Dendritic Cells Link Innate and Adaptive Immunity
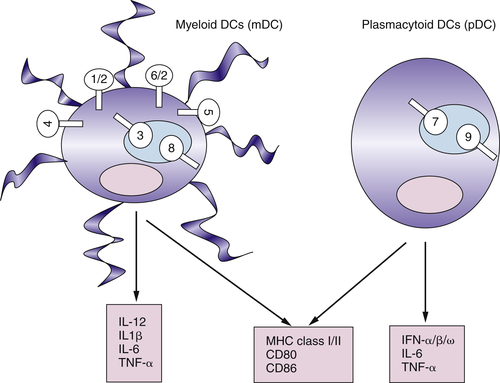
Myeloid DCs and Plasmacytoid DCs Express Different Sets of TLRs
Functional Plasticity of DCs
Targeting TLRs on DCs to Induce Effective Antitumor Immunity
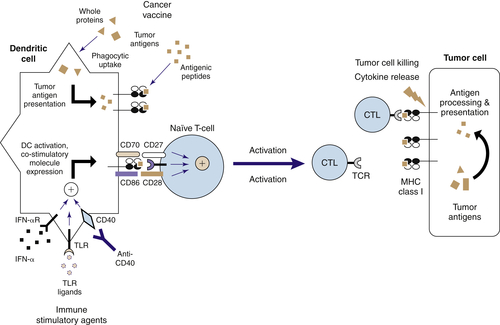
The Nature of Antitumor Immunity
T Cells Can Recognize Self-Antigens Expressed by Tumors
Autoimmune Vitiligo and Response to Immunotherapy
Cancer Vaccines, Cytokines, and Immunotherapy
Cytokine Therapy of Cancer
Current Status of Cancer Vaccines
Whole-Cell Cancer Vaccine Strategies
Antigen-Specific Vaccine Approaches
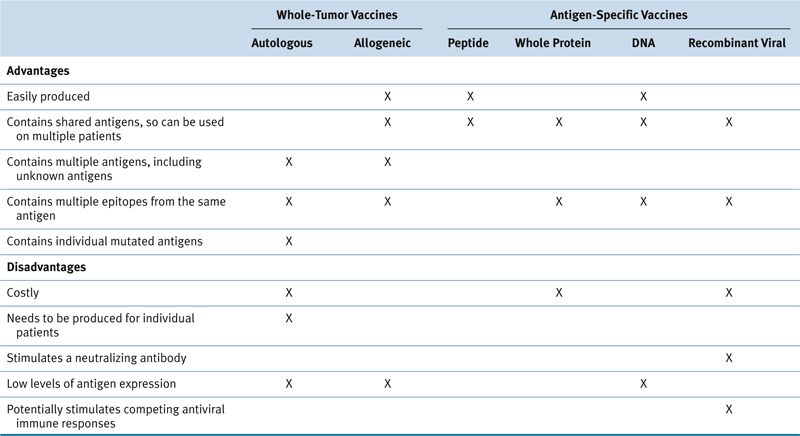
Table 51-1
Advantages and Disadvantages of Various Cancer Vaccine Approaches
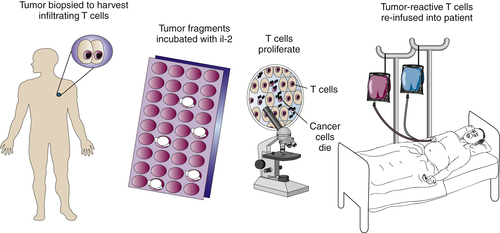
Adoptive Immunotherapy of Cancer
Tumor-Infiltrating Lymphocytes
Gene-Modified T Lymphocytes
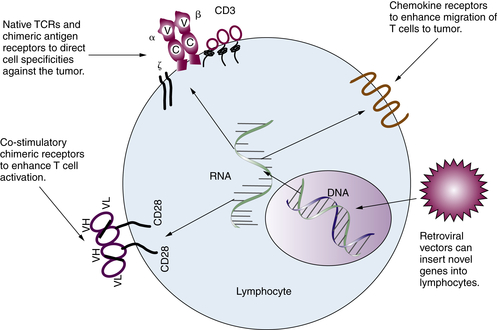
Immune Regulatory Cells and Molecules in Human Cancer
Regulatory T Lymphocytes in Human Cancer
Table 51-2
Potentially Targetable Immunoregulatory Molecules
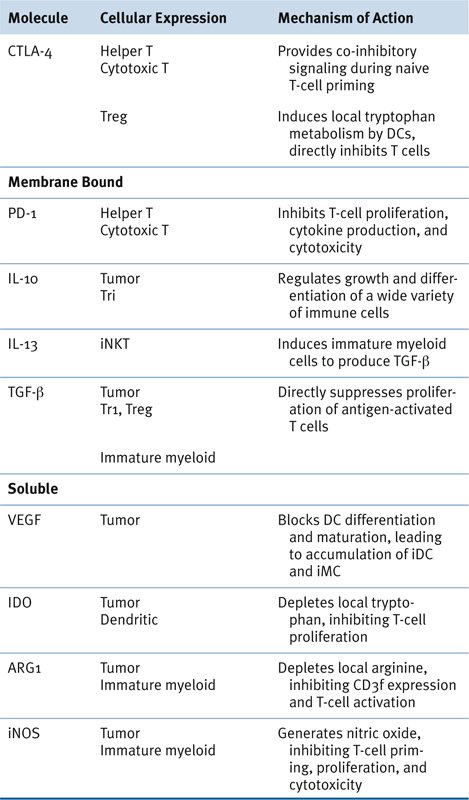
Myeloid-Derived Suppressor Cells in Human Cancer
Immune Checkpoint Blockade
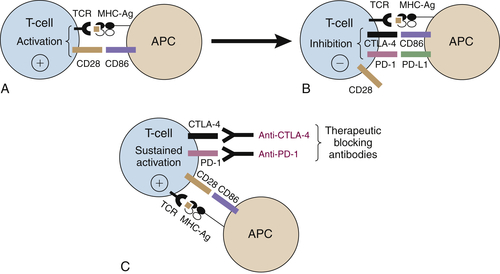
CTLA-4 Blockade
PD-1/PD-L1 Blockade
Checkpoint Blockade and the Induction of Autoimmunity
Future Cancer Immunotherapies Will Use Basic Principles of Cellular Immunity
1. Cancer vaccines . Immunol Today . 1993 ; 14 : 310 – 316 .
2. Progress in the development of immunotherapy for the treatment of patients with cancer . J Intern Med . 2001 ; 250 : 462 – 475 .
3. Adoptive T cell therapy of tumors: mechanisms operative in the recognition and elimination of tumor cells . Adv Immunol . 1991 ; 49 : 281 – 355 .
4. Raising the bar: the curative potential of human cancer immunotherapy . Sci Transl Med . 2012 ; 4 127ps8 .
5. A genomic view of immunology . Nature . 2001 ; 409 : 836 – 838 .
6. Innate immune recognition . Annu Rev Immunol . 2002 ; 20 : 197 – 216 .
7. Toll-like receptors and innate immunity . Adv Immunol . 2001 ; 78 : 1 – 56 .
8. Toll-like receptors and innate immunity . Nat Rev Immunol . 2001 ; 1 : 135 – 145 .
9. Toll-like receptors in the induction of the innate immune response . Nature . 2000 ; 406 : 782 – 787 .
10. Subsets of human dendritic cell precursors express different toll-like receptors and respond to different microbial antigens . J Exp Med . 2001 ; 194 : 863 – 869 .
11. Dendritic cells and the control of immunity . Nature . 1998 ; 392 : 245 – 252 .
12. Dendritic cell subsets and lineages, and their functions in innate and adaptive immunity . Cell . 2001 ; 106 : 259 – 262 .
13. Active immunization against cancer with dendritic cells: the near future . Int J Cancer . 2001 ; 94 459-173 .
14. The dorsoventral regulatory gene cassette spatzle/Toll/cactus controls the potent antifungal response in Drosophila adults . Cell . 1996 ; 86 : 973 – 983 .
15. Innate immunity: impact on the adaptive immune response . Curr Opin Immunol . 1997 ; 9 : 4 – 9 .
16. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene . Science . 1998 ; 282 : 2085 – 2088 .
17. Thymic stromal lymphopoietin: master switch for allergic inflammation . J Exp Med . 2006 ; 203 : 269 – 273 .
18. Microbial compounds selectively induce Th1 cell-promoting or Th2 cell-promoting dendritic cells in vitro with diverse Th cell-polarizing signals . J Immunol . 2002 ; 168 : 1704 – 1709 .
19. Triggering TLR signaling in vaccination . Trends Immunol . 2006 ; 27 : 49 – 55 .
20. IPC: professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors . Annu Rev Immunol . 2005 ; 23 : 275 – 306 .
21. Plasmacytoid dendritic cells induce NK cell-dependent, tumor antigen-specific T cell cross-priming and tumor regression in mice . J Clin Invest . 2008 ; 118 : 1165 – 1175 .
22. Human tumor antigens for cancer vaccine development . Immunol Rev . 1999 ; 170 : 85 – 100 .
23. Vitiligo in patients with melanoma: normal tissue antigens can be targets for cancer immunotherapy . J Immunother Emphasis Tumor Immunol . 1996 ; 19 : 81 – 84 .
24. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993 . J Clin Oncol . 1999 ; 17 : 2105 – 2116 .
25. Update on adjuvant interferon therapy for high-risk melanoma . Oncology (Williston Park) . 2002 ; 16 : 1177 – 1187 discussion 90-92, 97 .
26. Prognostic significance of autoimmunity during treatment of melanoma with interferon . N Engl J Med . 2006 ; 354 : 709 – 718 .
27. Vaccination with irradiated tumor cells engineered to secrete murine granulocyte-macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity . Proc Natl Acad Sci U S A . 1993 ; 90 : 3539 – 3543 .
28. Role of bone marrow-derived cells in presenting MHC class I-restricted tumor antigens . Science . 1994 ; 264 : 961 – 965 .
29. GM-CSF-secreting melanoma vaccines . Oncogene . 2003 ; 22 : 3188 – 3192 .
30. MHC class I and class II presentation of tumor antigen in retrovirally and adenovirally transduced dendritic cells . Cancer Gene Ther . 2002 ; 9 : 946 – 950 .
31. Vaccination for leukemia . Biol Blood Marrow Transplant . 2006 ; 12 : 13 – 18 .
32. Impact of cytokine administration on the generation of antitumor reactivity in patients with metastatic melanoma receiving a peptide vaccine . J Immunol . 1999 ; 163 : 1690 – 1695 .
33. Cancer immunotherapy: moving beyond current vaccines . Nat Med . 2004 ; 10 : 909 – 915 .
34. Donor lymphocyte infusions to treat hematologic malignancies in relapse after allogeneic blood or marrow transplantation . Cancer Control . 2002 ; 9 : 123 – 137 .
35. Adoptive immunotherapy of EBV-associated malignancies with EBV-specific cytotoxic T-cell lines . Curr Top Microbiol Immunol . 2001 ; 258 : 221 – 229 .
36. In vitro predictors of therapeutic response in melanoma patients receiving tumor-infiltrating lymphocytes and interleukin-2 . J Clin Oncol . 1994 ; 12 : 1475 – 1483 .
37. Shedding light on immunotherapy for cancer . N Engl J Med . 2004 ; 350 : 1461 – 1463 .
38. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes . Science . 2002 ; 298 : 850 – 854 .
39. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma . J Clin Oncol . 2005 ; 23 : 2346 – 2357 .
40. Dendritic cells strongly boost the antitumor activity of adoptively transferred T cells in vivo . Cancer Res . 2004 ; 64 : 6783 – 6790 .
41. Cancer regression in patients after transfer of genetically engineered lymphocytes . Science . 2006 ; 314 : 126 – 129 .
42. Lysis of ovarian cancer cells by human lymphocytes redirected with a chimeric gene composed of an antibody variable region and the Fc receptor gamma chain . J Exp Med . 1993 ; 178 : 361 – 366 .
43. kat: a high-efficiency retroviral transduction system for primary human T lymphocytes . Blood . 1994 ; 83 : 43 – 50 .
44. Targeting of T lymphocytes to Neu/HER2-expressing cells using chimeric single chain Fv receptors . J Immunol . 1993 ; 151 : 6577 – 6582 .
45. Recognition of human colon cancer by T cells transduced with a chimeric receptor gene . Cancer Gene Ther . 2000 ; 7 : 284 – 291 .
46. Redirecting migration of T cells to chemokine secreted from tumors by genetic modification with CXCR2 . Hum Gene Ther . 2002 ; 13 : 1971 – 1980 .
47. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta/CD28 receptor . Nat Biotechnol . 2002 ; 20 : 70 – 75 .
48. Naturally arising CD4+ regulatory t cells for immunologic self-tolerance and negative control of immune responses . Annu Rev Immunol . 2004 ; 22 : 531 – 562 .
49. Regulatory T cells in autoimmmunity∗ . Annu Rev Immunol . 2000 ; 18 : 423 – 449 .
50. In vivo sites and cellular mechanisms of T reg cell-mediated suppression . J Exp Med . 2006 ; 203 : 489 – 492 .
51. Thymic selection of CD4+CD25+ regulatory T cells induced by an agonist self-peptide . Nat Immunol . 2001 ; 2 : 301 – 306 .
52. Origin of regulatory T cells with known specificity for antigen . Nat Immunol . 2002 ; 3 : 756 – 763 .
53. Regulatory T cells and Toll-like receptors in tumor immunity . Semin Immunol . 2006 ; 18 : 136 – 142 .
54. Tumor-specific human CD4+ regulatory T cells and their ligands: implications for immunotherapy . Immunity . 2004 ; 20 : 107 – 118 .
55. Recognition of a new ARTC1 peptide ligand uniquely expressed in tumor cells by antigen-specific CD4+ regulatory T cells . J Immunol . 2005 ; 174 : 2661 – 2670 .
56. Regulatory T-cell response and tumor vaccine-induced cytotoxic T lymphocytes in human melanoma . Hum Immunol . 2004 ; 65 : 794 – 802 .
57. Foxp3 expressing CD4+CD25(high) regulatory T cells are overrepresented in human metastatic melanoma lymph nodes and inhibit the function of infiltrating T cells . J Immunol . 2004 ; 173 : 1444 – 1453 .
58. The role of IL-10 and TGF-beta in the differentiation and effector function of T regulatory cells . Int Arch Allergy Immunol . 2002 ; 129 : 263 – 276 .
59. Type 1 T regulatory cells . Immunol Rev . 2001 ; 182 : 68 – 79 .
60. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer . N Engl J Med . 2003 ; 348 : 203 – 213 .
61. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival . Nat Med . 2004 ; 10 : 942 – 949 .
62. The expression of the regulatory T cell-specific forkhead box transcription factor FoxP3 is associated with poor prognosis in ovarian cancer . Clin Cancer Res . 2005 ; 11 : 8326 – 8331 .
63. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer . Proc Natl Acad Sci U S A . 2005 ; 102 : 18538 – 18543 .
64. Immature myeloid cells and cancer-associated immune suppression . Cancer Immunol Immunother . 2002 ; 51 : 293 – 298 .
65. Mechanisms and functional significance of tumour-induced dendritic-cell defects . Nat Rev Immunol . 2004 ; 4 : 941 – 952 .
66. Myeloid-derived suppressor cell heterogeneity and subset definition . Curr Opin Immunol . 2010 ; 22 : 238 – 244 .
67. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells . Nat Med . 1996 ; 2 : 1096 – 1103 .
68. Vascular endothelial growth factor impairs the functional ability of dendritic cells through Id pathways . Biochem Biophys Res Commun . 2005 ; 334 : 193 – 198 .
69. Antibodies to vascular endothelial growth factor enhance the efficacy of cancer immunotherapy by improving endogenous dendritic cell function . Clin Cancer Res . 1999 ; 5 : 2963 – 2970 .
70. Minimal recruitment and activation of dendritic cells within renal cell carcinoma . Clin Cancer Res . 1998 ; 4 : 585 – 593 .
71. Human sunlight-induced basal-cell-carcinoma-associated dendritic cells are deficient in T cell co-stimulatory molecules and are impaired as antigen-presenting cells . Am J Pathol . 1997 ; 150 : 641 – 651 .
72. Decreased antigen presentation by dendritic cells in patients with breast cancer . Clin Cancer Res . 1997 ; 3 : 483 – 490 .
73. Alterations in the frequency of dendritic cell subsets in the peripheral circulation of patients with squamous cell carcinomas of the head and neck . Clin Cancer Res . 2002 ; 8 : 1787 – 1793 .
74. Altered maturation of peripheral blood dendritic cells in patients with breast cancer . Br J Cancer . 2003 ; 89 : 1463 – 1472 .
75. Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer . J Immunol . 2001 ; 166 : 678 – 689 .
76. Resection of solid tumors reverses T cell defects and restores protective immunity . J Immunol . 2000 ; 164 : 2214 – 2220 .
77. Surgical removal of primary tumor reverses tumor-induced immunosuppression despite the presence of metastatic disease . Cancer Res . 2004 ; 64 : 2205 – 2211 .
78. Mechanism of immune dysfunction in cancer mediated by immature Gr-1+ myeloid cells . J Immunol . 2001 ; 166 : 5398 – 5406 .
79. Activated granulocytes and granulocyte-derived hydrogen peroxide are the underlying mechanism of suppression of t-cell function in advanced cancer patients . Cancer Res . 2001 ; 61 : 4756 – 4760 .
80. Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species . J Immunol . 2004 ; 172 : 989 – 999 .
81. Oxidative stress by tumor-derived macrophages suppresses the expression of CD3 zeta chain of T-cell receptor complex and antigen-specific T-cell responses . Proc Natl Acad Sci U S A . 1996 ; 93 : 13119 – 13124 .
82. Suppressed T-cell receptor zeta chain expression and cytokine production in pancreatic cancer patients . Clin Cancer Res . 2001 ; 7 : 933s – 939s .
83. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses . Cancer Res . 2004 ; 64 : 5839 – 5849 .
84. Arginase-producing myeloid suppressor cells in renal cell carcinoma patients: a mechanism of tumor evasion . Cancer Res . 2005 ; 65 : 3044 – 3048 .
85. Novel cancer immunotherapy agents with survival benefit: recent successes and next steps . Nat Rev Cancer . 2011 ; 11 : 805 – 812 .
86. Cancer immunotherapy: co-stimulatory agonists and co-inhibitory antagonists . Clin Exp Immunol . 2009 ; 157 : 9 – 19 .
87. CTLA-4 suppresses the pathogenicity of self antigen-specific T cells by cell-intrinsic and cell-extrinsic mechanisms . Nat Immunol . 2010 ; 11 : 129 – 135 .
88. Cell-autonomous and -non-autonomous roles of CTLA-4 in immune regulation . Trends Immunol . 2011 ; 32 : 428 – 433 .
89. Cutting edge: CTLA-4 on effector T cells inhibits in trans . J Immunol . 2012 ; 189 : 1123 – 1127 .
90. Enhancement of antitumor immunity by CTLA-4 blockade . Science . 1996 ; 271 : 1734 – 1736 .
91. Improved survival with ipilimumab in patients with metastatic melanoma . N Engl J Med . 2010 ; 363 : 711 – 723 .
92. Anti-CTLA-4 antibody therapy: immune monitoring during clinical development of a novel immunotherapy . Semin Oncol . 2010 ; 37 : 473 – 484 .
93. Ipilimumab monotherapy in patients with pretreated advanced melanoma: a randomised, double-blind, multicentre, phase 2, dose-ranging study . Lancet Oncol . 2010 ; 11 : 155 – 164 .
94. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma . N Engl J Med . 2011 ; 364 : 2517 – 2526 .
95. PD-1 and its ligands in tolerance and immunity . Annu Rev Immunol . 2008 ; 26 : 677 – 704 .
96. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor . Immunity . 1999 ; 11 : 141 – 151 .
97. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice . Science . 2001 ; 291 : 319 – 322 .
98. PD-1 blockade inhibits hematogenous spread of poorly immunogenic tumor cells by enhanced recruitment of effector T cells . Int Immunol . 2005 ; 17 : 133 – 144 .
99. Clinical significance and therapeutic potential of the programmed death-1 ligand/programmed death-1 pathway in human pancreatic cancer . Clin Cancer Res . 2007 ; 13 : 2151 – 2157 .
100. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade . Proc Natl Acad Sci U S A . 2002 ; 99 : 12293 – 12297 .
101. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer . N Engl J Med . 2012 ; 366 : 2443 – 2454 .
102. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer . N Engl J Med . 2012 ; 366 : 2455 – 2465 .
103. CTLA-4 blockade increases IFNgamma-producing CD4+ICOShi cells to shift the ratio of effector to regulatory T cells in cancer patients . Proc Natl Acad Sci U S A . 2008 ; 105 : 14987 – 14992 .
104. Preoperative CTLA-4 blockade: tolerability and immune monitoring in the setting of a presurgical clinical trial . Clin Cancer Res . 2010 ; 16 : 2861 – 2871 .
105. Oncogenic BRAF(V600E) promotes stromal cell-mediated immunosuppression via induction of interleukin-1 in melanoma . Clin Cancer Res . 2012 ; 18 : 5329 – 5340 .






