the consequent risk for heterozygotes of developing cancer approaching 100%. This level of risk means that more invasive means of screening with more frequent and earlier initiation of screening protocols are justified than would be acceptable for the population in general (Table 14.10).
Table 14.10 Conditions in which Prophylactic Surgery is an Accepted Treatment, and Treatments that are under Evaluation, as an Option for the Familial Cancer-Predisposing Syndromes or Individuals at Increased Risk for the Common Cancers
| Disorder | Treatment |
|---|---|
| Accepted Treatment | |
| Familial adenomatous polyposis Ovarian cancer families Breast cancer families MEN2 |
Total colectomy Oophorectomy Bilateral mastectomy Total thyroidectomy |
| Under Evaluation | |
| Familial adenomatous polyposis | Non-digestible starch—to delay onset of polyposis Sulindac—to reduce rectal and duodenal adenomas |
| Breast cancer families | Tamoxifen—to prevent development of breast cancer Avoidance of oral contraceptives and hormone replacement therapy |
Inherited Susceptibility for the Common Cancers
Screening for the common cancers arising from inherited susceptibility has only relatively recently been established and by its very nature involves a long-term undertaking for the individual at risk as well as his or her physician or surgeon. It is important to emphasize that the natural enthusiasm for screening needs to be balanced with the paucity of hard data in many instances on the relative benefits and risks. However, recommended screening protocols are increasingly evidence based as more data become available (Box 14.2).
Box 14.2
Requirements of a Screening Test for Persons at Risk for a Familial Cancer-Predisposing Syndrome or at Increased Risk for the Common Cancers
What Sites Should Be Screened?
Familial cancer-predisposing syndromes
This can be a very difficult problem with some of the family cancer syndromes, such as the Lynch type II form of HNPCC, in which a person at risk can develop cancer at a number of different sites. Screening for every possible cancer that can occur would mean frequent investigation by a variety of different specialists and/or investigations. This would result in an unwieldy and unpleasant protocol. People at risk for HNPCC should have regular colonoscopy, and females may be offered pelvic screening for gynecological malignancies, although the efficacy is very debatable. Some of the other cancers that can occur in those at risk for Lynch type II, such as stomach cancer, are not seen in every family, and so screening is usually restricted to people from those families in which these cancers have affected at least one family member. In those at risk for Li-Fraumeni syndrome, a wide spectrum of cancers can occur. However, apart from regular mammography, no satisfactory screening is available for the other malignancies (see Table 14.9).
Inherited Susceptibility for the Common Cancers
Colorectal cancer
Breast cancer
It is also argued that the radiation exposure associated with annual mammography could be detrimental if started at an early age, leading to an increased risk of breast cancer through screening when carried out over a long period. This is of particular concern in families with Li-Fraumeni syndrome, because mutations in the TP53 gene have been shown experimentally in vitro to impair the repair of DNA damaged by X-irradiation. However, most experts believe that there is a greater relative benefit than risk in identifying and treating breast cancer in women from this high-risk group, although formal evaluation of such screening programs continues.
Ovarian cancer
Measuring the levels of CA125, an antigenic determinant of a glycoprotein that is present in increased levels in the blood of women with ovarian cancer, can be also be used as a screening test for women at increased risk of developing ovarian cancer. CA125 levels are not specific to ovarian cancer, as they are also increased in women with a number of other disorders, such as endometriosis. In addition, there are problems with sensitivity (p. 319), because CA125 levels are not necessarily increased in all women with ovarian cancer. Because of the problems outlined with these various screening modalities, many women with an increased risk of developing ovarian cancer choose to have their ovaries removed prophylactically after their family is complete. However, this in turn raises the issue of the benefits and risks associated with taking hormone replacement therapy.
What Treatment Is Appropriate?
Surgical intervention is the treatment of choice for persons at risk for some of the familial cancer-predisposing syndromes—e.g., prophylactic thyroidectomy in MEN type 2 (especially MEN2B) or colectomy in FAP. For those with a high risk from an inherited susceptibility for one of the common cancers (e.g., colon or breast/ovary), prophylactic surgery is also an accepted option, but the decision is more complex and dependent on the individual patient’s choice. The option of prophylactic mastectomy in women at high risk of developing breast cancer is very appealing to some patients but totally abhorrent to others, and alternative management in the form of frequent surveillance, and possibly drugs such as the anti-estrogen tamoxifen, can be offered. For patients at high risk of colonic cancer, dietary modification such as the use of non-digestible starch, or the use of drugs such as the aspirin-like non-steroidal anti-inflammatory sulindac, may have value (see Table 14.10).
Those at an increased risk of developing cancer, especially if it is one of the single-gene dominantly inherited cancer-predisposing syndromes, or one of the single-gene causes of the common cancers, find themselves in an unenviable situation concerning both their health and the possibility of transmitting the condition to their children. Unfortunately, they are also likely in future to experience increasing difficulties in other areas of life, such as insurance and employment (p. 366).
Cowell JK, editor. Molecular genetics of cancer. Oxford: Bios Scientific, 1995.
A multiauthor text covering the cancer family syndromes and the common cancers.
Eeles RA, Ponder BAJ, Easton DF, Horwich A, editors. Genetic predisposition to cancer. London: Chapman & Hall, 1996.
Harris H, Miller OJ, Klein G, Worst P, Tachibam T. Suppression of malignancy by cell fusion. Nature. 1969;350:377-378.
Studies that eventually led to the concept of tumor suppressor genes.
Hodgson SV, Maher ER. A practical guide to human cancer genetics, 3rd ed. Cambridge: Cambridge University Press; 2007.
An up-to-date second edition of this text covering the developing field of human cancer genetics.
King RA, Rotter JI, Motulsky AG, editors. The genetic basis of common diseases. Oxford: Oxford University Press, 1992.
Six chapters of this text cover the basic biology, epidemiology, and familial aspects of cancer.
Knudson AG. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci. 1971;68:820-823.
Proposal of the ‘two-hit’ hypothesis for the development of retinoblastoma.
Lalloo F, Kerr B, Friedman J, Evans G. Risk assessment and management in cancer genetics. Oxford: Oxford University Press; 2005.
Li FP, Fraumeni JF. Soft tissue sarcomas, breast cancer, and other neoplasms: a familial syndrome? Ann Intern Med. 1969;71:747-752.
The original description of the Li-Fraumeni syndrome.
Lynch HT. ‘Cancer families’: adenocarcinomas (endometrial and colon carcinoma) and multiple primary malignant neoplasms. Recent Results Cancer Res. 1967;12:125-142.
The description of the cancer family syndrome now known as Lynch II.
Offit K. Clinical cancer genetics. Chichester: Wiley-Liss; 1998.
Volgelstein B, Kinzler KW. The genetic basis of human cancer. London: McGraw-Hill; 2002.
Elements