[level-membership-for-dermatology-category]24
Bullous Pemphigoid, Mucous Membrane Pemphigoid, and Epidermolysis Bullosa Acquisita
This chapter, in addition to Chapters 23 and 25, cover the autoimmune bullous diseases. The concepts of direct immunofluorescence (DIF) and indirect immunofluorescence (IIF) are reviewed in Chapter 23 (see Fig. 23.2), as are the recommended sites for performing skin biopsies for DIF (see Fig. 23.1).
Bullous Pemphigoid (BP)
• Both pruritic fixed urticarial plaques and tense bullae are seen (Figs. 24.1 and 24.2); the latter can develop within normal skin or areas of erythematous skin and produce erosions when they rupture; oral lesions are much less common than in pemphigus vulgaris (10–30% of patients).
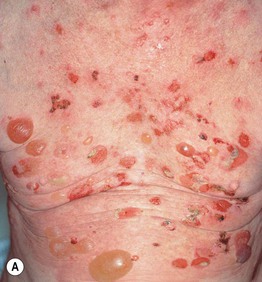
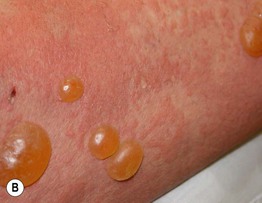
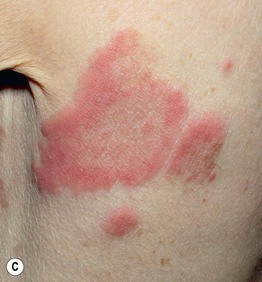
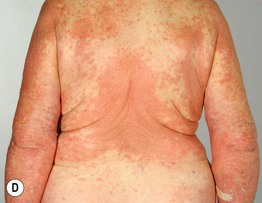
Fig. 24.1 Bullous pemphigoid. A Classic presentation with multiple tense bullae arising on normal and erythematous skin; several of the bullae have ruptured, leaving circular erosions. B Urticarial papules and plaques, along with bullae containing serous fluid. C Firm annular urticarial plaques. D Eczematous presentation with large dermatitic plaques. C, D, Courtesy, Philippe Bernard, MD, and Luca Borradori, MD.
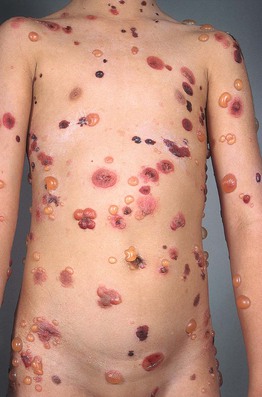
Fig. 24.2 Childhood bullous pemphigoid. Generalized bullae, both tense (fresh) and flaccid (old with re-epithelialization), as well as erosions with hemorrhagic crusts.
• Pruritus and nonspecific eczematous (see Fig. 24.1D) or papular lesions can precede the more characteristic cutaneous lesions and may be the predominant finding; unusual variants include dyshidrosiform (palms and soles), vegetans (major body folds), and localized (e.g. pretibial in adults; vulvar in children, acral in infants), as well as those that mimic prurigo nodularis and toxic epidermal necrolysis (Fig. 24.3).
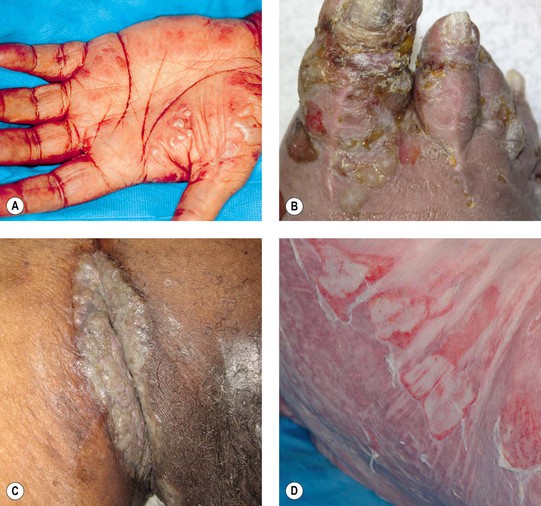
Fig. 24.3 Bullous pemphigoid – unusual clinical variants. Grouped vesicles and bullae on the palms (A) and toes (B) that can resemble pompholyx (dyshidrosiform pemphigoid). C Vegetating plaque in the inguinal crease (pemphigoid vegetans). D Toxic epidermal necrolysis-like lesions with large erosions. A, D, Courtesy, Philippe Bernard, MD, and Luca Borradori, MD.
• Histologically, a subepidermal bulla plus an infiltrate of eosinophils is seen when the lesions are bullous; DIF demonstrates immunodeposits of IgG and/or C3 in a linear array along the basement membrane zone (see Fig. 23.3B); in general, by salt-split skin immunofluorescence studies, the immunodeposits are in the roof (epidermal side) of the blister (Fig. 24.4).
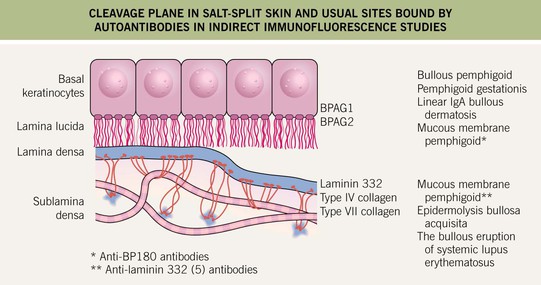
Fig. 24.4 Cleavage plane in salt-split skin and usual sites bound by autoantibodies in indirect immunofluorescence studies. The cleavage plane in 1 M NaCl salt-split skin is in the lower portion of the lamina lucida. Circulating autoantibodies from patients with various subepidermal immunobullous diseases bind to different sites, e.g. the epidermal versus dermal side of the split. Courtesy, Kim Yancey, MD.
• Both enzyme-linked immunosorbant assay and IIF can detect circulating autoantibodies in the sera in at least 70–80% of patients (see Table 23.1).
• At least 50% of patients have a peripheral eosinophilia.
Table 24.1
Therapeutic ladder for bullous pemphigoid.
Key to evidence-based support: (1) prospective controlled trial; (2) retrospective study or large case series; (3) small case series or individual case reports.
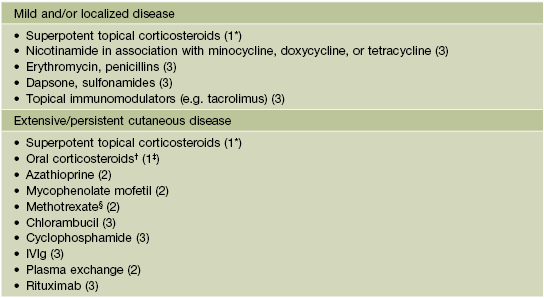
* Validated.
† Prednisone doses of at least 0.5–0.75 mg/kg/day seem to be necessary to control extensive disease but increase serious side effects, including mortality.
‡ Validated for prednisone.
§ In elderly patients, low-dose regimen (2.5–10 mg/week) can be effective.
Note: Superpotent topical corticosteroids should be considered in any patient and may be combined with a systemic therapy.
Mucous Membrane (Cicatricial) Pemphigoid
• The mucous membranes represent the major site of involvement, in particular conjunctival (erosions, scarring with symblepharon formation, blindness) and oral mucosae (persistent erosions of the buccal and palatal mucosae, desquamative gingivitis) (Figs. 24.5 and 24.6).
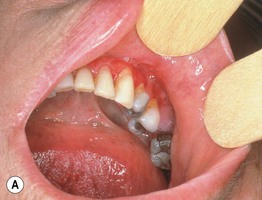
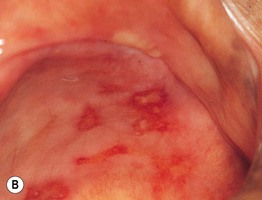
Fig. 24.5 Mucous membrane (cicatricial) pemphigoid – mucosal involvement. A Erythema and erosions of the gingival margins (desquamative or erosive gingivitis). B Chronic erosions on the hard palate. B, Courtesy, C. Prost, MD.
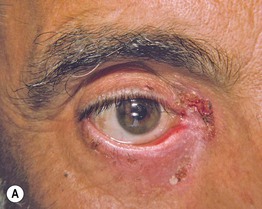
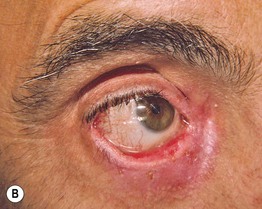
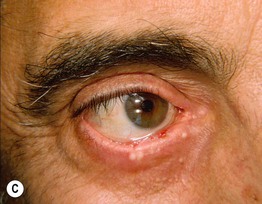
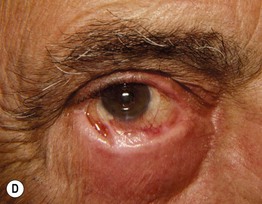
Fig. 24.6 Progression of mucous membrane (cicatricial) pemphigoid. A Erosion of the lower eyelid margin and erythema plus scale-crust of the inner canthus and lower eyelid. B Three months later, ectropion and thickening of the lower eyelid in addition to erosions. C Six months later, smaller erosions but scarring and milia formation. D Seven years later, further scarring with significant shortening of the inferior fornix due to symblepharon. Courtesy, Louis Fragola, Jr., MD.
• Cutaneous lesions are seen in a quarter of patients and favor the head and neck region and upper trunk; erythematous plaques that develop vesicles, erosions, and scarring are characteristic (Fig. 24.7).
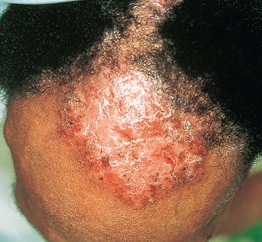
Fig. 24.7 Mucous membrane (cicatricial) pemphigoid – cutaneous involvement. An erythematous plaque with atrophy and crusting; note the scarring alopecia. In the Brunsting–Perry variant, there are cutaneous lesions in the head and neck region but minimal or no mucosal involvement. Courtesy, Philippe Bernard, MD, and Luca Borradori, MD.
• DDx: pemphigus vulgaris, occasionally BP, EBA, and LABD; if limited to oral mucosa, lichen planus or pemphigus vulgaris (see Fig. 59.5); if limited to scalp, consider other causes of scarring alopecia.
• Rx: potent topical or intralesional CS, dapsone, cyclophosphamide (severe or progressive ocular disease), alone or in combination with systemic CS, and rituximab.
Epidermolysis Bullosa Acquisita (EBA)
• Two major clinical forms: (1) mechanobullous which resembles the genetic disorder epidermolysis bullosa; and (2) inflammatory which resembles bullous pemphigoid (Fig. 24.8).
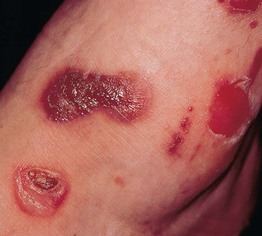
Fig. 24.8 Epidermolysis bullosa acquisita – inflammatory presentation. Bullous and erosive lesions in a patient with multiple myeloma. This form resembles bullous pemphigoid. Courtesy, Philippe Bernard, MD, and Luca Borradori, MD.
• In the more common mechanobullous form, bullae arise in sites of friction and are followed by erosions, scarring, and milia formation (Fig. 24.9); oral lesions can also be seen (Fig. 24.10).
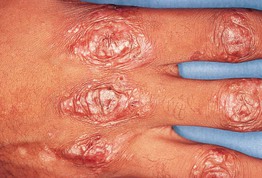
Fig. 24.9 Epidermolysis bullosa acquisita – mechanobullous presentation. Milia and scarring that favor sites of trauma overlying joints, in association with skin fragility.
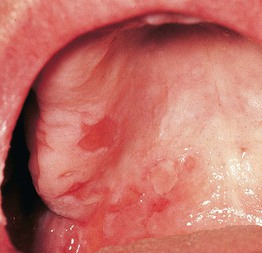
Fig. 24.10 Epidermolysis bullosa acquisita – oral lesions. Multiple erosions of the palate reminiscent of mucous membrane pemphigoid. Courtesy, C. Prost, MD.
• Histological and DIF features are similar to BP, but there is usually a sparser infiltrate and few eosinophils in the mechanobullous form; DIF studies demonstrate linear IgG > C3 or IgA at the basement membrane zone; circulating autoantibodies are detected in ~50% of patients and by salt-split skin immunofluorescence studies, the immunodeposits are in the floor (dermal side) of the blister (see Fig. 24.4).
For further information see Chs. 28 and 30. From Dermatology, Third Edition.
[/level-membership-for-dermatology-category][not-level-membership-for-dermatology-category]24
Bullous Pemphigoid, Mucous Membrane Pemphigoid, and Epidermolysis Bullosa Acquisita
This chapter, in addition to Chapters 23 and 25, cover the autoimmune bullous diseases. The concepts of direct immunofluorescence (DIF) and indirect immunofluorescence (IIF) are reviewed in Chapter 23 (see Fig. 23.2), as are the recommended sites for performing skin biopsies for DIF (see Fig. 23.1).
Bullous Pemphigoid (BP)
• Both pruritic fixed urticarial plaques and tense bullae are seen (Figs. 24.1 and 24.2); the latter can develop within normal skin or areas of erythematous skin and produce erosions when they rupture; oral lesions are much less common than in pemphigus vulgaris (10–30% of patients).
Fig. 24.1 Bullous pemphigoid. A Classic presentation with multiple tense bullae arising on normal and erythematous skin; several of the bullae have ruptured, leaving circular erosions. B Urticarial papules and plaques, along with bullae containing serous fluid. C Firm annular urticarial plaques. D Eczematous presentation with large dermatitic plaques. C, D, Courtesy, Philippe Bernard, MD, and Luca Borradori, MD.
Fig. 24.2 Childhood bullous pemphigoid. Generalized bullae, both tense (fresh) and flaccid (old with re-epithelialization), as well as erosions with hemorrhagic crusts.
• Pruritus and nonspecific eczematous (see Fig. 24.1D) or papular lesions can precede the more characteristic cutaneous lesions and may be the predominant finding; unusual variants include dyshidrosiform (palms and soles), vegetans (major body folds), and localized (e.g. pretibial in adults; vulvar in children, acral in infants), as well as those that mimic prurigo nodularis and toxic epidermal necrolysis (Fig. 24.3).
Fig. 24.3 Bullous pemphigoid – unusual clinical variants. Grouped vesicles and bullae on the palms (A) and toes (B) that can resemble pompholyx (dyshidrosiform pemphigoid). C Vegetating plaque in the inguinal crease (pemphigoid vegetans). D Toxic epidermal necrolysis-like lesions with large erosions. A, D, Courtesy, Philippe Bernard, MD, and Luca Borradori, MD.
• Histologically, a subepidermal bulla plus an infiltrate of eosinophils is seen when the lesions are bullous; DIF demonstrates immunodeposits of IgG and/or C3 in a linear array along the basement membrane zone (see Fig. 23.3B); in general, by salt-split skin immunofluorescence studies, the immunodeposits are in the roof (epidermal side) of the blister (Fig. 24.4).
Fig. 24.4
[/not-level-membership-for-dermatology-category]
