Calcitonin
Synthesis, Secretion, and Cells of Origin
Peptides Related to Calcitonin
Calcitonin in the Central Nervous System
Calcitonin and Its Receptors in Cancer
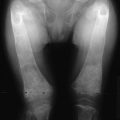
In the course of experiments seeking a factor that, in addition to parathyroid hormone (PTH), might contribute to the tight control of serum calcium in mammals, Copp and colleagues1 discovered calcitonin. By perfusing the thyroparathyroid glands of dogs and sheep with high calcium concentrations, they obtained evidence for the secretion of a factor that rapidly lowered the blood calcium; they called this factor calcitonin and suggested that it was produced by the parathyroid gland. Subsequently, calcitonin was found by others to be produced by the thyroid in mammals. After it was noted that parathyroidectomy by cautery in the rat resulted in much greater calcium lowering than that resulting from surgery (Fig. 2-1),2 it was found that acid extracts of rat thyroid injected into young rats caused a lowering of serum calcium, and the hypocalcemic factor was called thyrocalcitonin.3 MacIntyre and co-workers,4 using thyroparathyroid perfusions in dogs and goats, also established the thyroidal origin of the hypocalcemic agent. It was by then apparent that calcitonin and thyrocalcitonin were identical. The accepted nomenclature became calcitonin (CT), which described a new hormone of thyroid gland origin that was likely to be important in calcium homeostasis.
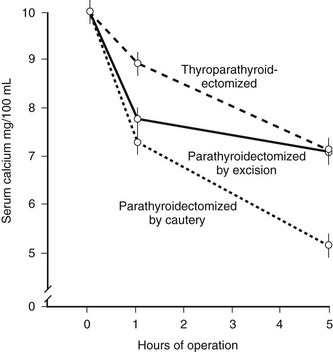
FIGURE 2-1 Comparison of the effects of surgical thyroparathyroidectomy with parathyroidectomy by cautery or surgery in the rat. (Data from Hirsch PF et al, Endocrinology 73:244–251, 1963.)
Synthesis, Secretion, and Cells of Origin
CT is produced by the C cells of the mammalian thyroid, with its secretion dependent on serum calcium levels.5 Although the dominant site of production of CT in mammals is the thyroid C cell, the distribution of these cells throughout the thyroid gland varies considerably among mammalian species, and there is evidence that in some animals CT-producing cells might be found in other parts of the neck, including the thymus. In fish and in most birds, CT is produced by the ultimobranchial glands. Whereas in mammalian development the ultimobranchial bodies fuse with the posterior lobes of the developing thyroid to become the C cells, in submammalian vertebrates these bodies remain separate, and the ultimobranchial glands constitute a separate endocrine system. The CTs of ultimobranchial origin are highly potent in their actions upon mammalian targets. The physiologic significance of CT in fish and birds remains uncertain, although it is interesting to note that CT has been reported to suppress osteoclastic activity in the scales of freshwater and seawater teleosts.6 Although there is little doubt that increased serum calcium is an important secretagogue for CT in normal and malignant C cells, the exact mechanisms by which calcium provokes exocytosis of CT have not been fully elucidated. The same extracellular calcium–sensing receptor that mediates decreased PTH secretion from parathyroid cells7 is also found in C cells and is likely to represent the primary molecular entity through which C cells detect changes in extracellular calcium and control CT release.
Agents that elevate C cell cyclic adenosine monophosphate (cAMP) may stimulate CT secretion, since cAMP analogues have this effect both in vivo and in vitro. Probably the most important CT secretagogues, apart from calcium, are the gastrointestinal hormones. In the pig, gastrin appears to be an effective physiologic secretagogue, suggesting that CT may have a physiologic role postprandially as a hormone that assists uptake of calcium after a calcium-rich meal by preventing the efflux of calcium from bone into blood.8 Although there is some evidence for this role in pigs and rats, studies in humans remain to be performed. Other gastrointestinal hormones, including glucagon, cholecystokinin, and secretin, are also capable of promoting CT secretion. The gastrin analogue, pentagastrin, has been used clinically as a provocative test for CT secretion in patients with medullary carcinoma of the thyroid. Other hormones that influence calcium homeostasis may also directly or indirectly influence CT secretion. 1,25-Dihydroxyvitamin D3 (1,25[OH]2D3) administration has been reported to increase plasma CT levels; this was suggested to occur via specific thyroid C cell receptors for 1,25(OH)2D3, which modify secretion of CT.9 Both CT and 1,25(OH)2D3 levels are raised in pregnancy and lactation, leading to the suggestion that CT may act to protect the skeleton in the face of increased calcium demand by the fetus.
Serum and thyroid concentrations of CT increase markedly with age in the rat, in association with substantial increases in thyroid content of CT mRNA.10 In normal rats subjected to acute calcium stimulation in vivo, thyroid CT mRNA is increased. On current evidence it seems that calcium can stimulate both synthesis and secretion of CT by thyroid C cells.
CT secretion has been studied extensively in patients with medullary carcinoma of the thyroid, who have elevated CT levels (see later). However, circulating CT levels in normal human subjects are very low and their measurement requires sensitive and specific assays. The level of CT in normal human blood appears to be less than 10 pg/mL (3 picomolar). Circulating levels of CT are increased in several pathologic states, such as CT-secreting tumors.11,12 In addition to the CT monomer (≈3500 Daltons), high molecular weight forms circulate, and elevated levels of these molecules can be useful diagnostically in certain situations, such as acute pancreatitis13,14 and infection/inflammatory conditions.15 In fact, it has been reported that ProCT is toxic and that immunoneutralization with immunoglobulin (Ig)G that is reactive to this molecule significantly improves survival in animal models of sepsis.16
Chemistry
The CT sequence has been determined for many species, the common features being that it is a 32 amino acid peptide with a carboxyterminal proline amide and a disulfide bridge between cysteine residues at positions 1 and 7.17 Based on their amino acid sequence homologies, the different CTs (Fig. 2-2) are classified into three groups: (1) artiodactyl, which includes porcine, bovine, and ovine; (2) primate/rodent, which includes human and rat CT; and (3) teleost/avian, which includes salmon, eel, goldfish, and chicken. The common structural features of the CT molecule contribute importantly to biological activity, with the standard assay that has been used since the discovery of CT being one that measures the hypocalcemic response in young rats. Subsequently, receptor-based assays have been used also, and structure/function relationships are largely shared in these various assays. The order of biological potency of the CTs is, in general, teleost≥artiodactyl≥human, although absolute biological activities vary considerably among CT receptors of different species and receptor isoforms within species. Studies of substituted, deleted, and otherwise modified CTs have provided considerable information regarding structure/activity relationships of the CT molecule, showing, for example, that the ring structure serves to stabilize the molecule. The disulfide bridge of the ring can be chemically substituted by an N-N bond, as in aminosuberic eel CT, and this modification yields an extremely stable and fully potent CT variant.18 The sequence differences among species are concentrated in the middle portion of the molecule, and these differences contribute to the wide variations in biological potencies. However, the outcomes of studies of structural requirements for biological activity have varied with the different biological assays used, and the type of receptor used is able to profoundly influence the results. For example, residues in the carboxyterminal half of salmon CT are more important for binding competition with the two rat receptor isoforms and the human receptor, whereas residues in the aminoterminus are more important for interaction with the porcine receptor.5

FIGURE 2-2 Alignment of CT sequences from different species. The shaded amino acids indicate identity with salmon CT. All CTs have a disulphide-bridged loop between cysteines (C) at position 1 and 7, a glycine at position 28, and a proline amide at position 32. Amino acids 4, 5, and 6 are also conserved across all species.
Calcitonin Gene
As with other hormonal peptides, CT is synthesized as a larger precursor molecule, which is processed by cleavage and amidation before secretion. CT is synthesized as a large molecular weight precursor (136 amino acids), with a leader sequence at the aminoterminus that is cleaved during transport of the molecule into the endoplasmic reticulum. A potentially important posttranslational modification of CT is that of glycosylation. It had been noted that the tripeptide sequence, Asn-Leu-Ser, found within the aminoterminal ring structure of CT is invariate among the CTs of different species. This sequence is an acceptor site for N-linked glycosylation. This, together with evidence for glycosylation of tumor CT, led to detailed studies showing that the CT precursor is indeed a glycoprotein, and that the only N-linked glycosylation site in the entire precursor was within the CT portion itself.19 The biological significance of CT glycosylation has yet to be determined.
The complete sequences of the cDNA for human, rat, mouse, chicken, sheep, dog,20 and various species of fish CTs and the DNA sequence of the full human CT gene have been determined.21–23 These show that the hormone is flanked in the precursor by N- and C-terminal peptides, but the biological significance of these peptides is unknown. The human CT gene has been located in the p14-qter region of chromosome 11.24
Alternative Gene Product—CGRP
The CT gene transcript actually encodes a second distinct peptide known as CT gene-related peptide (CGRP), which is produced by tissue-specific alternative splicing of the gene (Fig. 2-3). The mature CGRP and CT mRNAs predict proteins that share sequence identity in the aminoterminal regions, but in the carboxyterminal regions the nucleotide sequences are almost entirely different. The mature, secreted 32 and 37 amino acid CT and CGRP peptides, respectively, result from cleavage of both aminoterminal and carboxyterminal flanking sequences at specific cleavage sites, as depicted in Fig. 2-3.24 CT mRNA is found largely in the thyroid, and CGRP mRNA is found primarily in the nervous system.25 However, aberrant expression of CGRP may be seen in medullary thyroid carcinoma.26 Two different CT/CGRP genes, α and β, have been identified in man and rat.
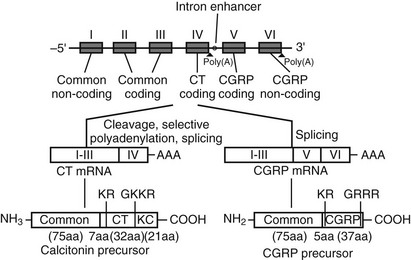
FIGURE 2-3 Organization of the CT/CGRP gene illustrating alternative patterns of processing of the primary transcript and subsequent protein processing. The exons are denoted I through VI in Roman numerals, introns are represented by a single line (not to scale).
Processing of the pre-mRNA to the CT mRNA transcript involves usage of exon 4 as a 3′-terminal exon with concomitant polyadenylation at the end of exon 4. Processing to produce the CGRP mRNA involves the exclusion of exon 4 and direct ligation of exon 3 to exon 5, with polyadenylation at the end of exon 6. The hCT/CGRP exon 4, like many differentially incorporated exons, has been characterized as having weak processing signals. Weak differential exons are frequently associated with special enhancer sequences that facilitate exon recognition in the presence of accessory factors that bind to the enhancer. Indeed, such an enhancer, located in the intron downstream of exon 4, has been described for the CT/CGRP gene. In addition, sequences within exon 4 are necessary for the inclusion of exon 4.27
Physiology—Bone and Kidney
The physiologic role of CT is not fully understood. It is currently viewed as an inhibitor of bone resorption, whose function is to prevent bone loss at times of stress on skeletal calcium conservation, particularly pregnancy, lactation, and growth. Earlier concepts of CT as a regulator of extracellular fluid calcium are probably relevant only in young and growing animals, in which rapid bone modeling and turnover are required for development of the skeleton. In the rat, for example, which is used for the in vivo assay of CT, the calcium-lowering effect of the hormone is less marked with increasing age of the animal (Fig. 2-4).28 However, the ability of CT to counteract the effects of a calcium load was shown not to be impaired in older animals, at least in the rat29—an observation that has not been explained and that has not been extended to other species.
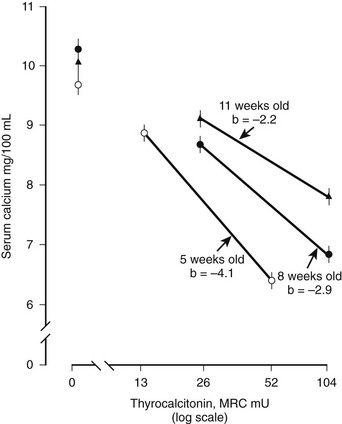
FIGURE 2-4 Decreasing hypocalcemic response to calcitonin with increasing age of the rat. (Data from Cooper CW et al, Endocrinology 81:610–617, 1967.)
In normal adult human subjects, even quite large doses of CT have little effect on serum calcium levels. In those subjects in whom bone turnover is increased (e.g., in thyrotoxicosis, Paget’s disease), CT treatment acutely inhibits bone resorption and lowers the serum calcium.30 Given that the acute effect of CT on serum calcium is related to the prevailing rate of bone resorption, it is not surprising that CT has little or no effect on serum calcium in the mature animal or human subject, since the rate of bone resorption is slow in maturity. The physiologic function of CT in maturity nevertheless may be to regulate the bone resorptive process in a continuous or intermittent manner. It follows that CT should not necessarily be regarded as a “calcium-regulating hormone” in maturity, but may yet be shown to be such in stages of rapid growth (e.g., in the young, in states of increased bone turnover). It is nevertheless important that bone resorption be regulated, and CT is the only hormone known to be capable of carrying out this function through a direct action on bone. Physiologic roles for other members of the CT family, such as amylin and CGRP, in the regulation of resorption have not yet been substantiated. An antiresorptive role for CT in maturity might become more important in circumstances in which skeletal integrity is at particular risk (e.g., in pregnancy and lactation). Evidence in support of such an important physiologic role for endogenous CT in protecting against bone loss is provided by experiments showing that cancellous bone loss in thyroparathyroidectomized rats treated with PTH was greater than that in similarly treated sham-operated controls.31 In addition, mice in which the CT/CGRP gene was ablated showed a severe drop in bone mineral content during lactation, although the maternal skeleton recovered to baseline thereafter.32,33
Calcitonin Actions In Bone
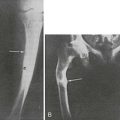
The first evidence of the mechanism of action of CT was obtained by showing in organ culture of bone that CT inhibited bone resorption.34 Inhibition of resorption appeared to be explained by a direct action on osteoclasts. CT treatment of resorbing bone in vitro resulted in rapid loss of osteoclast ruffled borders and decreased release of lysosomal enzymes. In vivo evidence was also consistent with an inhibitory action upon bone resorption. Loss of ruffled borders in osteoclasts was seen in patients with Paget’s disease, in whom bone biopsies were taken before and 30 minutes after an injection of CT.35 In the same clinical study, CT was noted to decrease the number of osteoclasts, in addition to altering their ultrastructure. CT infusion in rats led to an immediate reduction in the rate of excretion of hydroxyproline, consistent with inhibition of breakdown of bone collagen.36 Other studies led to similar conclusions, with no evidence to suggest any increase in the active uptake of calcium by bone.37
Studies of the actions of hormones on isolated bone cell populations established that CT acts directly on osteoclasts, with receptor autoradiography showing osteoclasts as the only discernible bone cell targets.38 Mammalian osteoclasts possess abundant, specific, high-affinity receptors for CT (Fig. 2-5), and CT stimulates cAMP formation in a sensitive and dose-dependent manner,38 as well as increasing intracellular free calcium levels and protein kinase C activity.39 As stated, the direct effect of CT upon the osteoclast was found to result in rapid inhibition of activity, reflected in cessation of motility and contraction of the cell. Although isolated osteoclasts remained quiescent in CT as long as the hormone was present, they regained activity when osteoblasts were added to the culture.40 This escape of osteoclasts from inhibition by CT took place at a rate proportional to the number of osteoblasts with which they were in contact. CT reduced the cytoplasmic spreading of isolated osteoclasts in a dose-dependent manner. PTH had no effect unless osteoblasts were co-cultivated with the osteoclasts, in which case addition of PTH resulted in a marked increase in cytoplasmic spreading of osteoclasts. It cannot be assumed that these phenomena reflect the responses of cells in bone in vivo, but this work provided for the first time some useful direct observations of actions of hormones on isolated bone cell preparations containing osteoclasts. These observations though may be relevant to our interpretation of recent findings in mice rendered null for the CT/CGRP gene and in those haploinsufficient for the CT receptor (vide infra).
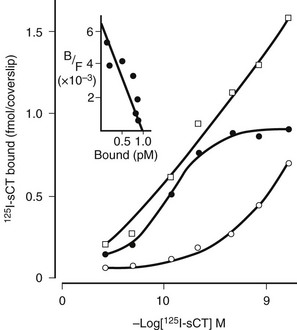
FIGURE 2-5 Saturation analysis of labeled calcitonin binding to rat osteoclasts. Scatchard analysis of specific binding (inset) shows receptor number of 4.8 × 106 per cell. (Data from Nicholson GC et al, J Clin Invest 78:355–360, 1986.)
The molecular mechanisms by which CT decreases osteoclast function have yet to be fully defined. The rapid effects of the hormone may be brought about through actions on a cytoskeletal function of osteoclasts, after initial events involving generation of several intracellular second messengers. Early events in CT signal transduction have been studied in a variety of cell types and are described in greater detail later. The other means by which CT could inhibit resorption is through inhibition of osteoclast formation. In vivo data and results from CT inhibition of resorption in organ culture are suggestive of this. The development of methods of studying osteoclast formation in vitro from hemopoietic precursor cells has allowed this question to be addressed directly. Several reports have described CT inhibiting osteoclast-like cell formation in bone marrow cultures of human, baboon, and mouse origin.41–44 However, these experiments were all conducted at relatively high CT concentrations, and the effects were small. In other studies, in which lower concentrations of CT were used, which nevertheless reduced CT receptor mRNA expression in developing mouse osteoclasts, no reduction in osteoclast formation was observed.45–47 The multinucleated osteoclasts that formed in the continuous presence of exogenous CT had fewer nuclei though, and the osteoclasts generated under these conditions were deficient in CT receptor mRNA and protein but nevertheless capable of resorbing bone. In elegant studies of CT administration to mice, Ikegame et al48 showed that the CT-induced drop in serum calcium was linked temporally to the loss of osteoclast ruffled borders. Further, frequent dosing of the animals resulted in insensitivity to CT in terms of recovery of osteoclast ruffled borders and return of serum calcium to control levels. It was significant that treatment of mice with CT initially rendered osteoclasts unable to bind 125I-sCT, which recovered after a single treatment but not with repeated treatment.48 These findings may be relevant to the mechanism of “escape” from CT that is observed clinically.
Osteoblasts
Although the best understood action of CT in bone is as an antiresorptive agent, numerous reports have described actions also on cells of the osteoblast lineage, as well as direct and indirect effects on bone formation. CT increased [3H]thymidine incorporation in embryonic chicken calvariae, in the transformed murine calvarial cell lines MMB and MC-3T3-E1 and in primary cultures of cells prepared from newborn mouse calvaria.49 CT was also shown to stimulate [3H]thymidine incorporation in primary human osteoblasts50 and to increase expression of insulin-like growth factors (IGFs) by human SaOS-2 cells.51 None of these observations has been confirmed, and indeed data failing to show such effects of CT have been published.52 In addition to the fact that relatively high concentrations of CT were used in these early experiments, results with primary osteoblasts should be interpreted with caution at present, because it is known that calvarial osteoblast preparations are contaminated with osteoclast precursors, and a recent report provides evidence that primary osteoblast preparations contain a substantial proportion of bone-specific macrophages, which have been termed “osteomacs.”53 This raises the possibility that the presumed osteoblast responses may actually be mediated by cells of the monocyte-macrophage lineage. Furthermore, no published evidence is convincing of specific, functional CT receptors in any cells of the osteoblast lineage. Inadequate criteria were used in a claim that 125I-sCT was bound specifically to osteocyte-like cells, MLO-Y4.54 In the same work, CT at high concentrations was associated with a small increase in cAMP and with protection of the cells from apoptosis induced by etoposide, tumor necrosis factor (TNF)-α or dexamethasone. It has been claimed that CT can hasten and improve the process of fracture healing in normal55 and osteoporotic rats.56 If this proves to be the case, the mechanism could be similar to that of bisphosphonates, which have also been shown to enhance the strength of the healed fracture, apparently by modulating resorption during the remodeling phase of bone repair.57 It could also relate to possible stimulation of angiogenesis by CT, which has been shown for human microvascular endothelial cells, albeit at supraphysiologic concentrations.58
New Understanding of The Role of Calcitonin In Bone: Studies In Genetically Manipulated Mice
The preceding discussion of the physiology of CT and its action on bone reflects views that have remained largely unaltered over many years, with few new data to change them. The data on which they are based, particularly since normal and low circulating CT levels cannot be measured with confidence (vide supra), do not provide a convincing argument for a specific physiologic role for calcitonin. Indeed, in a recent review, one of the co-discoverers of calcitonin59 argued the case that calcitonin is not involved in calcium homeostasis or in any other important physiologic function, except possibly in protection of the skeleton under conditions of calcium stress. Some recent work has changed this situation. Ablation of the CT/CGRP gene in mice resulted in viable and fertile mice with no production of CT or CGRPα.32 As expected, these mice were much less able than wild-type mice to overcome the hypercalcemia induced by a calcium load,32 and they lost excessive bone during lactation.33 The great surprise with these mice, however, was that they had increased bone mass, with histomorphometric parameters showing increased bone formation.32 This suggested that a normal role for calcitonin might be that of an inhibitor of bone formation. The finding was unexpected and counterintuitive, and it was possible that the dual ablation of CT and CGRPα might explain it, although there was no obvious mechanism for this. Indeed, the increased bone formation phenotype was not found when CGRPα-deficient mice were examined,60 thereby making calcitonin deficiency likely responsible for the increased bone formation. In reviewing the role of calcitonin, Hirsch59 had considered the results obtained with the CT/CGRP−/− mice but regarded them as inconclusive; as information accumulates, the new physiology of calcitonin is becoming more apparent. The CT/CGRP-deficient mice were protected against ovariectomy-induced bone loss,32,60 and, it was striking that after the age of 6 months these mice showed severe cortical porosity, even though indices of increased bone formation were maintained.61,62 Taken together, the observations are indicative of an inhibitory effect of calcitonin on bone formation, most likely an indirect one, and a direct effect on bone resorption through action on the osteoclast.
The significance and importance of these findings were enhanced however with the outcome of the studies of Dacquin et al63 in mice in which the CT receptor (CTR) was genetically manipulated. CTR−/− mice were embryologically lethal, and this was thought to be due to a placental effect. The CTR+/− mice, however, exhibited a bone phenotype virtually indistinguishable from the CT/CGRP−/− mice, with increased bone mass and increased bone formation on histomorphometry. Thus, the conclusion is that in mice the removal of calcitonin production or action results in an increased amount of bone, implying a physiologic role for calcitonin as a tonic inhibitor of bone formation. The same group prepared mice at least 94% deficient in the CTR and again found evidence of increased indices of bone formation.64 How the effect on bone formation comes about remains to be determined. The lack of evidence for specific calcitonin receptors and responses in osteoblasts is compelling, so it is very likely that the physiologic roles of calcitonin in bone are brought about through two pathways: a direct effect on osteoclasts to inhibit resorption and an indirect one resulting in the elaboration of a critical, locally active factor that is necessary for bone formation. This could result from signals through receptors in the osteoclast or in the hypothalamus.62,65 Resolution of this question will be a matter of very great interest, especially in light of ample recent evidence for central regulation of bone metabolism.66
Renal Actions of Calcitonin
When infused into thyroparathyroidectomized rats, CT caused a dose-dependent phosphaturia, but the effect on phosphate excretion was only a minor one in comparison with the phosphaturic effect of PTH.67 Although this was demonstrated in human subjects also, in several species CT failed to have any effect on phosphate excretion. Thus, it seems unlikely that the phosphaturic effect is of any major physiologic significance.
A number of other renal effects of CT, including a transient increase in calcium excretion due probably to inhibition of renal tubular calcium reabsorption, have been noted.68 Although this has not usually been regarded as an important effect of CT, it has been linked to the calcium-lowering effect of CT in hypercalcemic patients with metastatic bone disease. The use of CT in the treatment of hypercalcemia due to cancer has been based exclusively on the inhibition of osteolysis by CT. Some evidence has been produced that failure of the kidneys to excrete the calcium load derived from bone breakdown is a major contributor to the hypercalcemia. This has prompted careful study of the relative contributions to the hypocalcemic effect of CT of its renal and skeletal components. It was concluded that inhibition of renal tubular reabsorption by CT can induce a rapid fall in serum calcium, and that the magnitude of this effect depends upon the correction of volume depletion, which inevitably accompanies hypercalcemia.69 Thus, the calciuretic action of CT may assume greater importance than was hitherto suspected.
CT receptors are present in rat kidney,70 and the action of CT upon adenylate cyclase activity has been localized in the human nephron, predominantly to the medullary and cortical portions of the thick ascending limb and to the early portion of the distal convoluted tubule. The co-localization of the CT receptor mRNA expression and cell surface receptors with G protein–sensitive adenylate cyclase is consistent with cAMP being an important mediator of CT action in this organ. A possible role for CT in the kidney is to regulate 1,25(OH)2D3 levels, with an original observation of enhanced 1-hydroxylation of 25(OH)D in the proximal straight tubule of the kidney by CT stimulation of the expression of 25(OH)D 1α-hydroxylase.71 Subsequently, Shinki et al72 showed that CT administration to rats induced renal CYP27B1 when serum calcium levels were normal or high, and this was supported by a report that CT treatment in rats increased renal production of CYP27B1 mRNA.73 CT stimulation of the expression of CYP24 in CTR-transfected HEK-293 cells has also been reported.74 The authors speculated that, since 1,25(OH)2D3 and CT synergistically stimulate CYP24 mRNA production in kidney cells, this latter action of CT could be part of the process by which it regulates serum calcium by controlling renal production of 1,25(OH)2D3.
Peptides Related to Calcitonin
Amylin is a 37 amino acid peptide that is co-secreted with insulin from pancreatic β cells following nutrient ingestion. Amylin at physiologic concentrations is important in the integrated control of nutrient influx with potent actions, including inhibition of gastric emptying, gastric acid secretion, food intake, digestive enzyme secretion, and glucagon secretion.75 Amylin at higher concentrations also acts to inhibit insulin secretion from the pancreas and to promote glycogen breakdown and to decrease insulin-stimulated incorporation of glucose into glycogen in skeletal muscle. Thus, amylin is thought to act as a partner to insulin in metabolic regulation, although this effect may not occur at normal circulating levels of the peptide.76 In the kidney, amylin is proposed to have a diverse range of actions, including modulation of Ca2+ excretion and thiazide receptor levels, proliferative effects on tubule epithelium, and increasing renin activity.77,78 Amylin−/− mice have been shown to have less bone as the result of increased bone resorption, and in vitro tests indicated that this may be due to release of an amylin-mediated attenuator of osteoclastogenesis.63 These authors speculate that the receptor for this effect is independent of the CTR gene, which, with receptor activity modifying proteins (RAMPs), forms the basis of characterized amylin receptor phenotypes (see later). However, their conclusion is based on studies in animals with the amylin−/+, CTR−/+ genotype, and therefore RAMP/CTR-based amylin receptors cannot be excluded as the target for amylin action.
Amylin receptors are also widely expressed in brain, where administered peptide induces many potent effects. These include decreased appetite and gastric acid secretion, hyperthermia, adipsia, and reduction in growth hormone–releasing hormone. Central amylin injection may also modulate memory and the extrapyramidal motor system.75,77 The molecular basis for amylin receptor phenotype is discussed later.
CGRP is a pleiotropic neuropeptide with a diverse range of actions including potent dilation of vascular beds, as well as relaxation of other smooth muscle, inotropy and chronotropy in the heart, and paracrine regulation of pituitary hormone release, and many central effects, such as suppression of appetite and gastric acid secretion, modulation of body temperature, and modulation of sympathetic outflow. CGRP also acts to modulate nicotinic acetylcholine receptor levels at neuromuscular junctions. CGRP weakly modulates calcium homeostasis, although this is likely to reflect its low affinity for interaction with CTRs. The actions of CGRP have been extensively reviewed elsewhere.78,79 Specific CGRP receptors have been characterized in many tissues and it is likely that more than one subtype of receptor exists. CGRP receptors arise from hetero-oligomerization of RAMP1 with either the calcitonin receptor-like receptor (CLR) or CTR,80,81 although weaker interactions are also seen with other RAMP/CLR- or RAMP/CTR-based receptors.82
Adrenomedullin was originally isolated from human pheochromocytoma and is abundant in the normal adrenal medulla, hence its name. The full-length peptide of ≈50 amino acids shares approximately 25% homology with CGRP across its N-terminal 37 amino acids.83 Adrenomedullin is a potent dilator of many vascular beds and is protective against conditions such as cardiac hypertrophy, perivascular fibrosis, renal damage, and pulmonary hypertension.84,85 Adrenomedullin receptors arise from heterodimers of CLR and RAMP2 or RAMP3.81 Both adrenomedullin and amylin can stimulate osteoblast proliferation at low concentration.86 Fig. 2-6 illustrates complex formations between the CTR and RAMPs 1, 2, and 3.
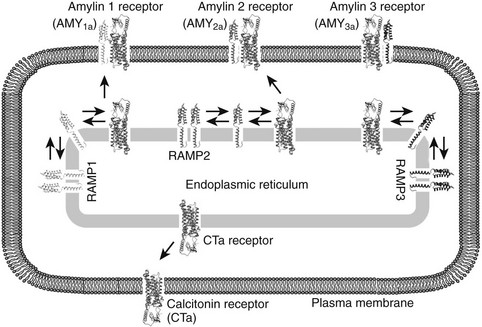
FIGURE 2-6 Representative illustration of complex formation between the calcitonin receptor (CT) and RAMPs 1, 2, and 3 in the endoplasmic recticulum (ER). All three RAMPs are postulated to exist as homodimers in the ER; however, in the presence of CTR an equilibrium is established in which hetero-oligomers between the receptor and each of the RAMPs are formed. To date, the stoichiometry of the oligomers remains unclear. AMY1a, AMY2a, and AMY3a receptors are generated when the CTa receptor isoform is complexed with RAMP 1, 2, and 3, respectively. In addition, although the oligomers between the CT receptor and RAMPs can then be transported to the cell surface, the CT receptor is transported to the cell surface in the absence of RAMPs. The human CT receptor has two major splice variants, CTa and CTb, that arise from the absence or presence of a 16 amino acid insert in intracellular domain 1. The nomenclature for amylin receptors formed by the different receptor isoforms is denoted by (a) or (b) in the name (e.g., AMY1a denotes an amylin receptor formed from the CTa receptor and RAMP 1). It has been hypothesized that RAMPs may reside in a homodimerized form in intracellular sorting compartments such as the endoplasmic reticulum.
Recently, new peptides related to the CT family of peptides have been identified. The calcitonin receptor–stimulating peptides (CRSPs) were originally isolated/cloned from porcine brain and, being localized principally to brain and pituitary, have been proposed as potential endogenous ligands for central CTRs.87–89 However, not all CRSPs activate CTRs or related receptors, suggesting that they may also stimulate other receptors.88 To date, homologous peptides have not been identified in humans or rodents, although bovine and canine homologues do exist.89 In addition, a second adrenomedullin-like peptide (adrenomedullin 2; also known as intermedin) has been identified in multiple species, including human. This peptide has similar affinity for both CGRP and adrenomedullin receptors, although the physiologic significance of this peptide is still unclear.82,90,91
Calcitonin in the Central Nervous System
Intraventricular administration of CT generates potent effects that include analgesia and inhibition of appetite and gastric acid secretion, as well as modulation of hormone secretion and the extrapyramidal motor system.77,79,81,92 Both immunoreactive CT-like peptide and CT receptors have been demonstrated in the brain and nervous system of rats, humans, and other species. Immunoreactive CT related antigenically to hCT (hCTI) has also been demonstrated in the nervous systems of protochordates, lizards, and pigeons, as well as at low levels in human and rat brain extracts. Furthermore, radioimmunoassay analyses point to the presence of sCT-like peptide material in human and rat brain.93
Low levels of hCTI occur in extracts of postmortem human brain. However, in addition to hCTI, low levels of material chromatographically and immunologically similar to sCT (sCTI) have been found, with concentrations in the brain ≈10 times greater than those in serum and cerebrospinal fluid. Similarly, extracts of rat brain diencephalon contain an sCT-like peptide.93 Although hCTI has been detected in rat brain, only limited evidence has been found for the presence of rat CT (rCT) mRNA. Immunoreactive CT-like material also occurs in the pituitary of both mammals and lower vertebrates, although the identity of the pituitary CT remains to be established. The physiologic significance of CT-like immunoreactivity in the pituitary remains to be fully elucidated; however, CT receptors are present in the intermediate pituitary, and therefore the CT-like material may act as a paracrine regulator of these receptors. CT has been recently described as participating in a complex cross-talk between extracellular signaling molecules in the pituitary.94 Treatment of pituitary cells with CT in culture modulates the secretion of prolactin,95,96 and treatment of the cultures with an anticalcitonin antibody enhances it.97 Targeted overexpression of calcitonin in gonadotrophs of mice leads to long-term hypoprolactinemia, decreased PRL gene expression, female subfertility, and a selective underdevelopment of lactotrophs.98,99
Calcitonin Receptors
Direct evidence for binding of CT to osteoclasts was obtained from the work of Nicholson et al,38 who used in vitro autoradiography to demonstrate 125I-sCT binding specifically to osteoclasts and their precursor cells. The CT receptor became a required phenotypic feature to establish cells as osteoclasts.100 Radioligand binding studies in tissue sections, membranes, and cultured cells have revealed an extensive extraskeletal distribution of CTRs. These sites include kidney, brain, pituitary, placenta, testis and spermatozoa, lung, and lymphocytes, as well as cancer-derived cells from lung, breast, pituitary, and embryonal carcinoma.77
The centrally mediated actions of CT correlate well with the locations of CT binding sites. Autoradiographic mapping revealed high binding densities associated with parts of the ventral striatum and amygdala, the hypothalamic and preoptic areas, as well as most of the circumventricular organs. High-density binding also occurs in parts of the periaqueductal gray, the reticular formation, most of the midline raphe nuclei, parabrachial nuclei, locus coeruleus, and solitary tract nucleus.92,101,102 CTR mRNA has been confirmed in many of these sites, including kidney, brain, lung, placenta, and osteoclasts, along with many cancer cell lines. However, CTR mRNA studies have also provided evidence for previously uncharacterized sites of CTR expression, including prostate, normal and neoplastic breast, and thyroid.77 As was discussed earlier, there is no evidence for CTR expression in osteoblasts.
Receptor Cloning
Our knowledge of the molecular basis of CT action, in terms of both ligand binding and postbinding events, was greatly augmented by the molecular cloning of CT receptors. The first cloned receptor was the porcine CTR, which was isolated by expression cloning from a cDNA library derived from the renal epithelial cell line, LLC-PK1.103 Subsequently, the sequences of CTR genes from man, rat, mouse, rabbit, guinea pig, and several species of fish have been reported, and the phylogenetic relationships between CTRs of mammalian and nonmammalian species have been well summarized.104 Analysis of the predicted protein translation product(s) revealed that these receptors comprise approximately 500 amino acids and belong to the class II (family B) subclass of G protein–coupled receptors, which include the receptors for other peptide hormones such as secretin, PTH, glucagon, glucagon-like peptide 1, vasoactive intestinal polypeptide, pituitary adenylate cyclase–activating peptide, and gastric inhibitory peptide.
Receptor Isoforms
Receptor cloning also provided direct evidence for receptor heterogeneity and the existence of multiple receptor isoforms that arise from alternative splicing of the CTR mRNA primary transcript. The human receptor, of which there are at least 5 splice variants, is the most extensively studied. The most common hCTR splice variant occurs in intracellular domain 1, generating a 16 amino acid insert in this domain (I1+).105–107 These are now termed CTa (insert negative) and CTb (insert positive).108 Additional splice variants have been identified in other species. In rodents, alternate splicing leads to two receptor isoforms (these have previously been termed C1a and C1b), which differ by the presence (C1b; E2+) or absence (C1a) of an additional 37 amino acids in the second extracellular domain. However, it is unlikely that expression of the rodent C1b isoform occurs in humans. In rabbits, an additional splice variant, in which the exon encoding transmembrane domain 7 is spliced out (ΔTM7; delta e13), has been isolated.
Investigation of the significance of the splice variants revealed that inserts or deletions in the CTR structure lead to alterations in ligand binding (E2+, Δamino47), signal transduction (I1+), or both (ΔTM7). For the E2+ variants, there is a loss of affinity for peptides exhibiting weak α-helical secondary structure, such as human CT, while peptides with strong helical secondary structure, such as salmon CT, maintain high affinity at this receptor.109 In contrast, the CTb (I1+) receptor isoform displays similar affinity to the CTa (I1−) variant for CT-like peptides but has markedly altered G protein–coupling efficiency. The presence of the 16 amino acid insert leads to complete loss of intracellular calcium mobilization. Activation by calcitonin of its receptor can result in coupling to multiple different G proteins (see Fig. 2-6). The effect on signaling via Gs is cell type dependent but overall is maintained with decreased efficiency, leading to a 10- to 100-fold reduction in the potency of peptides for stimulation of cAMP production.106 It is interesting to speculate about the functional significance of the different receptor isoforms, particularly those with apparently impaired signaling capacity. Accumulating evidence suggests that G protein–coupled receptors can form homodimers and heterodimers, and that these interactions can cross-modulate the activity of the partners. Indeed, in the case of the rabbit CTR (delta e13), Seck et al110,111 reported that this isoform can complex with the CTa isoform and inhibit its cell surface expression and activity, thus exerting an endogenous dominant negative effect on CTR signaling.
Receptor Gene
The CTR gene, like the genes for other class II G protein–coupled receptors, is complex, comprising at least 14 exons with introns ranging in size from 78 nucleotides to >20,000 nucleotides. The total receptor gene is estimated to exceed 70 kb in length.112 The organization and size of the human gene are similar to the pig CTR gene, although with some interspecies differences in organization. For instance, in the pig the I1 insert is generated by selective use of alternate splice sites located in exon 8. However, in humans, the I1 insert occurs on a separate exon, with at least one additional exon proximal to exon 7 in the pig.106 Isolation of the mouse CTR gene113 revealed that the locations of introns within the coding region of the mCTR gene (exons E3-E14) are identical to those of the porcine and human CTR genes. The predicted structure of the mefugu fish gene is more complex than the human gene, with an additional nine exons.104 It is interesting to note that both fish and mammalian genes code for micro-RNA (miR)-489 in intron 3, the function of which remains to be determined.
The regulatory portion of the gene contains three putative promoters (P1, P2, and P3), giving rise to multiple CTR isoforms, which differ in the 5′ region of the gene and generate 5′-untranslated regions of very different lengths, in a tissue specific manner.113 Promoters P1 and P2 are utilized in osteoclasts, brain, and kidney, and the proximal promoter of the human CTR (hCTRP1) was transcriptionally active in all cell lines tested, with high-level activity dependent on an 11 bp Sp1/Sp3 binding site.114 In contrast, promoter P3 appears to be osteoclast specific and is sensitive to the osteoclast-inducing cytokine, RANKL, as well as the RANKL-induced transcription factor, NFATc1, consistent with a role for the latter in regulating the CTR gene in osteoclasts.115 The human CTR gene is located on chromosome 7 at 7q21.3. In the mouse it is in the proximal region of chromosome 6, while the pig gene is located in chromosomal band 9q11-12. These pig and mouse chromosomal regions are homologous to 7q in humans. It is interesting to note that the mouse CTR gene is expressed preferentially from the maternal allele in brain, with no allelic bias detected in other tissues, indicating that the mouse CTR gene is imprinted in a tissue-specific manner.116
Receptor Polymorphisms and Osteoporosis
An interesting, but as yet unconfirmed, report describes an association between the expression of the CTa receptor mRNA in circulating monocytes of postmenopausal, but not premenopausal, women and their serum levels of bone alkaline phosphatase and urinary deoxypyridinolone.117 The authors therefore suggested a link between CTR expression and increased bone resorption postmenopausally.
Restriction fragment length polymorphism (RFLP) studies have identified a polymorphism within the CTR gene coding sequence; Nakamura et al,118 using the Alu I restriction enzyme, identified a polymorphism arising from a single nucleotide substitution, which leads to either a proline (CC genotype) or a leucine (TT genotype) at amino acid 447 in the human CTa receptor (amino acid 463 of the hCTb receptor), with heterozygotes designated TC. In this Japanese population, the proline heterozygote was the most prevalent (≈70%), with the leucine homozygote accounting for ≈10% of the population and the heterozygote for ≈20%. Additional analyses across different ethnic groups have revealed that the Leu homozygote is most prevalent in Caucasians, with decreased prevalence in African Americans and Hispanics and very low frequency in Asians (0% to 10%).119–122
The influence of the polymorphism on bone mineral density (BMD) has been studied by multiple investigators. Most studies have identified significant correlations between CTR genotype and osteoporotic markers (BMD, fracture risk); however there is considerable divergence among studies in regard to which genotype is more favorable. High incidence of the T allele (either Leu homozygotes or heterozygotes) has been correlated with both increased BMD119,121,123 and decreased BMD.119,124,125 The underlying basis for this divergence is unclear. It may be due to linkage disequilibrium of the CTR polymorphism with other genes involved in bone homeostasis. Alternatively, it may be related to the complex nature of action of CT and amylin (likely utilizing the same core receptor) alluded to above from gene knockout studies.63 This work indicates that CT can affect both bone formation and resorption, while amylin may be important for osteoclastogenesis.
The CTR genotype has also been linked to body weight in premenopausal Japanese women,87 and to incidence of kidney stones.126 To date, in vitro studies have not identified significant functional differences in ligand binding or cAMP generation between the two polymorphic variants.127 A large number of other, probably silent, polymorphisms in the hCTR, whose frequency varies among ethnic groups, have been identified.127
Amylin and Calcitonin Gene-Related Peptide Receptors
Recent work revealed that the CT receptor–like receptor (CLR), which has highest homology with the cloned CT receptor, is a CGRP receptor. Functionally, however, this receptor requires the coexpression of a novel protein, termed receptor activity modifying protein 1 (RAMP 1). RAMP 1 is a member of a family of three single transmembrane proteins and acts to modify the glycosylation of CLR, enhance the trafficking of the receptor protein to the cell surface and contributing to the cell surface ligand binding and specificity of the receptor. RAMP 2 and RAMP 3 also enhance the trafficking of CLR to the cell surface; association of CLR with RAMP 2 and RAMP 3 gives rise to adrenomedullin-like receptors.128,129 It has been demonstrated80,81 that the molecular identity of amylin receptors was also founded on RAMP-based hetero-oligomers. In this case, the RAMPs interacted with the CTR to form distinct amylin receptor phenotypes, depending on which RAMP was complexed with the receptor (see Fig. 2-6). It is intriguing that the CTR/RAMP 1 complex, in addition to being a high-affinity amylin receptor, potently interacts with CGRP.80 This behavior may contribute to some of the heterogeneity seen in CGRP receptor analyses. Unlike CLR, CTRs do not require RAMPs to translocate to the cell surface and exhibit classic CTR phenotype under these conditions. Both RAMPs and receptors are subject to dynamic regulation, although in the case of CLR-based receptors, RAMPs often appear to be the major component regulated.82,130
Signaling
Usage of the cAMP pathway in mediating CT action in osteoclasts was discussed earlier. CT also stimulates adenylate cyclase activity in the kidney, with the pattern of CT responsiveness paralleling the distribution of CTRs in this tissue. CT induction of cAMP has now been documented in a large number of cultured CTR-bearing cells that include LLC-PK1 pig kidney cells, and cancers of lung, breast, and bone. Receptor cloning and expression studies have confirmed that cAMP production is an important component of CTR-mediated signaling.103,105,106
It is now apparent that G protein–coupled receptors can interact with and signal through multiple G proteins (Fig. 2-7). Thus, CT action in osteoclasts is probably regulated by alternate signaling pathways, in apparently species-specific ways. In mice, this response appears to be mediated predominantly by the protein kinase A pathway,131,132 although inhibition of osteoclast-mediated bone resorption by CT can be mimicked by both dibutyryl cAMP and phorbol esters or blocked by protein kinase inhibitors.133 Coupling of the CTR to Gq can also activate phospholipase C, leading to increased cytosolic calcium and triphosphate levels and activation of protein kinase C.134 Thus, in human osteoclasts, activation of protein kinase C pathways appears to be the predominant mechanism of CT-mediated osteoclast inhibition,39 although PKA may be more important in human odontoclasts.135
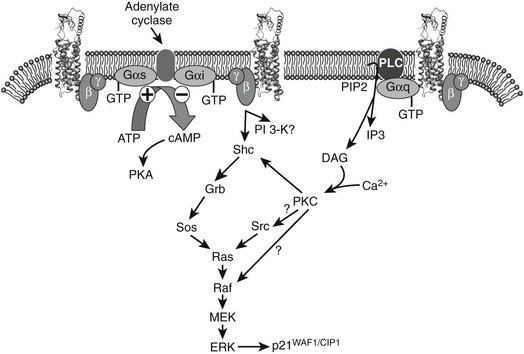
FIGURE 2-7 Calcitonin activation of the CTa receptor can lead to coupling to multiple different G proteins. Coupling to Gαi is seen less frequently and may be due, at least in part, to a PKC-dependent inhibition of Gi. The balance of activation of different G proteins and effectors is critical for the cell-specific actions of CT. Also illustrated is the current understanding of CT-induced MAPK activation, as delineated in stably transfected HEK-293 cells. ERK1/2 phosphorylation requires both PTX-sensitive G proteins (presumably Gi via βγ subunits) and Gq. The latter leads to rises in intracellular calcium and activation of PKC, and both contribute to ERK phosphorylation. At least part of the activation occurs following phosphorylation of Shc and mobilization of its downstream effectors. Activation of ERK increases phosphorylation of p21WAF/CIP1 and modulation of cell growth. Although strongly activated by CTa receptors, Gs is not involved in activation of MAPKs.
Calcitonin induction of interleukin (IL)-6 production in pituitary folliculo-stellate cells required both PKA and PKC signaling pathways.136 In hepatocytes, even at very low concentrations, CT is capable of increasing cytosolic calcium.137 CT-induced differentiation of early rat embryos is dependent upon intracellular calcium mobilization.138 It is interesting to note that although CT inhibits proton extrusion from osteoclasts,139 it can increase H+ efflux from nonosteoclastic cells, in a PKC-dependent manner.140 The significance of this activity is not clear. CT-induced changes mediated by cAMP or intracellular calcium in LLC-PK1 pig kidney cells are cell cycle dependent.141 Expression of cloned receptors in a variety of cell types has now conclusively shown that CT receptors of human, rat, and porcine origin are capable of signaling through both cAMP and calcium-activated second messenger systems. It is important to note that comparison of the calcium response in cell lines expressing different CT receptor levels has suggested that the magnitude of the response is proportional to the receptor density, and it is therefore possible that relative receptor density in target tissues may influence the signaling pathway(s) activated.
CT treatment of CTR-bearing cells, in the presence of extracellular calcium, initiates a sustained rise in intracellular calcium, the extent of which is dependent on the concentration of the extracellular calcium and is proportional to the receptor density.142 Because osteoclasts, which express high levels of CTR, are reportedly exposed to calcium concentrations as high as 26 mM during bone resorption,143 this phenomenon may have particular relevance for this cell type. CT and extracellular calcium both can cause intracellular calcium transients in isolated osteoclasts, and each agent greatly augments the signal produced by either agent alone.144
CTR-mediated activation of the MAPK pathway has been described,145–147 with a role for both Gq and Gi/o proteins—the latter principally via βγ G protein subunits, as is commonly the case for other G protein–coupled receptors. There is also evidence that CT can influence cell attachment by modulating components of focal adhesions and the cytoskeleton.146 CT treatment of osteoclasts disrupts the F-actin ring that is thought to correspond to the sealing zone64,148 and increases the tyrosine phosphorylation of paxillin and FAK. In rabbit osteoclasts, these actions were mediated by PKC.146 These actions of CT, together with decreased osteoclast motility, cytosol retraction, and disassembly of podosomes, have been linked to modulation of the activity and intracellular localization of Pyk2 and Src in osteoclasts.149 It has also been shown, in nonosteoclastic cells, that CT can cause cell death by aniokis, as the result of loss of cell attachment.150
It is interesting to note that recent work suggests that coexpression of RAMPs with the CTR leads to pathway-specific modulation of coupling, with strong augmentation of amylin-mediated Gs-coupling, but little effect on Gq- or Gi- mediated signaling, at least for RAMP 1– or RAMP 3–coupled receptors.151
Receptor Regulation
Regulation of the level and/or affinity of cell surface receptors is a key component in the physiologic and pathophysiologic responses to both endogenous and pharmacologically administered agents. The CTR is subject to both homologous (CT-induced) and heterologous regulation. CT-induced CTR downregulation was initially demonstrated in various transformed cell lines and subsequently in primary kidney cell cultures. The induction by CT of resistance to its own action in osteoclasts is such a specific process and correlates so closely in dose dependence to CT efficacy as an agonist,48 that it appears to be an important accompaniment of CT action. Proof of this might require genetic experiments, just as genetic manipulation has finally begun to cast light on the physiologic functions of CT.32,63
Receptor downregulation is mediated by specific loss of cell surface receptors, which occurs via an energy-dependent internalization of the ligand-receptor complex, in which the principal internalization pathway involves processing of the receptor-ligand complex into lysosomes and subsequent degradation of the receptor.152 Receptor regulatory responses to CT are likely to be cell or tissue dependent. For example, in mouse or rat osteoclasts, a potent downregulation of CTR mRNA appears to be mediated by a cAMP-dependent mechanism, in addition to downregulation of the receptor by internalization.153 Similar CT-mediated CTR regulation is also observed in human osteoclasts, but, as stated above, activation of protein kinase C pathways is suggested to be the predominant mechanism of CT-mediated osteoclast inhibition in human osteoclasts.39 In contrast to these observations in osteoclasts, downregulation of CTRs in UMR106-06 cells and T47D cells is not accompanied by changes in CTR mRNA levels.113 The CT-induced receptor mRNA loss in osteoclasts appears to be due principally to destabilization of receptor mRNA.46 The 3′-untranslated region of the mouse and rat CTR mRNA contains four AUUUA motifs, as well as other A/U-rich domains, which can function as signals for rapid mRNA inactivation. Thus, addition of A/U-rich CTR 3′UTR sequences considerably shortens the mRNA half-life of a reporter gene, and evidence shows the involvement of AUF1 p40, HuR in the regulation of CTR mRNA.154 It is also worth noting that the degree of internalization of human CTRs appears to be isoform specific, with the I1+ variant being resistant to internalization.106 Thus, the regulation of receptors and consequently peptide responses may vary according to the levels of specific receptor isoforms present in each tissue.
Data on regulation of CTR by other agents are limited. In mouse osteoclast cultures, the glucocorticoid dexamethasone increased the level of cell surface CTR following upregulation of receptor mRNA levels, the latter mediated at the level of transcription.46 Moreover, the CT-mediated decrease in cell surface receptor and mRNA was attenuated by dexamethasone. Increased production of CTR in response to glucocorticoid stimulation also occurs in the human T47D cell line, where cortisol is required for the expression of CTRs, suggesting that this may be a common regulatory mechanism for induction of CTR expression. It is worth noting the clinical evidence suggesting that glucocorticoids, given together with CT, might prevent to some extent the CT-induced resistance to its own action.155
Calcitonin and Its Receptors in Cancer
Medullary Carcinoma of The Thyroid
The classic syndrome of CT excess is medullary carcinoma of the thyroid (MCT). This tumor clearly differs in origin from all other thyroid cancers, since it is a tumor of the C cells, derived from the ultimobranchial bodies. MCT may occur as a sporadic or a genetic (familial) tumor.156–160 The only effective treatment for MCT is surgical. The earlier this can be performed in the course of this disease, the better is the likelihood of a cure. Once the tumor becomes palpable, any treatment is unlikely to be curative. Genetic testing for the locus of mutation in the responsible gene (the RET proto-oncogene) often has considerable clinical relevance in management, as early surgery is key to survival, especially in the syndrome known as multiple endocrine neoplasia type 2b (MEN-2b).
Calcitonin Receptors In Other Cancers
Specific high-affinity receptors for CT have been demonstrated in human lymphoid cell lines, in a human lung cancer cell line, in several human breast cancer cell lines, including MCF-7, T47D, and ZR-75, and in prostate cancer cells.152,153,161,162 Indeed, CTR mRNA appears to be a frequent accompaniment of human tumors. The first clinical study of CT receptors in surgically obtained human breast cancers identified receptor mRNA production in all of 18 cancers by reverse transcription/polymerase chain reaction (PCR).163
In situ hybridization in the same study showed that CT receptor expression was confined to tumor cells in the sections. In contrast, a subsequent study, using laser capture microdissection and PCR, found that the CTR in breast cancer tissue was actually reduced compared with adjacent normal breast tissue.164 The significance of the CTR in cancer cells is not known; however forced expression of human CT in LNCaP human prostate cancer cells was reported to dramatically enhance their oncogenic characteristics,165 and CT is expressed endogenously in the prostate.166 The same group also showed that CT increased hallmarks of invasiveness of prostate cancer cells, including the concentration and activity of MMP-2 and MMP-9.167 Similar results have been reported for MDA-MB-231 human breast cancer cells.168
Calcitonin in Growth and Development
The presence of CT and its receptors in a large number of cell types and tissue sites suggests multiple physiologic roles. CTRs have been identified in bone, kidney, brain, pituitary, testis, prostate, spermatozoa, breast, lung, and lymphocytes, as well as in cancer-derived cells from lung, breast, prostate, pituitary, bone (osteoclastoma, osteogenic sarcoma), and embryonal carcinoma.77 CT can potently modulate the growth of some CTR-bearing cells. Treatment with CT has been shown to stimulate the growth of human prostate cancer cells, in which the peptide increases both intracellular calcium and cAMP levels.169 Conversely, knock-down of calcitonin receptor expression in these cells induced apoptosis and growth arrest.162 The effects of CT on growth are clearly cell type dependent, since CT treatment repressed the growth of T47D human breast cancer cells, an action proposed to be mediated by the specific activation of the type II isoenzyme of the cAMP-dependent protein kinase.161 In cells overexpressing the human CTR, CT inhibited cell growth and caused an accumulation of cells in the G2 phase of the cell cycle, associated with a prolonged increase in p21WAF1/CIP1 expression170 and sustained activation of the p42/44 MAP kinase proteins.145
Evidence has been found to be consistent with the involvement of CT in cell growth161,169 and differentiation and in tissue development and remodeling.171 CT may be involved in both blastocyst implantation and development138,172 of the early blastocyst, and CT downregulates E-cadherin expression in rat uterine epithelium during implantation.130 It was shown recently that CT promotes the outgrowth of trophoblast cells on endometrial epithelial cells, perhaps thereby facilitating embryo implantation.173 Other reports indicate embryonic expression of the mouse CTR, suggesting that CTRs may play important roles in morphogenesis.171 It has been reported that CT is expressed in the pregnant mouse mammary gland, exclusively in and secreted from the luminal epithelial cells, and that its expression is progesterone dependent.174 It was found that CT induction spatiotemporally correlates with increases in progesterone-induced mammary gland proliferation and structural remodeling, suggesting that CT may be involved in one or both of these progesterone-dependent processes. As was discussed earlier, CT can inhibit cell proliferation161 or can have mitogenic actions.169 CT also may have a role in cell survival, and as discussed above has been claimed to be protective of drug-induced apoptosis in osteoblast-like and osteocyte-like cells54 and prostate cancer cell lines,175 and to promote the survival of osteoclasts.134 On the other hand, CT has been found to potentiate neuronal death due to oxygen and glucose deprivation,176 and to be pro-apoptotic in serum-deprived cells.150
Calcitonin as a Therapeutic
Calcitonin has been used in the treatment of hypercalcemia in many different conditions, especially malignancy, but its effective use is limited by rapid tachyphylaxis, often within 24 hours of use.177 The mechanism of resistance seems to involve receptor downregulation, which to some extent is blocked by corticosteroid use (see Receptor Regulation earlier).155
Osteoporosis and Paget’s Disease of Bone
The discussions in this chapter of the mechanism of action of CT provide some background for the use of CT in therapy. One of the most important unanswered questions concerning CT is whether its role as an inhibitor of bone resorption is such that CT deficiency can contribute to the development of osteoporosis. It is the antiresorptive action of CT that is the basis for its use clinically in osteoporosis, for which it is approved in several countries. Although its use remains somewhat controversial, CT administered by injection or by nasal spray has been found to decrease vertebral fracture risk in postmenopausal osteoporotic women.178,179 A reported advantage of calcitonin is its analgesic effect on bone pain, which probably is mediated centrally179; CT treatment has modest effects on bone quantity (BMD) or bone turnover markers, but new evidence suggests improvement in bone quality parameters after administration of salmon CT nasal spray over 2 years. The results of the QUEST study, in which effects of CT on bone structure were assessed by high-resolution MRI, suggest therapeutic benefit of CT-NS compared with placebo in maintaining trabecular microarchitecture at multiple skeletal sites.180 Similar protection by CT of bone structure was obtained in ovariectomized sheep, again with MRI used to assess bone changes.181 A problem with CT use clinically is its bioavailability, and attempts are now being made to produce orally bioavailable CT, with promising results.182,183
Osteoarthritis
A number of recent studies have suggested the potential benefit of CT in osteoarthritis. In both in vitro and in vivo animal models,184,185 CT was found to attenuate the indices of progression of osteoarthritis, including degradation of collagen type II, a hallmark of articular cartilage damage. In humans, CT treatment reduced circulating CTX-II186 and improved functional disability in patients, albeit in a small test group, selected for active disease.187 The mechanism of action of CT in this putative chondroprotection is not clear. It has been claimed, on the one hand, that CT has direct effects on bovine articular chondrocytes via the CTR,188 but a recent study was unable to find CTR expression or responsiveness to CT in human chondrocytes.189 The findings are nonetheless intriguing and benefits could result from the effects of CT on subchondral bone, as have been described for bisphosphonate treatment of animal models of osteoarthritis.190 Potential benefits of CT have also been suggested for inflammatory arthritis, since it was found to preserve bone morphology in a rat model of rheumatoid arthritis, particularly when used with prednisolone.191
References
1. Copp, DH, Cameron, EC, Cheney, BA, et al. Evidence for calcitonin—a new hormone from the parathyroid that lowers blood calcium. Endocrinology. 1962;70:638–649.
2. Hirsch, PF, Gauthier, GF, Munson, PL. Thyroid hypocalcemic principle and recurrent laryngeal nerve injury as factors affecting the response to parathyroidectomy in rats. Endocrinology. 1963;73:244–252.
3. Hirsch, PF, Voelkel, EF, Munson, PL. Thyrocalcitonin: hypocalcemic hypophosphatemic principle of the thyroid gland. Science. 1964;146:412–413.
4. Foster, GV, Baghdiantz, A, Kumar, MA, et al. Thyroid origin of calcitonin. Nature. 1964;202:1303–1305.
5. Pearse, AG, Carvalheira, AF. Cytochemical evidence for an ultimobranchial origin of rodent thyroid C cells. Nature. 1967;214(5091):929–930.
6. Suzuki, N, Suzuki, T, Kurokawa, T. Suppression of osteoclastic activities by calcitonin in the scales of goldfish (freshwater teleost) and nibbler fish (seawater teleost). Peptides. 2000;21(1):115–124.
7. Brown, EM, Gamba, G, Riccardi, D, et al. Cloning and characterization of an extracellular Ca(2+)-sensing receptor from bovine parathyroid. Nature. 1993;366(6455):575–580.
8. Care, AD, Bates, RF, Swaminathan, R, et al. The role of gastrin as a calcitonin secretagogue. J Endocrinol. 1971;51(4):735–744.
9. Freake, HC, MacIntyre, I. Specific binding of 1,25-dihydroxycholecalciferol in human medullary thyroid carcinoma. Biochem J. 1982;206(1):181–184.
10. Jacobs, JW, Simpson, E, Penschow, J, et al. Characterization and localization of calcitonin messenger ribonucleic acid in rat thyroid. Endocrinology. 1983;113(5):1616–1622.
11. Becker, KL, Snider, RH, Silva, OL, et al. Calcitonin heterogeneity in lung cancer and medullary thyroid cancer. Acta Endocrinol (Copenh). 1978;89(1):89–99.
12. Tobler, PH, Dambacher, MA, Born, W, et al. A new bioactive form of human calcitonin. Cancer Res. 1983;43(8):3793–3799.
13. Canale, DD, Donabedian, RK. Hypercalcitoninemia in acute pancreatitis. J Clin Endocrinol Metab. 1975;40(4):738–741.
14. O’Neill, WJ, Jordan, MH, Lewis, MS, et al. Serum calcitonin may be a marker for inhalation injury in burns. J Burn Care Rehabil. 1992;13(6):605–616.
15. Lind, L, Bucht, E, Ljunghall, S. Pronounced elevation in circulating calcitonin in critical care patients is related to the severity of illness and survival. Intensive Care Med. 1995;21(1):63–66.
16. Becker, KL, Nylen, ES, Snider, RH, et al. Immunoneutralization of procalcitonin as therapy of sepsis. J Endotoxin Res. 2003;9(6):367–374.
17. Martin, TJ, Findlay, DM, Moseley, JM, et al. Calcitonin. In: Avioli LV, Krane SM, eds. Metabolic Bone Disease and Clinically Related Disorders. ed 3. St. Louis: Academic Press; 1998:95–121.
18. Yamauchi, H, Shiraki, M, Otani, M, et al. Stability of [Asu1,7]-eel calcitonin and eel calcitonin in vitro and in vivo. Endocrinol Jpn. 1977;24(3):281–285.
19. Jacobs, JW, Lund, PK, Potts, JT, Jr., et al. Procalcitonin is a glycoprotein. J Biol Chem. 1981;256(6):2803–2807.
20. Mol, JA, Kwant, MM, Arnold, IC, et al. Elucidation of the sequence of canine (pro)-calcitonin. A molecular biological and protein chemical approach. Regul Pept. 1991;35(3):189–195.
21. Jacobs, JW, Goodman, RH, Chin, WW, et al. Calcitonin messenger RNA encodes multiple polypeptides in a single precursor. Science. 1981;213(4506):457–459.
22. Lasmoles, F, Jullienne, A, Day, F, et al. Elucidation of the nucleotide sequence of chicken calcitonin mRNA: direct evidence for the expression of a lower vertebrate calcitonin-like gene in man and rat. Embo J. 1985;4(10):2603–2607.
23. Steenbergh, PH, Hoppener, JW, Zandberg, J, et al. Calcitonin gene related peptide coding sequence is conserved in the human genome and is expressed in medullary thyroid carcinoma. J Clin Endocrinol Metab. 1984;59(2):358–360.
24. Rosenfeld, MG, Amara, SG, Evans, RM. Alternative RNA processing: determining neuronal phenotype. Science. 1984;225(4668):1315–1320.
25. Zaidi, M, Breimer, LH, MacIntyre, I. Biology of peptides from the calcitonin genes. Q J Exp Physiol. 1987;72(4):371–408.
26. Roos, BA, Yoon, MJ, Frelinger, AL, et al. Tumor growth and calcitonin during serial transplantation of rat medullary thyroid carcinoma. Endocrinology. 1979;105(1):27–32.
27. Lou, H, Gagel, RF, Berget, SM. An intron enhancer recognized by splicing factors activates polyadenylation. Genes Dev. 1996;10(2):208–219.
28. Cooper, CW, Hirsch, PF, Toverud, SU, et al. An improved method for the biological assay of thyrocalcitonin. Endocrinology. 1967;81(3):610–616.
29. Harper, C, Toverud, SU. Ability of thyrocalcitonin to protect against hypercalcemia in adult rats. Endocrinology. 1973;93(6):1354–1359.
30. Martin, TJ, Melick, RA. The acute effects of porcine calcitonin in man. Australas Ann Med. 1969;18(3):258–263.
31. Yamamoto, M, Seedor, JG, Rodan, GA, et al. Endogenous calcitonin attenuates parathyroid hormone-induced cancellous bone loss in the rat. Endocrinology. 1995;136(2):788–795.
32. Hoff, AO, Catala-Lehnen, P, Thomas, PM, et al. Increased bone mass is an unexpected phenotype associated with deletion of the calcitonin gene. J Clin Invest. 2002;110(12):1849–1857.
33. Woodrow, JP, Sharpe, CJ, Fudge, NJ, et al. Calcitonin plays a critical role in regulating skeletal mineral metabolism during lactation. Endocrinology. 2006;147(9):4010–4021.
34. Friedman, J, Raisz, LG. Thyrocalcitonin: inhibitor of bone resorption in tissue culture. Science. 1965;150(702):1465–1467.
35. Matthews, JL, Martin, JH. Immediate changes in the ultrastructure of bone cells following thyrocalcitonin administration. In: Talmadge RV, Munson PL, eds. Calcium, Parathyroid Hormone and the Calcitonins. Amsterdam: Excerpta Medica; 1972:375–382.
36. Martin, TJ, Robinson, CJ, MacIntyre, I. The mode of action of thyrocalcitonin. Lancet. 1966;1(7443):900–902.
37. Robinson, CJ, Martin, TJ, Matthews, EW, et al. Mode of action of thyrocalcitonin. J Endocrinol. 1967;39(1):71–79.
38. Nicholson, GC, Moseley, JM, Sexton, PM, et al. Abundant calcitonin receptors in isolated rat osteoclasts. Biochemical and autoradiographic characterization. J Clin Invest. 1986;78(2):355–360.
39. Samura, A, Wada, S, Suda, S, et al. Calcitonin receptor regulation and responsiveness to calcitonin in human osteoclast-like cells prepared in vitro using receptor activator of nuclear factor-kappaB ligand and macrophage colony-stimulating factor. Endocrinology. 2000;141(10):3774–3782.
40. Chambers, TJ, Athanasou, NA, Fuller, K. Effect of parathyroid hormone and calcitonin on the cytoplasmic spreading of isolated osteoclasts. J Endocrinol. 1984;102(3):281–286.
41. MacDonald, BR, Takahashi, N, McManus, LM, et al. Formation of multinucleated cells that respond to osteotropic hormones in long term human bone marrow cultures. Endocrinology. 1987;120(6):2326–2333.
42. Takahashi, N, Mundy, GR, Kuehl, TJ, et al. Osteoclast-like cell formation in fetal and newborn long-term baboon marrow cultures is more sensitive to 1,25-dihydroxyvitamin D3 than adult long-term marrow cultures. J Bone Miner Res. 1987;2(4):311–317.
43. Linkhart, TA, Linkhart, SG, Kodama, Y, et al. Osteoclast formation in bone marrow cultures from two inbred strains of mice with different bone densities. J Bone Miner Res. 1999;14(1):39–46.
44. Galvin, RJ, Bryan, P, Venugopalan, M, et al. Calcitonin responsiveness and receptor expression in porcine and murine osteoclasts: a comparative study. Bone. 1998;23(3):233–240.
45. Ikegame, M, Rakopoulos, M, Martin, TJ, et al. Effects of continuous calcitonin treatment on osteoclast-like cell development and calcitonin receptor expression in mouse bone marrow cultures. J Bone Miner Res. 1996;11(4):456–465.
46. Wada, S, Udagawa, N, Akatsu, T, et al. Regulation by calcitonin and glucocorticoids of calcitonin receptor gene expression in mouse osteoclasts. Endocrinology. 1997;138(2):521–529.
47. Wada, S, Udagawa, N, Nagata, N, et al. Calcitonin receptor down-regulation relates to calcitonin resistance in mature mouse osteoclasts. Endocrinology. 1996;137(3):1042–1048.
48. Ikegame, M, Ejiri, S, Ozawa, H. Calcitonin-induced change in serum calcium levels and its relationship to osteoclast morphology and number of calcitonin receptors. Bone. 2004;35(1):27–33.
49. Farley, JR, Tarbaux, NM, Hall, SL, et al. The anti-bone-resorptive agent calcitonin also acts in vitro to directly increase bone formation and bone cell proliferation. Endocrinology. 1988;123(1):159–167.
50. Farley, J, Dimai, HP, Stilt-Coffing, B, et al. Calcitonin increases the concentration of insulin-like growth factors in serum-free cultures of human osteoblast-line cells. Calcif Tissue Int. 2000;67(3):247–254.
51. Villa, I, Dal Fiume, C, Maestroni, A, et al. Human osteoblast-like cell proliferation induced by calcitonin-related peptides involves PKC activity. Am J Physiol Endocrinol Metab. 2003;284(3):E627–E633.
52. Naot, D, Bava, U, Matthews, B, et al. Differential gene expression in cultured osteoblasts and bone marrow stromal cells from patients with Paget’s disease of bone. J Bone Miner Res. 2007;22(2):298–309.
53. Chang, MK, Raggatt, LJ, Alexander, KA, et al. Osteal tissue macrophages are intercalated throughout human and mouse bone lining tissues and regulate osteoblast function in vitro and in vivo. J Immunol. 2008;181(2):1232–1244.
54. Plotkin, LI, Weinstein, RS, Parfitt, AM, et al. Prevention of osteocyte and osteoblast apoptosis by bisphosphonates and calcitonin. J Clin Invest. 1999;104(10):1363–1374.
55. Bulbul, M, Esenyel, CZ, Esenyel, M, et al. Effects of calcitonin on the biomechanics, histopathology, and radiography of callus formation in rats. J Orthop Sci. 2008;13(2):136–144.
56. Li, X, Luo, X, Yu, N, et al. Effects of salmon calcitonin on fracture healing in ovariectomized rats. Saudi Medical Journal. 2007;28(1):60–64.
57. Amanat, N, McDonald, M, Godfrey, C, et al. Optimal timing of a single dose of zoledronic acid to increase strength in rat fracture repair. J Bone Miner Res. 2007;22(6):867–876.
58. Chigurupati, S, Kulkarni, T, Thomas, S, et al. Calcitonin stimulates multiple stages of angiogenesis by directly acting on endothelial cells. Cancer Res. 2005;65(18):8519–8529.
59. Hirsch, PF, Baruch, H. Is calcitonin an important physiological substance? Endocrine. 2003;21(3):201–208.
60. Schinke, T, Liese, S, Priemel, M, et al. Decreased bone formation and osteopenia in mice lacking alpha-calcitonin gene-related peptide. J Bone Miner Res. 2004;19(12):2049–2056.
61. Huebner, AK, Schinke, T, Priemel, M, et al. Calcitonin deficiency in mice progressively results in high bone turnover. J Bone Miner Res. 2006;21(12):1924–1934.
62. Huebner, AK, Keller, J, Catala-Lehnen, P, et al. The role of calcitonin and alpha-calcitonin gene-related peptide in bone formation. Arch Biochem Biophys. 2008;473(2):210–217.
63. Dacquin, R, Davey, RA, Laplace, C, et al. Amylin inhibits bone resorption while the calcitonin receptor controls bone formation in vivo. J Cell Biol. 2004;164(4):509–514.
64. Davey, RA, Turner, A, McManus, JF, et al. The calcitonin receptor plays a physiological role to protect against hypercalcemia in mice. J Bone Miner Res. 2008.
65. Martin, TJ, Sims, NA. Osteoclast-derived activity in the coupling of bone formation to resorption. Trends Mol Med. 2005;11(2):76–81.
66. Takeda, S, Elefteriou, F, Levasseur, R, et al. Leptin regulates bone formation via the sympathetic nervous system. Cell. 2002;111(3):305–317.
67. Robinson, CJ, Martin, TJ, MacIntyre, I. Phosphaturic effect of thyrocalcitonin. Lancet. 1966;2(7454):83–84.
68. Williams, CC, Matthews, EW, Moseley, JM, et al. The effects of synthetic human and salmon calcitonins on electrolyte excretion in the rat. Clin Sci. 1972;42(2):129–137.
69. Hosking, DJ, Gilson, D. Comparison of the renal and skeletal actions of calcitonin in the treatment of severe hypercalcaemia of malignancy. Q J Med. 1984;53(211):359–368.
70. Sexton, PM, Adam, WR, Moseley, JM, et al. Localization and characterization of renal calcitonin receptors by in vitro autoradiography. Kidney Int. 1987;32(6):862–868.
71. Kawashima, H, Torikai, S, Kurokawa, K. Calcitonin selectively stimulates 25-hydroxyvitamin D3–1 alpha-hydroxylase in proximal straight tubule of rat kidney. Nature. 1981;291(5813):327–329.
72. Shinki, T, Ueno, Y, DeLuca, HF, et al. Calcitonin is a major regulator for the expression of renal 25-hydroxyvitamin D3–1alpha-hydroxylase gene in normocalcemic rats. Proc Natl Acad Sci U S A. 1999;96(14):8253–8258.
73. Murayama, A, Takeyama, K, Kitanaka, S, et al. Positive and negative regulations of the renal 25-hydroxyvitamin D3 1alpha-hydroxylase gene by parathyroid hormone, calcitonin, and 1alpha,25(OH)2D3 in intact animals. Endocrinology. 1999;140(5):2224–2231.
74. Gao, XH, Dwivedi, PP, Omdahl, JL, et al. Calcitonin stimulates expression of the rat 25-hydroxyvitamin D3–24-hydroxylase (CYP24) promoter in HEK-293 cells expressing calcitonin receptor: identification of signaling pathways. J Mol Endocrinol. 2004;32(1):87–98.
75. Young, A. Amylin and the integrated control of nutrient influx. Adv Pharmacol. 2005;52:67–77.
76. Pittner, RA, Albrandt, K, Beaumont, K, et al. Molecular physiology of amylin. J Cell Biochem. 1994;55(Suppl):19–28.
77. Sexton, PM, Findlay, DM, Martin, TJ. Calcitonin. Curr Med Chem. 1999;6(11):1067–1093.
78. Poyner, DR. Molecular pharmacology of receptors for calcitonin-gene-related peptide, amylin and adrenomedullin. Biochem Soc Trans. 1997;25(3):1032–1036.
79. Wimalawansa, SJ. Calcitonin gene-related peptide and its receptors: molecular genetics, physiology, pathophysiology, and therapeutic potentials. Endocr Rev. 1996;17(5):533–585.
80. Christopoulos, G, Perry, KJ, Morfis, M, et al. Multiple amylin receptors arise from receptor activity-modifying protein interaction with the calcitonin receptor gene product. Mol Pharmacol. 1999;56(1):235–242.
81. Morfis, M, Christopoulos, A, Sexton, PM. RAMPs: 5 years on, where to now? Trends Pharmacol Sci. 2003;24(11):596–601.
82. Hay, DL, Christopoulos, G, Christopoulos, A, et al. Pharmacological discrimination of calcitonin receptor: receptor activity-modifying protein complexes. Mol Pharmacol. 2005;67(5):1655–1665.
83. Wimalawansa, SJ. Amylin, calcitonin gene-related peptide, calcitonin, and adrenomedullin: a peptide superfamily. Crit Rev Neurobiol. 1997;11(2–3):167–239.
84. Niu, P, Shindo, T, Iwata, H, et al. Protective effects of endogenous adrenomedullin on cardiac hypertrophy, fibrosis, and renal damage. Circulation. 2004;109(14):1789–1794.
85. Kandler, MA, Von Der Hardt, K, Mahfoud, S, et al. Pilot intervention: aerosolized adrenomedullin reduces pulmonary hypertension. J Pharmacol Exp Ther. 2003;306(3):1021–1026.
86. Cornish, J, Reid, IR. Effects of amylin and adrenomedullin on the skeleton. J Musculoskelet Neuronal Interact. 2001;2(1):15–24.
87. Katafuchi, T, Hamano, K, Kikumoto, K, et al. Identification of second and third calcitonin receptor-stimulating peptides in porcine brain. Biochem Biophys Res Commun. 2003;308(3):445–451.
88. Katafuchi, T, Hamano, K, Minamino, N. Identification, structural determination, and biological activity of bovine and canine calcitonin receptor-stimulating peptides. Biochem Biophys Res Commun. 2004;313(1):74–79.
89. Katafuchi, T, Kikumoto, K, Hamano, K, et al. Calcitonin receptor-stimulating peptide, a new member of the calcitonin gene-related peptide family. Its isolation from porcine brain, structure, tissue distribution, and biological activity. J Biol Chem. 2003;278(14):12046–12054.
90. Roh, J, Chang, CL, Bhalla, A, et al. Intermedin is a calcitonin/calcitonin gene-related peptide family peptide acting through the calcitonin receptor-like receptor/receptor activity-modifying protein receptor complexes. J Biol Chem. 2004;279(8):7264–7274.
91. Takei, Y, Hyodo, S, Katafuchi, T, et al. Novel fish-derived adrenomedullin in mammals: structure and possible function. Peptides. 2004;25(10):1643–1656.
92. Sexton, PM. Central nervous system binding sites for calcitonin and calcitonin gene-related peptide. Mol Neurobiol. 1991;5(2–4):251–273.
93. Hilton, JM, Mitchelhill, KI, Pozvek, G, et al. Purification of calcitonin-like peptides from rat brain and pituitary. Endocrinology. 1998;139(3):982–992.
94. Denef, C. Paracrinicity: the story of 30 years of cellular pituitary crosstalk. J Neuroendocrinol. 2008;20(1):1–70.
95. Shah, GV, Wang, W, Grosvenor, CE, et al. Calcitonin inhibits basal and thyrotropin-releasing hormone-induced release of prolactin from anterior pituitary cells: evidence for a selective action exerted proximal to secretagogue-induced increases in cytosolic Ca2+. Endocrinology. 1990;127(2):621–628.
96. Judd, AM, Kubota, T, Kuan, SI, et al. Calcitonin decreases thyrotropin-releasing hormone-stimulated prolactin release through a mechanism that involves inhibition of inositol phosphate production. Endocrinology. 1990;127(1):191–199.
97. Shah, GV, Deftos, LJ, Crowley, WR. Synthesis and release of calcitonin-like immunoreactivity by anterior pituitary cells: evidence for a role in paracrine regulation of prolactin secretion. Endocrinology. 1993;132(3):1367–1372.
98. Yuan, R, Kulkarni, T, Wei, F, et al. Targeted overexpression of calcitonin in gonadotrophs of transgenic mice leads to chronic hypoprolactinemia. Mol Cell Endocrinol. 2005;229(1–2):193–203.
99. Sarkar, DK, Kim, KH, Minami, S. Transforming growth factor-beta 1 messenger RNA and protein expression in the pituitary gland: its action on prolactin secretion and lactotropic growth. Mol Endocrinol. 1992;6(11):1825–1833.
100. Takahashi, N, Yamana, H, Yoshiki, S, et al. Osteoclast-like cell formation and its regulation by osteotropic hormones in mouse bone marrow cultures. Endocrinology. 1988;122(4):1373–1382.
101. Paxinos, G, Chai, SY, Christopoulos, G, et al. In vitro autoradiographic localization of calcitonin and amylin binding sites in monkey brain. J Chem Neuroanat. 2004;27(4):217–236.
102. Hilton, JM, Chai, SY, Sexton, PM. In vitro autoradiographic localization of the calcitonin receptor isoforms, C1a and C1b, in rat brain. Neuroscience. 1995;69(4):1223–1237.
103. Lin, HY, Harris, TL, Flannery, MS, et al. Expression cloning of an adenylate cyclase-coupled calcitonin receptor. Science. 1991;254(5034):1022–1024.
104. Nag, K, Kato, A, Sultana, N, et al. Fish receptor has novel features. General and Comparative Endocrinology. 2007;154:48–58.
105. Gorn, AH, Lin, HY, Yamin, M, et al. Cloning, characterization, and expression of a human calcitonin receptor from an ovarian carcinoma cell line. J Clin Invest. 1992;90(5):1726–1735.
106. Moore, EE, Kuestner, RE, Stroop, SD, et al. Functionally different isoforms of the human calcitonin receptor result from alternative splicing of the gene transcript. Mol Endocrinol. 1995;9(8):959–968.
107. Albrandt, K, Brady, EM, Moore, CX, et al. Molecular cloning and functional expression of a third isoform of the human calcitonin receptor and partial characterization of the calcitonin receptor gene. Endocrinology. 1995;136(12):5377–5384.
108. Poyner, DR, Sexton, PM, Marshall, I, et al. International Union of Pharmacology. XXXII. The mammalian calcitonin gene-related peptides, adrenomedullin, amylin, and calcitonin receptors. Pharmacol Rev. 2002;54(2):233–246.
109. Houssami, S, Findlay, DM, Brady, CL, et al. Divergent structural requirements exist for calcitonin receptor binding specificity and adenylate cyclase activation. Mol Pharmacol. 1995;47(4):798–809.
110. Seck, T, Baron, R, Horne, WC. Binding of filamin to the C-terminal tail of the calcitonin receptor controls recycling. J Biol Chem. 2003;278(12):10408–10416.
111. Seck, T, Baron, R, Horne, WC. The alternatively spliced deltae13 transcript of the rabbit calcitonin receptor dimerizes with the C1a isoform and inhibits its surface expression. J Biol Chem. 2003;278(25):23085–23093.
112. Zolnierowicz, S, Cron, P, Solinas-Toldo, S, et al. Isolation, characterization, and chromosomal localization of the porcine calcitonin receptor gene. Identification of two variants of the receptor generated by alternative splicing. J Biol Chem. 1994;269(30):19530–19538.
113. Anusaksathien, O, Laplace, C, Li, X, et al. Tissue-specific and ubiquitous promoters direct the expression of alternatively spliced transcripts from the calcitonin receptor gene. J Biol Chem. 2001;276(25):22663–22674.
114. Pondel, MD, Partington, GA, Mould, R. Tissue-specific activity of the proximal human calcitonin receptor promoter is mediated by Sp1 and an epigenetic phenomenon. FEBS Lett. 2003;554(3):433–438.
115. Shen, Z, Crotti, TN, Flannery, MR, et al. A novel promoter regulates calcitonin receptor gene expression in human osteoclasts. Biochim Biophys Acta. 2007;1769(11–12):659–667.
116. Hoshiya, H, Meguro, M, Kashiwagi, A, et al. Calcr, a brain-specific imprinted mouse calcitonin receptor gene in the imprinted cluster of the proximal region of chromosome 6. J Hum Genet. 2003;48(4):208–211.
117. Beaudreuil, J, Taboulet, J, Orcel, P, et al. Calcitonin receptor mRNA in mononuclear leucocytes from postmenopausal women: decrease during osteoporosis and link to bone markers with specific isoform involvement. Bone. 2000;27(1):161–168.
118. Nakamura, M, Zhang, ZQ, Shan, L, et al. Allelic variants of human calcitonin receptor in the Japanese population. Hum Genet. 1997;99(1):38–41.
119. Zofkova, I, Zajickova, K, Hill, M, et al. Does polymorphism C1377T of the calcitonin receptor gene determine bone mineral density in postmenopausal women? Exp Clin Endocrinol Diabetes. 2003;111(7):447–449.
120. Tsai, FJ, Chen, WC, Chen, HY, et al. The ALUI calcitonin receptor gene polymorphism (TT) is associated with low bone mineral density and susceptibility to osteoporosis in postmenopausal women. Gynecol Obstet Invest. 2003;55(2):82–87.
121. Braga, V, Sangalli, A, Malerba, G, et al. Relationship among VDR (BsmI and FokI), COLIA1, and CTR polymorphisms with bone mass, bone turnover markers, and sex hormones in men. Calcif Tissue Int. 2002;70(6):457–462.
122. Nakamura, M, Morimoto, S, Zhang, Z, et al. Calcitonin receptor gene polymorphism in Japanese women: correlation with body mass and bone mineral density. Calcif Tissue Int. 2001;68(4):211–215.
123. Braga, V, Mottes, M, Mirandola, S, et al. Association of CTR and COLIA1 alleles with BMD values in peri- and postmenopausal women. Calcif Tissue Int. 2000;67(5):361–366.
124. Masi, L, Becherini, L, Gennari, L, et al. Allelic variants of human calcitonin receptor: distribution and association with bone mass in postmenopausal Italian women. Biochem Biophys Res Commun. 1998;245(2):622–626.
125. Masi, L, Cimaz, R, Simonini, G, et al. Association of low bone mass with vitamin D receptor gene and calcitonin receptor gene polymorphisms in juvenile idiopathic arthritis. J Rheumatol. 2002;29(10):2225–2231.
126. Chen, WC, Wu, HC, Lu, HF, et al. Calcitonin receptor gene polymorphism: a possible genetic marker for patients with calcium oxalate stones. Eur Urol. 2001;39(6):716–719.
127. Wolfe, LA, 3rd., Fling, ME, Xue, Z, et al. In vitro characterization of a human calcitonin receptor gene polymorphism. Mutat Res. 2003;522(1–2):93–105.
128. McLatchie, LM, Fraser, NJ, Main, MJ, et al. RAMPs regulate the transport and ligand specificity of the calcitonin-receptor-like receptor. Nature. 1998;393(6683):333–339.
129. Fraser, NJ, Wise, A, Brown, J, et al. The amino terminus of receptor activity modifying proteins is a critical determinant of glycosylation state and ligand binding of calcitonin receptor-like receptor. Mol Pharmacol. 1999;55(6):1054–1059.
130. Udawela, M, Hay, DL, Sexton, PM. The receptor activity modifying protein family of G protein coupled receptor accessory proteins. Semin Cell Dev Biol. 2004;15(3):299–308.
131. Suzuki, H, Nakamura, I, Takahashi, N, et al. Calcitonin-induced changes in the cytoskeleton are mediated by a signal pathway associated with protein kinase A in osteoclasts. Endocrinology. 1996;137(11):4685–4690.
132. Granholm, S, Lundberg, P, Lerner, UH. Calcitonin inhibits osteoclast formation in mouse haematopoietic cells independently of transcriptional regulation by receptor activator of NF-κB and c-Fms. J Endocrinol. 2007;195(3):415–427.
133. Zaidi, M, Datta, HK, Moonga, BS, et al. Evidence that the action of calcitonin on rat osteoclasts is mediated by two G proteins acting via separate post-receptor pathways. J Endocrinol. 1990;126(3):473–481.
134. Offermanns, S, Iida-Klein, A, Segre, GV, et al. G alpha q family members couple parathyroid hormone (PTH)/PTH-related peptide and calcitonin receptors to phospholipase C in COS-7 cells. Mol Endocrinol. 1996;10(5):566–574.
135. Takada, K, Kajiya, H, Fukushima, H, et al. Calcitonin in human odontoclasts regulates root resorption activity via protein kinase A. J Bone Miner Metab. 2004;22(1):12–18.
136. Kiriyama, Y, Tsuchiya, H, Murakami, T, et al. Calcitonin induces IL-6 production via both PKA and PKC pathways in the pituitary folliculo-stellate cell line. Endocrinology. 2001;142(8):3563–3569.
137. Yamaguchi, M. Stimulatory effect of calcitonin on Ca2+ inflow in isolated rat hepatocytes. Mol Cell Endocrinol. 1991;75(1):65–70.
138. Wang, J, Rout, UK, Bagchi, IC, et al. Expression of calcitonin receptors in mouse preimplantation embryos and their function in the regulation of blastocyst differentiation by calcitonin. Development. 1998;125(21):4293–4302.
139. Kajiya, H, Okamoto, F, Fukushima, H, et al. Calcitonin inhibits proton extrusion in resorbing rat osteoclasts via protein kinase A. Pflugers Arch. 2003;445(6):651–658.
140. Santhanagopal, A, Chidiac, P, Horne, WC, et al. Calcitonin (CT) rapidly increases NA(+)/H(+) exchange and metabolic acid production: effects mediated selectively by the C1A CT receptor isoform. Endocrinology. 2001;142(10):4401–4413.
141. Chakraborty, M, Chatterjee, D, Kellokumpu, S, et al. Cell cycle-dependent coupling of the calcitonin receptor to different G proteins. Science. 1991;251(4997):1078–1082.
142. Stroop, SD, Thompson, DL, Kuestner, RE, et al. A recombinant human calcitonin receptor functions as an extracellular calcium sensor. J Biol Chem. 1993;268(27):19927–19930.
143. Silver, IA, Murrills, RJ, Etherington, DJ. Microelectrode studies on the acid microenvironment beneath adherent macrophages and osteoclasts. Exp Cell Res. 1988;175(2):266–276.
144. Malgaroli, A, Meldolesi, J, Zallone, AZ, et al. Control of cytosolic free calcium in rat and chicken osteoclasts. The role of extracellular calcium and calcitonin. J Biol Chem. 1989;264(24):14342–14347.
145. Raggatt, LJ, Evdokiou, A, Findlay, DM. Sustained activation of Erk1/2 MAPK and cell growth suppression by the insert-negative, but not the insert-positive isoform of the human calcitonin receptor. J Endocrinol. 2000;167(1):93–105.
146. Zhang, Z, Baron, R, Horne, WC. Integrin engagement, the actin cytoskeleton, and c-Src are required for the calcitonin-induced tyrosine phosphorylation of paxillin and HEF1, but not for calcitonin-induced Erk1/2 phosphorylation. J Biol Chem. 2000;275(47):37219–37223.
147. Findlay, DM, Sexton, PM. Calcitonin. Growth Factors. 2004;22(4):217–224.
148. Okumura, S, Mizoguchi, T, Sato, N, et al. Coordination of microtubules and the actin cytoskeleton is important in osteoclast function, but calcitonin disrupts sealing zones without affecting microtubule networks. Bone. 2006;39(4):684–693.
149. Shyu, JF, Shih, C, Tseng, CY, et al. Calcitonin induces podosome disassembly and detachment of osteoclasts by modulating Pyk2 and Src activities. Bone. 2007;40(5):1329–1342.
150. Findlay, DM, Raggatt, LJ, Bouralexis, S, et al. Calcitonin decreases the adherence and survival of HEK-293 cells by a caspase-independent mechanism. J Endocrinol. 2002;175(3):715–725.
151. Morfis, M, Tilakaratne, N, Furness, SG, et al. Receptor activity modifying proteins differentially modulate the G protein-coupling efficiency of amylin receptors. Endocrinology. 2008;149:5423–5431.
152. Schneider, HG, Raue, F, Zink, A, et al. Down-regulation of calcitonin receptors in T47D cells by internalization of calcitonin-receptor complexes. Mol Cell Endocrinol. 1988;58(1):9–15.
153. Wada, S, Martin, TJ, Findlay, DM. Homologous regulation of the calcitonin receptor in mouse osteoclast-like cells and human breast cancer T47D cells. Endocrinology. 1995;136(6):2611–2621.
154. Yasuda, S, Wada, S, Arao, Y, et al. Interaction between 3′ untranslated region of calcitonin receptor messenger ribonucleic acid (RNA) and adenylate/uridylate (AU)-rich element binding proteins (AU-rich RNA-binding factor 1 and Hu antigen R). Endocrinology. 2004;145(4):1730–1738.
155. Binstock, ML, Mundy, GR. Effect of calcitonin and glucocorticoids in combination on the hypercalcemia of malignancy. Ann Intern Med. 1980;93(2):269–272.
156. Hazard, JB. The C cells (parafollicular cells) of the thyroid gland and medullary thyroid carcinoma. A review. Am J Pathol. 1977;88(1):213–250.
157. Sipple, JH. Multiple endocrine neoplasia type 2 syndromes: historical perspectives. Henry Ford Hosp Med J. 1984;32(4):219–221.
158. Block, MA, Jackson, CE, Greenawald, KA, et al. Clinical characteristics distinguishing hereditary from sporadic medullary thyroid carcinoma. Treatment implications. Arch Surg. 1980;115(2):142–148.
159. Wells, SA, Jr., Ontjes, DA. Multiple endocrine neoplasia type II. Annu Rev Med. 1976;27:263–268.
160. Melvin, KE, Tashjian, AH, Jr. The syndrome of excessive thyrocalcitonin produced by medullary carcinoma of the thyroid. Proc Natl Acad Sci U S A. 1968;59(4):1216–1222.
161. Ng, KW, Livesey, SA, Larkins, RG, et al. Calcitonin effects on growth and on selective activation of type II isoenzyme of cyclic adenosine 3′:5′-monophosphate-dependent protein kinase in T 47D human breast cancer cells. Cancer Res. 1983;43(2):794–800.
162. Thomas, S, Muralidharan, A, Shah, GV. Knock-down of calcitonin receptor expression induces apoptosis and growth arrest of prostate cancer cells. Int J Oncol. 2007;31(6):1425–1437.
163. Gillespie, MT, Thomas, RJ, Pu, ZY, et al. Calcitonin receptors, bone sialoprotein and osteopontin are expressed in primary breast cancers. Int J Cancer. 1997;73(6):812–815.
164. Wang, X, Nakamura, M, Mori, I, et al. Calcitonin receptor gene and breast cancer: quantitative analysis with laser capture microdissection. Breast Cancer Res Treat. 2004;83(2):109–117.
165. Thomas, S, Chigurupati, S, Anbalagan, M, et al. Calcitonin increases tumorigenicity of prostate cancer cells: evidence for the role of protein kinase A and urokinase-type plasminogen receptor. Mol Endocrinol. 2006;20(8):1894–1911.
166. Chien, J, Ren, Y, Qing Wang, Y, et al. Calcitonin is a prostate epithelium-derived growth stimulatory peptide. Mol Cell Endocrinol. 2001;181(1–2):69–79.
167. Sabbisetti, V, Chigurupati, S, Thomas, S, et al. Calcitonin stimulates the secretion of urokinase-type plasminogen activator from prostate cancer cells: its possible implications on tumor cell invasion. Int J Cancer. 2006;118(11):2694–2702.
168. Han, B, Nakamura, M, Zhou, G, et al. Calcitonin inhibits invasion of breast cancer cells: involvement of urokinase-type plasminogen activator (uPA) and uPA receptor. Int J Oncol. 2006;28(4):807–814.
169. Shah, GV, Rayford, W, Noble, MJ, et al. Calcitonin stimulates growth of human prostate cancer cells through receptor-mediated increase in cyclic adenosine 3′,5′-monophosphates and cytoplasmic Ca2+ transients. Endocrinology. 1994;134(2):596–602.
170. Evdokiou, A, Raggatt, LJ, Atkins, GJ, et al. Calcitonin receptor-mediated growth suppression of HEK-293 cells is accompanied by induction of p21WAF1/CIP1 and G2/M arrest. Mol Endocrinol. 1999;13(10):1738–1750.
171. Jagger, C, Chambers, T, Pondel, M. Transgenic mice reveal novel sites of calcitonin receptor gene expression during development. Biochem Biophys Res Commun. 2000;274(1):124–129.
172. Zhu, LJ, Cullinan-Bove, K, Polihronis, M, et al. Calcitonin is a progesterone-regulated marker that forecasts the receptive state of endometrium during implantation. Endocrinology. 1998;139(9):3923–3934.
173. Li, HY, Shen, JT, Chang, SP, et al. Calcitonin promotes outgrowth of trophoblast cells on endometrial epithelial cells: involvement of calcium mobilization and protein kinase C activation. Placenta. 2008;29(1):20–29.
174. Ismail, PM, DeMayo, FJ, Amato, P, et al. Progesterone induction of calcitonin expression in the murine mammary gland. J Endocrinol. 2004;180(2):287–295.
175. Salido, M, Vilches, J, Lopez, A. Neuropeptides bombesin and calcitonin induce resistance to etoposide induced apoptosis in prostate cancer cell lines. Histol Histopathol. 2000;15(3):729–738.
176. Asrari, M, Lobner, D. Calcitonin potentiates oxygen-glucose deprivation-induced neuronal death. Exp Neurol. 2001;167(1):183–188.
177. Potts, JT, Finkelstein, J, Medical treatment of hypercalcemia. Endocrinology. ed 5. DeGroot, LJ, Jameson, JL, eds. Endocrinology. Philadelphia: WB Saunders; 2006;vol 2.
178. Chesnut, CH, 3rd., Silverman, S, Andriano, K, et al. A randomized trial of nasal spray salmon calcitonin in postmenopausal women with established osteoporosis: the prevent recurrence of osteoporotic fractures study. PROOF Study Group. Am J Med. 2000;109(4):267–276.
179. Chesnut, CH, 3rd., Azria, M, Silverman, S, et al. Salmon calcitonin: a review of current and future therapeutic indications. Osteoporos Int. 2008;19(4):479–491.
180. Chesnut, CH, 3rd., Majumdar, S, Newitt, DC, et al. Effects of salmon calcitonin on trabecular microarchitecture as determined by magnetic resonance imaging: results from the QUEST study. J Bone Miner Res. 2005;20(9):1548–1561.
181. Jiang, Y, Zhao, J, Geusens, P, et al. Femoral neck trabecular microstructure in ovariectomized ewes treated with calcitonin: MRI microscopic evaluation. J Bone Miner Res. 2005;20(1):125–130.
182. Lee, YH, Sinko, PJ. Oral delivery of salmon calcitonin. Adv Drug Deliv Rev. 2000;42(3):225–238.
183. Komarova, SV, Shum, JB, Paige, LA, et al. Regulation of osteoclasts by calcitonin and amphiphilic calcitonin conjugates: role of cytosolic calcium. Calcif Tissue Int. 2003;73(3):265–273.
184. Behets, C, Williams, JM, Chappard, D, et al. Effects of calcitonin on subchondral trabecular bone changes and on osteoarthritic cartilage lesions after acute anterior cruciate ligament deficiency. J Bone Miner Res. 2004;19(11):1821–1826.
185. Sondergaard, BC, Oestergaard, S, Christiansen, C, et al. The effect of oral calcitonin on cartilage turnover and surface erosion in an ovariectomized rat model. Arthritis Rheum. 2007;56(8):2674–2678.
186. Bagger, YZ, Tanko, LB, Alexandersen, P, et al. Oral salmon calcitonin induced suppression of urinary collagen type II degradation in postmenopausal women: a new potential treatment of osteoarthritis. Bone. 2005;37(3):425–430.
187. Manicourt, DH, Azria, M, Mindeholm, L, et al. Oral salmon calcitonin reduces Lequesne’s algofunctional index scores and decreases urinary and serum levels of biomarkers of joint metabolism in knee osteoarthritis. Arthritis Rheum. 2006;54(10):3205–3211.
188. Sondergaard, BC, Wulf, H, Henriksen, K, et al. Calcitonin directly attenuates collagen type II degradation by inhibition of matrix metalloproteinase expression and activity in articular chondrocytes. Osteoarthritis Cartilage. 2006;14(8):759–768.
189. Lin, Z, Pavlos, NJ, Cake, MA, et al. Evidence that human cartilage and chondrocytes do not express calcitonin receptor. Osteoarthritis Cartilage. 2008;16(4):450–457.
190. Hayami, T, Pickarski, M, Wesolowski, GA, et al. The role of subchondral bone remodeling in osteoarthritis: reduction of cartilage degeneration and prevention of osteophyte formation by alendronate in the rat anterior cruciate ligament transection model. Arthritis Rheum. 2004;50(4):1193–1206.
191. Mancini, L, Paul-Clark, MJ, Rosignoli, G, et al. Calcitonin and prednisolone display antagonistic actions on bone and have synergistic effects in experimental arthritis. Am J Pathol. 2007;170(3):1018–1027.