Chapter 52E Cancer and the Nervous System
Management of Primary Nervous System Tumors in Infants and Children
Pediatric Primary Nervous System Tumors
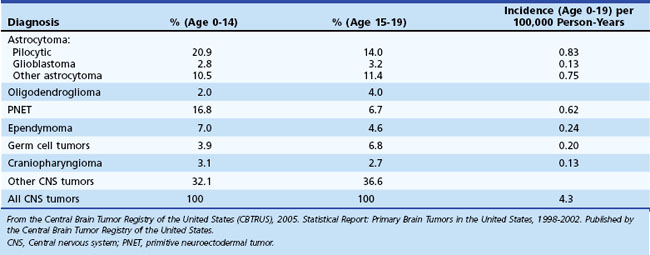
Primary brain tumors account for nearly 20% of all malignancies during childhood and adolescence in the United States (Ries et al., 1999). These tumors are second only to leukemia in frequency among all childhood cancers and are the most common solid tumor in children (Ries et al., 1999; Swensen and Bushhouse, 1998). The location, histological features, prognosis, and treatment of pediatric brain tumors are different from those of adult brain tumors and vary significantly according to age within the pediatric population (Table 52E.1). The location of approximately 85% of primary brain tumors in children aged 2 to 12 years is the posterior fossa; supratentorial brain tumors are more common in adolescents and children younger than age 2. Astrocytomas, the majority of which are low-grade, account for approximately half of pediatric central nervous system (CNS) tumors. Supratentorial high-grade astrocytomas represent only 6% to 12% of all primary pediatric brain tumors, and diffuse intrinsic brainstem gliomas represent 10%. Primitive neuroectodermal tumors (PNETs) (14.5%), other gliomas (15%), ependymomas (6.5%), germ cell tumors (4.6%), and craniopharyngiomas (3%) account for most of the other types (Central Brain Tumor Registry of the United States [CBTRUS], 2005). Despite significant improvements in prognosis for many pediatric cancers in recent decades, CNS malignancies remain a major cause of morbidity and mortality. However, the pace of both clinical and laboratory research has accelerated over the past decade, and management of this diverse group of tumors is undergoing a transformation owing to a new understanding of brain tumor biology and genetics on a molecular level.
Embryonal Tumors
Primitive Neuroectodermal Tumor
Background
Primitive neuroectodermal tumors, or embryonal CNS tumors, are the most common group of malignant brain tumors in children and include medulloblastomas, supratentorial PNETs, pineoblastomas, and atypical teratoid/rhabdoid tumors. There have been several changes to the categorization of embryonal tumors in the 2007 revised WHO classification of CNS tumors (Brat et al., 2008). Four distinct variants of medulloblastoma are now defined: large cell medulloblastoma, anaplastic medulloblastoma, medulloblastoma with extensive nodularity, and desmoplastic/nodular medulloblastoma. In addition, supratentorial PNETs, ependymoblastomas, and medulloepitheliomas have been consolidated under the same designation, CNS PNET. Medulloblastoma represents approximately 85% of intracranial PNETs and 15% of all pediatric brain tumors. The incidence in males is twice that of females, and the median age at diagnosis is 5 to 7 years. Taken as a group, 80% of all pediatric intracranial PNETs are diagnosed before age 10. According to the incidence data generated by the National Cancer Institute’s Surveillance, Epidemiology and End Results (SEER) registry, the incidence of primitive neuroectodermal tumors has remained fairly stable over the past 20 years (Linabery and Ross, 2008).
Etiology
Although histological similarity unites the various PNET subtypes, laboratory-based studies have identified a diverse and complex range of molecular biological subtypes of PNET. The regulation of signaling pathways that control brain development is abnormal in medulloblastoma and supratentorial PNET. In addition, the use of DNA microarray analysis indicates clear molecular distinctions between different embryonal CNS tumors (Pomeroy et al., 2002). This allows classification based on gene expression patterns, a more detailed understanding of the cellular origin of the various tumors in this class, and better prediction of outcome (Gilbertson and Ellison, 2008; Rossi et al., 2008).
Original reports by Bailey and Cushing in 1925 suggested that medulloblastomas originated from multipotential “medulloblasts” thought to be capable of generating both glial and neuronal cells. Experimental evidence now indicates that the two main histopathological subtypes of medulloblastoma, desmoplastic and nondesmoplastic (formerly “classic”), originate from two distinct germinal zones within the cerebellum—that is, the external granular layer, which contains committed granule cell precursors, and the ventricular zone, which contains multipotential stem cells that give rise to the majority of cerebellar neurons (Gilbertson and Ellison, 2008; Rossi et al., 2008). Whereas desmoplastic medulloblastoma tends to express markers of granule cell lineage, suggesting an origin from granule cell precursors, nondesmoplastic medulloblastoma more frequently expresses markers associated with nongranule neurons, suggesting an origin in the ventricular zone of the developing cerebellum (Read et al., 2006). Gene expression analysis supports these data by demonstrating separate expression profiles, with desmoplastic medulloblastoma cells expressing genes more closely associated with proliferating granule cell precursors, and the expression profiles of nondesmoplastic medulloblastoma revealing a more heterogeneous set of markers not clearly associated with any particular cerebellar cell type (Ehrbrecht et al., 2006; Northcott et al., 2010).
There is limited understanding of the etiology of the other embryonal CNS tumors. Supratentorial PNETs originate from multipotential cells that have the capacity for differentiation along multiple lineages (Li et al., 2005). However, specific markers for supratentorial PNETs have not yet been defined, making further histopathological classification difficult. Among the less common embryonal CNS tumors, medulloepitheliomas appear to be derived from primitive cells in the subependymal layer, whereas ependymoblastomas presumably arise from periventricular neuroepithelial precursor cells. There are no convincing data to support a dietary or environmental etiology of PNETs from various epidemiological studies. However, a genetic predisposition for the development of medulloblastoma, supratentorial PNET, and pineoblastoma exists in rare cancer-susceptibility syndromes such as Turcot syndrome, nevoid basal cell carcinoma syndrome (Gorlin syndrome), and Li-Fraumeni syndrome (Attard et al., 2007; Hottinger and Khakoo, 2009). These syndromes, along with observations from the genomic profiling of pediatric brain tumors, have shed light on the molecular pathways involved in tumorigenesis. Several groups have independently used this information to separate medulloblastomas into four or five different categories (Kool et al., 2008; Northcott et al., 2011; Thompson et al., 2006). These categories include medulloblastomas resulting from dysregulation along the wingless (WNT) pathway, abnormal sonic hedgehog (SHH) pathway signaling, and two or three additional subsets that are less well characterized.
The clinical manifestations of Turcot syndrome include the development of colon cancer and brain tumors. This syndrome has been further classified into type 1 and type 2. Patients with Turcot syndrome type 1 have mutations in the mismatch repair gene, bMLH1, which leads to the formation of gliomas. Those with type 2 disease have germline mutations in the adenomatous polyposis coli (APC) gene that leads to development of medulloblastomas through the WNT pathway (Attard et al., 2007). Patients with sporadic medulloblastomas have been found to have mutations in several genes involved in the wingless pathway, including inactivation of the tumor suppressor genes, APC (3%) and AXIN (2%), and/or activating mutations in CTNNB1 gene (8%) (Baeza et al., 2003; Yokoto et al., 2002).
Nevoid basal cell carcinoma syndrome, also known as Gorlin syndrome, is an autosomal dominant disorder characterized by the development of basal cell carcinomas, multiple bony cysts, and malignant tumors including medulloblastomas in young children (Kimonis et al., 1997). Patients with this disorder have mutations involving the ptc1 (Patched 1) gene (Hahn et al., 1996). This gene encodes for the protein which is the receptor of SHH. The SHH pathway is critically involved in the normal development of the cerebellum (Lewis et al., 2004). Approximately 10% to 20% of patients with sporadic medulloblastoma have mutations involving the SHH pathway. The ptc1 gene is the most commonly affected gene within this pathway, but mutations in other genes along this pathway including smoothened, fused, and suppressor of fused (SUFU) have also been identified (Raffel et al., 1997; Taylor et al., 2002; Zurawel et al., 2000). Although mutations in the WNT and SHH pathways are important in the development of medulloblastoma, they account for only 30% to 40% of the sporadic cases of medulloblastoma. Abnormal signaling along other pathways, such as Notch, has also been implicated in the development of medulloblastomas (Lasky and Wu, 2005).
Trilateral retinoblastoma is a well-recognized syndrome characterized by bilateral retinoblastomas occurring concurrently with a pineoblastoma with retinoblastic features (Finelli et al., 1995). Although approximately half of the cases of trilateral retinoblastoma are associated with the familial form of retinoblastoma, one analysis indicates that most children with trilateral retinoblastoma have ordinary hereditary retinoblastoma that is complicated by trilateral disease by chance, thus dispelling the notion that trilateral retinoblastoma is caused by a different allele than that which causes ordinary retinoblastoma (Kivela, 1999).
Although reports of supratentorial PNET and medulloblastomas have occurred in patients with the Li-Fraumeni syndrome (mutations in the TP53 gene on chromosome 17), no compelling evidence exists to suggest that TP53 mutation alone is sufficient to promote development of a PNET (Weber et al., 1998). In addition, isolated TP53 mutations appear to be less common in sporadic medulloblastoma than would be expected (Burns et al., 2002; Portwine et al., 2001). However, evidence exists that alterations in p53 function may influence the effect of PTCH1 mutations in promoting the growth of medulloblastoma (Wetmore et al., 2001). Although most medulloblastomas are sporadic, familial cases unrelated to the mentioned syndromes have also been reported (von Koch et al., 2002).
Diagnosis
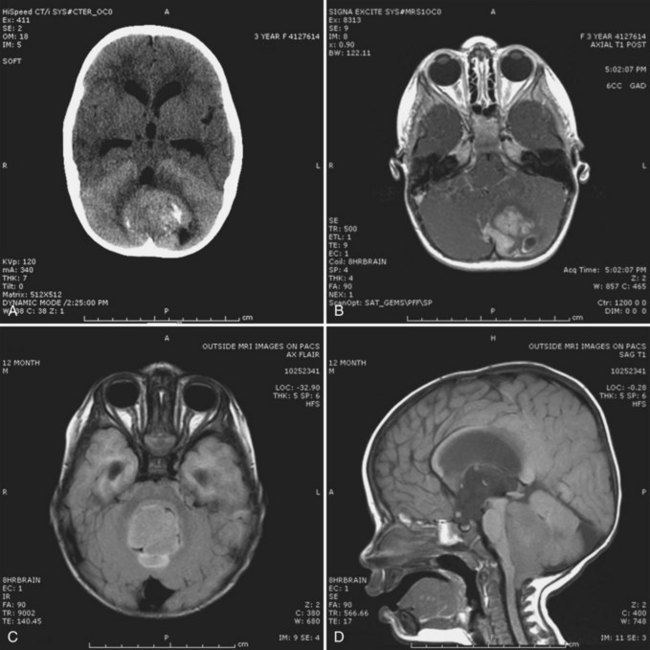
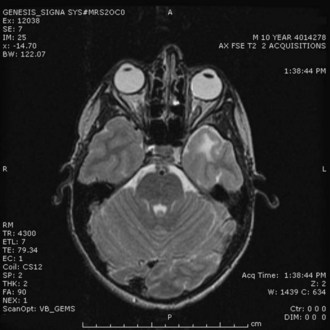
A high level of clinical suspicion is critical to make an early diagnosis of PNET. Neuroimaging is usually the first step, with computed tomography (CT) scan of the brain frequently obtained in the acute setting. Contrast-enhanced cranial MRI provides definitive imaging of the tumor. Certain MRI features may help distinguish the various types of posterior fossa tumors (medulloblastoma, ependymoma and pilocytic astrocytoma) of childhood. Although no single radiological feature is pathognomonic of a medulloblastoma, certain common characteristics exist. On CT, a medulloblastoma is generally hyperdense and homogeneously enhancing, filling the fourth ventricle (Fig. 52E.1, A). On contrast-enhanced cranial MRI, medulloblastomas are often isointense or hypointense to surrounding normal brain on T1-weighted images (see Fig. 52E.1, B). The uniform hypercellularity of most medulloblastomas typically results in a relatively homogeneous image appearance. On T2-weighted images, medulloblastoma can appear to be hyperintense or more frequently can display mixed signal characteristics indicative of small intratumoral cysts, calcification, or small areas of hemorrhage (see Fig. 52E.1, C). Because medulloblastoma typically arises in the roof of the fourth ventricle, a cleft of cerebrospinal fluid (CSF) beneath the tumor in the fourth ventricular canal helps distinguish this tumor from an ependymoma, which typically arises from the floor of the fourth ventricle. Ependymomas can fill the fourth ventricle and calcify more frequently than medulloblastomas. In addition, ependymomas often contain cysts, making their overall image appearance more heterogeneous. Because these tumors typically arise near the obex, ependymomas frequently extend out the foramen of Magendie over the dorsal surface of the cervical spinal cord or through the foramen of Luschka. Pilocytic astrocytomas typically arise in the cerebellar hemispheres and have the appearance of a cystic mass with an enhancing mural nodule. On T2-weighted MRI, these tumors often appear as areas of homogenous high signal intensity, with the fluid collections defining the less intense tissue components of the tumor (see Fig. 52E.1, D).
Management
In children in whom the diagnosis of intracranial PNET is suspected, a contrast-enhanced spinal MRI should be obtained at the time of diagnosis to assist in neurosurgical planning and staging (Cohen et al., 1995). After radiological diagnosis, corticosteroids are frequently used to control increased ICP. Most often the child will require admission for observation and pediatric neurosurgical consultation. If the child has unstable mental status or vital signs, emergency external ventricular drainage may be required before surgery. The goals of surgery are to control ICP, achieve a gross total resection (if feasible), establish a histological diagnosis, and bank frozen tumor for molecular analysis prior to protocol therapy. Placement of a ventriculoperitoneal (VP) shunt before surgery is no longer commonly practiced but may be necessary after surgery. A lumbar puncture for CSF cytological evaluation should only be performed after ICP has been relieved after surgery. Potential complications of posterior fossa surgery include cerebellar mutism and aseptic meningitis. The posterior fossa, or cerebellar mutism, syndrome may occur in as many as 40% of children undergoing posterior fossa surgery (Wells et al., 2010). The characteristics include reduced speech output or mutism, personality changes, hypotonia, ataxia, and reduced oral intake. Symptoms typically appear 1 to 2 days after surgery and may last for a few months, with varying degrees of recovery (Catsman-Berrevoets and Aarsen, 2010).
Risk stratification of PNET is currently based on clinical staging and certain neuropathological features, although molecular observations will most probably be incorporated into the next generation of clinical trials. Clinical staging follows the completion of perioperative brain and spine MRI and lumbar CSF cytological analysis. The M (metastasis) staging criteria include: M0, no metastases; M1, positive CSF cytology alone; M2, intracranial metastases; M3, spinal metastases; and M4, systemic metastases. The major determinants of clinical risk categorization are age at diagnosis, metastasis (M stage), primary tumor site, and volume of residual postoperative disease. In addition, the presence of large-cell anaplasia has recently emerged as a risk factor (Von Hoff et al., 2010). Distinction is made between age younger than 3 years versus older than 3 years, stage M0 versus stages higher than M0, cerebellar versus non-cerebellar primary tumor, postoperative residual tumor volume of less than 1.5 cm2 versus greater than 1.5 cm2, and presence or absence of large-cell anaplasia (Von Hoff et al., 2010; Zeltzer et al., 1999).
Patients are currently stratified into either standard-risk or high-risk prognostic groups. Standard-risk patients are those who are older than age 3 at diagnosis, have residual tumor volume of less than 1.5 cm2, have a primary tumor in the cerebellum, have no evidence of metastasis (M0), and do not have anaplastic medulloblastoma. Children younger than age 3 are always at high risk because of higher rates of recurrence (McNeil et al., 2002). In addition, patients with residual tumor volume greater than 1.5 cm2, any metastatic disease, or large-cell/anaplastic medulloblastoma are at high risk for recurrence. Treatment of intracranial PNETs usually consists of surgery, radiation therapy, and chemotherapy, with specific therapies guided primarily by age and risk stratification. The current recommended therapy for standard-risk patients includes craniospinal irradiation (23.4 Gy) with a boost to the primary tumor site to 54 Gy. Vincristine is usually administered weekly with radiation therapy. Following radiation therapy, adjuvant chemotherapy is given, with the most common regimen consisting of 8 cycles of cisplatin, lomustine, and vincristine (Packer et al., 2006). Current research in this group of patients consists of identifying adjuvant chemotherapy with less ototoxicity than cisplatin and less myelosuppression than lomustine.
In addition to developing effective chemotherapy regimens with less toxicity, several groups are trying to decrease the toxicity associated with radiation therapy. The use of proton-beam radiation therapy as an alternative to high-energy x-rays (photons) has been studied and found to have the potential to limit late effects of radiation therapy by reducing the exposure of normal tissue to radiation (Durante and Loeffler, 2010). The basis for the interest in proton irradiation in the treatment of pediatric malignancies is the differences between charged particle beams (photons) and proton beams. Standard photon (x-ray) beams deliver a maximum radiation dose near the surface, followed by a continuously reducing dose with increasing depth. As a result, tissues outside the target area receive an exit dose of the radiation, which can lead to significant morbidity in patients receiving craniospinal irradiation. In contrast, as protons move through tissue, they ionize particles and deposit a radiation dose along their path. The maximum dose, called the Bragg peak, occurs shortly before the point of greatest tissue penetration, which is dependent on the energy of the proton beam. Because the energy is precisely controlled, placement of the Bragg peak within the tumor and tissues targeted to receive the radiation dose is possible. Because the protons are absorbed at this point, normal tissues beyond the target receive very little radiation (MacDonald et al., 2006). Thus, the use of proton radiation can spare adjacent critical structures such as the optic apparatus, cochlea, and hypothalamus when they are not adjacent to tumor volume, potentially resulting in fewer long-term sequelae (Merchant et al., 2008b).
All newly diagnosed patients younger than 3 years of age at diagnosis are classified as high risk. The frequency of leptomeningeal dissemination is also increased at the time of diagnosis in young children (27%-43%) versus older children (20%-25%) with similar histological diagnoses (Heideman et al., 1997), and there is a higher incidence of aggressive tumor variants such as atypical teratoid/rhabdoid tumor (Hilden, 2004). After surgical resection, chemotherapy—alone or with involved-field radiation therapy—often follows in an effort to reduce the high incidence of developmental and neuropsychological sequelae in young children and infants treated with craniospinal irradiation. The risks of radiation therapy in infants and young children are significantly greater than the benefits of therapeutic response in terms of neurocognitive development. Consequently, several chemotherapy regimens designed to delay or eliminate the need for craniospinal irradiation are undergoing evaluation, including regimens employing intraventricular chemotherapy (Rutkowski et al., 2005) and high-dose chemotherapy with autologous stem cell rescue (Dhall et al., 2008; Fangusaro et al., 2008). Several different approaches are being used to try to improve survival in older children with high-risk disease (M+, >1.5 cm2 residual tumor volume, anaplasia, supratentorial location). These include the use of autologous hematopoietic progenitor cells to shorten the interval between chemotherapy cycles, as well as the addition of high-dose methotrexate (Chi et al., 2004; Gajjar et al., 2006). The Children’s Oncology Group is currently studying the use of carboplatin as a radiosensitizer, as well as 13-cis-retinoic acid during and following adjuvant chemotherapy.
Prognosis
Standard-risk medulloblastoma patients have a 5-year survival of approximately 80%; 5-year survival in high-risk medulloblastoma patients is approximately 30% to 40% (Oyharcabal-Bourden et al., 2005; Packer et al., 2006). The presence of CNS dissemination is the single most important factor that correlates with outcome (Helton et al., 2002). Most initial recurrences occur at the primary site. Salvage therapy is rarely curative, but long-term disease control may occur with high-dose myeloablative chemotherapy (Dunkel et al., 2010; Gardner, 2004). Lifelong serial surveillance imaging is required in anticipation of radiation-induced secondary malignancies such as meningioma or high-grade glioma (Kantar et al., 2004). Radiation therapy, particularly in young children, can also cause significant adverse late effects in cognitive development, growth, and endocrine function (Mulhern et al., 1999; 2005).
Atypical Teratoid/Rhabdoid Tumor
Atypical teratoid/rhabdoid tumor is an uncommon highly malignant tumor that was initially described in the mid 1980s (Briner et al., 1985). These tumors may arise anywhere in the body but are most common in the kidney where they are referred to as malignant rhabdoid tumors and in the CNS where they are classified as atypical teratoid/rhabdoid tumors (AT/RT). AT/RT occur primarily in the posterior fossa, either in isolation or in association with multiple prior tumors in other parts of the body such as the kidneys.
Owing to similar morphology, these tumors may be confused with primitive neuroectodermal tumors and choroid plexus carcinomas. They are often quite heterogeneous, with areas of primitive neuroectodermal, epithelial, and mesenchymal cells in addition to classic rhabdoid cells. However, AT/RT has recently been recognized as a distinct pathological entity. Unique molecular findings support this classification, including deletions or translocations at 22q11.2, the genetic site for the tumor-suppressor gene SMARCB1/hSNF5/INI1, and the identification of germline and somatic mutations of INI1 in approximately 75% of patients with CNS AT/RT (Biegel et al., 2002; Roberts and Biegel, 2009). AT/RTs are seen primarily in infants and young children (Bambakidis et al., 2002), with a peak incidence between birth and age 3. AT/RTs account for approximately 1% to 2% of all childhood brain tumors, but these neoplasms represent nearly 10% of CNS tumors in infants (Biegel et al., 2006). Children with AT/RT often present with signs and symptoms similar to those with medulloblastoma or PNET, including vomiting, loss of milestones, irritability, and increasing head circumference.
Despite the use of multimodality therapies, the prognosis for patients with AT/RT remains poor. Treatments have included various combinations of surgery, chemotherapy, and irradiation (Hilden et al., 2004). The role of each of these modalities is unclear, but a few long-term survivors have been reported with the use of high-dose chemotherapy, intrathecal chemotherapy, and treatments based upon protocols used in patients with rhabdomyosarcoma with parameningeal extension (Athale et al., 2009; Chi et al., 2009; Finkelstein-Shechter et al., 2010; Gardner et al., 2008; Olson et al., 1995). The Children’s Oncology Group is currently studying the use of multidrug chemotherapy including high-dose methotrexate, high-dose chemotherapy with autologous stem cell rescue, and radiation therapy.
Astrocytic Tumors
The spectrum of astrocytic tumors is broad and includes a wide range of glial neoplasms that differ in anatomical location, morphological features, degree of invasiveness, and clinical course. Grading of astrocytomas by the World Health Organization (WHO) is predictive of patient survival (Luis et al., 2007) (Table 52E.2). Astrocytomas can be classified as low-grade (WHO grade I and II) or high-grade (WHO grade III and IV). The most common pediatric subtypes recognized by the current WHO classification will be discussed.
Table 52E.2 WHO Classification and Grading of Low-Grade and High-Grade Glial and/or Neuronal Tumors
| Astrocytic Tumors | Oligodendroglial and Oligoastrocytic Tumors | Neuronal and Mixed Neuronal-Glial Tumors |
|---|---|---|
| Pilocytic astrocytoma (I) | Oligodendroglioma (II) | Gangliocytoma (I) |
| Subependymal giant cell astrocytoma (II) | Anaplastic oligodendroglioma (III) | Ganglioglioma (I) |
| Pilomyxoid astrocytoma (II) | Oligoastrocytoma (II) | Anaplastic ganglioglioma (III) |
| Diffuse astrocytoma (II) Pleomorphic xanthoastrocytoma (II) |
Anaplastic oligoastrocytoma (III) | Desmoplastic infantile astrocytoma and ganglioglioma (I) |
| Anaplastic Astrocytoma (III) Glioblastoma (IV) |
Dysembryoplastic neuroepithelial tumor (I) | |
| Central neurocytoma (II) |
Adapted from Luis, D.N. Ohgaki, H. Wiestler, O.D., et al. (Eds.), 2007. WHO Classification of Tumors of the Central Nervous System, fourth ed. International Agency for Research on Cancer, Lyon, France.
Pilocytic Astrocytoma
Background
Pilocytic astrocytomas (PAs) are well-circumscribed, slow-growing tumors classified as WHO grade I (Luis et al., 2007). These tumors are the most common gliomas found in children and represent approximately 20% of all childhood brain tumors. Pilocytic astrocytomas are typically diagnosed in the first 2 decades, with no clear gender predilection. Neurofibromatosis type 1 (NF1) is associated with an increased risk of PA (Rodriguez and Berthrong, 1996). No other definite predisposing factors are known. Histologically, PAs consist of bipolar cells with long “fiberlike” processes, hence the name pilocytic. Other distinctive histological features of PAs include Rosenthal fibers, eosinophilic granular bodies, and microcysts. Constitutive activation of the mitogen-activated protein kinase (MAPK) signaling pathway through specific gene fusions and activating mutations has recently been identified in the majority of PAs, as well as other pediatric low-grade astrocytomas (Tatevossian et al., 2010), and may provide an opportunity for the future development of molecular targeted therapies.
Clinical Presentation
Pilocytic astrocytomas can arise anywhere in the CNS, but most commonly occur in the optic pathways, the optic chiasm/hypothalamus, thalamus and basal ganglia, cerebral hemispheres, cerebellum, midbrain, and medulla. Pilocytic astrocytomas of the spinal cord are less common. The spectrum of clinical manifestations depends on the structures involved and may include visual deficits, obstructive hydrocephalus, macrocephaly in younger patients, ataxia, endocrine dysfunction, focal neurological deficits, long-tract signs, cranial nerve dysfunction, and seizures if the tumor involves cortical structures. Cerebellar PAs usually present with symptoms indicative of increased ICP, such as headache, nausea, and vomiting. Similarly, because brainstem PAs are usually dorsal and exophytic, obstructive hydrocephalus is often the presenting feature. The diencephalic syndrome is unique to low-grade gliomas, usually juvenile PAs arising in infants in the hypothalamus or optic pathways, and consists of emaciation with normal linear growth, frequently accompanied by hyperemesis, hyperkinesis, and nystagmus (Fleischman et al., 2005).
Diagnosis
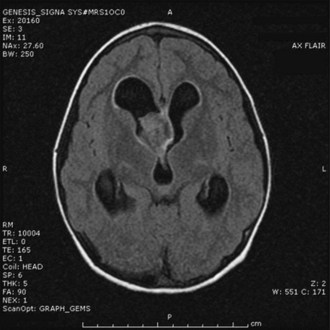
This diagnosis should be suspected when any of the mentioned features are observed in a young patient. The history of illness is usually long; other signs of chronicity such as bone remodeling, scoliosis, or hemihypotrophy may be present, depending on the primary tumor location. The typical MRI appearance is a homogeneously enhancing mass with only minimal associated edema. Intratumoral cysts are common depending on location—that is, cerebellum, cerebrum, midbrain, and spinal cord (Fig. 52E.2). Rarely, PAs present with diffuse leptomeningeal dissemination, especially the infant variant (pilomyxoid astrocytoma) (Komotar et al., 2005).
Management
Chemotherapy is assuming an increasingly important role in the management of diencephalic low-grade gliomas in younger patients and in children with unresectable and progressive tumors. Various regimens such as carboplatin and vincristine or thioguanine, procarbazine, lomustine, and vincristine (TPCV protocol), have produced consistent durable responses (Packer et al., 1997; Prados et al., 1997; Shaw and Wisoff, 2003). Both regimens were compared in a randomized phase III trial by the Children’s Oncology Group, and final study results are expected to be published in the near future. Temozolomide, an oral alkylating agent with a favorable side-effect profile, has been effective in uncontrolled studies as monotherapy in adult low-grade gliomas (Kesari et al., 2009; Quinn et al., 2003). Temozolomide also appears to be active in children with low-grade gliomas, achieving stable disease in 41% of patients in a Children’s Oncology Group study (Nicholson et al., 2007). Other regimens that have shown evidence of activity in small patient series include vinblastine (Lafay-Cousin et al., 2005) and irinotecan/bevacizumab (Packer et al., 2009). “Anti-angiogenic” regimens with oral medications given at a low dose on a daily “metronomic” schedule are also under investigation (Samuel et al., 2009). BRAF, which is constitutively active in the majority of PAs, has recently emerged as a novel, promising target for small-molecule inhibitors (Tatevossian et al., 2010). Because of concern for adverse effects including neurocognitive and endocrine effects as well as secondary malignancies, radiation therapy is usually deferred in children with low-grade gliomas, especially in those with diencephalic tumors and NF1. Additional late effects of radiation when used in younger children with diencephalic gliomas include strokes related to a moyamoya-like syndrome (Arita et al., 2003; Merchant et al., 2009, Serdaroglu et al., 2000).
Prognosis
The prognosis for resectable tumors is excellent, with 5-year recurrence-free survival of greater than 90% after gross total resection (Shaw and Wisoff, 2003). The most critical variable in the treatment of PAs is the anatomical location of the tumor. Complete resections are most difficult for tumors located in the brainstem, spinal cord, and hypothalamus. As such, the progression-free survival of children with centrally located tumors (e.g., optic chiasm, thalamus, hypothalamus) is reduced. The proliferative index of pediatric PAs does not appear to be associated with outcome (Horbinski et al, 2010). Given the risk of long-term sequelae of radiation therapy, especially in young children, chemotherapy is now usually preferred as the first therapeutic modality in most patients with progressive tumors not amenable to gross total resection (Sievert and Fisher, 2009). The use of chemotherapy as initial treatment in patients with centrally located or unresectable tumors allows for the delay of radiation therapy until the child is less likely to suffer major developmental and neuropsychological sequelae. Ultimately, the quality of survival depends on multiple factors including tumor location, extent of tumor resection, timing of any radiation therapy, and side effects of surgery, chemotherapy, and radiation. Malignant transformation of PA and other low-grade astrocytomas in children is rare (Broniscer et al., 2007).
Optic Pathway Glioma
Background
Optic pathway gliomas (OPG) may be considered a subset of PAs, but their unique features and management requirements warrant a separate discussion. OPG represent approximately 4% to 6% of all primary pediatric brain tumors. These tumors may involve various parts of the optic pathway, such as the optic nerves, optic chiasm, optic tract, and optic radiations. The tumor may also infiltrate the adjacent hypothalamus and temporal lobes. Optic nerve gliomas are strongly associated with NF1, and NF1 patients represent about 50% of patients treated for DPG. Although approximately 20% of NF1 children scanned prospectively from birth will acquire MRI abnormalities suggestive of an anterior or posterior OPG, less than half of those will develop progressive neurological and radiological disease, and the occasional tumor may undergo spontaneous regression (Parsa et al., 2001). OPGs in NF1 patients have been suggested to have a more indolent course than those arising in patients without NF1 (Deliganis et al., 1996). In a recent series however, progression-free survival for NF1 patients requiring treatment appeared similar to non-NF1 patients (Nicolin et al., 2009).
Clinical Presentation
Most OPGs are low-grade astrocytomas, primarily PAs (WHO grade I) (Cummings et al., 2000). Although most OPGs are of lower histological grade, the clinical course of these tumors may be aggressive when the optic pathways and hypothalamus are invaded. Age is an important prognostic factor: children younger than 5 years of age experience a more aggressive course. Unilateral optic nerve gliomas present with the classic triad of vision loss, proptosis, and optic atrophy. Chiasmatic involvement may lead to unilateral or bilateral vision loss, optic tract involvement may lead to a visual-field deficit, and large dorsally exophytic or hypothalamic components of the tumor may lead to obstructive hydrocephalus. Further invasion into brain parenchyma may result in visual-field defects and hemiparesis. The diencephalic syndrome is unique to infant presentations of OPG and hypothalamic PA and presents with irritability, failure to thrive, nystagmus, visual loss, and hydrocephalus in the first or second year of life (Fleischman et al., 2005).
Diagnosis and Management
The clinical diagnosis of an OPG should be suspected when a child presents with visual impairment associated with nystagmus and optic atrophy. It is very difficult to ascertain visual loss in younger children, but close behavioral observation during play may raise suspicion. Contrast-enhanced cranial or orbital MRI typically shows a solid, cystic, or solid and cystic tumor with enhancement (Fig. 52E.3). MRI studies and clinical presentation may distinguish an OPG from other childhood tumors that arise in the suprasellar location, such as a germ cell tumor or craniopharyngioma. The unpredictable clinical course of patients with OPGs has led to controversy regarding the optimal management of these tumors. The clinical course, age of onset, severity of symptoms, size and extent of the tumor, and the presence of NF1 may all impact management decisions. Treatment usually starts promptly in young patients, those with progressive symptoms, and those with extensive CNS involvement; preservation of vision is paramount. The initial treatment of choice is chemotherapy, which may cause stabilization or regression of the tumor (Silva et al., 2000). Combination therapy with carboplatin and vincristine (Kato et al., 1998; Packer et al., 1997) is most commonly used as first-line therapy, but the TPCV protocol (Lancaster et al., 2003) and other low-grade glioma protocols are also used. Although a definitive role for radiation therapy exists in the management of OPG, most favor a delay in initiating radiation in young patients (Jahraus and Tarbell, 2006). Recovery of visual deficits is unpredictable even if a radiographic tumor response is observed (Moreno et al., 2010).
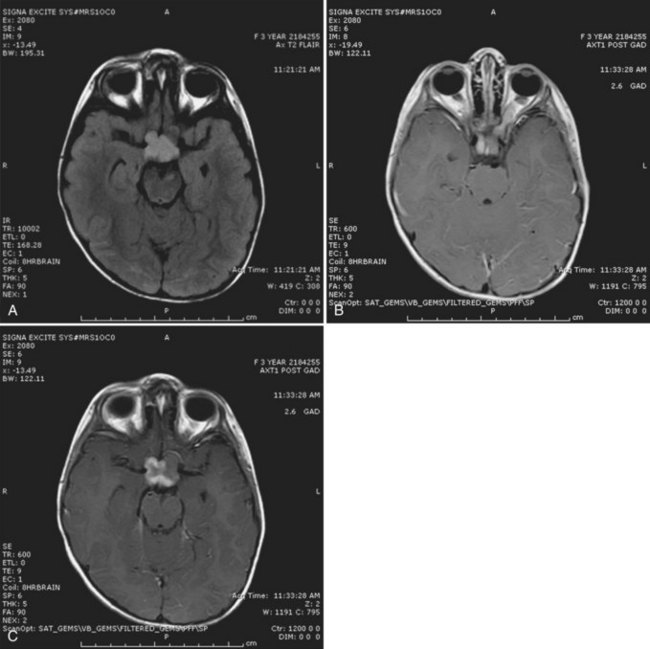
 -year-old girl with a history of neurofibromatosis type 1 (NF1) who was seen after an ophthalmological evaluation revealed a left eye deviation and visual-field loss. A, Magnetic resonance imaging (MRI) reveals T2 prolongation in the optic chiasm. B, There is significant enlargement of the left optic nerve. C, Coronal T1 postgadolinium imaging shows the lesion to be contrast enhancing. The combined findings were highly suggestive of an optic pathway glioma.
-year-old girl with a history of neurofibromatosis type 1 (NF1) who was seen after an ophthalmological evaluation revealed a left eye deviation and visual-field loss. A, Magnetic resonance imaging (MRI) reveals T2 prolongation in the optic chiasm. B, There is significant enlargement of the left optic nerve. C, Coronal T1 postgadolinium imaging shows the lesion to be contrast enhancing. The combined findings were highly suggestive of an optic pathway glioma.Prognosis
Although OPGs are usually low-grade tumors, the location of these neoplasms often results in serious morbidity. The growth rate of OPGs often slows in older children and young adults. The most robust adverse prognostic factor across multiple studies has been age younger than 1 year at diagnosis, although some suggest that children with NF1 may have a longer progression-free survival (Opocher et al., 2006). Patients with OPGs associated with NF1 may remain stable for several years. Close observation and symptomatic management are appropriate for this subpopulation.
Subependymal Giant Cell Astrocytoma
Background
Subependymal giant cell astrocytomas (SEGA) usually originate in the ependymal lining of the lateral ventricles and are associated almost exclusively with tuberous sclerosis (TS) (Kumar and Singh, 2004). Sporadic SEGAs are rare. TS is an autosomal dominant genetic syndrome caused by mutations in either TSC1 (hamartin) or TSC2 (tuberin), which are regulators of key cellular signaling pathways including the mammalian target of rapamycin (mTOR) pathway (Jozwiak et al., 2008).
Diagnosis and Management
Contrast-enhanced cranial CT and MRI are essential for early and accurate diagnosis. Whereas the former is better for detecting small calcified lesions, MRI is superior to CT in identifying areas of gliosis, heterotopia, and SEGA, which gives the typical radiographic appearance of candle dripping (Nabbout et al., 1999). SEGAs typically demonstrate diffuse contrast enhancement on both CT and MRI studies. Gross total resection is the treatment of choice for SEGAs that are large, progressive, and causing obstructive hydrocephalus. This can be accomplished by a transcallosal craniotomy and rarely after an endoscopic procedure. The main surgical risk is injury to the forniceal columns, resulting in memory disturbance. Alternatively, a VP shunt can provide symptomatic relief. The benign behavior of these tumors warrants re-operation in the case of recurrence or progression after subtotal resection. Pharmacological inhibition of mTOR with rapamycin is emerging as a promising alternative to surgery for these and other TS-related conditions (Franz et al., 2006). SEGA may respond dramatically to mTOR inhibitors, but the tumors will recur once mTOR inhibition is terminated. The long-term use of these inhibitors, which are also immunosuppressive, is unknown and will require further study.
Prognosis
Subependymal giant cell astrocytomas are usually benign tumors, and gross total resection may be curative. However, multiple tumors may arise in some patients with TS. The overall prognosis for patients with TS is good, despite increased susceptibility to other tumor types including rhabdomyomas of the myocardium and angiomyomas of the kidney, liver, adrenals, and pancreas (Crino et al., 2006).
Diffuse Astrocytoma
Background
Diffuse astrocytomas (DAs, WHO grade II) are a common variant of low-grade astrocytoma (LGA), composed primarily of fibrillary neoplastic astrocytes and distinct from PAs. The most typical histological subtype is also termed fibrillary astrocytoma. DAs represent 12% to 18% of all pediatric intracranial tumors. No gender predilection exists, and the peak age at diagnosis is 6 to 10 years. DAs may arise in any location within the CNS but are most commonly diagnosed in the frontal and temporal lobes. Brainstem and spinal cord are less common locations, and cerebellar DAs are rare. TP53 gene mutations are a genetic hallmark of DAs and can be found in more than 60% of tumors (Okamoto et al., 2004).
Clinical Presentation
Initial symptoms vary depending on the location of the tumor and may exist for months to years before a definitive diagnosis (Table 52E.3). DAs within the brainstem have a different clinical course compared to DAs in other locations and will be discussed under Diffuse Intrinsic Pontine Glioma. Brainstem DAs often undergo malignant transformation within months. This transformation is much less likely in other locations. Patients with medullary tumors may present with a long history of dysphagia, hoarseness, ataxia, and hemiparesis. Cervicomedullary tumors may cause medullary or upper cervical symptoms such as neck discomfort, weakness or numbness of the hands, and an asymmetrical quadriparesis. Patients with midbrain tumors such as a tectal glioma often present with signs and symptoms of increased ICP. Other symptoms include diplopia and hemiparesis.
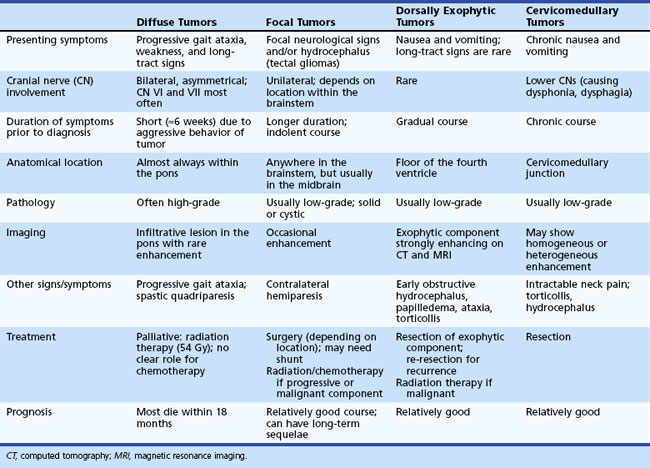
Diagnosis and Management
Most DAs appear isodense on CT scan, without significant contrast enhancement. The tumor is hypointense on T1-weighted and hyperintense on T2-weighted MRI, with minimal or no contrast enhancement with the exception of dorsally exophytic brainstem tumors (Fig. 52E.4). The risk of malignant transformation appears to be much higher in adults than in children, although the molecular abnormalities observed during transformation in children are similar to those observed in adult high-grade gliomas. These abnormalities include p53 overexpression, as well as RB1 and/or CDKN2A and PTEN deletions. Somatic mutations of the isocitrate dehydrogenase 1 (IDH1) gene, which is found in more than 80% of adult DAs (Yan et al., 2009), appears to be rare in pediatric DAs. Management of DAs depends on the clinical prodrome and location of the primary tumor (Ernestus et al., 1996). Rapidly evolving clinical symptoms in the setting of a resectable tumor usually warrant prompt neurosurgical intervention. In patients with incidental diagnoses or lesions with a long history of indolent and mild symptoms, deferral of radical surgery is an option, with close MRI and clinical surveillance. If the tumor is surgically well accessible, some physicians and patients prefer a preemptive strategy with the hope of averting ultimate neoplastic transformation. Patients who show progressive neurological symptoms or MRI evidence of tumor growth require therapeutic intervention (Jallo et al., 2001). The likelihood of gross total resection of a diffuse fibrillary astrocytoma is low, especially when the tumor is located in an eloquent location such as the medulla. However, in patients with supratentorial tumors, radical resection may confer a long symptom-free interval. Chemotherapy or radiation therapy is indicated when radical resection is not feasible (Sievert and Fisher, 2009) or the tumor shows early signs of neoplastic transformation.
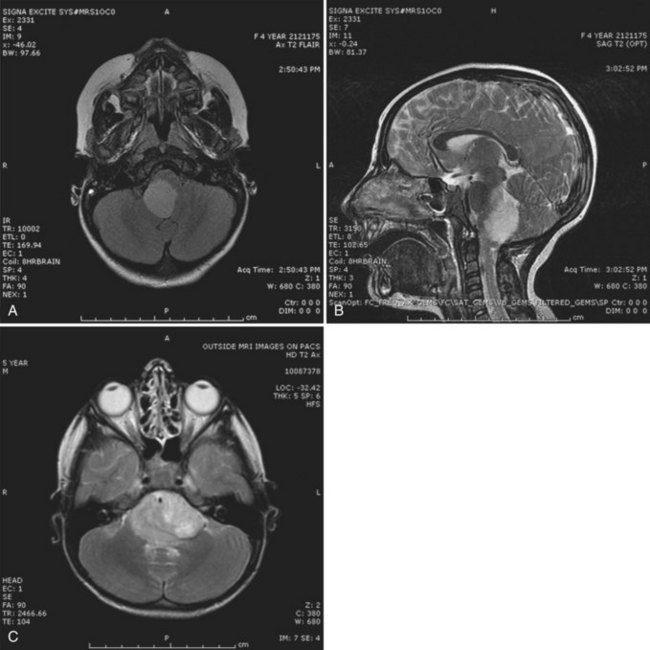
Prognosis
Long-term survival is possible for children with completely resected supratentorial DAs, with the notable exception of those with diffuse pontine gliomas, in whom the prognosis is poor (Jallo et al., 2004; Mauffrey, 2006). Gemistocytic astrocytoma, a variant of DA characterized by the presence of gemistocytic neoplastic astrocytes with eosinophilic cytoplasm, has a less favorable clinical course because of the propensity of these tumors to rapidly progress to higher-grade lesions such as anaplastic astrocytoma (WHO grade III) and glioblastoma (WHO grade IV). The prognosis for focal midbrain tumors is also favorable in spite of the fact that complete resection is not possible (Hamilton et al., 1996; Stark et al., 2005). No convincing evidence of a prognostic value of molecular markers such as TP53 mutations or the proliferative index has been shown for pediatric DAs to date.
Diffuse Intrinsic Pontine Glioma
Background
Diffuse intrinsic pontine gliomas (DIPGs) represent up to 75% of all pediatric brainstem tumors and account for approximately 10% of all childhood brain tumors. Mean age at diagnosis is approximately 8 years, and there is no gender predilection (Albright and Pollack, 2004).
Clinical Presentation
Most DIPG patients present with a relatively short (weeks to months) history of cranial nerve deficits, long-tract signs, and ataxia. A more protracted history may be seen in other brainstem tumors such as juvenile pilocytic astrocytomas. On MRI, DIPGs appear as infiltrative tumors within an enlarged pons. They are typically hypointense on T1-weighted imaging and hyperintense on T2-weighted imaging. Contrast enhancement is variable but usually absent to minimal at initial presentation. No specific imaging characteristics on conventional MRI at diagnosis are predictive of survival (Hargrave et al., 2008). Leptomeningeal spread may already be present at time of diagnosis, and MRI of the entire spine is therefore recommended initially as well as at follow-up (Sethi et al., 2010).
Diagnosis and Management
In patients with compatible history and MRI findings, the diagnosis of DIPG can be established without biopsy. In patients with a prolonged history, unusual neurological signs, or atypical MRI findings, diagnoses other than DIPG (e.g., juvenile pilocytic astrocytoma [JPA]) should be entertained and a biopsy strongly considered. Brainstem JPAs usually display an anterior exophytic growth pattern. PNETs and AT/RTs rarely occur in the brainstem. Alexander disease, a neurodegenerative disorder caused by mutations of the GFAP gene on chromosome 17q21, may mimic brainstem gliomas (Van Poppel et al., 2009). Genetic testing is available for this disease. Demyelinating conditions may also resemble DIPG radiographically, and in addition to CSF analysis, diffusion tensor imaging fiber tracking has been suggested to aid in the diagnosis (Giussani et al., 2010). Although biopsies are not performed routinely, DIPG tissue samples obtained at initial diagnosis can usually be classified as WHO grade II-IV astrocytomas (Gilbertson et al., 2003). In contrast, specimens obtained from autopsies invariably show progression to glioblastomas (i.e., WHO grade IV astrocytomas). A recent genomic analysis of DIPGs showed recurrent genomic alterations that were distinct compared to pediatric supratentorial high-grade gliomas, highlighting a different biology underlying the two entities (Zarghooni et al., 2010).
Prognosis
Radiotherapy remains the mainstay of therapy, and although the majority of patients will initially respond radiographically and/or clinically, nearly all patients develop subsequent tumor progression, often with disseminated disease . Despite numerous attempts at improving survival with addition of chemotherapy or biological modifiers, no therapy to date has resulted in significantly improved outcome compared to radiotherapy alone (Hargrave et al., 2006). Treatment of recurrent disease with bevacizumab and irinotecan appears largely ineffective (Gururangan et al., 2010; Narayana et al., 2010). Preliminary results from clinical trials suggest that nimotuzumab, a monoclonal antibody against epidermal growth factor receptor (EGFR), may have some activity in the recurrent setting (Lam et al., 2009). Overall, the outcome for patients with DIPGs remains dismal, with a median time to progression of approximately 6 months, median overall survival of less than 12 months, and only rare survivors beyond 2 years after initial diagnosis. DIPG remains one of the most frustrating entities in pediatric oncology, and novel treatment approaches are urgently needed.
Pleomorphic Xanthoastrocytoma
Clinical Presentation
PXAs are typically large and superficially located, especially in the temporal lobes. Seizures are the most common initial symptom. Contrast-enhanced MRI typically shows a large, enhancing tumor with occasional cystic components and calcification. On histopathological assessment, the proliferative indices are usually low, although necrosis, endothelial proliferation, and mitoses are observed in some patients. Consequently, PXAs may be confused with glioblastoma because of the presence of multinucleated cells and occasional foci of necrosis (Giannini et al., 1999). Prognosis appears to correlate with the mitotic index (Sugita et al., 2000).
Diagnosis and Management
PXA should be suspected in children presenting with new-onset seizures, focal deficits, and a large enhancing cortical mass on contrast-enhanced cranial MRI. The goal of surgery is to achieve a gross total resection, which is curative in the majority of patients (Rao et al., 2010). Adjuvant therapy should be deferred while the patient is monitored with serial MRI scans.
Prognosis
The 5-year progression-free survival rate for patients with PXAs is better than 70%. However, the presence of mitoses, endothelial proliferation, or necrosis on the pathological specimen, although very rare, may significantly alter the clinical behavior and prognosis (Sugita et al., 2000). Patients with anaplastic PXA with malignant transformation have been reported (Tekkok and Sav, 2004). Radiation therapy or chemotherapy does not appear to significantly alter the poor outcome in these patients.
Anaplastic Astrocytoma and Glioblastoma
Background
Anaplastic astrocytoma (WHO grade III) and glioblastoma (formerly glioblastoma multiforme, WHO grade IV) are the most common high-grade astrocytomas (HGA). The current WHO classification lists giant cell glioblastoma and gliosarcoma as histological subtypes. Overall, HGA are much less common in children than in adults. Supratentorial high-grade astrocytomas represent only 6% to 12% of all pediatric brain tumors, and diffuse intrinsic brainstem gliomas represent 10%. Most of these tumors arise in the cerebral hemispheres or within deeper midline structures such as the midbrain and pons. A number of genetic abnormalities have been implicated in HGA in both adults and children, including overexpression of the EGFR (Bredel et al., 1999) and EGFRvIII deletion mutations (Bax et al., 2009), TP53 mutations, PTEN tumor suppressor gene mutations (Thorarinsdottir et al., 2008), and mutations in DNA repair pathways (Giunti et al., 2009). More recently, somatic mutations of the isocitrate dehydrogenase 1 and 2 genes (IDH1, IDH2) have been identified in a subset of adult HGAs, most frequently in tumors that evolved from lower-grade gliomas (i.e., secondary glioblastomas) (Yan et al., 2009). In pediatric brain tumors, however, IDH mutations are rare. It is becoming increasingly clear that despite histological similarities, key biological differences exist between adult HGAs, pediatric HGAs, and diffuse intrinsic brainstem gliomas (Bax et al., 2009; Paugh et al., 2010; Zarghooni et al., 2010).
Genetic syndromes involving DNA mismatch repair (MMR) genes, including Turcot syndrome, are associated with HGA (Felton et al., 2004). Affected individuals often display NF1-like manifestations such as café-au-lait spots, and it has been hypothesized that MMR deficiency may lead to secondary NF1 gene inactivation in affected tissues. With regard to TP53 mutations, Li-Fraumeni syndrome has also been associated with gliomas of different grades, and overexpression of p53 has been associated with an adverse outcome independent of histological findings and clinical prognostic factors (Pollack et al., 2002).
Clinical Presentation
The clinical manifestations of HGA depend on the anatomical location of the tumor as well as on the age of the patient. The clinical prodromes are usually short and rapidly evolving, with symptoms or signs of increased ICP or focal neurological dysfunction, although in some patients, an HGA may arise in the setting of prolonged symptoms from a low-grade fibrillary astrocytoma (Tamber and Rutka, 2003).
Diagnosis and Management
The basis for suspicion of HGA is the clinical presentation and the contrast-enhanced cranial MRI. The MRI features are a combination of diffuse nonenhancing signal abnormalities and focal enhancing solid lesions. Intratumoral cysts often correlate with spontaneous necrosis. The T2 signal is often more diffuse, consistent with both infiltrative tumor and vasogenic tumor-associated edema (Fig. 52E.5). Significant mass effect, hydrocephalus, and intratumoral hemorrhage may be present. Gross total resection is the initial treatment goal (Pollack, 1999). This facilitates a more accurate diagnosis and makes subsequent radiation therapy tolerable. The established role of radiation in the treatment of older children has developed over the past 25 years. Chemotherapy has an emerging role, both alone and in combination with radiation. Treatment approaches primarily based on chemotherapy, including high-dose chemotherapy and autologous bone marrow transplantation, have been mainly used in an attempt to delay radiation in infants and young children, who are more prone to radiation-induced neurocognitive effects (Finlay and Zacharoulis, 2005). Building on the results of a trial in adult glioblastoma patients that demonstrated a survival benefit with combined radiation and temozolomide (Stupp et al., 2005), chemoradiation strategies for high-grade gliomas in children are being explored in clinical trials.
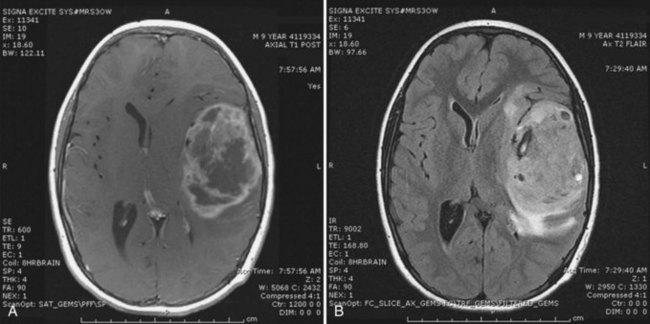
Prognosis
Children with high-grade gliomas continue to have a poor prognosis, despite the use of multimodal therapy. Age younger than 3 years, radical resection, WHO grade III histology, and the absence of TP53 mutations are favorable risk factors (Pollack et al., 2002; Qaddoumi et al., 2009). Patients with diffuse unresectable thalamic and pontine HGAs have the worst prognosis (Reardon et al., 1998). For diffuse intrinsic brainstem tumors, no adjuvant treatment has shown a benefit over conventional radiotherapy to date, and the prognosis remains dismal. New treatment approaches under investigation include the upfront use of bevacizumab, although its activity in the recurrent setting in children has been largely disappointing (Gururangan et al., 2010; Narayana et al., 2010). Multiple experimental agents are currently undergoing phase I and phase II evaluation in children, such as the EGFR antibody, nimotuzumab (Lam et al., 2009).
Neuronal and Mixed Neuronal-Glial Tumors
Ganglioglioma
Clinical Presentation
Seizures are the initial manifestation in approximately 50% of ganglioglioma patients. Complex partial seizures are common because the typical tumor location is in the medial temporal lobe (Luyken et al., 2004).
Diagnosis and Management
Contrast-enhanced cranial MRI often reveals a supratentorial cystic mass. The MRI appearance of these tumors is variable, but the lesion is frequently hypointense on T1-weighted sequences and hyperintense on T2-weighted images (Fig. 52E.6, A). Contrast enhancement varies in intensity from marked to absent and may be nodular, solid, or circumferential. A large size and a supratentorial location characterize an infantile variant of ganglioglioma, desmoplastic infantile ganglioglioma (see Fig. 52E.6, B) (Bachli et al., 2003). Gross total resection is the treatment of choice for gangliogliomas. Unresectable or recurrent tumors may be treated with radiation therapy (Johnson et al., 1997; Liauw et al., 2007; Rades et al., 2010).
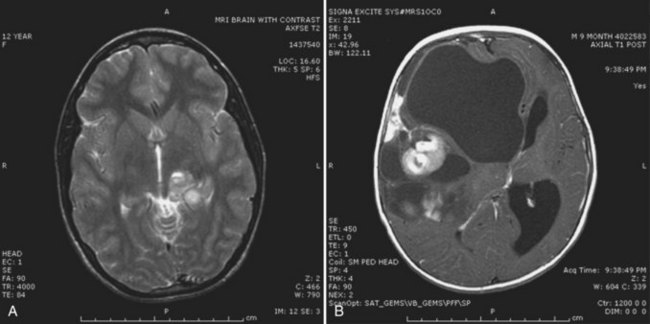
Prognosis
Gross total resection is often curative. Thus, location and extent of resection are the most important prognostic factors. In patients with recurrent or unresectable ganglioglioma, response to chemotherapy has been demonstrated (Johnson et al., 1997). These tumors may undergo malignant transformation over time, similar to a diffuse astrocytoma (WHO grade II) (Liauw et al., 2007). Similarly, desmoplastic infantile gangliogliomas, although often cured by resection, may undergo rapid progression and metastasis consistent with malignant transformation (De Munnynck et al., 2002).
Desmoplastic Infantile Astrocytoma or Ganglioglioma
Characteristic of desmoplastic infantile astrocytomas (DIA) or ganglioglioma (DIG) are their large size and superficial cortical location at presentation. These tumors may involve an entire hemisphere and are usually diagnosed in infants and children younger than age 2 years. The most common presentation consists of seizures, although progressive focal deficits may also be a first manifestation. The tumor is revealed by contrast enhancement on MRI and usually has a large cystic component. Histological features include collagenized regions with astrocytes and admixed small, primitive-appearing cells in nodular aggregates, hence the name desmoplastic. Regional leptomeningeal dissemination is common despite classification as a WHO grade I lesion. Nevertheless, patients with DIA often have a good prognosis if gross total resection can be achieved (Sugiyama et al., 2002).
Dysembryoplastic Neuroepithelial Tumor
Background
Dysembryoplastic neuroepithelial tumors (DNETs) represent approximately 1% of all neuroepithelial brain tumors in patients younger than 20 years of age (Rosemberg and Vieira, 1998). Two-thirds of DNETs are located in the temporal lobes, and 5% to 15% of temporal lobe resections for intractable epilepsy show DNETs. These lesions are classified as WHO grade I tumors (Luis et al., 2007).
Diagnosis and Management
Contrast-enhanced cranial MRI shows absence of edema and only minimal if any enhancement (Fig. 52E.7). Pathological findings include a specific neuronal-glial element manifested by glial fibrillary acidic protein (GFAP)-negative oligodendroglia-like cells and neurons in a mucinous eosinophilic background that give the appearance of floating neurons. Because histopathological analysis shows oligodendrocytes, astrocytes, or both, the differential diagnosis often includes oligodendroglioma, mixed oligoastrocytoma, and ganglioglioma. DNETs have a benign course, and gross total resection is often curative (Sandberg et al., 2005). Adjuvant chemotherapy and radiation therapy are not recommended.
Central Neurocytoma
Background
Central neurocytoma is a rare tumor of neuronal lineage that usually arises in a periventricular location. The diagnosis of a central neurocytoma should be a consideration in young patients with an intraventricular mass. Hydrocephalus may arise from obstruction of CSF flow at the level of the foramina of Munro (Schmidt et al., 2004).
Diagnosis and Management
Contrast-enhanced cranial MRI typically shows an isointense mass with minimal enhancement (Fig. 52E.8). A tumor arising in the posterior third ventricular region may represent a pineocytoma, but a pineoblastoma must be excluded (Hirato and Nakazato, 2001). On pathological study, the presence of perinuclear halos on light microscopy may lead to a mistaken diagnosis of oligodendroglioma. Immunohistochemical staining with neuronal markers such as synaptophysin helps distinguish this tumor from an oligodendroglioma. Very rare atypical forms with mitoses, necrosis, and endothelial proliferation have been reported (Mackenzie, 2000; Soylemezoglu et al., 1997). Patients with a mitotic index greater than 3% have a worse prognosis (Rades et al., 2004). Gross total resection can be curative for central neurocytomas. Radiation and chemotherapy may be deferred in patients undergoing radical resections, but they should be monitored closely. This emphasizes the importance of differentiating central neurocytoma from oligodendroglioma, because the latter tumor may require adjuvant radiation and chemotherapy. Radiation therapy is a consideration only for atypical forms with a high mitotic index (>3%) and for recurrent unresectable tumors.
Prognosis
The outcome in atypical central neurocytomas is difficult to predict accurately because of the rarity of the tumor. However, a meta-analysis has shown that lesions with a mitotic index less than 3% have a less than 15% risk of recurrence and a 95% 5-year overall survival, compared to 38% and 66%, respectively, for tumors with a mitotic index greater than 3% (Rades et al., 2004). Both chemotherapy and radiotherapy have been used in the recurrent setting and may produce prolonged disease stabilization and occasionally partial responses (Leenstra et al., 2007; Sharma et al., 2006). The role of adjuvant therapy after incomplete resection, however, is not well defined.
Other Central Nervous System Tumors
Oligodendroglioma
Background
Oligodendrogliomas are rare, accounting for approximately 2% of pediatric brain tumors, and are diffusely infiltrative tumors. They are most commonly found in the cerebral cortex but may arise anywhere in the CNS. Depending on tumor location, most patients present with seizures or signs of raised ICP (Peters et al., 2004). Oligodendrogliomas are typically hypointense on T1-weighted MRI sequences and hyperintense on T2-weighted sequences. The tumor may appear partially contrast enhancing, cystic, hemorrhagic, and/or calcified. Histologically, the tumor cells resemble oligodendroglial cells. Oligodendrogliomas are graded as either WHO grade II or III, (i.e., anaplastic oligodendroglioma [AO]). AO features frequent mitotic figures, nuclear atypia, microvascular proliferation, and occasional necrosis. Mixed tumors with both oligodendroglial and astrocytic components exist and are classified as oligoastrocytoma or anaplastic oligoastrocytoma. There is no specific immunohistochemical marker that unequivocally differentiates oligodendroglial from astrocytic tumors. Molecularly, oligodendroglial tumors are characterized by an unbalanced translocation of chromosomes 1 and 19, which results in a co-deletion of 1p and 19q. Several studies have established the 1p/19q co-deletion as a molecular signature for oligodendrogliomas in adults, although the prevalence in pediatric oligodendroglial tumors is rare (Kreiger et al., 2005; Raghavan et al., 2003).
Management
Whenever feasible, gross total resection should be attempted. For residual, progressive, or recurrent tumors, optimal therapy in the pediatric population has not been established. Oligodendrogliomas are generally sensitive to chemotherapy and radiotherapy. Based on adult experiences, chemotherapy such as PCV (procarbazine/CCNU/vincristine) and temozolomide have been used in pediatric patients in an attempt to delay radiotherapy. In adult studies, both chemotherapy and radiotherapy have been shown to prolong progression-free survival (Bromberg and van den Bent, 2009), but the optimal treatment sequence is subject to investigation in ongoing studies.
Prognosis
In pediatric series, the most relevant prognostic factors for improved progression-free and overall survival were gross total resection and low tumor grade. For low-grade (i.e., non-anaplastic) oligodendrogliomas, 5-year progression-free survival and overall survival of 81% and 84%, respectively, have been reported (Peters et al., 2004). The 1p/19q co-deletion, which has been associated with a favorable prognosis and sensitivity to chemotherapy and radiotherapy in adult tumors, is rare in children and of unclear prognostic significance.
Ependymoma
Background
Ependymomas are glial tumors that arise from ependymal cells within the CNS. This tumor represents approximately 10% of all childhood intracranial neoplasms, constituting the third most common pediatric brain tumor after astrocytoma and medulloblastoma. Some 90% of pediatric ependymomas are intracranial; 75% arise in the posterior fossa. Most supratentorial ependymomas are located in the brain parenchyma away from the ependymal surface, in contrast to infratentorial ependymomas. Spinal cord ependymomas represent less than 10% of pediatric intramedullary spinal tumors, but ependymomas represent more than 50% of intramedullary spinal tumors in adults (Lee et al., 2006). Although morphologically these tumors appear similar, they have a distinct gene expression profile based upon their anatomical location (Taylor et al., 2005). The most consistent genetic defects in ependymoma have been either monosomy 22 or structural abnormalities of 22q, raising the possibility of a tumor suppressor gene on chromosome 22 (Hulsebos et al., 1999; Suarez-Merino et al., 2005). Other molecular defects that have been described include abnormal expression of ERBB2 and ERBB4 receptors (Gilbertson et al., 2002), the p53 homologue p73 (Kamiya and Nakazato, 2002), vascular endothelial growth factor protein (Korshunov et al., 2002), or the p53 regulator MDM2 (Suzuki and Iwaki, 2000). Individuals with NF-2 have an increased susceptibility to intramedullary spinal cord ependymomas (Pollack and Mulvill, 1997). Although the NF2 gene is located at 22q12, mutations in NF2 are rarely found in sporadic ependymomas. A recent microarray analysis of pediatric ependymomas has identified a cluster of genes distinct from NF2 that may be involved in ependymoma tumorigenesis (Suarez-Merino et al., 2005). Moreover, expression profiling indicates that histologically similar ependymomas from different parts of the CNS are in fact molecularly and clinically distinct disease subgroups (Ebert et al., 1999; Taylor et al., 2005).
Clinical Presentation
The presenting symptoms of infratentorial ependymomas relate to their origin from ependymal tissue lining the fourth ventricle. Hydrocephalus results when the tumor fills the fourth ventricle, causing headache, irritability, nausea, vomiting, ataxia, and papilledema. A common sign of the tumor in infants is increased head circumference. Tumors that extend out one of the foramina of Luschka compromise lower cranial nerves and cause hoarseness and dysphagia. If the tumor extends through the foramen of Magendie, the patient may complain of neck discomfort and have torticollis. Spinal cord ependymomas are typically located in the cervical region. The most common presenting symptom is localized pain at the level of the lesion. The typical description of pain is that it is worse at night, presumably due to congestion of the spinal venous plexus in the recumbent position. The second most common symptom is radicular dysesthesias, and a late manifestation of this symptom is progressive spastic quadriparesis. Thoracic ependymomas are associated with scoliosis. Myxopapillary ependymomas of the conus medullaris and filum terminale may present with low back pain, radicular pain, saddle anesthesia, and sphincter dysfunction (Nagib and O’Fallon, 1997).
Diagnosis
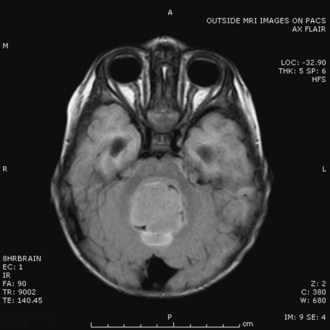
A typical MRI appearance of a fourth ventricular ependymoma is that of a homogeneously enhancing solid mass extending out one of the foramina of Luschka or the foramen of Magendie, with associated obstructive hydrocephalus (Fig. 52E.9). Although there is no formal staging system for ependymomas, the WHO classification system recognizes three grades: grade I, subependymoma and myxopapillary ependymoma; grade II, classic ependymoma; and grade III, anaplastic ependymoma. Because ependymomas typically arise in the ependymal linings of ventricles, tumors may spread through the entire neuraxis. Evaluation of patients should include contrast-enhanced MRI scans of the brain and entire spinal cord, as well as cytological evaluation of the CSF. Several factors have been associated with an unfavorable outcome in patients with ependymomas, including a younger age at diagnosis, anaplastic histology, subtotal resection, and a high mitotic index. Of these, the single most important factor in determining prognosis appears to be the degree of resectability (Perilongo et al., 1998).
Management
The first line of treatment is surgery, with a goal of gross total resection. Complete resection of spinal cord and supratentorial ependymomas is feasible. Technological advances such as the operating microscope, Cavitron ultrasonic aspirator, intraoperative ultrasound, MRI, and electrophysiological monitoring have reduced operative morbidity and allowed more complete tumor resection. Overall, spinal cord ependymomas are more easily resectable than astrocytomas owing to the presence of a better demarcated cleavage plane. Historically, the use of radiation in young children with ependymomas was avoided because the risks of cognitive, endocrine, and developmental side effects would be highest. Previous recommendations had been to reserve radiation for patients in whom a gross total resection was not possible or for patients with recurrent disease. However, the failure of adjuvant chemotherapy to delay radiation therapy for a significant time in younger patients with ependymomas of the fourth ventricle, as well as the advent of conformal radiation, has made postoperative radiation therapy an attractive option for pediatric patients (Mansur et al., 2004; Merchant, 2002). Several studies suggest that radiation therapy prolongs progression-free survival after subtotal resection of an ependymoma (McLaughlin et al., 1998). As such, there is now growing evidence supporting the use of adjuvant radiation for spinal cord and supratentorial ependymomas (Merchant et al., 2004).
As noted, no clear role exists for adjuvant chemotherapy in the management of ependymomas (Siffert and Allen, 1998). Nonetheless, several small series in patients with newly diagnosed and recurrent ependymoma have reported objective responses to carboplatin, cisplatin, ifosfamide, and etoposide. Chemotherapy is used more often for infants and younger children with incompletely resected or disseminated disease (Duffner et al., 1998). In one study, chemotherapy was associated with a 40% complete response rate in patients who received pre-irradiation chemotherapy because of residual postoperative tumor (Grill et al., 2001). Unfortunately, treatment options for children with recurrent ependymoma are quite limited. However, a small number of children, including those with metastases at recurrence, have had prolonged survival with re-resection and re-irradiation (Merchant et al., 2008).
Prognosis
The most important prognostic factors for both intracranial and spinal cord ependymoma are age, tumor grade, and extent of surgical resection (Paulino et al., 2002; Pollack et al., 1995b). Children younger than age 3, those with WHO grade III disease, or those with less than a gross total resection have lower rates of survival (Horn et al., 1999). The 5-year progression-free and overall survival rates for patients with subtotal versus total resections of posterior fossa ependymomas are 25% and 66%, respectively. Similarly, the prognosis for patients with disseminated disease is much worse (Ernestus et al., 1996). The expression of human telomerase reverse transcriptase correlates with progression-free and overall survival of pediatric patients with intracranial ependymoma (Tabori et al., 2006).
Germ Cell Tumor
Background
Germ cell tumors are the most prevalent tumor of the pineal region and represent approximately 3% to 5% of intracranial childhood malignances in the United States (Keene et al., 2007). These tumors are morphologically homologous to the germ cell tumors that arise in the gonads. Germ cell tumors of the CNS are divided into two clinical grades that correlate with response to adjuvant chemotherapy: pure germinomas (60%) and nongerminomatous germ cell tumors (40%). Nongerminomatous germ cell tumors (NGGCT) include embryonal cell carcinoma, immature and mature teratomas, endodermal sinus tumor, choriocarcinoma, and mixed germ cell tumors. The majority of germ cell tumors (95%) arise in midline CNS structures, with approximately 40% in the suprasellar cistern, 50% in the pineal region, and 5% involving both sites at diagnosis. Germinoma is the most common tumor arising in the pineal region; NGGCTs and germinomas arise with equal frequency in the suprasellar region. Intracranial germ cell tumors occur primarily in the second and third decades of life. Pineal region tumors are more common in males, whereas there is an equal sex distribution in the suprasellar region. The incidence of CNS germ cell tumors in Asian populations, such as in Korea and Japan, appears to be higher than in other ethnic groups (Packer et al., 2000).
Clinical Presentation
Patients diagnosed with suprasellar germ cell tumors may have an unusually long prodrome, often years in duration. The earliest symptoms usually involve endocrine dysfunction, most frequently diabetes insipidus, with patients suffering from polyuria and polydipsia. Eventually other endocrine manifestations arise, such as growth impairment, precocious puberty, and hypothyroidism. Vision loss and symptoms of increased ICP are late manifestations when the tumor has either reached a large size or has spread in a periventricular distribution to the third and lateral ventricles. Patients with tumors arising in the pineal region usually present with a shorter prodrome consisting of symptoms of raised ICP—that is, headache, nausea, and vomiting due to obstructive hydrocephalus. Limitation of vertical gaze, convergence nystagmus, impaired pupillary reflexes, and double vision may be apparent on neurological examination, owing to compression of the midbrain tectum (Parinaud syndrome). Papilledema due to obstructive hydrocephalus is observed. Germ cell tumors can also cause precocious puberty when they release large concentrations of β-human chorionic gonadotropin (β-hCG) (Ogino et al., 2005).
Diagnosis and Management
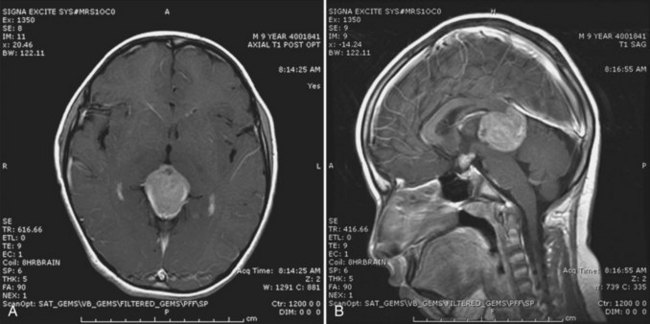
Contrast-enhanced cranial MRI is necessary for the evaluation of pineal region tumors. Pure germinomas are typically isointense on T1-weighted images and slightly hyperintense on T2-weighted images, with intense homogeneous contrast enhancement (Fig. 52E.10). Intratumoral cysts or calcification may be present. NGGCTs (e.g., mixed germ cell tumors or teratomas) often have a more heterogeneous appearance due to a mixture of benign and malignant components. The specific type of pineal region tumor cannot be determined from the radiographic appearance alone. Patients typically require histological or biochemical confirmation of specific tumor histology at diagnosis for optimal management because of the range of tumor types that arise in these areas. Histological confirmation may not be necessary in patients with elevated lumbar CSF concentrations of tumor markers (α-fetoprotein [AFP] or β-hCG) consistent with an NGGCT (Seregni et al., 2002). There is a high correlation between the tumor marker profile in CSF and types of NGGCT. Elevations of AFP occur with endodermal sinus tumors and embryonal carcinomas, whereas a high level (>2000 mIU/mL) of β-hCG alone is consistent with choriocarcinoma. Pure germinoma may have modest elevations of CSF β-hCG up to 50 mIU/mL. Elevations of lactate dehydrogenase isoenzymes and placental alkaline phosphatase are also detectable in CSF in germinoma patients. Tumor markers are also useful in monitoring response to treatment and surveillance for early signs of recurrence.
Radiation is frequently employed in the treatment of CNS germ cell tumors, with radiosensitivity determined by tumor histology. Germinomas are the most radiosensitive type, with 90% progression-free survival at 5 years, whereas NGGCTs have a 5-year survival rate of 30% to 40% (Ogawa et al., 2004). Several treatment alternatives exist regarding the management of intracranial germinomas. Radiation alone is usually administered in relatively high doses and large volumes (whole ventricular or craniospinal) even for localized disease. Although the 10-year survival ranges from 80% to 90%, children often suffer from the late consequences of radiation. Alternatively, the use of chemotherapy followed by response-based radiation therapy permits a selective reduction not only in dose but also in volume of radiation in patients whose tumors completely regress after two to four courses of chemotherapy (Kretschmar et al., 2007). For NGGCT, more aggressive chemotherapy (Kellie et al., 2002; 2004) and high-dose/high-volume radiation therapy are required to provide improved survival.
Prognosis
The prognosis for patients with pineal region tumors depends on histology. Their sensitivity to radiation and chemotherapy give germinomas an excellent prognosis. NGGCTs, on the other hand, tend to have a less favorable prognosis, but more recent treatment protocols that use combinations of chemotherapy and radiation have markedly improved outcome (Echevarria et al., 2008). Recurrent germ cell tumors may respond to salvage chemotherapy or additional radiation therapy.
Craniopharyngioma
Diagnosis and Management
The typical appearance on cranial MRI (Fig. 52E.11, A-B) is a multicystic and solid enhancing suprasellar mass, which if large enough, results in hydrocephalus and forward displacement and stretching of the optic nerves and chiasm (Brunel et al., 2002). The cystic component is often bright on the T1 images prior to contrast. An important additional radiographic diagnostic sign is intratumoral calcifications on a nonenhanced CT scan (see Fig. 52E.11, C).
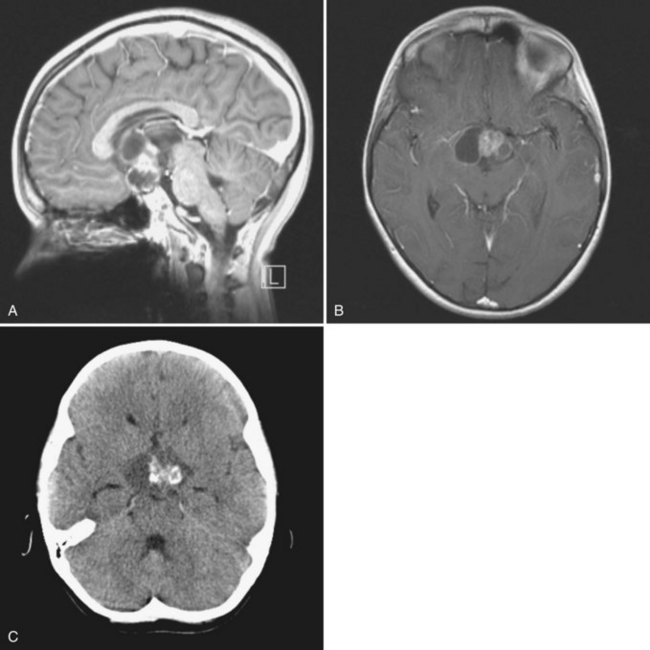
Surgical removal of the tumor is the most effective treatment, and complete microsurgical resection in experienced hands, when feasible, is the treatment of choice for newly diagnosed craniopharyngiomas (Elliott et al., 2010). Transcranial or transsphenoidal surgical approaches are commonly used. Transsphenoidal resection is the preferred method for tumors in a subdiaphragmatic location and is associated with a lower incidence of postoperative diabetes insipidus. Despite the surgical accessibility of many of these tumors, radical resection does not always guarantee recurrence-free survival. Moreover, aggressive resection can cause multiple hormonal deficiencies, visual-field and acuity deficits, and symptoms of hypothalamic injury such as eating disorders, altered sleep/wake rhythms, memory impairment, and loss of impulse control. Another approach to craniopharyngioma combines incomplete surgical resection followed by radiation therapy in an attempt to preserve quality of life (Marchal et al., 2005; Saint-Rose et al., 2005). Radiosurgery may be used for small areas of residual or recurrent disease. Long-term complications of radiation for craniopharyngiomas include cognitive and endocrine deficits, secondary malignancies, optic neuropathy, and vascular injury leading to Moyamoya disease. A temporizing approach is the instillation of sclerosing agents such as bleomycin or 32P into the tumoral cysts (Kim et al., 2007). However, the solid tumor usually progresses, and subsequent external beam radiation may be difficult to administer because of the unpredictable dosimetry of 32P (Hasegawa et al., 2004). Treatment of recurrent craniopharyngiomas often involves repeat resection or radiation therapy (Kalapurakal, 2005; Takahashi et al., 2005). Very limited information is available on the use of chemotherapy, and most agents have been administered into tumor-associated cysts via an Ommaya device (Cavalheiro et al., 2010).
Prognosis
The most important factors that correlate with progression-free survival are extent of resection and administration of postoperative radiation. Recurrence occurs in 30% of cases after total resection and in 57% of cases after subtotal resection. The recurrence rate drops to 30% when radiation follows subtotal resection. Unfortunately, most long-term survivors experience significant morbidity related to panhypopituitarism, cognitive impairment, and obesity (Karavitaki et al., 2005; Kendall-Taylor et al., 2005; Pedreira et al., 2006).
Choroid Plexus Tumors
Background
Choroid plexus tumors are rare tumors of neuroectodermal origin arising from the epithelium of the choroid plexus of the cerebral ventricles. Although tumors of the choroid plexus represent only 2% to 4% of pediatric brain tumors, this group represents 10% to 20% of tumors that develop in infancy. Three histological variants are described: choroid plexus papillomas (WHO grade I), atypical papillomas (WHO grade II), and choroid plexus carcinomas (WHO grade III) (Fuller, 2008). Clear diagnostic criteria for atypical papillomas are not established; however, the presence of mitotic activity (≥2 mitoses per 10 high-power fields) is the sole histological feature independently associated with recurrence (Jeibmann et al., 2006). Choroid plexus papillomas outnumber choroid plexus carcinomas by a ratio of at least 5 : 1. Choroid plexus tumors are typically located in areas where choroid plexus tissue normally occurs. Most tumors arise from the lateral ventricles (50% of cases) and the fourth ventricle (40%). Choroid plexus carcinoma is associated with the Li-Fraumeni syndrome, in which patients have TP53 germline mutations (Gonzalez et al., 2009). DNA sequences from the human neurotropic JC virus occur in choroid plexus tumors (Okamoto et al., 2005).
Diagnosis and Management
The diagnosis is suspected when a large enhancing tumor in the lateral ventricle is visualized on contrast-enhanced cranial MRI (Fig. 52E.12). Multilobular calcified contrast-enhancing intraventricular masses are characteristic of choroid plexus tumors. The primary treatment objective for both low-grade and high-grade choroid plexus tumors is gross total resection (Lafay-Cousin and Strother, 2007).
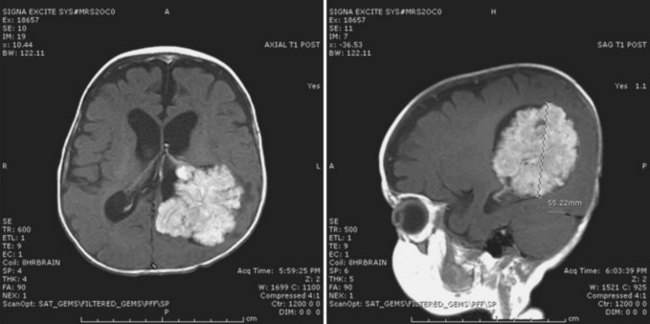
The extent of surgical resection is the single most important factor that determines the prognosis of a choroid plexus papilloma. However, a major obstacle to surgical removal of choroid plexus tumors is the rich vascular network within these tumors. The choroid plexus receives its blood supply from the anterior and posterior choroidal arteries, branches of the internal carotid artery, and the posterior cerebral artery (Wolburg and Paulus, 2009). For patients who undergo gross total resection of their choroid plexus carcinomas, the role of adjuvant therapy is unclear. However, there does appear to be a role for adjuvant therapy (chemotherapy and/or radiation therapy) for patients who have residual disease. Preliminary evidence suggests that choroid plexus carcinomas are chemosensitive tumors (Wrede et al., 2007). The role of adjuvant radiation is controversial. Radiation is reserved for children older than age 3, those who have had a subtotal resection, and those with malignant features within the tumor or dissemination of the tumor along the neuraxis. Although radiation oncologists have used several different approaches to treat children with choroid plexus carcinoma, there is a suggestion that children who received craniospinal irradiation fared better than those who were treated with whole-brain or involved-field irradiation (Mazloom et al., 2010).
Prognosis
Gross total resection is often curative for choroid plexus papilloma. Although these benign tumors may disseminate into the CSF, such cases tend to be clinically asymptomatic. The prognosis is significantly worse for choroid plexus carcinomas, particularly those patients with p53 mutations (Tabori et al., 2010). The latter tumors may produce nodular metastases along CSF pathways. Although several adjuvant chemotherapy regimens have been proposed to treat choroid plexus carcinomas, no standard therapy has been established (Gopal et al., 2008).
Treatment-Related Complications in Infants and Children with Primary Nervous System Tumors
Surgery
The cerebellar mutism or posterior fossa syndrome (PFS) has a wide range of expression and severity. The majority of patients develop diminished speech output within 24 to 48 hours of posterior fossa surgery, usually to resect a tumor that has presented with raised ICP. When severe, the mutism is often accompanied by emotional lability, profound hypotonia, dysphagia, and ataxia. Short-term interventions such as corticosteroids or VP shunts do not seem to have a beneficial role, but minor or major tranquilizers may mollify the severity of the behavioral effects. Improvement usually begins within several weeks, but there are often long-lasting if not permanent consequences including dysarthria, ataxia, emotional lability, and learning difficulties. Although PFS was once thought to be a fairly rare occurrence, with better methods of detection, signs of PFS may be found in as many as 40% of patients (Wells et al., 2010). Aseptic meningitis syndrome often emerges within 1 to 2 weeks of surgery during a corticosteroid taper. It is thought to be related to the presence of blood and tissue contents mixing with CSF after surgery. Patients developed fever, increased nuchal rigidity, and signs of raised ICP. A pseudomeningocele may evolve, and if the ICP is not relieved, CSF leakage from the operative incision may ensue, leading to a septic meningitis. The simplest remedy is to rule out a septic meningitis by culturing the CSF, increasing the corticosteroid dose, and prolonging its taper (Carmel and Greif, 1993).
Radiation Therapy
Radiation therapy may produce subacute and late effects on the CNS. Subacute effects include radiation somnolence syndrome (RSS) and Lhermitte sign (see later discussion). The RSS typically emerges within 1 to 2 months of initiating large-volume high-dose cranial irradiation. Patients typically become lethargic and anorexic and may develop symptoms that recapitulate the symptoms at presentation of the initial tumor. RSS spontaneously resolves within 3 months in most cases, but occasionally low-dose corticosteroids are required to ameliorate the anorexia (Ryan, 2000). Lhermitte sign consists of a sensation of electric shocks traveling down the spine on neck flexion and usually arises within several months of cervicothoracic spinal radiation (Lewanski et al., 2000). It usually resolves spontaneously within months, without any apparent serious sequelae. Both of these syndromes are probably related to transient parenchymal edema induced by radiation therapy.
Long-term consequences of radiation therapy are becoming more apparent as survival rates increase; some of the more significant include endocrine effects, decrease in cognitive function, vascular effects, and secondary malignancies (Hoffman and Yock, 2009). Endocrine issues including growth hormone and thyroid hormone deficiency result from irradiation to the hypothalamus and pituitary glands (Heikens et al., 1998). Linear growth retardation may also result from irradiation to the spine. Neurocognitive studies in children with medulloblastoma treated with craniospinal irradiation have disclosed progressive loss of cognition and short-term memory and slower processing speeds (Mulhern et al., 2005). These deficits are more pronounced in children treated at a younger age and with higher doses of irradiation.
The use of concomitant or adjuvant chemotherapy may increase the acute and delayed neurotoxicity of CNS radiation therapy or independently produce CNS injury. For example, a progressive leukoencephalopathy may emerge following administration of parenteral or intrathecal methotrexate, usually in the management of childhood acute lymphoid leukemia, and these effects are exacerbated by prior cranial radiation therapy (Mahoney et al., 1998). More severe hearing impairment may occur when cisplatin is used with whole-brain radiation therapy than when cisplatin is given alone (Walker et al., 1989). Pseudoprogression on MRI is more common following CNS radiation therapy when temozolomide and radiation therapy are given concomitantly than when radiation therapy is given alone (Brandsma et al., 2008; Clarke and Chang, 2009).
Radiation necrosis may occur after radiation delivery to the tumor and surrounding brain. The necrotic brain and tumor tissue can cause mass effect, edema, and contrast enhancement on MRI. This complication of radiation may be indistinguishable from progressive or recurrent tumor on conventional neuroimaging studies. In fact, cerebral radionecrosis can produce symptoms identical to those of an expanding tumor, including progressive focal neurological deficits, seizures, and increased ICP. Treatment of cerebral radionecrosis with corticosteroids may result in improvement of clinical symptoms and reduction in contrast enhancement on CT or MRI studies. Bevacizumab, a monoclonal antibody against the vascular EGFR, decreases cerebrovascular permeability and confers some clinical benefit in adults with cerebral radionecrosis (Gonzalez et al., 2007), but surgical debulking is often necessary to reduce mass effect and increased ICP. Involvement of small cerebral blood vessels may lead to ischemic strokes. Eventually the development of multiple small collateral vessels may result in a moyamoya pattern of vascular abnormality that in turn increases the risk for stroke or cerebral hemorrhage.
Secondary malignancies, including high-grade gliomas, atypical meningiomas (Santoro et al., 2002), and schwannomas, have been observed within the treatment field several years after the completion of radiation therapy. Patients with Turcot syndrome, Gorlin syndrome, and NF1 are more likely to develop a secondary malignant glioma after radiation therapy, compared to patients without these conditions (Stavrou et al., 2001). This provides additional rationale to defer radiation therapy (when possible) for low-grade gliomas.
As noted earlier in the chapter, in an effort to decrease radiation injury to normal tissues adjacent to a CNS target, protons rather than photons are being used with increasing frequency. Protons deposit their energy at the prescribed depth without the exit dose seen with photon radiation, owing to the Bragg peak effect (Hoffman and Yock, 2009). Although the number of children with CNS tumors treated with protons is still fairly small, the preliminary data as well as models comparing proton and photon fields suggest that this approach may result in similar tumor control with greater sparing of uninvolved normal tissue (MacDonald and Yock, 2010; Merchant et al., 2008).
Chemotherapy
Peripheral neuropathy is a common late effect of several chemotherapies (Quasthoff and Hartung, 2002). Cisplatin and carboplatin mainly affect proprioception and spare pain and temperature sensation. The usual presenting symptoms are painful dysesthesias and tingling sensations in the toes and later in the fingers but sparing motor fibers. In contrast, vincristine produces a sensorimotor neuropathy. The first symptoms are usually tingling in the toes and fingers; loss of ankle jerk is typically the first objective sign. Continued use leads to areflexia and motor weakness involving the dorsiflexors of the feet (foot drop). Patients with preexisting neuropathies may become quadriparetic after treatment with vincristine. Cerebellar syndromes of acute onset may be seen with high-dose cytarabine and occasionally with 5-fluorouracil. These complications are usually reversible within 2 weeks, but severe irreversible damage to Purkinje cells may occur with chronic use of the drug (several months or if the drug is reintroduced) (Friedman and Shetty, 2001). Transverse myelopathy occurs with prolonged treatments with intrathecal methotrexate or cytarabine. The risk is higher when combined with spinal irradiation.
Albright A.L., Pollack I.F. Brainstem gliomas. In: Winn H.R., editor. Youmans Neurological Surgery, vol. 3. Philadelphia: Saunders; 2004:3663-3669.
Arita K., Kurisu K., Sugiyama K., et al. Long-term results of conventional treatment of diencephalic pilocytic astrocytoma in infants. Childs Nerv Syst. 2003;19:145-151.
Athale U.H., Duckworth J., Odame I., et al. Childhood atypical teratoid rhabdoid tumor of the central nervous system. A Meta-analysis of observational studies. J Pediatr Hematol Oncol. 2009;31:651-663.
Attard T.M., Giglio P., Koppula S., et al. Brain tumors in individuals with familial adenomatous polyposis: a cancer registry experience and pooled case report analysis. Cancer. 2007;109:761-766.
Bachli H., Avoledo P., Gratzl O., et al. Therapeutic strategies and management of desmoplastic infantile ganglioglioma: two case reports and literature overview. Childs Nerv Syst. 2003;19:359-366.
Baeza N., Masuoka J., Kleihues P., et al. AXIN1 mutations but not deletions in cerebellar medulloblastomas. Oncogene. 2003;22:632-636.
Bambakidis N.C., Robinson S., Cohen M., et al. Atypical teratoid/rhabdoid tumors of the central nervous system: clinical, radiographic and pathologic features. Pediatr Neurosurg. 2002;37:64-70.
Bax D.A., Gaspar N., Little S.E., et al. EGFRvIII deletion mutations in pediatric high-grade glioma and response to targeted therapy in pediatric glioma cell lines. Clin Cancer Res. 2009;15:5753-5761.
Biegel J.A. Molecular genetics of atypical teratoid/rhabdoid tumors. Neurosurg Focus. 2006;20:E11.
Biegel J.A., Tan L., Zhang F., et al. Alterations of the hSNF5/INI1 gene in central nervous system atypical teratoid/rhabdoid tumors and renal and extrarenal rhabdoid tumors. Clin Cancer Res. 2002;8:3461-3467.
Brandsma D., Stalpers L., Taal W., et al. Clinical features, mechanisms, and management of pseudoprogression in malignant gliomas, [review]. Lancet Oncol, 9. 2008;5:453-461.
Brat D.J., Parisi J.E., Kleinschmidt-DeMasters B.K., et al. Surgical neuropathology update: a review of changes introduced by the WHO classification of tumors of the central nervous system, fourth ed. Arch Pathol Lab Med. 2008;132:993-1007.
Bredel M., Pollack I.F., Hamilton R.L., et al. Epidermal growth factor receptor expression and gene amplification in high-grade non-brainstem gliomas of childhood. Clin Cancer Res. 1999;5:1786-1792.
Briner J., Bannwart F., Kleihues P., et al. Malignant small cell tumor of the brain with intermediate filaments-a case of a primary cerebral rhabdoid tumor. Pediatr Pathol. 1985;3:117-118.
Bromberg J.E., van den Bent M.J. Oligodendrogliomas: molecular biology and treatment. Oncologist. 2009;14:155-163.
Broniscer A., Baker S.J., West A.N., et al. Clinical and molecular characteristics of malignant transformation of low-grade glioma in children. J Clin Oncol. 2007;20:682-689.
Brunel H., Raybaud C., Peretti-Viton P., et al. [Craniopharyngioma in children: MRI study of 43 cases]. Neurochirurgie. 2002;48:309-318.
Burns A.S., Jaros E., Cole M., et al. The molecular pathology of p53 in primitive neuroectodermal tumours of the central nervous system. Br J Cancer. 2002;87:1117-1123.
Carmel P.W., Greif L.K. The aseptic meningitis syndrome: a complication of posterior fossa surgery. Pediatr Neurosurg. 1993;19(5):276-280.
Catsman-Berrevoets C.E., Aarsen F.K. The spectrum of neurobehavioural deficits in the posterior fossa syndrome in children after cerebellar tumour surgery. Cortex. 2010;46:933-946.
Cavalheiro S., Di Rocco C., Vealenzuela S., et al. Craniopharyngiomas: intratumoral chemotherapy with interferon-alpha: a multicenter preliminary study with 60 cases. Neurosurg Focus. 2010;28(4):E12.
Central Brain Tumor Registry of the United States (CBTRUS), 2005. Statistical Report: Primary Brain Tumors in the United States. 1998-2002, CBTRUS, Hinsdale, IL.
Chi S.N., Gardner S.L., Levy S.K., et al. Feasibility and response to induction chemotherapy intensified with high-dose methotrexate for young children with newly diagnosed high-risk disseminated medulloblastoma. J Clin Oncol. 2004;22:4881-4887.
Chi S.N., Zimmerman M.A., Yao X., et al. Intensive multimodality treatment for children with newly diagnosed CNS atypical teratoid rhabdoid tumor. J Clin Oncol. 2009;27:385-389.
Clarke J.L., Chang S. Pseudoprogression and pseudoresponse: challenges in brain tumor imaging. Curr Neurol Rep. 2009;9:241-246.
Cohen B.H., Zeltzer P.M., Boyett J.M., et al. Prognostic factors and treatment results for supratentorial primitive neuroectodermal tumors in children using radiation and chemotherapy: a Children’s Cancer Group randomized trial. J Clin Oncol. 1995;13:1687-1696.
Crino P.B., Nathanson K.L., Henske E.P. The tuberous sclerosis complex. N Engl J Med. 2006;355:1345-1356.
Cummings T.J., Provenzale J.M., Hunter S.B., et al. Gliomas of the optic nerve: histological, immunochemical (MIB-1 and p53), and MRI analysis. Acta Neuropathol (Berl). 2000;99:563-570.
Deliganis A.V., Geyer J.R., Berger M.S. Prognostic significance of type 1 neurofibromatosis (von Recklinghausen disease) in childhood optic glioma. Neurosurgery. 1996;38:1114-1118.
De Munnynck K., Van Gool S., Van Calenbergh F., et al. Desmoplastic infantile ganglioglioma: a potentially malignant tumor? Am J Surg Pathol. 2002;26:1515-1522.
Dhall G., Grodman H., Ji L., et al. Outcome of children less than three years old at diagnosis with non-metastatic medulloblastoma treated with chemotherapy on the “Head Start” I and II protocols. Pediatr Blood Cancer. 2008;50:1169-1175.
Duffner P.K., Krischer J.P., Sanford R.A., et al. Prognostic factors in infants and very young children with intracranial ependymomas. Pediatr Neurosurg. 1998;28:215-222.
Dunkel I.J., Gardner S.L., Garvin J.H., et al. High-dose carboplatin, thiotepa and etoposide with autologous stem cell rescue for patients with previously irradiated recurrent medulloblastoma. Neuro Oncol. 2010;12:297-303.
Durante M., Loeffler J.S. Charged particles in radiation oncology. Nat Rev Clin Oncol. 2010;7:37-43.
Ebert C., von Haken M., Meyer-Puttlitz B., et al. Molecular genetic analysis of ependymal tumors. Am J Pathol. 1999;155:627-632.
Echevarria M.E., Fangusaro J., Goldman S. Pediatric central nervous system germ cell tumors: a review. Oncologist. 2008;13(6):690-699.
Ehrbrecht A., Muller U., Wolter M., et al. Comprehensive genomic analysis of desmoplastic medulloblastomas: identification of novel amplified genes and separate evaluation of the different histological components. J Pathol. 2006;208:554-563.
Elliott R.E., Hsieh K., Hochm T., et al. Efficacy and safety of radical resection of primary and recurrent craniopharyngiomas in 86 children. J Neurosurg Pediatr. 2010;5(1):27-29.
Ernestus R.I., Schroder R., Stutzer H., et al. Prognostic relevance of localization and grading in intracranial ependymomas of childhood. Childs Nerv Syst. 1996;12:522-526.
Fangusaro J., Finlay J., Sposto R., et al. Intensive chemotherapy followed by consolidative myeloablative chemotherapy with autologous hematopoietic cell rescue (AuHCR) in young children with newly diagnosed supratentorial primitive neuroectodermal tumors (sPNET’s): a report of the Head Start I and II experience. Pediatr Blood Cancer. 2008;50:312-318.
Felton K.E., Gilchrist D.M., Andrew S.E. Constitutive deficiency in DNA mismatch repair. Clin Genet. 2004;71:483-498.
Finelli D.A., Shurin S.B., Bardenstein D.S. Trilateral retinoblastoma: two variations. AJNR Am J Neuroradiol. 1995;16:166-170.
Finkelstein-Shechter T., Gassas A., Mabbott D., et al. Atypical teratoid or rhabdoid tumors: improved outcome with high dose chemotherapy. J Pediatr Hematol Oncol. 2010;32:e182-e186.
Finlay J.L., Zacharoulis S. The treatment of high grade gliomas and diffuse intrinsic pontine tumors of childhood and adolescence: a historical—and futuristic—perspective. J Neurooncol. 2005;75:253-266.
Fleischman A., Brue C., Poussaint T.Y., et al. Diencephalic syndrome: a cause of failure to thrive and a model of partial growth hormone resistance. Pediatrics. 2005;115:e742-e748.
Franz D.N., Leonard J., Tudor C., et al. Rapamycin causes regression of astrocytomas in tuberous sclerosis complex. Ann Neurol. 2006;59:490-498.
Friedman J.H., Shetty N. Permanent cerebellar toxicity of cytosine arabinoside (AraC) in a young woman. Mov Disord. 2001;16:575-577.
Fuller G.N. The WHO classification of tumors of the central nervous system, fouth ed. Arch Pathol Lab Med. 2008;132:906.
Gajjar A., Chintagumpala M., Ashley D., et al. Risk-adapted craniospinal radiotherapy followed by high-dose chemotherapy and stem-cell rescue in children with newly diagnosed medulloblastoma (St Jude Medulloblastoma- 96): long term follow up results from a prospective multicenter trial. Lancet Oncol. 2006;7:813-820.
Gardner S.L. Application of stem cell transplant for brain tumors. Pediatr Transplant. 2004;8(Suppl 5):28-32.
Gardner S.L., Asgharzadeh S., Green A., et al. Intensive induction chemotherapy followed by high dose chemotherapy with autologous hematopoietic progenitor cell rescue in young children newly diagnosed with central nervous system atypical teratoid rhabdoid tumors. Pediatr Blood Cancer. 2008;51:235-240.
Giannini C., Scheithauer B.W., Burger P.C., et al. Pleomorphic xanthoastrocytoma: what do we really know about it? Cancer. 1999;85:2033-2045.
Gilbertson R.J., Bentley L., Hernan R., et al. ERBB receptor signaling promotes ependymoma cell proliferation and represents a potential novel therapeutic target for this disease. Clin Cancer Res. 2002;8:3054-3064.
Gilbertson R.J., Ellison D.W. The origins of medulloblastoma subtypes. Annu Rev Pathol. 2008;3:341-365.
Gilbertson R.J., Hill D.A., Hernan R., et al. ERBB1 is amplified and overexpressed in high-grade diffusely infiltrative pediatric brain stem glioma. Clin Cancer Res. 2003;9:3620-3624.
Giunti L., Cetica V., Ricci U., et al. Type A microsatellite instability in pediatric gliomas as an indicator of Turcot syndrome. Eur J Hum Genet. 2009;17:919-927.
Giussani C., Poliakov A., Ferri R.T. DTI fiber tracking to differentiate demyelinating diseases from diffuse brain stem glioma. Neuroimage. 2010;52:217-223.
Gonzalez J., Kumar A.J., Conrad C.A., et al. Effect of bevacizumab on radiation necrosis of the brain. Int J Radiat Oncol Biol Phys. 2007;67:323-326.
Gonzalez K.D., Noltner K.A., Buzin C.H., et al. Beyond Li Fraumeni Syndrome: clinical characteristics of families with p53 germline mutations. J Clin Oncol. 2009;27:1250-1260.
Gopa 1 P., Parker J.R., Debski R., et al. Choroid plexus carcinoma. Arch Pathol Lab Med. 2008;132:1350-1354.
Grill J., Le Deley M.C., Gambarelli D., et al. Postoperative chemotherapy without irradiation for ependymoma in children under 5 years of age: a multicenter trial of the French Society of Pediatric Oncology. J Clin Oncol. 2001;19:1288-1296.
Gururangan S., Chi S.N., Young Poussaint T., et al. Lack of efficacy of bevacizumab plus irinotecan in children with recurrent malignant glioma and diffuse brainstem glioma: a Pediatric Brain Tumor Consortium study. J Clin Oncol 2010. 2010;28:3069-3075.
Hahn H., Wicking C., Zaphiropoulous P.G., et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell. 1996;85:841-851.
Hamilton M.G., Lauryssen C., Hagen N. Focal midbrain glioma: long term survival in a cohort of 16 patients and the implications for management. Can J Neurol Sci. 1996;23:204-207.
Hargrave D., Bartels U., Bouffet E. Diffuse brainstem glioma in children: critical review of clinical trials. Lancet Oncol. 2006;7:241-248.
Hargrave D., Chuang N., Bouffet E. Conventional MRI cannot predict survival in childhood diffuse intrinsic pontine glioma. J Neurooncol. 2008;86:313-319.
Hasegawa T., Kondziolka D., Hadjipanayis C.G., et al. Management of cystic craniopharyngiomas with phosphorous-32 intracavitary irradiation. Neurosurgery. 2004;54:813-820.
Heideman R.L., Packer R.J., Albright A.L., et al. Tumors of the central nervous system. In: Pizza P.A., Poplack D.G. Principles and Practice of Pediatric Oncology. third ed. New York: Lippincott-Raven; 1997:633-697.
Heikens J., Michiels E.M., Behrendt H., et al. Long-term neuro-endocrine sequelae after treatment for childhood medulloblastoma. Eur J Cancer. 1998;34:1592-1597.
Helton K.J., Gajjar A., Hill D.A., et al. Medulloblastoma metastatic to the suprasellar region at diagnosis: a report of six cases with clinicopathologic correlation. Pediatr Neurosurg. 2002;37:111-117.
Hilden J.M., Meerbaum S., Burger P., et al. Central nervous system atypical teratoid/rhabdoid tumor: results of therapy in children enrolled in a registry. J Clin Oncol. 2004;22:2877-2884.
Hirato J., Nakazato Y. Pathology of pineal region tumors. J Neurooncol. 2001;54:239-249.
Hoffman K.E., Yock T.I. Radiation therapy for pediatric central nervous system tumors. J Child Neurol. 2009;24:1387-1396.
Horbinski C., Hamilton R.L., Lovell C., et al. Impact of morphology, MIB-1, p53 and MGMT on outcome in pilocytic astrocytomas. Brain Pathol. 2010;20:581-588.
Horn B., Heideman R., Geyer R., et al. A multi-institutional retrospective study of intracranial ependymoma in children: identification of risk factors. J Pediatr Hematol Oncol. 1999;21:203-211.
Hottinger A.F., Khakoo Y. Neurooncology of familial cancer syndromes. J Child Neurol. 2009;24:1526-1535.
Hulsebos T.J.M., Oskam N.T., Bijleveld E.H., et al. Evidence for an ependymoma tumour suppressor gene in chromosome region 22pter–22q11.2. Nature. 1999;81:1150-1154.
Jahraus C.D., Tarbell N.J. Optic pathway gliomas. Pediatr Blood Cancer. 2006;46:586-596.
Jallo G.I., Biser-Rohrbaugh A., Freed D. Brainstem gliomas. Childs Nerv Syst. 2004;20:143-153.
Jallo G.I., Danish S., Velasquez L., et al. Intramedullary low-grade astrocytomas: long-term outcome following radical surgery. J Neurooncol. 2001;53:61-66.
Jeibmann A., Hasselblastt M., Gerss J., et al. Prognostic implications of atypical histologic features in choroid plexus papilloma. J Neuropathol Exp Neurol. 2006;65:1069-1073.
Johnson J.H.,Jr., Hariharan S., Berman J., et al. Clinical outcome of pediatric gangliogliomas: 99 cases over 20 years. Pediatr Neurosurg. 1997;27:203-207.
Jozwiak J., Jozwiak S., Wlodarski P. Possible mechanisms of disease development in tuberous sclerosis. Lancet Oncol. 2009;9:73-79.
Kalapurakal J.A. Radiation therapy in the management of pediatric craniopharyngiomas—a review. Childs Nerv Sys. 2005;21:808-816.
Kamiya M., Nakazato Y. The expression of p73, p21 and MDM2 proteins in gliomas. J Neurooncol. 2002;59:143-149.
Kantar M., Cetingul N., Savas K., et al. Radiotherapy-induced secondary cranial neoplasms in children. Childs Nerv Syst. 2004;20:46-49.
Karavitaki N., Brufani C., Warner J.T., et al. Craniopharyngiomas in children and adults: systematic analysis of 121 cases with long-term follow-up. Clin Endocrinol. 2005;62:397-409.
Kato T., Sawamura Y., Tada M., et al. Cisplatin/vincristine chemotherapy for hypothalamic/visual pathway astrocytomas in young children. J Neurooncol. 1998;37:263-270.
Keene D., Johnston D., Strother D., et al. Epidemiological survey of central nervous system germ cell tumors in Canadian children. J Neurooncol. 2007;82(3):289-295.
Kellie S.J., Boyce H., Dunkel I.J., et al. Primary chemotherapy for intracranial nongerminomatous germ cell tumors: results of the Second International CNS Germ Cell Study Group protocol. J Clin Oncol. 2004;22:846-853.
Kellie S.J., Wong C.K., Pozza L.D., et al. Activity of postoperative carboplatin, etoposide, and high-dose methotrexate in pediatric CNS embryonal tumors: results of a phase II study in newly diagnosed children. Med Pediatr Oncol. 2002;39:168-174.
Kendall-Taylor P., Jonsson P.J., Abs R., et al. The clinical, metabolic and endocrine features and the quality of life in adults with childhood-onset craniopharyngioma compared with adult-onset craniopharyngioma. Eur J Endocrinol. 2005;152:557-567.
Kesari S., Schiff D., Drappatz J., et al. Phase II Study of Protracted Daily Temozolomide for Low-Grade Gliomas in Adults. Clin Cancer Res. 2009;15:330-337.
Kim S.D., Park J.Y., Park J., et al. Radiological findings following postsurgical intratumoral bleomycin injection for cystic craniopharyngioma. Clin Neurol Neurosurg. 2007;109:236-241.
Kimonis V.E., Goldstein A.M., Pastakia B., et al. Clinical manifestations in 105 persons with nevoid basal cell carcinoma syndrome. Am J Med Genet. 1997;69:299-308.
Kivela T. Trilateral retinoblastoma: a meta-analysis of hereditary retinoblastoma associated with primary ectopic intracranial retinoblastoma. J Clin Oncol. 1999;17:1829-1837.
Komotar R.J., Mocco J., Jones J.E. Pilomyxoid astrocytoma: diagnosis, prognosis, and management. Neurosurg Focus. 2005;15:1-4. E7
Kool M., Koster J., Bunt J., et al. Integrated genomics identifies five medulloblastoma subtypes with distinct genetic profiles pathway signatures and clinicopathological features. PLoS One. 2008;3:1-14.
Korshunov A., Golanov A., Timirqaz V. Immunohistochemical markers for prognosis of ependymal neoplasms. J Neurooncol. 2002;58:255-270.
Kreiger P.A., Okada Y., Simon S., et al. Losses of chromosomes 1p and 19q are rare in pediatric oligodendrogliomas. Acta Neuropathol. 2005;109:387-392.
Kretschmar C., Kleinberg L., Greenberg M., et al. Pre-radiation chemotherapy with response-based radiation therapy in children with central nervous system germ cell tumors: a report from the Children’s Oncology Group. Pediatr Blood Cancer. 2007;48:285-291.
Kumar R., Singh V. Subependymal giant cell astrocytoma: a report of five cases. Neurosurg Rev. 2004;27:274-280.
Lafay-Cousin L., Holm S., Qaddoumi I., et al. Weekly vinblastine in pediatric low-grade glioma patients with carboplatin allergic reaction. Cancer. 2005;103:2636-2642.
Lafay-Cousin L., Strother D. Current treatment approaches for infants with central nervous system tumors. Oncologist. 2007;14:433-444.
Lam C., Bouffet E., Bartels U. Nimotuzumab in pediatric glioma. Future Oncol. 2009;5:1349-1361.
Lancaster D.L., Hoddes J.A., Michalski A. Tolerance of nitrosurea-based multiagent chemotherapy regime for low-grade pediatric gliomas. J Neurooncol. 2003;63:289-294.
Lasky J.L., Wu H. Notch signaling, brain development and human disease. Pediatr Res. 2005;57(5 Pt 2):104R-109R.
Lee J., Parsa A.T., Ames C.P., et al. Clinical management of intramedullary spinal ependymomas in adults. Neurosurg Clin N Am. 2006;17:21-27.
Leenstra J.L., Rodriguez F.J., Frechette C.M., et al. Central neurocytoma: Management recommendations based on a 35-year experience. Int J Radiat Oncol Biol Phys. 2007;67:1145-1154.
Lewanski C.R., Sinclair J.A., Stewart J.S. Lhermitte’s sign following head and neck radiotherapy. Clin Oncol (R Coll Radiol). 2000;12:98-103.
Lewis P.M., Gritili-Linde A., Seine R., et al. Sonic hedgehog signaling is required for expansion of granule neuron precursors and patterning of the mouse cerebellum. Dev Biol. 2004;270:393-410.
Li M.H., Bouffet E., Hawkins C.E., et al. Molecular genetics of supratentorial primitive neuroectodermal tumors and pineoblastoma. Neurosurg Focus. 2005;19:E3.
Liauw S.L., Byer J.E., Yachnis A.T., et al. Radiotherapy after subtotally resected or recurrent ganglioglioma. Int J Radiat Oncol Biol Phys. 2007;67:244-247.
Linabery A.M., Ross J.A. Trends in childhood cancer incidence in the U.S. (1992-2004). Cancer. 2008;112:416-432.
Luis D.N., Ohgaki H., Wiestler O.D., et al. WHO Classification of Tumours of the Central Nervous System. Lyon, France: International Agency for Research on Cancer, 2007.
Luyken C., Blumcke I., Fimmers R., et al. Supratentorial gangliogliomas: histopathologic grading and tumor recurrence in 184 patients with median follow-up of 8 years. Cancer. 2004;101:146-155.
MacDonald S.M., DeLaney T.F., Loeffler J.S. Proton beam radiation therapy. Cancer Invest. 2006;24:199-208.
MacDonald S.M., Yock T.I. Proton beam therapy following resection for childhood ependymoma. Childs Nerv Syst. 2010;26:285-291.
Mackenzie I.R.A. Central neurocytoma: histologic atypia, proliferation potential, and clinical outcome. Cancer. 2000;85:1606-1610.
Mahoney D.H., Shuster J.J., Nitschke R., et al. Acute neurotoxicity in children with B-precursor acute lymphoid leukemia: an association with intermediate-dose intravenous methotrexate and intrathecal triple therapy—a Pediatric Oncology Group study. J Clin Oncol. 1998;16:1712-1722.
Mansur D.B., Drzymala R.E., Rich K.M., et al. The efficacy of stereotactic radiosurgery in the management of intracranial ependymoma. J Neurooncol. 2004;66:187-190.
Marchal J.C., Klein O., Thouvenot P., et al. Individualized treatment of craniopharyngioma in children: ways and means. Childs Nerv Syst. 2005;21:665-669.
Mauffrey C. Pediatric brainstem gliomas: prognostic factors and management. J Clin Neurosci. 2006;13:431-437.
Mazloom A., Wolff J.E., Paulino A.C. The impact of radiotherapy fields in the treatment of patients with choroids plexus carcinoma. Int J Radiat Oncol Biol Phys. 2010. [epub ahead of print]
McLaughlin M.P., Marcus R.B., Buatti J.M., et al. Ependymoma: results, prognostic factors and treatment recommendations. Int J Radiat Oncol Biol Phys. 1998;40:845-850.
McNeil D.E., Cote T.R., Clegg L., et al. Incidence and trends in pediatric malignancies medulloblastoma/primitive neuroectodermal tumor: a SEER update. Surveillance Epidemiology and End Results. Med Pediatr Oncology. 2002;39:190-194.
Merchant T.E. Current management of childhood ependymoma. Oncology. 2002;16:629-642.
Merchant T.E., Boop F.A., Kun L.E., et al. A retrospective study of surgery and re-irradiation for recurrent ependymoma. Int J Radiat Oncol Biol Phys. 2008;71:87-97.
Merchant T.E., Hua C.E., Shukla H., et al. Proton versus photon radiotherapy for common pediatric brain tumors: comparison of models of dose characteristics and their relationship to cognitive function. Pediatr Blood Cancer. 2008;51:110-117.
Merchant T.E., Kun L.E., Wu S., et al. Phase II trial of conformal radiation therapy for pediatric low-grade glioma. J Clin Oncol. 2009;27:3598-3604.
Merchant T.E., Mulhern R.K., Krasin M.J., et al. Preliminary results from a phase II trial of conformation radiation therapy and evaluation of radiation-related CNS effects for pediatric patients with localized ependymoma. J Clin Oncol. 2004;22:3156-3162.
Moreno L., Bautista F., Ashley S., et al. Does chemotherapy affect the visual outcome in children with optic pathway glioma? A systematic review of the evidence. Eur J Cancer. 2010;46:2253-2259.
Mulhern R.K., Palmer S.L., Merchant T.E., et al. Neurocognitive consequences of risk-adapted therapy for childhood medulloblastoma. J Clin Oncol. 2005;23:5511-5519.
Mulhern R.K., Reddick W.E., Palmer S.L., et al. Neurocognitive deficits in medulloblastoma survivors and white matter loss. Ann Neurol. 1999;46:834-841.
Nabbout R., Santos M., Rolland Y., et al. Early diagnosis of subependymal giant cell astrocytoma in children with tuberous sclerosis. J Neurol Neurosurg Psych. 1999;66:370-375.
Nagib M.G., O’Fallon M.T. Myxopapillary ependymoma of the conus medullaris and filum terminale in the pediatric age group. Pediatr Neurosurg. 1997;26:2-7.
Narayana A., Kunnakkat S., Chacko-Mathew J., et al. Bevacizumab in recurrent high-grade pediatric gliomas. Neuro Oncol. 2010;12:985-990.
Nicholson H.S., Kretschmar C.S., Krailo M., et al. Phase 2 study of temozolomide in children and adolescents with recurrent central nervous system tumors: a report from the Children’s Oncology Group. Cancer. 2007;110:1542-1550.
Nicolin G., Parkin P., Mabbott D., et al. Natural history and outcome of optic pathway gliomas in children. Pediatr Blood Cancer. 2009;53:1231-1237.
Northcott P.A., Korshunov A., Witt H., et al. Medulloblastoma comprises four distinct molecular variants. J Clin Oncol. 2011;29:1408-1414.
Northcott P.A., Rutka J.T., Taylor M.D. Genomics of medulloblastoma: from Giemsa-banding to next-generation sequencing in 20 years. Neurosurg Focus. 2010;28:E6.
Ogawa K., Shikama N., Toita T., et al. Long-term results of radiotherapy for intracranial germinoma: a multi-institutional retrospective review of 126 patients. Int J Radiat Oncol Biol Phys. 2004;58:705-713.
Ogino H., Shibamoto Y., Takanaka T., et al. CNS germinoma with elevated serum human chorionic gonadotropin level: clinical characteristics and treatment outcome. Int J Radiat Oncol Biol Phys. 2005;62:803-808.
Okamoto H., Di Patre P.L., Burkhard C., et al. Population-based study on incidence, survival rates, and genetic alterations of low-grade diffuse astrocytomas and oligodendrogliomas. Acta Neuropathol. 2004;108:49-56.
Okamoto H., Mineta T., Ueda S., et al. Detection of JC virus DNA sequences in brain tumors in pediatric patients. J Neurosurg. 2005;102:294-298.
Olson T.A., Bayar E., Kosnik E., et al. Successful treatment of disseminated central nervous system malignant rhabdoid tumor. J Pediatr Hematol Oncol. 1995;17:71-75.
Opocher E., Kremer L.C.M., Da Dalt L., et al. Prognostic factors for progression of childhood optic pathway glioma: a systematic review. Eur J Cancer. 2006;42:1807-1816.
Oyharcabal-Bourden V., Kalifa C., Gentet J.C., et al. Standard-risk medulloblastoma treated by adjuvant chemotherapy followed by reduced-dose craniospinal radiation therapy: a French Society of Pediatric Oncology study. J Clin Oncol. 2005;23:4726-4734.
Packer R.J., Alter J., Allen J., et al. Carboplatin and vincristine chemotherapy for children with newly diagnosed progressive low-grade gliomas. J Neurosurg. 1997;86:747-754.
Packer R.J., Cohen B.H., Coney K. Intracranial germ cell tumors. Oncologist. 2000;5:312-329.
Packer R.J., Gajjar A., Vezina G., et al. Phase III study of craniospinal radiation therapy followed by adjuvant chemotherapy for newly diagnosed average-risk medulloblastoma. J Clin Oncol. 2006;24:4202-4208.
Packer R.J., Jakacki R., Horn M., et al. Objective response of multiply recurrent low-grade gliomas to bevacizumab and irinotecan. Pediatr Blood Cancer. 2009;52:791-795.
Parsa C.F., Hoyt C.S., Lesser R.L., et al. Spontaneous regression of optic gliomas: thirteen cases documented by serial neuroimaging. Arch Ophthalmol. 2001;119:516-529.
Paugh B.S., Qu C., Jones C., et al. Integrated molecular genetic profiling of pediatric high-grade gliomas reveals key differences with the adult disease. J Clin Oncol. 2010;28:3061-3068.
Paulino A.C., Wen B.C., Buatti J.M., et al. Intracranial ependymomas: an analysis of prognostic factors and patterns of failure. Am J Clin Oncol. 2002;25:117-122.
Pedreira C.C., Stargatt R., Maroulis H., et al. Health related quality of life and psychological outcome in patients treated for craniopharyngioma in childhood. J Pediatr Endocrinol Metab. 2006;19:15-24.
Perilongo G., Massimino M., Sotti G., et al. Analyses of prognostic factors in a retrospective review of 92 children with ependymoma: Italian Pediatric Neuro-Oncology Group. Med Pediatr Oncol. 1998;29:79-85.
Peters O., Gnekow A.K., Rating D., et al. Impact of location on outcome in children with low-grade oligodendroglioma. Pediatr Blood Cancer. 2004;43:250-256.
Pollack I.F. The role of surgery in pediatric gliomas. J Neurooncol. 1999;42:271-288.
Pollack I.F., Finkelstein S.D., Woods J., et alfor the Children’s Cancer Group. Expression of p53 and prognosis in children with malignant gliomas. N Engl J Med. 2002;346:420-427.
Pollack I.F., Gerszten P.C., Martinez A.J., et al. Intracranial ependymomas of childhood: long-term outcome and prognostic factors. Neurosurgery. 1995;37:655-666.
Pollack I.F., Mulvill J.J. Neurofibromatosis 1 and 2. Brain Pathol. 1997;7:823-836.
Pomeroy S.L., Tomayo P., Gaasenbeek M., et al. Prediction of central nervous system embryonal tumour outcome based on gene expression. Nature. 2002;415:436-442.
Portwine C., Chilton-MacNeill S., Brown C., et al. Absence of germline and somatic p53 alterations in children with sporadic brain tumors. J Neurooncol. 2001;52:227-235.
Prados M.D., Edwards M.S., Rabbitt J., et al. Treatment of pediatric low-grade gliomas with a nitrosurea-based multiagent chemotherapy regimen. J Neurooncol. 1997;32:235-241.
Qaddoumi I., Sultan I., Gajjar A. Outcome and prognostic features in pediatric gliomas. Cancer. 2009;115:5761-5770.
Quasthoff S., Hartung H.P. Chemotherapy-induced peripheral neuropathy. J Neurol. 2002;249:9-17.
Quinn J.A., Reardon D.A., Friedman A.H., et al. Phase II trial of temozolomide in patients with progressive low-grade glioma. J Clin Oncol. 2003;21:646-651.
Rades D., Schild S., Fehlauer F. Prognostic value of the MIB-1 labeling index for central neurocytomas. Neurology. 2004;62:987-989.
Rades D., Zwick L., Leppert J. The role of postoperative radiotherapy for the treatment of gangliogliomas. Cancer. 2010;116:432-442.
Raffel C., Jenkins R.B., Frederick L., et al. Sporadic medulloblastomas contain PTCH mutations. Cancer Res. 1997;57:842-845.
Raghavan R., Balani J., Perry A. Pediatric oligodendrogliomas: a study of molecular alterations on 1p and 19q using fluorescence in situ hybridization. Neuropathol Exp Neurol. 2003;62:530-537.
Rao A.A., Laack N.N., Giannini C., et al. Pleomorphic xanthoastrocytoma in children and adolescents. Pediatr Blood. Cancer. 2010;55:290-294.
Read T.A., Hegedus B., Wechsler-Reya R., et al. The neurobiology of neuro-oncology. Ann Neurol. 2006;60:3-11.
Reardon D.A., Gajjar A., Sanford R.A., et al. Bithalamic involvement predicts poor outcome among children with thalamic glial tumors. Pediatr Neurosurg. 1998;29:29-35.
National Cancer Institute, SEER Program. NIH Pub. No. 99-4649, Bethesda, MD, Ries L.A.G., Smith M.A., Gurney J.G. Cancer Incidence and Survival among Children and Adolescents: United States SEER Program 1975-1995. 1999.
Roberts C.W.M., Biegel J.A. The role of SMARCB1/INI1 in development of rhabdoid tumor. Cancer Biol Ther. 2009;8:412-416.
Rodriguez H.A., Berthrong M. Multiple primary intracranial tumors in von Recklinghausen’s neurofibromatosis. Arch Neurol. 1996;14:467-475.
Rosemberg S., Vieira G.S. Dysembryoplastic neuroepithelial tumor: an epidemiologic study from a single institution. Arq Neuropsiquiatr. 1998;56:223-238.
Rossi A., Caracciolo V., Russo G., et al. Medulloblastoma: from molecular pathology to therapy. Clin Cancer Res. 2008;14:971-976.
Rutkowski S., Bode U., Deinlein F., et al. Treatment of early childhood medulloblastoma by postoperative chemotherapy alone. N Engl J Med. 2005;352:978-986.
Ryan J. Radiation somnolence syndrome. J Pediatr Oncol Nurs. 2000;17:50-53.
Saint-Rose C., Puget S., Wray A., et al. Craniopharyngioma: the pendulum of surgical management. Childs Nerv Syst. 2005;21:691-695.
Samuel D.P., Wen P.Y., Kieran M.W. Antiangiogenic (metronomic) chemotherapy for brain tumors: current and future perspectives. Expert Opin Invest Drugs. 2009;18:973-983.
Sandberg D.I., Ragheb J., Dunover C., et al. Surgical outcomes and seizure control rates after resection of dysembryoplastic neuroepithelial tumors. Neurosurg Focus. 2005;18:1-4.
Santoro A., Minniti G., Paolini S., et al. Atypical tentorial meningioma 30 years after radiotherapy for a pituitary adenoma. Neurol Sci. 2002;22:463-467.
Schmidt M.H., Gottfried O.N., von Koch C.S., et al. Central neurocytoma: a review. J Neuro-oncol. 2004;66:377-384.
Serdaroglu A., Simsek F., Gucuyener K. Moyamoya syndrome after radiation therapy for optic pathway glioma: case report. J Child Neurol. 2000;15:765-767.
Seregni E., Massimino M., Nerini Molteni S., et al. Serum and cerebrospinal fluid human chorionic gonadotropin (hCG) and alpha-fetoprotein (AFP) in intracranial germ cell tumors. Int J Biol Markers. 2002;17:112-118.
Sethi R., Allen J., Donahue B., et al. Prospective neuraxis MRI surveillance reveals a high risk of leptomeningeal dissemination in diffuse intrinsic pontine glioma. Neurooncol. 2010. Jul 10 [Epub ahead of print]
Sharma M.C., Deb P., Sharma S., et al. Neurocytoma: a comprehensive review. Neurosurg Rev. 2006;29:270-285.
Shaw E.G., Wisoff J.H. Prospective clinical trials of intracranial low-grade glioma in adults and children. Neuro Oncol. 2003;5:153-160.
Sievert A.J., Fisher M.J. Pediatric low-grade gliomas. J Child Neurol. 2009;24:1397-1408.
Siffert J., Allen J.C. Chemotherapy in recurrent ependymoma. Pediatr Neurosurg. 1998;28:314-319.
Silva M.M., Goldman S., Keating G., et al. Optic pathway hypothalamic gliomas in children under three years of age: the role of chemotherapy. Pediatr Neurosurg. 2000;33:151-158.
Soylemezoglu F., Scheithauer B.W., Esteve J., et al. Atypical central neurocytoma. J Neuropathol Exp Neurol. 1997;56:551-556.
Stark A., Fritsch M., Claviez A., et al. Management of tectal glioma in childhood. Pediatr Neurol. 2005;33:33-38.
Stavrou T., Bromley C.M., Nicholson H.S., et al. Prognostic factors and secondary malignancies in childhood medulloblastoma. J Pediatr Hematol Oncol. 2001;23:431-436.
Stupp R., Mason W.P., van den Bent M.J., et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987-996.
Suarez-Merino B., Hubank M., Revesz T., et al. Microarray analysis of pediatric ependymoma identifies a cluster of 112 candidate genes including four transcripts at 22q12.1-q13.3. Neuro Oncol. 2005;7:20-31.
Sugita Y., Shigemori M., Okamoto K., et al. Clinicopathological study of pleomorphic xanthoastrocytoma: correlation between histological features and prognosis. Pathol Int. 2000;50:703-708.
Sugiyama K., Arita K., Shima T., et al. Good clinical course in infants with desmoplastic cerebral neuroepithelial tumor treated by surgery alone. J Neurooncol. 2002;59:63-69.
Suzuki S.O., Iwaki T. Amplification and overexpression of mdm2 gene in ependymomas. Mod Pathol. 2000;13:548-553.
Swensen A.R., Bushhouse S.A. Childhood cancer incidence and trends in Minnesota, 1988-1994. Minn Med. 1998;81:27-32.
Tabori U., Ma J., Carter M., et al. Human telomerase reverse transcriptase expression predicts progression and survival in pediatric intracranial ependymoma. J Clin Oncol. 2006;24:1522-1528.
Tabori U., Shlien A., Baskin B., et al. TP53 alterations determine clinical subgroups and survival of patients with choroid plexus tumors. J Clin Oncol. 2010;28:1995-2001.
Takahashi H., Yamaguch F., Teramoto A. Long-term outcome and reconsideration of intracystic chemotherapy with bleomycin for craniopharyngioma in children. Childs Nerv Syst. 2005;21:701-704.
Tamber M.S., Rutka J.T. Pediatric supratentorial high-grade gliomas. Neurosurg Focus. 2003;14:e1.
Tatevossian R.G., Lawson A.R., Forshew T., et al. MAPK pathway activation and the origins of pediatric low-grade astrocytomas. J Cell Physiol. 2010;222:509-514.
Taylor M.D., Liu L., Raffel C., et al. Mutations in SUFU predispose to medulloblastoma. Genet. 2002;31:306-310.
Taylor M.D., Poppleton H., Fuller C., et al. Radial glial cells are candidate stem cells of ependymoma. Cancer Cell. 2005;8:323-335.
Tekkok I.H., Sav A. Anaplastic pleomorphic xanthoastrocytomas. Review of the literature with reference to malignancy potential. Pediatr Neurosurg. 2004;40:171-181.
Thompson M.C., Fuller C., Hogg T.L., et al. Genomics identifies medulloblastoma subgroups that are enriched for specific genetic alterations. J Clin Oncol. 2006;24:1924-1931.
von Koch C.S., Gulati M., Aldape K., et al. Familial medulloblastoma: case report of one family and review of the literature. Neurosurgery. 2002;51:227-233.
Thorarinsdottir H.K., Santi M., McCarter R., et al. Protein expression of platelet-derived growth factor receptor correlates with malignant histology and PTEN with survival in childhood gliomas. Clin Cancer Res. 2008;14:3386-3394.
Von Hoff K., Hartmann W., Van Bueren A.O., et al. Large cell/anaplastic medulloblastoma: outcome according to myc status, histopathological and clinical risk factors. Pediatr Blood Cancer. 2010;54:369-376.
Van Poppel K., Broniscer A., Patay Z., et al. Alexander disease: an important mimicker of focal brainstem glioma. Pediatr Blood Cancer. 2009;53:1355-1356.
Walker D.A., Pillow J., Waters K.D., et al. Enhanced cisplatinum ototoxicity in children with brain tumours who have received simultaneous or prior cranial irradiation. Med Pediatr Oncol. 1989;17(1):48-52.
Weber R.G., Bridger J.M., Benner A., et al. Centrosome amplification as a possible mechanism for numerical chromosome aberrations in cerebral primitive neuroectodermal tumors with TP53 mutations. Cytogenet Cell Genet. 1998;83:266-269.
Wells E.M., Khademian Z.P., Walsh K.S., et al. Postoperative cerebellar mutism syndrome following treatment of medulloblastoma: neuroradiographic features and origin. J Neurosurg Pediatrics. 2010;5:329-334.
Wetmore C., Eberhart D.E., Curran C. Loss of p53 but not ARF accelerates medulloblastoma in mice heterozygous for patched. Cancer Res. 2001;61:513-516.
Wolburg H., Paulus W. Choroid plexus: biology and pathology. Acta Neuropath. 2009;119:75-88.
Wrede B., Liu P., Wolff J.E., et al. Chemotherapy improves the survival of patients with choroid plexus carcinoma: a meta-analysis of individual cases with choroid plexus tumors. J Neurooncol. 2007;85:345-351.
Yokoto N., Nishizawa S., Ohta S., et al. Role of WNT pathway in medulloblastoma oncogenesis. Int J Cancer. 2002;101:198-201.
Yan H., Parsons D.W., Jin G., et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360:765-773.
Zarghooni M., Bartels U., Lee E., et al. Whole-genome profiling of pediatric diffuse intrinsic pontine gliomas highlights platelet-derived growth factor receptor alpha and poly (ADP-ribose) polymerase as potential therapeutic targets. J Clin Oncol. 2010;28:1337-1344.
Zeltzer P.M., Boyett J.M., Finlay J.L., et al. Metastasis stage, adjuvant treatment, and residual tumor are prognostic factors for medulloblastoma in children: conclusions from the Children’s Cancer Group 921 randomized phase II study. J Clin Oncol. 1999;17:832-845.
Zurawel R.H., Allen C., Chiappa S., et al. Analysis of PTCH/SMO/SHH pathway genes in medulloblastoma. Genes Chromos Cancer. 2000;27:44-51.