[level-membership-for-neurosurgery-category]
CHAPTER 97 Brain Tumor Immunology and Immunotherapy
An Immune Primer
The function of the immune system is to protect the body.1 Immunotherapy involves the administration of an agent to an individual that stimulates the immune system to react against something foreign or harmful, such as a tumor or an infection. The end result of a vaccine is to develop protective immunity against the foreign agent or tumor. This defensive function is performed by leukocytes (white blood cells) and a number of accessory cells distributed throughout the body. Lymphocytes are the key cells controlling the immune response. They specifically recognize foreign material and distinguish it from the body’s “self” components. Immunity may be cell mediated (T cells, natural killer [NK] cells, and phagocytes) or humoral (B cells, antibody, complement). When stimulated, cytokines are a group of molecules, other than antibodies, produced by lymphocytes that are involved in regulating the immune system. They include the interleukins, the interferons, tumor necrosis factor (TNF), and colony-stimulating factor (CSF). There are two main types of lymphocytes: B cells, which produce antibodies; and T cells, which have a number of functions including helping B cells to make antibodies (CD4 helper T cells, TH2) or helping cytotoxic T-cell responses (CD4 helper T cells, TH1); recognizing and destroying virus-infected cells (CD8 effector T cells); and controlling the level and quality of the immune response (regulatory T cells).
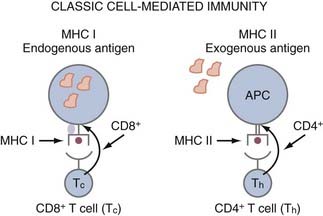
APCs are a group of cells that are capable of taking up antigens, partially degrading them, and presenting them to T cells in a form they can recognize. Although B cells recognize antigen in its native form, T cells only recognize antigenic peptide derivatives of complex antigens that have become associated with major histocompatibility complex (MHC) molecules. Thus, MHC molecules present antigen (i.e., peptides) to T cells. MHC class I molecules are found on all nucleated cells and platelets. MHC class II molecules (Ia antigens) required for helping B cells or making antibodies are expressed on B cells, macrophages, monocytes, APCs, and some T cells. CD8 cells (cytotoxic T lymphocytes [CTLs], killer) are class I restricted, meaning they only recognize antigen presented in the context of MHC class I molecules, whereas CD4 (helper T) cells are MHC class II restricted (Fig. 97-1). Antigens synthesized within a cell, such as viral polypeptides, associate preferentially with MHC class I molecules and present antigen directly to CD8 cells (“direct pathway”). In contrast, antigens that are taken up by an APC and partially degraded (processed) and returned to the cell surface associated with MHC class II molecules are recognized by CD4 cells (“indirect pathway”) (see Fig. 97-1). Syngeneic implies the same MHC as the organism or patient, and allogeneic implies a different MHC; hence the term allograft, which is rejection due to differences in MHC.
T cells in the CNS of healthy humans are a rare finding. However, during inflammatory responses, T cells are evident within the CNS. T cells require activation before entry into the CNS,2 but antigen specificity is not necessary for entry. T-cell infiltrates are commonly identified within human gliomas,3 and multiple studies have attempted to correlate the intensity of infiltration with survival,3–5 but this has not been consistently seen.6 One should bear in mind that these types of immunohistochemical assays do not take into account the functional activity of these cells or the contribution of inhibitor factors such as T regs. Thus, although these T cells have been activated in the systemic circulation, their functional activity has likely become impaired on entry into the tumor microenvironment,7 and it is not surprising that their presence in the tumor is not a definitive prognostic marker.
One concern about the use of immunotherapy in glioma patients is the induction of fatal autoimmunity. However, in previous immunotherapy trials of humans with brain tumors, there were only two possible cases of autoimmunity,8,9 so this remains a consideration but is likely related to the use of strong adjuvants.10 The rationale for selecting a tumor-specific antigen approach in immunotherapy is based on the facts that the targeting antigen preparation does not contain CNS antigens capable of inducing an autoimmune response and that the tumor-specific immune response can be monitored. However, the disadvantage of using a tumor-specific antigen is secondary to the heterogeneity of gliomas—one single antigen is unlikely to produce a durable response because a clonal negative population is likely to arise.11
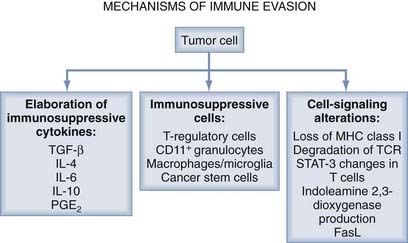
It is widely recognized that tumors evade the host immune response through a number of mechanisms, including the elaboration of immunosuppressive cytokines, the production of immunosuppressive immune cells (T regs), and signal transduction disruption (Fig. 97-2).12
Mechanisms of Immune Suppression and Therapeutic Agents
Overview: Mechanisms of Immune Evasion
The secretion of immunosuppressive factors by the tumors themselves has recently been identified as a further target for immunotherapy. In particular, interleukin-10 (IL-10) and transforming growth factor-β (TGF-β) are two examples of such immunosuppressive cytokines secreted by tumor cells. Furthermore, there is increased expression of FasL and galectin by glioma that triggers T-cell depletion and T-cell apoptosis.13
Recently, a subset of regulatory T cells (T regs) have been identified in the local tumor environment and in the draining lymph nodes of systemic tumors, and they are elevated in the CD4 compartment of the systemic blood circulation. These cells coexpress CD4 or CD25, or both, and display potent immunosuppressive T-cell activity.14 In vaccines used for tumors outside the CNS, monoclonal antibody inhibition of these T regs has been associated with enhanced antitumor immunogenicity (details provided later). The inhibitory effects of these suppressor cells may also be mediated by cytokines. Interestingly, IL-2–mediated immune regulation involves not only the stimulation of effector cells but also the regulation of T regs.15
Regulatory T Cells
T regs inhibit T-cell activation and effector function by downregulating IL-2 production, inhibiting interferon-γ (IFN- γ) production, and increasing TH2 cytokine production,16 thereby inhibiting the expression of MHC class II molecules, CD40, CD80, and CD86 and suppressing antigen presentation by monocytes and macrophages.17,18 The presence of T reg–mediated suppression has been documented in a variety of human cancers.19 Several studies on immunogenic murine tumors have shown that adoptively transferred T regs are responsible for inhibition of tumor-reactive effector T cells and that the elimination of T regs enhances antitumor immunity.16 Within glioblastoma multiforme (GBM) patients, although reduced in absolute number, T regs comprise a disproportionately large fraction of the patient’s peripheral CD4 T-cell compartment. This increase corresponds with a decrease in T-cell effector functions. Furthermore, in vitro depletion of peripheral blood T reg cells results in successful reversal of effector T-cell function, including increased proliferation and a switch to a TH1 (IL-2–positive, tumor necrosis factor-α [TNF-α]–positive, IFN-γ–positive) cytokine profile. Within murine tumor challenge models of established gliomas, in vivo depletion of CD4+ and CD25+ cells resulted in significant long-term survival.20,21
Within the glioma microenvironment, the host-generated effector T cells can be critically suppressed or overwhelmed by the T reg cells. Tissues from glioma patients obtained after surgical resection have been dissociated and stained for the CD8+ and CD4+ subsets. The tumor-infiltrating CD8+ T cells were phenotypically CD8+ and CD25−, indicating that these cells were not activated or proliferating. CD4+ T cells were more numerous than CD8+ T cells within glioma tissue and the majority of CD4+ T cells were T regs, as evident by positive intracellular staining of FoxP3, a transcription factor that is elevated only in T regs and not in activated effector CD4+ T cells.22 These data indicate that T regs not only can inhibit initial systemic immune activation but also can prevent the effector responses in the tumor microenvironment. Of note, lower grade gliomas have only rare T regs, and oligodendrogliomas none at all. Even within the GBM patients’ specimens, the presence of T reg infiltration is highly variable and not an independent predictor of survival, emphasizing the redundancy of immunosuppressive pathways. Human T regs have been shown to accumulate in tumors and in ascites induced by the tumor and macrophages by the chemokine CCL22, which mediates trafficking of T regs to the tumor.19 Similarly, gliomas elaborate the chemokine CCL2, which induced the intratumoral migration of T regs.23
Regulatory T-Cell Inhibition by Chemotherapeutic Agents
Overcoming T reg immunosuppression is an ongoing immune therapeutic approach that can be achieved through a variety of approaches, including the use of denileukin diftitox (Ontak),24 cyclophosamide,25 or signal transducer and activator of transcription type 3 (STAT-3) blockade agents,26 or by inhibiting the T reg trafficking (i.e., inhibition of CCL2) with temozolomide.27 Inactivation of T regs with an anti-CD25 antibody in murine glioma models has been shown to enhance vaccination-induced antitumor immune responses and to result in the eradication of intracerebral astrocytomas without induction of autoimmunity.28 Similar results were also obtained in this murine model system with systemic cytotoxic T-lymphocyte–associated protein type 4 (CTLA-4) blockade. The CTLA-4 blockade reversed the CD4+ T-cell deficit, similarly seen in malignant glioma patients, and normalized the ratio of T regs in tumor-bearing mice.29 Although eliminating the suppression of endogenous antitumor immune responses through the elimination of regulatory T cells may enhance tumor immune clearance, there is a potential risk for inducing autoimmunity; however, that was not found in the murine model. It is likely that the induction of TH17 responses, and not necessarily the inhibition of T regs, is the primary mechanism of inducing CNS autoimmunity.30,31 In patients with metastatic renal carcinoma, Ontak specifically depleted the T regs without inducing toxicity and significantly improved the stimulation of tumor-specific T-cell responses when those patients were stimulated with RNA-transfected DCs.32 Without vaccination, Ontak has been shown to be efficacious as a single agent in relapsed and refractory B-cell non-Hodgkin’s lymphoma33 and in cutaneous T-cell lymphoma.34 However, Ontak was not particularly effective in clearing the T reg cells in melanoma patients.35 No published studies to date have evaluated this approach in malignant glioma patients. Another set of potential T reg modulation includes agents that block or disrupt Notch signaling. Notch signaling has also been identified as playing a role in regulating the responses of T cells and can affect the differentiation of CD4+ T cells into T regs.36 However, it is unclear which members of the Notch family need to be blocked, and inhibitors are currently under development.
Lymphodepletion to augment immunologic responses has been described in both murine model systems37,38 and human cancer patients.39 These enhanced antitumor responses after lymphodepletion may be secondary to the removal of competition at the surface of APCs,40 enhanced availability of cytokines that augment T-cell activity (e.g., IL-7 and IL-15),41 or depletion of immune inhibitory T regs.42 Cyclophosphamide (CTX), an alkylating agent with a therapeutic effect against tumors at high doses, has preferential effects on inhibiting T regs at lower doses. CTX can abolish the function of CD4+, CD25+, FoxP3+ T cells and enhance cytotoxic T-cell responses.43 Pretreatment with CTX before antitumor vaccination results in the activation of CD8 tumor-specific T cells.44 When CTX is administered at subtumoricidal doses, there is improved immune response and tumor eradication of large established murine tumors (sarcoma) when treated in combination with IL-12. This combination enhances CD4, CD8 T cells and macrophage infiltration within the tumor and skews the immune responses to the TH1 phenotype.45 Similar findings have been made in the murine system treating established melanoma with IL-2, IL-1, and CTX.46 In addition to its induction of immunity to new antigens, CTX can also overcome tolerance. For example, the administration of CTX to mice bearing an established plasmacytoma resulted in the cure of 92% of mice, and further studies demonstrated that the cured mice rejected a subsequent tumor challenge.47 The mechanism appeared to involve both the generation of CD8+ cytotoxic T cells and the upregulation of B7-1 (costimulatory molecule) on tumor cells.48
Multiple clinical trials have demonstrated enhanced immune responses and improved clinical efficacy when CTX was administered before immunotherapy.49,50 Administration of CTX to patients with advanced melanoma and colorectal cancer resulted in enhanced delayed-type hypersensitivity (DTH) reactions and the development of antibodies.51 When CTX was administered to augment autologous melanoma vaccine in patients with metastatic melanoma, the inclusion of CTX resulted in enhanced DTH responses.52 Similar potentiation of immune responses was seen in metastatic breast cancer and renal cell carcinoma. CTX has also been used in a phase I clinical trial of glioma patients and demonstrated some evidence of clinical efficacy. Plautz and colleagues53 treated newly diagnosed glioma patients with a single dose of CTX before the administration of adoptively transferred T cells harvested from the patients’ lymph nodes. This study was conducted before our current understanding of the influence of CTX on T regs, and thus the contribution of the CTX to the clinical efficacy is unknown.
Nevertheless, when combining the different forms of treatment, all aspects of treatment must be taken into account. Different dosing schedules and methods of vaccination can abrogate the desired therapeutic response. The use of low-dose doxorubicin after vaccination with a granulocyte-macrophage colony-stimulating factor (GM-CSF)–secreting cancer cell vaccine in a CT26 colon carcinoma murine model showed a significant increase in CD8+ T-cell response compared with the use of vaccine alone. Yet, when low-dose CTX was administered after or concurrently with a GM-CSF–secreting cancer cell vaccine, the desired immune response was inhibited, and the result was equivalent to that seen in mice treated only with chemotherapy.54 Thus, with CTX administered before vaccination, the reduction of suppressive immunity increased the immunotherapy-mediated tumor clearance. Similar results were also obtained with paclitaxel.55
Another agent that is capable of suppressing T regs is temozolomide (TMZ), the standard of care for GBM. TMZ is an alkylating agent that causes cell death by inducing G2/M-phase cell cycle arrest and likely autophagy without apoptosis.56 TMZ can inhibit the proliferation of lymphocytes, but it also depletes T regs27,57 and the trafficking of T regs into the glioma microenvironment.23 This mechanism may prove beneficial in combination with vaccine therapy and was recently investigated in a phase II clinical trial (ACT II) in combination with PEP-3-KLH vaccination.
In murine models of established intracerebral tumors, the combination of IL-2 immunotherapy and intratumoral administration of bischloroethylnitrosourea (BCNU) polymers enhanced survival.58 The investigators suggested that the cytotoxic agents might increase the number of tumor peptide antigens released or abrogate tumor-derived T-cell suppressor factors, or both. When mice with established tumors (lymphoma) were treated with BCNU, they rejected secondary tumor challenges, indicating the induction of immunologic memory. Furthermore, mice treated with BCNU demonstrated high cytotoxic activity against tumor cells and low T-cell suppressor activity. Thus, although BCNU was directly tumoricidal, it is also possible that the BCNU inhibited the T regs, resulting in antitumor immunity.59 The combination of BCNU with the newer efficacious immunotherapies has not been explored in animal models or clinical trials.
Immuno suppressive Cytokines
Human glioblastoma cells secrete a variety of factors, such as prostaglandin E (PGE), IL-10, and TGF-β2 that suppress multiple immune functions. For example, TGF-β2 inhibits cytotoxic responses of T cells against tumor targets, PGE2 regulates the generation of immune responses, IL-10 downregulates MHC class II expression on monocytes and suppresses T-cell proliferation,60–62 and vascular endothelial growth factor (VEGF) inhibits the maturation of DCs and subsequently downregulates MHC class II molecules. Suppression of TGF-β with an antisense TGF-β has been shown to be efficacious in 9L and C6 glioma intracerebral models.53,63 Other attempts at TGF-β inhibition in murine models have included SD-208, a TGF-βRI kinase inhibitor, which slightly increased median survival,64 and Tranilast, a TGF-β1 antagonist, which resulted in a 50% reduction in intracranial tumor volume.65 A phase I/II clinical trial using an antisense oligodeoxynucleotide (AP 12009) targeted to suppress TGF-β2 in high-grade glioma patients demonstrated an increase in median survival after recurrence that exceeded the current data for chemotherapy.66 The success of this type of approach is dependent on the dependency of the glioma on this particular mechanism of immune suppression and is compounded by the fact that gliomas usually express a variety of immunosuppressive cytokines; the blockade of any one cytokine may not be expected to significantly impact the overall immunosuppressive milieu.
The taxanes, paclitaxel and docetaxel, bind to ß-tubulin, stabilizing microtubules, and resulting in G2/M-phase mitotic arrest. Paclitaxel can diminish the cytotoxicity of NK and T cells by interfering with IL-2–mediated activation. However, the best-known immunomodulatory effect of paclitaxel is its ability to trigger macrophages to secrete a variety of proinflammatory factors, such as IL-1ß, GM-CSF, tumor necrosis factor-α (TNF-α), and IL-12. This lipopolysaccharide-like activity enables paclitaxel, in the presence of a priming signal, to induce the tumoricidal activity of macrophages.67 To date, there have been no reports of combining paclitaxel treatment with cancer immunotherapy.
Clearly, further studies investigating the cytokine profile before chemotherapy are warranted to enhance our understanding of the endogenous immune response. For example, in patients with breast cancer that were evaluated before treatment with chemotherapy, there was a significant decrease in immune cells such as NK and LAK cells and immunostimulatory cytokines such as IL-2, GM-CSF, and interferon compared with healthy individuals. After the administration of docetaxel and paclitaxel, there was a significant increase in the number of immune cells and a decrease in PGE2 and TNF.68 Doxorubicin has also been shown to have immunomodulating effects, including increasing the activity of monocytes and increasing the secretion of IL-1 and IL-2.69
Immune Inhibitory Molecules
To initiate an immune response, T cells must receive two signals: one through the T-cell receptor (TCR), which recognizes antigen presented within the context of MHC; and the second through the costimulatory receptor CD28, which recognizes the costimulatory molecules CD80 and CD86 expressed on the surface of the APC. In response to antigen-specific stimulation, activated T cells produce IL-2 and proliferate. B7-H1 is a negative costimulatory molecule that inhibits CD4+ and CD8+ T-cell activation, can induce T-cell apoptosis,70,71 and is upregulated within the tumor microenvironment72 and specifically in gliomas.70 Blockade of B7-H1 with a monoclonal antibody can activate T cells, which are then able to inhibit tumor.73 This represents a novel mechanism by which glioma cells evade immune recognition and destruction. Blockade of B7-H1 with antibodies in combination with adoptive T-cell transfer has been shown to enhance cancer clearance73 and enhance myeloid DC-mediated immunotherapy72; however, whether sufficient quantities of these types of antibodies can be achieved in the CNS remains to be seen. T-cell anergy can also be induced if the inhibitory molecule CTLA-4 is present on the gliomas.74 CTLA-4 blockade has had some success in promoting antitumor T-cell responses in both preclinical and clinical settings,29,75 but it is not clear whether this approach would be successful against primary brain tumors or CNS metastasis.
HLA-G is a nonclassic MHC molecule with limited tissue distribution that has an immune regulatory function. Soluble HLA-G (sHLA-G) has been identified in the plasma of patients with melanoma, glioma, breast cancer, or ovarian cancer. sHLA-G can inhibit the functions of T and NK cells at high concentration76; thus, at levels within the tumor microenvironment, it would be anticipated that there is reduced immunologic responsiveness. Gene transfer of HLA-G1 or HLA-G5 into HLA-G–negative glioma cells (U87-MG) rendered these cells highly resistant to direct cytolytic clearance, inhibited T-cell proliferation, and prevented efficient priming of cytotoxic T cells. The inhibitory effects of HLA-G were directed against CD8 and CD4 T cells, but appeared to be NK-cell independent.77 Only a few HLA-G–positive cells within the tumor microenvironment are necessary to exert significant immune inhibitory effects77; thus, the inhibition of HLA-G blockade would need to be comprehensive. However, the frequency of expression of HLA-G and B7-H1 has not been assessed across glioma grades or types on tissue arrays; thus, the universality of this type of immune evasion is not currently known.
Phenomenon of Antigen-Loss Variants in Gliomas
Although tumor specificity has the advantage of not inducing potentially fatal autoimmunity, the intrinsic antigenic heterogeneity will prevent clearance of all cancer cells and the development of antigen-loss variants, especially for those antigens whose expression is not required by tumor cells for the maintenance of a transformed phenotype. The propagation of such antigen-loss variants is secondary to epitope immunodominance, that is, the preferential immunodetection of one or a few epitopes among many expressed on a given target.78 The parental tumor cells carrying the immune dominant epitope can be eliminated, and the formerly immune recessive epitopes then become dominant, allowing unrestricted tumor growth. It is well documented that tumor antigen expression is heterogeneous, even within the same tumor,79 and antigen loss has been observed with a variety of immune therapeutic approaches in various cancers.80,81
The epidermal growth factor receptor (EGFR) is often amplified and structurally rearranged in malignant gliomas, with the most common mutation being EGFRvIII. Treatment in a murine model with a vaccine consisting of a peptide encompassing the tumor-specific mutated segment of EGFRvIII (PEP-3) conjugated to keyhole limpet hemocyanin (KLH [PEP-3-KLH]) resulted in the loss of antigen expression. Specifically, EGFRvIII expression in mice that failed to respond to the PEP-3-KLH vaccination was lost in 80% of relapsing tumors, indicating that EGFRvIII-negative escape variants were a potential mechanism of treatment failure in active immunotherapy.11 Although EGFRvIII appears to represent a nearly terminal branch of tumor progression for malignant brain tumors and is expressed clonally within this lineage, antigen-specific tumor targeting might confer a selective growth advantage on neoplastic cells not expressing this epitope. It has been shown previously that treatment failure with antigen-specific passive humoral immunotherapy is not a result of antigen-loss variants,82 suggesting that antigen-negative escape variants are an etiology of treatment failure in active immunotherapy. In two separate phase II clinical trials (ACTIVATE and ACT II) in newly diagnosed EGFRvIII-expressing GBM patients who received the PEP-3-KLH, on tumor progression, the EGFRvIII expression was lost.57,84 To overcome this obstacle, cocktails of different antigens or whole-tumor lysates can be used as immunogens85; however, with nonspecific antigen approaches, there is a risk for autoimmune responses.10 To circumvent this, future approaches may involve screening the patient’s tumor for a panel of antigens86 and targeted immunotherapy selected based on these parameters—similar to selecting antibiotics for a given bacterial infection. Alternatively, upregulating the expression of these antigens with some forms of chemotherapy may restore the ability of the immune system to clear these cancer cells.87
A variety of agents have been shown to be capable of inducing or upregulating antigen expression. For example, gemcitabine, a nucleoside analogue, can increase tumor antigen cross-presentation and T-cell infiltration of the tumor.88 The demethylating agent DAC is capable of inducing the tumor-associated antigen MAGE-1 on melanoma cells with subsequent cytolytic T-cell clearance.89 Irinotecan (CPT-11) has been shown to induce the LYGD/E48 antigen, which resulted in synergistic tumor regression with a cytotoxin-armed antibody in a colorectal xenograft murine model.90 The combination of 5-fluorouracil and cisplatin (CDDP) can upregulate the antigen CEA and MHC class I in human colorectal cancer cell lines91 and was functionally relevant as evidenced by increased CEA-specific, MHC-restricted cytotoxic T-cell killing.87 Thus, many different types of chemotherapy can enhance antigen expression; however, none of these agents has been tested to determine whether it can restore antigen expression after clonal elimination with immunotherapy.
An alternative therapeutic approach for the issue of antigen heterogeneity would be to selectively screen each patient’s tumor for a panel of potential antigens, but this would be somewhat labor intensive and costly at present. Another alternative approach would be to vaccinate cancer patients with a panel of tumor-associated or tumor-specific antigens. Tissue arrays of gliomas from a large population of patients could be used to determine the frequency of the most common tumor antigens. This strategy would be very similar to that used in manufacturing flu vaccines annually, in which the most common virulent strains are incorporated as immunologic targets. Finally, exploration has begun on immunologic approaches for inducing immune responses against immune escape variants based on defects in the transporter associated with antigen processing,92 but these are not yet sufficiently developed for clinical trial application.
STAT-3—A Key Switch Mediating Immuno suppression
The ability of tumor cells to proliferate uncontrollably, resist apoptosis, sustain angiogenesis, and evade immune surveillance is regulated, in part, by the signal transducer and activator of transcription type 3 (STAT-3), which acts as a cytoplasmic signaling protein and nuclear transcription factor. Under normal physiologic conditions, latent STAT-3 activation is dependant on ligand-receptor interaction, primarily under the control of growth factor receptor tyrosine kinases, cytokines, and G-protein receptors.93 For example, EGFR and IL-6 activate Jak2, which then activates STAT-3 by phosphorylation of the tyrosine residue in the transactivation domain of STAT-3.94 In cancer cells, these tyrosine kinases are among the most frequently activated oncogenic proteins. STAT-3 has been shown to be persistently activated in most human cancers, including gliomas.95,96 The phosphorylation of STAT-3 has been attributed to EGFRvIII.97 IL-6 is expressed in the CNS, especially by reactive astrocytes, under a wide variety of conditions, such as hypoxia,98 traumatic and metabolic injury,99 and inflammation,100 which in turn also induces expression of p-STAT-3. The use of decoy antisense STAT-3 oligonucleotides and dominant-negative vectors has provided convincing evidence that STAT-3 is highly relevant to the growth and survival of many tumor types, including gliomas.101–103
STAT-3 has also emerged as a key switch that drives the underlying immuno suppression104 identified in cancer patients by preventing maturation of DCs and inhibiting the proliferation and activation of immune effector populations. STAT-3 has been shown to be a potent regulator of these anti-inflammatory responses by suppressing macrophage activation and limiting inflammatory responses.105,106 It also has been shown to become constitutively active in diverse tumor-infiltrating immune cells.107 For example, STAT-3 activity within NK cells and neutrophils directly reduces their cytotoxicity, whereas STAT-3 activity in DCs reduces the expression of MHC class II, CD80, CD86, and IL-12 in these cells, rendering them unable to stimulate T cells and generate antitumor immunity. Investigators have recently shown that by ablating STAT-3 in only the hematopoietic cells in mice resulted in marked enhancement of activated and functional T cells, NK cells, and DCs in tumor-bearing mice. This ablation of STAT-3 in only the hematopoietic cells also resulted in marked antitumor effects in vivo, indicating that STAT-3 expression in immune cells restrains antitumor immune eradication.107 The tumor microenvironment induces STAT-3 activity in tumor-associated immune cells.104,107,108
The blockade of STAT-3 with small molecular inhibitors in microglia obtained from the CNS of patients with GBM resulted in the upregulation of costimulatory molecules (e.g., CD80, CD86), induced systemic APCs to elaborate proinflammatory cytokines essential for T-cell effector responses, and induced activation and proliferation of T cells, indicating that STAT-3 blockade is a potent approach to modulating both the systemic and local tumor immune microenvironments. Of note, this potent immune activation occurred in immune cells obtained from patients with disease refractory to other conventional immune activators, such as toll-like receptor agonists. When the immune cells from immunosuppressed GBM patients were treated with a STAT-3 inhibitor, Western blot analysis demonstrated phosphorylation of ZAP-70 in T cells, thus bypassing the impairment of signal transduction typically identified in the immune systems of patients with cancer.109 The mechanism of T-cell activation is likely secondary to the inhibition of T regs present in these immune preparations, thus allowing immune activation (i.e., releasing the “brake” on immune cell activation and proliferation). IL-2 has been shown to regulate FoxP3 expression in human CD4+, CD25+ T regs by inducing STAT-3 binding of the first intron of the FoxP3 gene.110 Suppressor of cytokine signaling type 3 has been shown to be an inhibitor of STAT-3 signaling111 and transcriptional activity112,113 but is deficient in T regs.114 STAT-3 inhibitors should have a multiplicity of immune modulatory activities, such as allowance of expression of costimulatory molecules in the tumor microenvironment, inhibition of immune suppressive cytokines, maturation of DCs, and inhibition of T regs, and represents a newly emerging class of drugs that will be used for immune therapeutic purposes.
Immune Modulation by Central Nervous System Antigen-Presenting Cells
T-cell activation requires signals through both the MHC and costimulatory molecules, and the expression of MHC alone results in T-cell anergy.115 Low levels of expression of CD80 provide an immune escape advantage to cancer cells, and CD28-mediated costimulatory signals are essential for differentiation of functional tumor-specific cytotoxic CD8+ effector T cells.116,117 In tumor-bearing animals, repeated stimulation of the T cells is necessary, especially at the effector site, to generate tumor responses,118 indicating that if the APC failed to provide appropriate stimulation, the APC may contribute to immune suppression. Although microglia may be able to act as APCs during autoimmunity (multiple sclerosis) and infection (HIV),119,120 glioma-associated microglia are functionally impaired, are not able to stimulate T-cell responses, and are refractory to activation stimulation (i.e., toll-like receptor agonists).7 Intratumoral DCs may be able to provide these immune-stimulating conditions, but these are rarely present in malignant gliomas.7 Additionally, CD11b+ inhibitory macrophages (iMACs) have been described, which induce apoptotic death in CD8+ T cells. This inhibitory phenotype could be changed into one reflective of highly activated DCs by culturing the iMACs with IL-4 and GM-CSF,121 suggesting that the tumor microenvironment can be manipulated for therapeutic purposes. However, the delivery of cytokines directly to the CNS is problematic. Costimulatory molecules on glioma-associated microglia and macrophages have been shown to be lost or significantly downregulated. In attempts to overcome the failure of APCs, and specifically the lack of sufficient costimulation, vaccines with enhanced costimulatory capacity have been tested and demonstrated antitumor effects that have maintained high avidity effector-memory CD8+ T cells. Other approaches for potentially upregulating the costimulator molecules on APCs include CpG DNA,122 chemotherapy with cytosine arabinoside,123 transfection with poxviruses expressing the costimulatory molecules,124 or transfection of the replication-defective fowlpox recombinant vector rF-TRICOM (TrIad of COstimulatory Molecules).125
An emerging strategy for upregulating costimulatory molecules in the glioma microenvironment is with STAT-3 blockade agents. A small molecular inhibitor (WP1066) of STAT-3 has been shown to upregulate costimulatory molecules and proinflammatory cytokines on microglia obtained from the CNS of patients with GBM that were refractory to modulation by other conventional immune activators, such as toll-like receptor agonists. Furthermore, WP1066 can modulate enhanced T-cell functional activities in physiologic ranges that can be obtained within the CNS.109 These types of agents are anticipated to be in clinical trials of human patients within the next several years.
Immune Modulation by Glioma-Infiltrating Lymphocytes and TH2 Lymphocytes
After activation, CD4+ helper T cells are classified, based on their elaborated cytokine responses, as TH1 (promoting cell-mediated immunity) or TH2 (promoting humoral immunity), which predominantly secrete either interferon-γ (IFN-γ) or IL-4, respectively. A predominance of TH2-type cytokine response has been correlated with enhanced tumor growth.126,127 This TH2 polarization has been demonstrated in gliomas128 and is likely mediated by microglia-secreted macrophage-derived chemokine (MDC)129 or neuropeptides, or both.130 One therapeutic approach altering the TH2 intratumoral skewing would be to limit the chemokines that direct migration of the immunosuppressive immune cells to the tumor microenvironment, such as with a STAT-3 inhibitor.26 Alternatively, preferential TH1 responses could be induced with cyclooxygenase-2 (COX-2) inhibition.131
Impaired Immune Responses
The proliferative response of stimulated T cells, along with the levels of the IL-2 receptor and tyrosine phosphorylation in response to IL-2, from malignant glioma patients are reduced compared with healthy donors.132 These results suggest a defect in the expression of functional IL-2 receptor and T cells isolated directly from human gliomas that are not activated as reflected by expression of the IL-2 receptor CD25.7 Soluble products from tumors may suppress T-cell proliferation through a mechanism that involves downregulation of Jak3 expression and inhibition of IL-2–dependent signaling pathways.133 A potential mechanism for overcoming this is the constitutive activation of the T cells by transfection with CD3 or CD28, or both, for adoptive transfer therapeutic purposes. An antibody, r28M, directed against a melanoma-associated proteoglycan expressed on glioblastoma cells activates T cells through the CD28 molecule without additional stimulus from the TCR-CD3 complex and induces T-cell–mediated killing of glioblastoma cells in vitro and in vivo.134
Doxorubicin, an anthracycline antibiotic, can enhance the cytotoxic T-cell compartment. Pretreatment of mice with doxorubicin produced enhanced cytotoxic T-cell responses and activation of macrophages, as reflected by the secretion of IL-1 and IL-2.51 There was a twofold increase in the number of macrophages that could support the generation of the cytotoxic T cells. Ultimately, the macrophages became tumoricidal, and the effect persisted for 14 to 18 days. Further studies demonstrated that the administration of doxorubicin 5 days after a GM-CSF–secreting vaccine injection potently induced cytotoxic T-cell activity.54 Cancer patients treated with doxorubicin at a dose of 25 mg/m2 developed an increase in the percentage of circulating CD8+ T cells having twofold higher cytotoxic activity.135 Ideally, the chemotherapy selected for use in combination with immunotherapy should have some intrinsic tumoricidal activity. Although doxorubicin has little clinical efficacy against gliomas, this approach could potentially be used to boost the T-cell responses in glioma patients with further clinical development.
T-Cell Apoptosis
T-cell apoptosis has been observed frequently within malignant gliomas.136 The induction of apoptosis involves the activation of caspases, leading to nuclear fragmentation and apoptosis, and is triggered by deprivation of survival stimuli such as costimulators7 or by binding of ligands to death-inducing membrane receptors.137 Both of these mechanisms are operational within gliomas. FasL is predominantly expressed on T cells after activation by antigen and IL-2. When mature T cells are repeatedly stimulated by antigens, they coexpress Fas and FasL.137 Gliomas can evade killing by tumor-infiltrating T cells by overexpressing FasL, rendering the tumor resistant to apoptosis138 while delivering a death signal to Fas-expressing cells, which include the activated cancer-infiltrating T cells. However, high-grade gliomas also express the apoptotic receptors Fas and CD95, rendering them susceptible to antibody-stimulated Fas- and CD95-mediated apoptosis and CD8+ cytotoxic killing.139 Alternatively, agents such as topotecan can be administered to patients to upregulate Fas on gliomas that renders the tumor more susceptible to cytotoxic immune cell clearance.139–141
Topotecan penetrates the blood-brain barrier but has demonstrated only modest clinical efficacy in glioma patients, probably owing to failure to achieve in vivo levels sufficient to induce apoptosis of the tumor. Nevertheless, topotecan has been shown to upregulate Fas and CD95 in high-grade gliomas, rendering them more susceptible to immune clearance.139,141 Thus, although levels achieved are insufficient for direct glioma clearance within the CNS, this agent potentially could be exploited as an immune modulator. Other chemotherapeutics, such as doxorubicin142 (Adriamycin) and its analogues (epirubicin and pirarubicin),143,144 VP-16 (etoposide), cisplatin,144 and the combination of cisplatin and 5-fluorouracil145 can also increase cytotoxicity mediated by T cells and NK cells by upregulating Fas.
A synergistic antitumor effect was observed with paclitaxel and FasL in two human malignant glioma cell lines, T98G and LN-229.146 The synergy of paclitaxel and FasL was achieved independently of either the G2/M-phase cell cycle arrest or p53 activation. At low concentrations of paclitaxel, Bcl-2 phosphorylation was induced in the glioma cells, which in turn interfered with the heterodimerization of Bcl-2 with Bax and the inhibition of Fas-induced apoptosis by Bcl-2. The investigators hypothesized that the synergistic antiproliferative effect of paclitaxel and FasL on malignant glioma cells stemmed from the upregulation of the Bcl-2/Bax rheostat in favor of Bax, thereby sensitizing the tumor cells to apoptosis. Interestingly, this mechanistic synergy was not observed with other agents such as teniposide.147
T-cell apoptosis is also induced by glioma-expressed CD70 and gangliosides. The glucosylceramide synthase inhibitor (PPPP) has been shown to reduce the ability of GBM cell lines to induce apoptosis in T cells.148 Alternative strategies for blocking the T-cell death pathway include proteosome inhibitors, antisense oligonucleotide inhibitors, and other small molecule inhibitors. Although preclinical trials using some of these compounds have been successful in regressing established tumors, their therapeutic efficacy in cancer patients has yet to be established. Furthermore, apoptotic or necrotic tumor death is a desirable effect, and specific agents that block only T-cell apoptosis have yet to be devised.
Resistance to Innate Immune Clearance
The innate immune system includes NK cells, macrophages, granulocytes, and the complement system and is triggered by the recognition of microbial nonself molecules. Human tumor cells lose the expression of the ligands for NK activation, and this constitutes a tumor escape mechanism for NK cell–mediated responses. Accordingly, activating the innate immune system by forcing glioma cells to express danger signals such as NKG2D ligands is a potential strategy for immunotherapy for these tumors.149 However, the use of adenoviral vectors to express a variety of markers has been problematic in human clinical trials. NK inhibitory receptors can also regulate NK cell cytotoxicity and T cells. The nonclassic MHC class I molecule HLA-E, expressed on gliomas,150 can transmit either an activating signal through CD94/NKG2C or an inhibitory signal mediated by CD94/NKG2A. By small interfering RNA (siRNA)-mediated silencing of HLA-E or blocking of CD94/NKG2A, NK and CTL activity could be enhanced, and this represents a potential therapeutic strategy; however, the prevalence of this type of immune evasion within gliomas has not been clarified.
Overview: Strategies to Augment the Immune Response
Today, three main immunotherapeutic approaches for glioma have demonstrated the most promising clinical results: peptide vaccination strategies, DC vaccinations, and adoptive T-cell transfers. The determination of efficacy has been confounded by the lack of appropriate control groups, unaccounted for prognostic variables, or insufficient numbers of treated patients to draw conclusions.150a
There are many approaches to immunotherapy, and the strategies that are further discussed here include cytokine-mediated cellular therapies, antibody-directed therapies, tumor vaccines, and combination therapies. Cellular immunotherapy involves the isolation of immune effector cells from the patient, expansion and activation in vitro, and then infusion into the patient; examples include cytokine-activated NK cells, cytokine-activated lymphocytes (adoptive T-cell transfer) or tumor-infiltrating lymphocytes, and DCs. The use of monoclonal antibodies has predominately been for delivery of radioactivity or a toxin; however, unconjugated antibodies169 may have future clinical trial potential if delivered locally. Vaccination strategies ultimately would induce the patient’s own immune system to react against these tumors and represents a theoretical ideal of immunologic approaches. Finally, the immune modulatory approaches, which consist of inhibiting T regs or altering the local tumor microenvironment with agents such as STAT-3 blockade, are forthcoming. Many of the newer clinical trials are employing a combination of approaches, including concurrently administered chemotherapeutics with immune modulatory effects.
A variety of strategies have been used to increase the immunogenetic properties of various therapies for brain tumors. The immune response can be augmented by genetic modification of immune cells to secrete cytokines, including IL-2, IL-4, IL-12, GM-CSF, and IFN-γ.151–161 One can also alter the MHC of the cells to express allogeneic determinants or genetically modify the tumor cells to express costimulatory molecules such as B7.153,162 In some instances, objective evidence of tumor regression has been observed in patients receiving immunizations only with tumor cell immunogens, suggesting the potential effectiveness of this type of cellular immunotherapy for malignant neoplasms. Further genetic modification of cells through transduction with tumor RNA or DNA may result in the expression of specific tumor-associated antigens and enhanced immunogenicity.159,162–165 In addition, modification of delivery techniques used to treat intracerebral tumors has included intrathecal, intralymphatic, subcutaneous, and intratumoral injections of treatment cells.152,163 Most recently, the inhibition of immunosuppressive cells or factors induced by the tumors has been a target for enhancing the immunogenicity of treatment strategies.28,29,63,166 These techniques to enhance the immune response have been used in the development of current cellular vaccine studies. However, the major questions in immunotherapeutic development remain:
Immunotherapeutic Approaches and Recent Clinical Trial Results
Cytokine-Mediated Strategies
Recent advances in our understanding of the biology of the immune system have led to the identification of numerous cytokines that modulate immune responses (Tables 97-1 and 97-2).167–169 These agents mediate many of the immune responses involved in antitumor immunity. Several of these cytokines have been produced by recombinant DNA methodology and evaluated for their antitumor effects. In experimental clinical trials, the administration of cytokines and related immunomodulators has resulted in objective tumor responses in some patients with various types of neoplasms.170–172
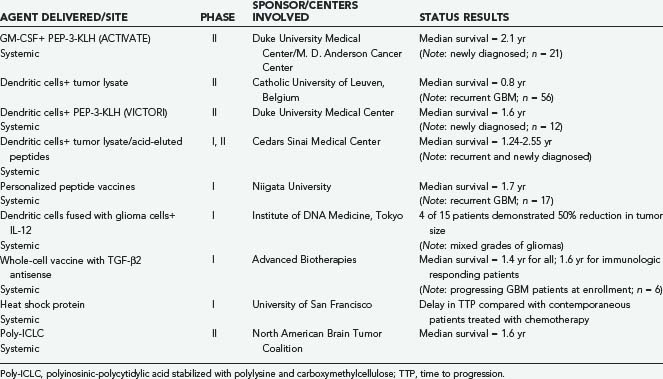
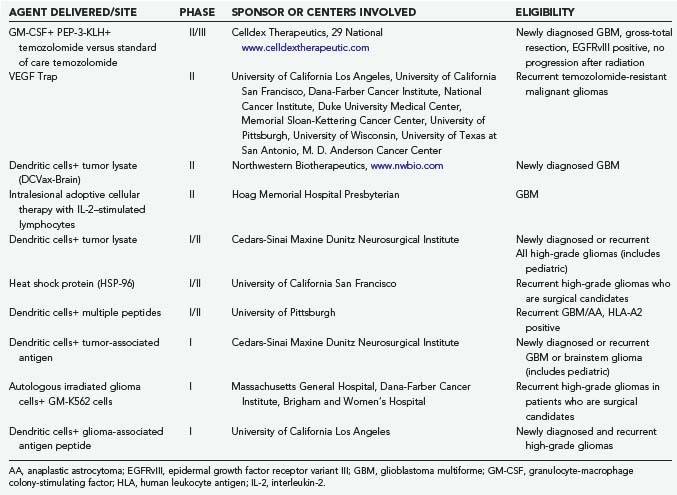
IL-2 is an important cytokine in the generation of antitumor immunity.172 IL-2 has no direct toxic effect on cancer cells but mediates its antitumor activity by the modulation of the host’s immune response to the neoplasm. In response to tumor antigens, the helper T-cell subset of lymphocytes secretes small quantities of IL-2. This IL-2 acts locally at the site of tumor antigen presentation to activate cytotoxic T cells and NK cells that mediate systemic tumor cell destruction. In preclinical animal studies, cytotoxic immune responses were induced in animals with an intracerebral glioma treated with allogeneic IL-2–secreting fibroblasts administered intracerebrally.156 In addition, it has recently been shown that IL-2 regulatory effects involve T regs.173 Intravenous, intralymphatic, or intralesional administration of IL-2 has resulted in clinically significant responses in several types of cancer.170–172174 However, severe toxicities (hypotension and edema) limit the dose and efficacy of intravenous and intralymphatic IL-2 administration.170,171,174 The toxicity of systemically administered cytokines is not surprising because these agents mediate local cellular interactions, and they are normally secreted in quantities too small to have systemic effects. To circumvent the toxicity of systemic IL-2 administration, several investigators have examined intralesional injection of IL-2.152,163,175–177 This approach eliminates the toxicity associated with systemic IL-2 administration. However, multiple intralesional injections are required to optimize therapeutic efficacy.175,176 These injections are impractical for many patients without potential significant morbidity, particularly when tumor sites are not accessible for direct injection.
Another cytokine that has been investigated for immunotherapeutic use in brain tumors is IFN-γ. IFN-γ induces the expression of MHC class I determinants, augments the sensitivity of tumor cells to cytotoxic T-lymphocyte–mediated lysis, and may make weakly antigenic neoplastic cells more immunogenic. Transfection of the IFN-γ gene into cells has led to an inhibition of tumor growth.156,159
Similarly, studies using IL-4–secreting cells demonstrated increased survival and antitumor cytotoxic responses, suggesting that an immune response can take place against a tumor of the central nervous system in situ.158,178,179 In other preclinical animal studies, a specific and significant peripheral systemic immunocytotoxic response (by cromium-51 release assay) was induced in animals with an intracerebral glioma treated with allogeneic IL-2–secreting fibroblasts administered intracerebrally.156 Thus, the secretion of IL-2, or other cytokines, by the cellular immunogen, or an immunogenic derivative of the cells, may have altered the blood-brain barrier, enabling the immunogen to reach the spleen and lymph nodes in the periphery.180,181
Cytokine gene transfer has resulted in significant antitumor immune responses in several animal tumor models.158,159,182,183 In these studies, the transfer of cytokine genes into tumor cells has reduced or abrogated the tumorigenicity of the cells after implantation into syngeneic hosts. The transfer of genes for IL-2,153,182,183 IFN-γ,156,159 and IL-4157,158 significantly reduced or eliminated the growth of several different histologic types of murine tumors. Other cytokines capable of producing similar results include GM-CSF160 and IL-12.161,184 In the studies employing IL-2 gene transfer, the treated animals also developed systemic antitumor immunity and were protected against subsequent tumor challenges with the unmodified parental tumor.182,183,185 Similar inhibition of tumor growth and protective immunity were also demonstrated when immunizations were performed with a mixture of unmodified parental tumor cells and genetically modified tumor cells engineered to express the IL-2 gene. No toxicity associated with expression of the cytokine transgenes was reported in these animal tumor studies.159,182,183,186 An alternative strategy is to genetically modify tumor cells to express an antisense gene to TGF-β, which is a cytokine highly expressed in glioma cells that acts to inhibit the function of cytotoxic T cells.63
Although preclinical studies with cytokine gene therapy appear promising,157,160,162,187–189 clinical trials for brain tumors have been limited. Some of these trials have involved immunization with tumor cells modified with the IL-2 gene,190 IL-4 gene,179 or TGF-β2 antisense gene.166 The toxic effects of cytokines in the CNS may limit the quantity that can be administered.191,192 Neurologic effects and cerebral edema have been seen in animals injected intracranially with syngeneic cytokine-secreting cells.177 In contrast, however, studies using allogeneic cytokine-secreting cells demonstrated an increase in survival and no long-term or short-term toxicity.163,186
The toxicity of a cellular-based cytokine gene therapy for tumors is likely to depend in part on the survival of the genetically modified cells in the CNS. In studies investigating the survival of an allogeneic IL-2–secreting vaccine in the CNS by two different means, polymerase chain reaction and bioassay,186 it was shown that allogeneic cells survive in the CNS less than 28 days. The cells, like other allografts, were rejected. The cells were well tolerated, and the animals did not demonstrate any significant neurologic or systemic toxicity. This suggests that cytokine-secreting allogeneic cells may serve as a safe and useful vehicle for the safe delivery of cytokines into brain tumors and supports the possibility and safety of using a monthly retreatment schedule in a clinical protocol.
Cellular Immunotherapy
Previous immunotherapy strategies initially used classic immunologic cell types, including activated lymphocytes and LAK cells. More recently, a variety of cells have been investigated for their usefulness in tumor oncology, including tumor cells themselves (syngeneic or allogeneic), DCs, and fibroblasts (syngeneic or allogeneic). Although syngeneic tumor cells have the advantage that they express most of the appropriate antigens needed for targeted therapy, many types of tumors are difficult to establish in culture. It is also conceivable that a subpopulation of the primary tumor, selected for its capacity to grow in vitro, may not reflect the tumor cell population as a whole, especially because tumors such as gliomas are known to be heterogeneous. In addition, cytokine gene therapies requiring the transduction of autologous tumor cells may not be practical for many cancer patients. Modification of neoplastic cells taken directly from tumor-bearing patients may be difficult. In particular, a primary tumor cell line, required for retroviral modification, has to be established. An alternative cell type that can be used for therapeutic immunizations is the DC, which is a specialized APC. Preclinical studies have indicated that immunizing either mice or rats with DC pulsed using tumor cell antigens can stimulate a cytotoxic T-cell response that is tumor specific and that engenders protective immunity against CNS tumor in the treated animals.193,194
There are a number of advantages to using genetically modified allogeneic cells, such as an allogeneic fibroblast cytokine-secreting cell line as an “off-the-shelf” cellular vaccine. Fibroblasts obtained from established allogeneic fibroblast cell lines may be readily cultured in vitro and genetically modified to express and secrete cytokines.6,154,161,162,165,190 The cells can be subsequently injected directly into the tumor bed. The use of allogeneic rather than syngeneic cells was initially based on evidence that allogeneic MHC determinants augment the immunogenic properties of the tumor vaccine.155,165 Application of genetically modified fibroblasts in therapeutic vaccines facilitates titration of single or multiple cytokine doses independent of tumor cell doses. Like other allografts, the allogeneic cytokine-secreting cells are rejected. Furthermore, the number of cells can be expanded as desired for multiple rounds of therapy. In addition, the slow continuous release of cytokines and the eventual rejection of the allograft may be a useful advantage in the treatment of brain tumors in which long-term secretion of high concentrations of certain cytokines may be associated with increased morbidity. Thus, an allogeneic cytokine-secreting vaccine is readily available, easily expanded, possibly less toxic, and more immunogenic. These considerations provide the rationale for examining the use of allogeneic fibroblasts genetically modified to secrete cytokines in further studies as a means of enhancing antitumor immune responses in treatment of malignant intracerebral tumors.154,156,161,162,165,190
Two of the more promising cellular immune therapeutic approaches are DC immunotherapy and adoptive T-cell transfers. Earlier cellular-based studies included the use of alloreactive cytotoxic lymphocytes195 or LAK cells196 combined with IL-2. More recently, therapeutic immunizations have been with DCs. Several clinical trials have been completed with promising results at the University of California Los multiple centers. The DCs are loaded with acid-eluted peptides, tumor homogenates, or the tumor-specific antigen EGFRvIII after being stimulated and matured with GM-CSF and IL-4. The patients are then systemically vaccinated with the DC preparations. In one of the phase I clinical trials,197 nine newly diagnosed high-grade glioma patients received three separate vaccinations spaced 2 weeks apart. A robust infiltration of T cells was detected in tumor specimens, and median survival was 15.1 months (compared with 8.6 months for a control population). A subsequent report198 involving eight GBM patients produced a median survival time of 33.3 months, compared with a median survival of 7.5 months for a comparable group of patients receiving other treatment protocols. Currently, a phase II multi-institutional clinical trial is under way that is using DCs pulsed with tumor homogenate in GBM patients sponsored by Northwest Biotherapeutics. To generate this vaccine, the patients must undergo surgery to obtain the tumor to generate homogenate and have sufficient tumor volume.
In a phase I clinical trial (VICTORI) conducted at Duke University Medical Center employing an antigen-specific approach to EGFRvIII with DCs, 15 newly diagnosed glioma patients were enrolled (World Health Organization [WHO]; WHO grade IV, n = 12). After surgical resection, each patient underwent leukapheresis to obtain peripheral blood mononuclear cells (PBMCs) to generate the autologous DCs and for baseline immunologic monitoring. The first three patients received 3 × 107 mature DCs per vaccination intradermally beginning 2 weeks after completion of radiation therapy, and the remaining patients received one third of their total generated DCs per injection (up to 1.1 × 108 cells). Patients underwent magnetic resonance imaging (MRI) every 2 months for evidence of toxicity or tumor progression. Enrolled patients demonstrated immunologic responses without any evidence of adverse events aside from grade I and grade II local reactions at the vaccine injection site. Humoral and cellular immune responses that were specific for EGFRvIII were detected ex vivo after vaccination. Two patients were allowed on the trial with MRI enhancement at the time of vaccination, and both these patients had near-complete resolution of radiographic enhancement after the vaccine. Median time to progression (TTP) in patients with GBM (n = 12) was 6.8 months, and median survival was 18.7 months weeks.199
The research group at Cedars-Sinai Medical Center in Los Angeles reported the results of a clinical trial using chemotherapy after patients received the vaccine protocol. Mean survival for patients receiving only the vaccine was 18 months, with a 2-year survival rate of 8%, whereas those receiving both the vaccine and chemotherapy had a mean survival time of 26 months with a 2-year survival rate of 42%.200 The authors of the study hypothesized that the improved outcome was due to the vaccine having primed the apoptotic machinery of the cancer cells so that chemotherapy was then able to trigger the apoptotic pathway. However, an alternative explanation exists—the immunotherapy may be selecting the more aggressive, infiltrative tumor cells, leaving behind more chemosensitive cells. Regardless, this study indicates that a synergy may exist between immunotherapy and chemotherapy that needs to be further explored.
An alternative cellular vaccine approach is the amplification of the T cells that are generated by the individual cancer patient in response to tumor cells. Glioblastoma tumor cells gathered during surgery were cultured in the presence of growth factors and then injected subcutaneously back into the patients. After development of an immune reaction, the lymph nodes draining the location of the injection were resected to obtain lymphocytes directed against the tumor cells and were stimulated with staphylococcus toxin and IL-2. This generated a large number of activated T cells, which were infused back into the patient. Of 10 patients, 2 had tumor regression, but 8 patients had progressive disease.53 It is unknown whether these T cells are functional once they enter into the tumor microenvironment. Techniques to maintain T-cell activation despite immunosuppressive factors are under development. Another inherent problem with cellular immunotherapy is the ex vivo manipulation of the cells—these techniques can be labor intensive, require special facilities for generating a product that can be infused into human patients, and will probably only be available at specialty centers.
Antibody Therapy
Monoclonal antibodies can be used to induce apoptosis, to mediate immune responses, or as a delivery vehicle for chemotherapeutics, toxins, or radionucleotides. One of the most exploited targets has been tenascin, an extracellular matrix glycoprotein, ubiquitously expressed in malignant gliomas but not normal brain. The monoclonal antibody 81C6 binds an epitope within the alternatively spliced fibronectin type III region of tenascin. In a phase II clinical trial of newly diagnosed GBM patients, the iodine-131–labeled murine 81C6 was injected into the patient’s surgically created cavity. Median survival times were 19.9 months for newly diagnosed GBM patients and 12 months for recurrent GBM patients, which significantly surpassed the historical controls even when accounting for established prognostic factors, including age and Karnofsky performance score.201,202 Patient-specific dosing in newly diagnosed GBM patients further pushed median survival to 23 months.
Targeting cell surface receptors with immunotoxins provides another strategy in the antibody-directed immunotherapeutic treatment of brain tumors. The targeted agents are composed of a toxin derived from bacteria, like pseudomonas endotoxin or diphtheria toxin, coupled to either an antibody specific for a membrane antigen (immunotoxin) or a ligand specific for cell surface receptors (cytotoxin).203 The specificity of immunotoxin therapy depends on the presence or overexpression of unique receptors or antigens on tumor cells and not normal cells. Specific ligands used for this type of therapy have included the transferrin receptor,204 the epidermal growth factor receptor,205 and two cytokine receptors, IL-4206 and IL-13.207,208 To target IL-4 and IL-13 receptors, recombinant fusion cytotoxins composed of IL-4 or IL-13 and a mutated form of pseudomonas exotoxin (PE) have been engineered. It has been shown that these chimeric cytotoxins are highly toxic in vitro to glioma cell lines, whereas normal cells are spared their cytotoxic effects.
An important consideration that arises in treating malignant gliomas with these agents is that the blood-brain barrier does not allow delivery of large molecules to intracranial tumors. Thus, several routes of administration have been tried, including intrathecal, intravenous, intra-arterial, and intratumoral. However, the development of convection-enhanced intratumoral delivery of macromolecules into the brain209 may eventually overcome this challenge. In initial clinical trials using IL-13 PE administered to patients with malignant gliomas using a convection-enhanced delivery system, the patients tolerated the infusion of this drug, and there was some modest prolongation of survival.209,210 Ultimately, optimization of delivery will be needed to bring this approach into the realm of standard care.
Another related treatment strategy has been used that takes advantage of the fact that EGFR is overexpressed in a significant number of malignant brain tumors, and recombinant toxins targeting this growth factor receptor have therefore been developed.206 Once again, convection-enhanced delivery techniques are needed to aid in drug delivery, although problems involving ineffective infusion are a recurrent theme in patients treated with these agents. Nevertheless, some durable radiographic responses have been reported, suggesting the potential efficacy of these drugs in treatment of patients with malignant brain tumors.
Vaccination Strategies
The genetic alterations occurring within gliomas210 can provide specific targets for immunotherapy, but none are universally ubiquitous or maintain expression throughout the disease course. The implications of tumor heterogeneity must be recognized in the design of rational therapeutic approaches and ultimately may require either an individualized approach or targeting with multiple agents. A number of different vaccination strategies are being evaluated211,212 and include approaches to vaccination with tumor-associated antigens (TAAs), for example, those based on (1) defined antigens or antigenic peptides, (2) tumor cell lysates or lysate fractions, and (3) whole irradiated tumor cells or apoptotic tumor cell bodies. Vaccines prepared using TAAs or TAA-derived epitopes presented by APCs are loaded onto DCs and used in clinical trials for patients with gliomas.178,197,213,214 However, these TAAs have to first be identified and then purified, requiring a tremendous effort requiring an “antigen discovery” step. Often the quantity of purified antigen must be increased to enable multiple immunizations of the cancer patient. While new TAAs are being discovered, the question of which TAA to be used in the vaccine is uncertain and extensively debated. The choice of TAA is not a trivial decision. Not only are isolation and purification of TAAs or antigenic peptide highly labor intensive, but it also remains uncertain whether TAA- or peptide-based vaccines are superior to tumor cell vaccines. Selection of the immunizing antigen is generally based on its abundant expression in the tumor and lack of expression in normal tissues. Antigens that do meet this criterion may not always be immunogenic. Furthermore, the heterogeneity of antigen expression in the tumor cell population is likely to be a concern. Some tumor cells may not express the antigen chosen for therapy. For example, de Vries80 found that expression of known tumor antigens such as gp100 and tyrosinase was variable in different melanoma lesions in the same patient. Not all the malignant cells in the patient’s neoplasm expressed these antigenic determinants. Because the tumor cell population is heterogeneous, tumor cells that fail to express the defined antigen chosen for therapy are likely to escape destruction by the activated immune system and are likely the source of recurrent tumor.
The use of tumor lysates, lysate fractions, or apoptotic tumor bodies for vaccination overcomes some of the tumor heterogeneity limitations. However, preparation of the vaccine requires the availability of autologous tumor, often in substantial quantities. Vaccines have been prepared by culturing patient-derived DCs and then pulsing or “feeding” the cells tumor lysates or apoptotic bodies. However, DC-based vaccines are laborious and costly to prepare. Their efficacy in generating antitumor immune responses capable of tumor rejection remains unproved. In all cases, the optimal adjuvant, protein or peptide concentration, ratios of DCs or apoptotic bodies and routes of delivery, and immunization schedules remain undefined. They may be critical for success. Immunization by injection of “naked” plasmid DNA or RNA encoding a tumor antigen is also currently under evaluation. However, as for immunization with defined antigens, there is a danger that the antigen specified by the polynucleotide chosen may not be the most appropriate for CTL or helper T-cell generation. Anichini and associates215 found that CTLs in melanoma patients are not always directed toward known melanoma antigens, such as Melan-A/Mart-1, MAGE-3, gp100, or tyrosinase. It is likely that there are multiple other tumor antigens in addition to those previously identified that are expressed by different cells that comprise the malignant cell population. Only certain of these TAAs are able to induce tumor-specific immune responses. The identification of the most “clinically relevant” tumor antigen cannot always be accomplished a priori, without extensive preclinical studies. Even then, subsequent validation in the patient may not confirm that these are “tumor rejection” antigens.
Another strategy involves preparation of vaccines by transfer of tumor DNA or RNA into an immunizing cell. The major advantage of this approach is that TAAs do not have to be purified or produced in large quantities. DNA-based vaccines are able to elicit robust and long-lasting activation of the immune system, which results in tumor rejection.163,164 In comparison with protein vaccines, DNA-based vaccines provide prolonged expression and direct presentation of tumor antigens. This offers an opportunity for the development of effector as well as memory immune responses to many different epitopes encoded by the tumor-derived DNA. From a practical point of view, these vaccines are easy and relatively inexpensive to prepare. Unlike the tumor lysate and homogenate strategies, these vaccines can be prepared from only a limited quantity of tumor DNA, which can be obtained from small surgical specimens or even biopsies. In the case of a recipient fibroblast, these cells can be selected to meet the requirement for rapid expansion in culture and MHC restriction. DNA-based vaccines offer a number of important advantages, which is greatly encouraging for their further development for cancer immunotherapy. The use of tumor vaccines as a “protective treatment” introduced after tumor resection may play an important role in delaying tumor recurrence in the clinical setting.
An ideal immunologic approach to cancer treatment is an off-the-shelf approach, similar to the commercially available vaccines. One such immunotherapeutic approach for newly diagnosed GBM is the vaccine CDX-110 (PEP-3-KLH), which is a peptide-based vaccination approach, again targeting the tumor-specific antigen EGFRvIII. Preclinical data demonstrated that when mice with established intracerebral tumors were treated with a single dose of PEP-3-KLH, there was a significant increase in long-term and median survival.11 A phase II multicenter trial (ACTIVATE) was conducted at Duke University Medical Center and the University of Texas M. D. Anderson Cancer Center and enrolled 18 newly diagnosed GBM patients with tumor expression of EGFRvIII.84 After completion of radiation therapy, patients received three vaccinations at 2-week intervals of PEP-3-KLH with GM-CSF. Subsequent vaccinations were continued monthly until tumor progression was evident. Adverse events consisted mostly of grade I and grade II vaccine site reactions. Both humoral and cellular immune responses to the EGFRvIII were enhanced after vaccination. Median PFS in this trial was 14.2 months and not significantly different between sites (P = .3445) and compared favorably to an institutional historical cohort matched for eligibility criteria and adjusted for age and Karnofsky performance score (n = 17), in which the median PFS was only 6.3 months. The median survival time for all study subjects was 26 months, which again compared well with the historical data, even after adjustments for age and Karnofsky performance score. Further evidence for efficacy was provided by the observation that among the patients with recurrent tumors in whom pathologic material could be obtained at recurrence, 82% lost EGFRvIII expression, suggesting a possible mechanism of failure involving antigen escape. The results are also notable for the fact that the expression of EGFRvIII on the GBM is a negative prognosticator in patients surviving more than 1 year. Typically, the median survival is 12.8 months for patients with EGFRvIII-expressing tumors, and no patient survives more than 18 months.216 The ACTIVATE clinical trial was conducted before temozolomide became the standard of care,217 so a second phase II clinical trial (ACT II) combined the CDX-110 vaccine with temozolomide.
One issue that arises for the peptide vaccine strategies is that the EGFRvIII is not ubiquitously expressed, and therefore many GBM patients are not candidates. In a phase I/II clinical trial at the University of California San Francisco, the vaccination strategy circumvented the identification of a precise tumor antigen by vaccinating with heat shock proteins, which act as immune chaperones for the tumor antigens.217a The heat shock proteins are purified from the resected tumor by antigenics and then subsequently administered back to the patient. The patients who received the heat shock protein had induced immune response. Efficacy data are pending; however, this type of approach is limited logistically by the volume of tumor that needs to be resected to generate a vaccine.
Synergy of Chemotherapy with Immunotherapy
Conventional wisdom has been that chemotherapy and immunotherapy are two antagonistic forms of therapy. Model systems have demonstrated that the depletion of immune cell subsets can abrogate the efficacy of several types of immunotherapeutic approaches,11,82 indicating that chemotherapy administered during the effector stages of immunotherapy may be deleterious to efficacy. However, this does not preclude using chemotherapeutic agents when appropriately timed with immunotherapy or at lower doses to minimize the aforementioned effects. Certain chemotherapy drugs do, in fact, have positive immunomodulatory properties that stimulate the production of proinflammatory cytokines (TNF, IL-12, and IL-1) and increase NK cell antitumor and LAK cell activity.218 Furthermore, the depletion of certain effector cells, such as T regs, may be a highly desirable outcome of chemotherapy, yielding greater immunotherapeutic efficacy, or may promote a desirable cytokine profile for adequate tumor control. One particular area of investigation that has been underexplored is an analysis of the specific immunologic effects, or the lack thereof, of various chemotherapeutic agents. Of note, the immune modulatory effects of a particular chemotherapeutic agent cannot be extrapolated to all, and caution needs to be employed in using combination therapy. For example, the number of patients able to generate an immune response to a peptide was diminished and toxicity increased when they were vaccinated with IL-2 during chemotherapy.219,220 Recent studies by Ahmadzadeh and coworkers221 have shown that the administration of IL-2 in patients with metastatic melanoma and renal cell carcinoma leads to a significant increase in CD4+, CD25hi, FoxP3+ T regs. Patients were infused with a high-dose bolus of IL-2 (720,000 IU/kg) intravenously every 8 hours to a maximum of 15 doses. About 20% of the patients demonstrated a clinical response, although the mechanism of action remains unclear. Moreover, there was a significant in vivo expansion of the T regs, and patients experiencing that fared much worse. Therefore, chemotherapies that induce strong IL-2 responses may not be desirable.
In recent animal studies, the combination of a cellular vaccine consisting of cytokine-secreting allogeneic fibroblasts and the drug thiazolidinedione (a nuclear hormone receptor involved in metabolism, signal transduction, apoptosis, and differentiation) led to increased survival and specific cytotoxic responses.225 The improved efficacy of combination drug with vaccine therapy suggests that these drugs may enhance immunotherapy by inhibiting immunosuppression (as described previously) or may actually augment the immunogenicity of the tumor itself, through either apoptosis or alteration of membranes or proteins, changing the antigenicity of the tumor. In the clinical trials that have attempted to address the mechanisms of immunologic failure, certain mechanisms predominate. In the case of adoptive T-cell transfers, many effector cells are administered, which can potentially overwhelm the T reg population. Clinical responses to this have been documented, especially in melanoma patients. Nevertheless, eventually the disease recurs, which has been attributed to the T regs regaining hemostasis and eventually predominating. Several agents have been identified that can reduce the relative T reg population. These agents include temozolomide, CTX, and denileukin diftitox. Thus, with clinical trials that are using adoptive T-cell transfer, it would be logical to determine when T regs become prominent. When a patient experiences an elevation of the T reg population, an agent that depletes the T regs can be administered, which may further enhance and prolong the clinical efficacy observed in such patients. A greater understanding of how these cells are involved in the basic biology of cancer patients is needed. In the specific circumstances of glioma patients, we do not know how the various phases of treatment, such as surgical resection, radiation, or recurrence, influence the T reg population, but we do know that the influx of T regs is not ubiquitous across all types of gliomas and is heterogeneous even within GBMs.223
One could consider that part of the observed clinical efficacy of temozolomide may be attributable to the trafficing to the glioma micro-environment rather than solely to its direct antiglioma effects. When a vaccination is administered during the nadir of the temozolomide level, there are likely enhanced effector responses.57 Such effector responses may be secondary to a lag in the recovery of T regs, allowing a greater clonotypic expansion than would otherwise occur without the temozolomide. Given the fact that temozolomide has been established as a standard of care for glioblastoma patients, in future trials, it is difficult to justify failing to administer this to patients.
A phase II clinical trial was undertaken at University of Texas M. D. Anderson Cancer Center and Duke University Medical Center to assess the immunogenicity and efficacy of an EGFRvIII-specific peptide vaccine in patients with newly diagnosed EGFRvIII-expressing GBM in combination with simultaneous standard or continuous temozolomide.57 After resection and radiation and concurrent temozolomide, consecutive cohorts received subsequent monthly cycles of 200 mg/m2 or continuous 100 mg/m2 temozolomide simultaneous with intradermal vaccinations with an EGFRvIII-specific peptide (PEP-3) conjugated to keyhole limpet hemocyanin (KLH) until tumor progression or death. Twenty-two patients were enrolled. There were no significant differences in vaccine immunogenicity, progression-free survival, or overall survival between temozolomide regimens. Although temozolomide induced grade II lymphopenia in 53.8% of patients, the coadministration of temozolomide with the EGFRvIII vaccine (CDX-110) resulted in strong sustained immune responses to EGFRvIII in 100% of evaluated patients. Median progression-free survival was 15.2 months.224 Median survival was 23.6 months. These data indicate that the concurrent administration of temozolomide with immunotherapy does not ablate immunologic or clinical efficacy and is the current design of the registration trial by Celldex.
Conclusion
Future immunotherapies will likely incorporate the following components:
Abdeljabar E, Han Y, Lesniak M. Prolongation of survival following depletion of CD4+CD25+ regulatory T cells in mice with experimental brain tumors. J Neurosurg. 2006;105:430-437.
Barba D, Saris SC, Holder C, et al. Intratumoral LAK cell and interleukin-2 therapy of human gliomas. J Neurosurg. 1989;70:175-182.
Dudley ME, Wunderlich JR, Robbins PF, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298:850.
Ehtesham M, Kabos P, Kabosova A, et al. The use of interleukin 12-secreting neural stem cells for the treatment of intracranial glioma. Cancer Res. 2002;62:5657.
Fakhrai H, Dorigo O, Shawler DL, et al. Eradication of established intracranial rat gliomas by transforming growth factor beta antisense gene therapy. Proc Natl Acad Sci U S A. 1996;93:2909.
Fecci PE, Mitchell DA, Whitesides JF, et al. Increased regulatory T-cell fraction amidst a diminished CD4 compartment explains cellular immune defects in patients with malignant glioma. Cancer Res. 2006;66:3294.
Fecci PE, Ochiai H, Mitchell DA, et al. Systemic CTLA-4 blockade ameliorates glioma-induced changes to the CD4+ T cell compartment without affecting regulatory T-cell function. Clin Cancer Res. 2007;13:2158.
Giezeman-Smits KM, Okada H, Brissette-Storkus CS, et al. Cytokine gene therapy of gliomas: induction of reactive CD4+ T cells by interleukin-4-transfected 9L gliosarcoma is essential for protective immunity. Cancer Res. 2000;60:2449.
Glick RP, Lichtor T, de Zoeten E, et al. Prolongation of survival of mice with intracerebral glioma treated with semi-allogeneic fibroblasts secreting IL-2. Neurosurgery. 1999;45:867-874.
Heimberger AB, Hussain SF, Aldape K, et al. Tumor-specific peptide vaccination in newly-diagnosed patients with GBM. J Clin Oncol. 2006;24:1.
Hussain SF, Kong L-Y, Jordan J, et al. A novel small molecule inhibitor of signal transducers and activators of transcription 3 reverses immune tolerance in malignant glioma patients. Cancer Res. 2007;67:9630.
Hussain SF, Yang D, Suki D, et al. The role of human glioma-infiltrating microglia/macrophages in mediating antitumor immune responses. Neuro Oncol. 2006;8:261.
Kong LY, Abou-Ghazal MK, Wei J, et al. A novel inhibitor of STAT3 activation is efficacious against established central nervous system melanoma and inhibits regulatory T cells. Clin Cancer Res. 2008;14:5759-5768.
Kruse C, Cepeda L, Owens B, et al. Treatment of recurrent glioma with intracavitary alloreactive cytotoxic lymphocytes and interleukin-2. Cancer Immunol Immunother. 1997;45:77-87.
Liau LM, Prins RM, Kiertscher SM, et al. Dendritic cell vaccination in glioblastoma patients induces systemic and intracranial T-cell responses modulated by the local central nervous system tumor microenvironment. Clin Cancer Res. 2005;11:5515.
Lichtor T, Glick RP, Kim TS, et al. Prolonged survival of mice with glioma injected intracerebrally with double cytokine-secreting cells. J Neurosurg. 1995;83:1038.
Lichtor T, Glick RP, Lin H, et al. Intratumoral injection of IL-secreting syngeneic/allogeneic fibroblasts transfected with DNA from breast cancer cells prolongs the survival of mice with intracerebral breast cancer. Cancer Gene Ther. 2005;12:708.
Okada H, Pollack IF, Lieberman F, et al. Gene therapy of malignant gliomas: a pilot study of vaccination with irradiated autologous glioma and dendritic cells admixed with IL-4 transduced fibroblasts to elicit an immune response. Human Gene Ther. 2001;12:575.
Reardon DA, Akabani G, Coleman RE, et al. Phase II trial of murine131I-labeled antitenascin monoclonal antibody 81C6 administered into surgically created resection cavities of patients with newly diagnosed malignant gliomas. J Clin Oncol. 2002;20:1389.
Sampson Sampson JH, Heimberger AB, Archer GE, et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J Clin Oncology. 2010;28:4722-4729.
Sampson JH, Aldape KD, Gilbert MR, et al. Temozolomide as a vaccine adjuvant in GBM. J Clin Oncol. 2007;25:2020.
Sampson Sampson JH, Aldape KD, Archer GE, et al. Greater chemotherapy-induced lymphopenia enhances tumor-specific immune responses that eliminate EGFRvIII-expressing tumor cells in patients with glilblastoma. Neuro-Oncology. 2010 Dec. 10. [Epub ahead of print]
Spagnolo A, Glick RP, Lin H, et al. Prolonged survival of mice with an established intracerebral glioma receiving combined treatment with PPAR-γ thiazolidinedione agonists and IL-2 secreting syngeneic/allogeneic fibroblasts. J Neurosurg. 2007;106:299-305.
Tada M, de Tribolet N. Recent advances in immunobiology of brain tumors. J Neurooncol. 1993;17:261.
Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol. 2007;7:41.
Yu JS, Burwick JA, Dranoff G, et al. Gene therapy for metastatic brain tumors by vaccination with granulocyte-macrophage colony-stimulating factor-transduced tumor cells. Human Gene Ther. 1997;8:1065.
Yu J, Liu G, Ying H, et al. Vaccination with tumor lysate-pulsed dendritic cells elicits antigen-specific, cytotoxic T-cells in patients with malignant glioma. Cancer Res. 2004;64:4973.
1 Male D. Immunology. London: Grover Med Pub; 1991.
2 Hickey WF, Hsu BL, Kimura H. T-lymphocyte entry into the central nervous system. J Neurosci Res. 1991;28:254.
3 von Hanwehr RI, Hofman FM, Taylor CR, et al. Mononuclear lymphoid populations infiltrating the microenvironment of primary CNS tumors. Characterization of cell subsets with monoclonal antibodies. J Neurosurg. 1984;60:1138.
4 Brooks WH, Markesbery WR, Gupta GD, et al. Relationship of lymphocyte invasion and survival of brain tumor patients. Ann Neurol. 1978;4:219.
5 Palma L, Di Lorenzo N, Guidetti B. Lymphocytic infiltrates in primary glioblastomas and recidivous gliomas. Incidence, fate, and relevance to prognosis in 228 operated cases. J Neurosurg. 1978;49:854.
6 Safdari H, Hochberg FH, Richardson EPJr. Prognostic value of round cell (lymphocyte) infiltration in malignant gliomas. Surg Neurol. 1985;23:221.
7 Hussain SF, Yang D, Suki D, et al. The role of human glioma-infiltrating microglia/macrophages in mediating antitumor immune responses. Neuro Oncol. 2006;8:261.
8 Bloom HJ, Peckham MJ, Richardson AE, et al. Glioblastoma multiforme: a controlled trial to assess the value of specific active immunotherapy in patients treated by radical surgery and radiotherapy. Br J Cancer. 1973;27:253.
9 Trouillas P. Immunology and immunotherapy of cerebral tumors. Current status. Rev Neurol (Paris). 1973;128:23.
10 Wikstrand CJ, Bigner DD. Hyperimmunization of non-human primates with BCG-CW and cultured human glioma-derived cells. Production of reactive antisera and absence of EAE induction. J Neuroimmunol. 1981;1:249.
11 Heimberger AB, Crotty LE, Archer GE, et al. Epidermal growth factor receptor VIII peptide vaccination is efficacious against established intracerebral tumors. Clin Cancer Res. 2003;9:4247.
12 Weiner L. Cancer immunotherapy: the endgame begins. N Engl J Med. 2008;358:2664-2666.
13 Bower R, Lim M, Harsh G. Immunotherapy for gliomas. Contemp Neurosurg. 2007;29:1-5.
14 Wang HY, Lee DA, Peng G, et al. Tumor specific human CD4+ regulatory T cells and their ligands: implications for immunotherapy. Immunity. 2004;20:107-118.
15 Refaeli Y, Van Parijs L, London CA, et al. Biochemical mechanisms of IL-2-regulated Fas-mediated T cell apoptosis. Immunity. 1998;8:615-623.
16 Sakaguchi S. Naturally arising CD4+ regulatory T cells for immunologic self-tolerance and negative control of immune responses. Annu Rev Immunol. 2004;22:531.
17 Misra N, Bayry J, Lacroix-Desmazes S, et al. Cutting edge: human CD4+CD25+ T cells restrain the maturation and antigen-presenting function of dendritic cells. J Immunol. 2004;172:4676.
18 Taams LS, van Amelsfort JM, Tiemessen MM, et al. Modulation of monocyte/macrophage function by human CD4+CD25+ regulatory T cells. Hum Immunol. 2005;66:222.
19 Liyanage UK, Moore TT, Joo HG, et al. Prevalence of regulatory T cells is increased in peripheral blood and tumor microenvironment of patients with pancreas or breast adenocarcinoma. J Immunol. 2002;169:2756.
20 Abdeljabar E, Han Y, Lesniak M. prolongation of survival following depletion of CD4+CD25+ regulatory T cells in mice with experimental brain tumors. J Neurosurg. 2006;105:430-437.
21 Fecci PE, Mitchell DA, Whitesides JF, et al. Increased regulatory T-cell fraction amidst a diminished CD4 compartment explains cellular immune defects in patients with malignant glioma. Cancer Res. 2006;66:3294.
22 Fontenot JD, Rudensky AY. A well adapted regulatory contrivance: regulatory T cell development and the forkhead family transcription factor Foxp3. Nat Immunol. 2005;6:331.
23 Jordan JT, Sun WH, Hussain SF, et al. Preferential migration of regulatory T cells mediated by glioma-secreted chemokines can be blocked with chemotherapy. Cancer Immunol Immunother. 2008;57:123.
24 Foss F. Clinical experience with denileukin diftitox (ONTAK). Semin Oncol. 2006;33:11.
25 Berd D, Mastrangelo MJ. Effect of low dose cyclophosphamide on the immune system of cancer patients: reduction of T-suppressor function without depletion of the CD8+ subset. Cancer Res. 1987;47:3317.
26 Kong LY, Abou-Ghazal MK, Wei J, et al. A novel inhibitor of STAT3 activation is efficacious against established central nervous system melanoma and inhibits regulatory T cells. Clin Cancer Res. 2008;14:5759-5768.
27 Su YB, Sohn S, Krown SE, et al. Selective CD4+ lymphopenia in melanoma patients treated with temozolomide: a toxicity with therapeutic implications. J Clin Oncol. 2004;22:610.
28 Fecci PE, Ochiai H, Mitchell DA, et al. Systemic CTLA-4 blockade ameliorates glioma-induced changes to the CD4+ T cell compartment without affecting regulatory T-cell function. Clin Cancer Res. 2007;13:2158.
29 Fecci PE, Sweeney AE, Grossi PM, et al. Systemic anti-CD25 monoclonal antibody administration safely enhances immunity in murine glioma without eliminating regulatory T cells. Clin Cancer Res. 2006;12:4294.
30 Bailey SL, Schreiner B, McMahon EJ, et al. CNS myeloid DCs presenting endogenous myelin peptides ‘preferentially’ polarize CD4+ T(H)-17 cells in relapsing EAE. Nat Immunol. 2007;8:172.
31 Oukka M. Interplay between pathogenic Th17 and regulatory T cells. Ann Rheum Dis. 2007;66(suppl III):iii87.
32 Dannull J, Su Z, Rizzieri D, et al. Enhancement of vaccine-mediated antitumor immunity in cancer patients after depletion of regulatory T cells. J Clin Invest. 2005;115:3623.
33 Dang NH, Hagemeister FB, Pro B, et al. Phase II study of denileukin diftitox for relapsed/refractory B-Cell non-Hodgkin’s lymphoma. J Clin Oncol. 2004;22:4095.
34 Olsen E, Duvic M, Frankel A, et al. Pivotal phase III trial of two dose levels of denileukin diftitox for the treatment of cutaneous T-cell lymphoma. J Clin Oncol. 2001;19:376.
35 Attia P, Maker AV, Haworth LR, et al. Inability of a fusion protein of IL-2 and diphtheria toxin (Denileukin Diftitox, DAB389IL-2, ONTAK) to eliminate regulatory T lymphocytes in patients with melanoma. J Immunother. 2005;28:582.
36 Hoyne GF, Le Roux I, Corsin-Jimenez M, et al. Serrate1-induced notch signalling regulates the decision between immunity and tolerance made by peripheral CD4+ T cells. Int Immunol. 2000;12:177.
37 Cheever MA, Greenberg PD, Fefer A. Specificity of adoptive chemoimmunotherapy of established syngeneic tumors. J Immunol. 1980;125:711.
38 North RJ. Cyclophosphamide-facilitated adoptive immunotherapy of an established tumor depends on elimination of tumor-induced suppressor T cells. J Exp Med. 1982;155:1063.
39 Dudley ME, Wunderlich JR, Robbins PF, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298:850.
40 Kedl RM, Rees WA, Hildeman DA, et al. T cells compete for access to antigen-bearing antigen-presenting cells. J Exp Med. 2000;192:1105.
41 Gattinoni L, Finkelstein SE, Klebanoff CA, et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J Exp Med. 2005;202:907.
42 Anthony PA, Piccirillo CA, Akpinarli A, et al. CD8+ T cell immunity against a tumor/self-antigen is augmented by CD4+ T helper cells and hindered by naturally occurring T regulatory cells. J Immunol. 2005;174:2591.
43 Taieb J, Chaput N, Schartz N, et al. Chemoimmunotherapy of tumors: cyclophosphamide synergizes with exosome based vaccines. J Immunol. 2006;176:2722.
44 Ercolini AM, Ladle BH, Manning EA, et al. Recruitment of latent pools of high-avidity CD8+ T cells to the antitumor immune response. J Exp Med. 2005;201:1591.
45 Tsung K, Meko JB, Tsung YL, et al. Immune response against large tumors eradicated by treatment with cyclophosphamide and IL-12. J Immunol. 1998;160:1369.
46 Cao X, Zhang W, Wan T, et al. Enhanced antitumor immune responses of IL-2 gene-modified tumor vaccine by combination with IL-1 and low dose cyclophosphamide. J Exp Clin Cancer Res. 1999;18:173.
47 Hengst JCD, Mokyr MB, Dray S. Cooperation between cyclophosphamide tumoricidal activity and host antitumor immunity in the cure of mice bearing large MOPC-315 tumors. Cancer Res. 1981;41:2163.
48 Sojka DK, Donepudi M, Bluestone JA, et al. Melphalan and other anticancer modalities up-regulate B7-B1 gene expression in tumor cells. J Immunol. 2000;164:6230.
49 Holtl L, Ramoner R, Zelle-Rieser C, et al. Allogeneic dendritic cell vaccination against metastatic renal cell carcinoma with or without cyclophosphamide. Cancer Immunol Immunother. 2005;54:663.
50 MacLean GD, Miles DW, Rubens RD, et al. Enhancing the effect of THERATOPE STn-KLH cancer vaccine in patients with metastatic breast cancer by pretreatment with low-dose intravenous cyclophosphamide. J Immunother Emphasis Tumor Immunol. 1996;19:309.
51 Ehrke MJ, Mihich E, Berd D, et al. Effects of anticancer drugs on the immune system in humans. Semin Oncol. 1989;16:230.
52 Berd D, Maguire HC, McCue P, et al. Treatment of metastatic melanoma with an autologous tumor-cell vaccine: clinical and immunological results in 64 patients. J Clin Oncol. 1990;8:1858.
53 Plautz GE, Miller DW, Barnett GH, et al. T cell adoptive immunotherapy of newly diagnosed gliomas. Clin Cancer Res. 2000;6:2209.
54 Nigam A, Yacavone RF, Zahurak ML, et al. Immunomodulatory properties of antineoplastic drugs administered in conjunction with GM-CSF-secreting cancer cell vaccines. Int J Oncol. 1998;12:161.
55 Chu Y, Wang LX, Yang G, et al. Efficacy of GM-CSF-producing tumor vaccine after docetaxel chemotherapy in mice bearing established Lewis lung carcinoma. J Immunother. 2006;29:367.
56 Kanzawa T, Germano IM, Komata T, et al. Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ. 2004;11:448.
57 Sampson JH, Aldape KD, Archer GE, et al. Greater chemotherapy-induced lymphopenia enhances tumor-specific immune responses that eliminate EGFRvIII-expressing tumor cells in patients with glioblastoma. Neuro Oncol. 2010 Dec 10. [Epub ahead of print]
58 Sampath P, Hanes J, DiMeco F, et al. Paracrine immunotherapy with interleukin-2 and local chemotherapy is synergistic in the treatment of experimental brain tumors. Cancer Res. 1999;59:2107.
59 Nagarkatti M, Kaplan AM. The role of suppressor T-cells in BCNU-mediated rejection of a syngeneic tumor. J Immunol. 1985;135:1510.
60 Platten M, Wick W, Weller M. Malignant glioma biology: role for TGF-beta in growth, motility, angiogenesis, and immune escape. Microsc Res Tech. 2001;52:401.
61 Roszman T, Elliott L, Brooks W. Modulation of T-cell function by gliomas [see comments]. Immunol Today. 1991;12:370.
62 Tada M, de Tribolet N. Recent advances in immunobiology of brain tumors. J Neurooncol. 1993;17:261.
63 Fakhrai H, Dorigo O, Shawler DL, et al. Eradication of established intracranial rat gliomas by transforming growth factor beta antisense gene therapy. Proc Natl Acad Sci U S A. 1996;93:2909.
64 Uhl M, Aulwurm S, Wischhusen J, et al. SD-208, a novel transforming growth factor B receptor I kinase inhibitor, inhibits growth and invasiveness and enhances immunogenicity of murine and human glioma cells in vitro and in vivo. Cancer Res. 2004;64:7954.
65 Platten M, Wild-Bode C, Wick W, et al. N-[3,4-dimethoxycinnamoyl]-anthranilic acid (Tranilast) inhibits transforming growth factor-B release and reduces migration and invasiveness of human malignant glioma cells. Int J Cancer. 2001;93:53.
66 Schlingensiepen KH, Schlingensiepen R, Steinbrecher A, et al. Targeted tumor therapy with the TGF-beta2 antisense compound AP 12009. Cytokine Growth Factor Rev. 2006;17:129.
67 Chan OTM, Yang LX. The immunological effects of taxanes. Cancer Immunol Immunother. 2000;49:181.
68 Tsavaris N, Kosmas C, Vadiaka M, et al. Immune changes in patients with advanced breast cancer undergoing chemotherapy with taxanes. Br J Cancer. 2002;87:21.
69 Ehrke MJ, Maccubbin D, Ryoyama K, et al. Correlation between Adriamycin-induced augmentation of interleukin 2 production and of cell-mediated cytotoxicity in mice. Cancer Res. 1986;46:54.
70 Wintterle S, Schreiner B, Mitsdoerffer M, et al. Expression of the B7-related molecule B7-H1 by glioma cells: a potential mechanism of immune paralysis. Cancer Res. 2003;63:7462.
71 Dong H, Strome SE, Salomao DR, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8:793.
72 Curiel TJ, Wei S, Dong H, et al. Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat Med. 2003;9:562.
73 Strome SE, Dong H, Tamura H, et al. B7-H1 blockade augments adoptive T-cell immunotherapy for squamous cell carcinoma. Cancer Res. 2003;63:6501.
74 Contardi E, Palmisano GL, Tazzari PL, et al. CTLA-4 is constitutively expressed on tumor cells and can trigger apoptosis upon ligand interaction. Int J Cancer. 2005;117:538.
75 Kwon ED, Hurwitz AA, Foster BA, et al. Manipulation of T cell costimulatory and inhibitory signals for immunotherapy of prostate cancer. Proc Natl Acad Sci U S A. 1997;94:8099.
76 Rebmann V, Regel J, Stolke D, et al. Secretion of sHLA-G molecules in malignancies. Semin Cancer Biol. 2003;13:371.
77 Wiendl H, Mitsdoerffer M, Hofmeister V, et al. A functional role of HLA-G expression in human gliomas: an alternative strategy of immune escape. J Immunol. 2002;168:4772.
78 Schreiber H, Wu TH, Nachman J, et al. Immunodominance and tumor escape. Semin Cancer Biol. 2002;12:25.
79 Carlsson J, Daniel-Szolgay E, Frykholm G, et al. Homogeneous penetration but heterogeneous binding of antibodies to carcinoembryonic antigen in human colon carcinoma HT-29 spheroids. Cancer Immunol Immunother. 1989;30:269.
80 de Vries TJ, Fourkour A, Wobbes T, et al. Heterogeneous expression of immunotherapy candidate proteins gp100, MART-1, and tyrosinase in human melanoma cell lines and in human melanocytic lesions. Cancer Res. 1997;57:3223.
81 Liu G, Ying H, Zeng G, et al. HER-2, gp100, and MAGE-1 are expressed in human glioblastoma and recognized by cytotoxic T cells. Cancer Res. 2004;64:4980.
82 Sampson JH, Crotty LE, Lee S, et al. Unarmed, tumor-specific monoclonal antibody effectively treats brain tumors. Proc Natl Acad Sci U S A. 2000;97:7503.
83 Heimberger AB, Hussain SF, Aldape K, et al. Tumor-specific peptide vaccination in newly-diagnosed patients with GBM. J Clin Oncol. 2006;24:2529.
84 Sampson JH, Heimberger AB, Archer GE, et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J Clin Oncol. 2010;28:4722-4729.
85 Mitchell MS. Perspective on allogeneic melanoma lysates in active specific immunotherapy. Semin Oncol. 1998;25:623.
86 Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997.
87 Correale P, Cusi MG, Tsang KY, et al. Chemo-immunotherapy of metastatic colorectal carcinoma with gemcitabine plus FOLFOX 4 followed by subcutaneous granulocyte macrophage colony-stimulating factor and interleukin-2 induces strong immunologic and antitumor activity in metastatic colon cancer patients. J Clin Oncol. 2005;23:8950.
88 Nowak AK, Robinson BW, Lake RA. Gemcitabine exerts a selective effect on the humoral immune response: implications for combination chemo-immunotherapy. Cancer Res. 2002;62:2353.
89 Weber J, Salgaller M, Samid D, et al. Expression of the MAGE-1 tumor antigen is up-regulated by the demethylating agent 5-aza-2’-deoxycytidine. Cancer Res. 1994;54:1766.
90 Rubinfeld B, Upadhyay A, Clark SL, et al. Identification and immunotherapeutic targeting of antigens induced by chemotherapy. Nat Biotechnol. 2006;24:205.
91 Ohtsukasa S, Okabe S, Yamashita H, et al. Increased expression of CEA and MHC class I in colorectal cancer cell lines exposed to chemotherapy drugs. J Cancer Res Clin Oncol. 2003;129:719.
92 van Hall T, Wolbert EZ, van Veelen P, et al. Selective cytotoxic T-lymphocyte targeting of tumor immune escape variants. Nat Med. 2006;12:417.
93 Winston LA, Hunter T. JAK2, Ras, and Raf are required for activation of extracellular signal-regulated kinase/mitogen-activated protein kinase by growth hormone. J Biol Chem. 1995;270:30837.
94 Li B, Chang CM, Yuan M, et al. Resistance to small molecule inhibitors of epidermal growth factor receptor in malignant gliomas. Cancer Res. 2003;63:7443.
95 Rahaman SO, Harbor PC, Chernova O, et al. Inhibition of constitutively active Stat3 suppresses proliferation and induces apoptosis in glioblastoma multiforme cells. Oncogene. 2002;21:8404.
96 Lo HW, Hsu SC, Ali-Seyed M, et al. Nuclear interaction of EGFR and STAT3 in the activation of the iNOS/NO pathway. Cancer Cell. 2005;7:575.
97 Mizoguchi M, Betensky RA, Batchelor TT, et al. Activation of STAT3, MAPK, and AKT in malignant astrocytic gliomas: correlation with EGFR status, tumor grade, and survival. J Neuropathol Exp Neurol. 2006;65:1181.
98 Tarkowski E, Rosengren L, Blomstrand C, et al. Early intrathecal production of interleukin-6 predicts the size of brain lesion in stroke. Stroke. 1995;26:1393.
99 Lau LT, Yu AC. Astrocytes produce and release interleukin-1, interleukin-6, tumor necrosis factor alpha and interferon-gamma following traumatic and metabolic injury. J Neurotrauma. 2001;18:351.
100 Oh JW, Van Wagoner NJ, Rose-John S, et al. Role of IL-6 and the soluble IL-6 receptor in inhibition of VCAM-1 gene expression. J Immunol. 1998;161:4992.
101 Chan KS, Sano S, Kiguchi K, et al. Disruption of Stat3 reveals a critical role in both the initiation and the promotion stages of epithelial carcinogenesis. J Clin Invest. 2004;114:720.
102 Leong PL, Andrews GA, Johnson DE, et al. Targeted inhibition of Stat3 with a decoy oligonucleotide abrogates head and neck cancer cell growth. Proc Natl Acad Sci U S A. 2003;100:4138.
103 Xi S, Gooding WE, Grandis JR. In vivo antitumor efficacy of STAT3 blockade using a transcription factor decoy approach: implications for cancer therapy. Oncogene. 2005;24:970.
104 Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol. 2007;7:41.
105 Lang R, Patel D, Morris J, et al. Shaping gene expression in activated and resting primary macrophages by IL-10. J Immunol. 2002;169:2253.
106 O’Farrell AM, Liu YW, Moore KW, et al. IL-10 inhibits macrophage activation and proliferation by distinct signaling mechanisms: evidence for Stat3-dependent and -independent pathways. EMBO J. 1998;17:1006.
107 Kortylewski M, Kujawski M, Wang T, et al. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat Med. 2005;11:1314.
108 Kortylewski M, Yu H. Stat3 as a potential target for cancer immunotherapy. J Immunother. 2007;30:131.
109 Hussain SF, Kong L-Y, Jordan J, et al. A novel small molecule inhibitor of signal transducers and activators of transcription 3 reverses immune tolerance in malignant glioma patients. Cancer Res. 2007;67:9630.
110 Zorn E, Nelson EA, Mohseni M, et al. IL-2 regulates FOXP3 expression in human CD4+CD25+ regulatory T cells through a STAT-dependent mechanism and induces the expansion of these cells in vivo. Blood. 2006;108:1571.
111 Yoshimura A, Naka T, Kubo M. SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol. 2007;7:454.
112 Qin H, Roberts KL, Niyongere SA, et al. Molecular mechanism of lipopolysaccharide-induced SOCS-3 gene expression in macrophages and microglia. J Immunol. 2007;179:5966.
113 Starr R, Willson TA, Viney EM, et al. A family of cytokine-inducible inhibitors of signalling. Nature. 1997;387:917.
114 Pillemer BB, Xu H, Oriss TB, et al. Deficient SOCS3 expression in CD4+CD25+FoxP3+ regulatory T cells and SOCS3-mediated suppression of Treg function. Eur J Immunol. 2007;37:2082.
115 Yi-qun Z, Lorre K, de Boer M, et al. B7-blocking agents, alone or in combination with cyclosporin A, induce antigen-specific anergy of human memory T cells. J Immunol. 1997;158:4734.
116 Tirapu I, Huarte E, Guiducci C, et al. Low surface expression of B7-B1 (CD80) is an immunoescape mechanism of colon carcinoma. Cancer Res. 2006;66:2442.
117 Voigt H, Schrama D, Eggert AO, et al. CD28-mediated costimulation impacts on the differentiation of DC vaccination-induced T cell responses. Clin Exp Immunol. 2006;143:93.
118 Gabrilovich DI, Ciernik IF, Carbone DP. Dendritic cells in antitumor immune responses. I. Defective antigen presentation in tumor-bearing hosts. Cell Immunol. 1996;170:101.
119 Dangond F, Windhagen A, Groves CJ, et al. Constitutive expression of costimulatory molecules by human microglia and its relevance to CNS autoimmunity. J Neuroimmunol. 1997;76:132.
120 Satoh J, Lee YB, Kim SU. T-cell costimulatory molecules B7-1 (CD80) and B7-B2 (CD86) are expressed in human microglia but not in astrocytes in culture. Brain Res. 1995;704:92.
121 Bronte V, Apolloni E, Cabrelle A, et al. Identification of a CD11b+/Gr-1+/CD31+ myeloid progenitor capable of activating or suppressing CD8+ T cells. Blood. 2000;96:3838.
122 Jahrsdorfer B, Hartmann G, Racila E, et al. CpG DNA increases primary malignant B cell expression of costimulatory molecules and target antigens. J Leukoc Biol. 2001;69:81.
123 Vereecque R, Saudemont A, Quesnel B. Cytosine arabinoside induces costimulatory molecule expression in acute myeloid leukemia cells. Leukemia. 2004;18:1223.
124 Kaufman HL. Manipulating the local tumor microenvironment with poxviruses expressing costimulatory molecules. Ann N Y Acad Sci. 2005;1062:41.
125 Palena P, MinaZhu Z, Schlom J, et al. Human B cells that hyperexpress a triad of costimulatory molecules via avipox-vector infection: an alternative source of efficient antigen-presenting cells. Blood. 2004;104:192.
126 Bellone G, Turletti A, Artusio E, et al. Tumor-associated transforming growth factor-beta and interleukin-10 contribute to a systemic Th2 immune phenotype in pancreatic carcinoma patients. Am J Pathol. 1999;155:537.
127 Osawa E, Nakajima A, Fujisawa T, et al. Predominant T helper type 2-inflammatory responses promote murine colon cancers. Int J Cancer. 2006;118:2232.
128 Roussel E, Gingras MC, Grimm EA, et al. Predominance of a type 2 intratumoural immune response in fresh tumour-infiltrating lymphocytes from human gliomas. Clin Exp Immunol. 1996;105:344.
129 Columba-Cabezas S, Serafini B, Ambrosini E, et al. Induction of macrophage-derived chemokine/CCL22 expression in experimental autoimmune encephalomyelitis and cultured microglia: implications for disease regulation. J Neuroimmunol. 2002;130:10.
130 Delgado M, Gonzalez-Rey E, Ganea D. VIP/PACAP preferentially attract Th2 effectors through differential regulation of chemokine production by dendritic cells. FASEB J. 2004;18:1453.
131 Akasaki Y, Liu G, Chung NH, et al. Induction of a CD4+ T regulatory type 1 response by cyclooxygenase-2-overexpressing glioma. J Immunol. 2004;173:4352.
132 Ashkenazi E, Deutsch M, Tirosh R, et al. A selective impairment of the IL-2 system in lymphocytes of patients with glioblastomas: increased level of soluble IL-2R and reduced protein tyrosine phosphorylation. Neuroimmunomodulation. 1997;4:49.
133 Kolenko V, Wang Q, Riedy MC, et al. Tumor-induced suppression of T lymphocyte proliferation coincides with inhibition of Jak3 expression and IL-2 receptor signaling: role of soluble products from human renal cell carcinomas. J Immunol. 1997;159:3057.
134 Grosse-Hovest L, Wick W, Minoia R, et al. Supraagonistic, bispecific single-chain antibody purified from the serum of cloned, transgenic cows induces T-cell-mediated killing of glioblastoma cells in vitro and in vivo. Int J Cancer. 2005;117:1060.
135 Arinaga S, Akiyoshi T, Tsuji H. Augmentation of the generation of cell-mediated cytotoxicity after a single dose of Adriamycin in cancer patients. Cancer Res. 1986;46:4213.
136 Weller M, Fontana A. The failure of current immunotherapy for malignant glioma. Tumor-derived TGF-beta, T-cell apoptosis, and the immune privilege of the brain. Brain Res Brain Res Rev. 1995;21:128.
137 Siegel RM, Chan FK, Chun HJ, et al. The multifaceted role of Fas signaling in immune cell homeostasis and autoimmunity. Nat Immunol. 2000;1:469.
138 Walker PR, Saas P, Dietrich PY. Tumor expression of Fas ligand (CD95L) and the consequences. Curr Opin Immunol. 1998;10:564.
139 Ciusani E, Perego P, Carenini N, et al. Fas/CD95-mediated apoptosis in human glioblastoma cells: a target for sensitisation to topoisomerase I inhibitors. Biochem Pharmacol. 2002;63:881.
140 Dauer M, Herten J, Bauer C, et al. Chemosensitization of pancreatic carcinoma cells to enhance T cell-mediated cytotoxicity induced by tumor lysate-pulsed dendritic cells. J Immunother. 2005;28:332.
141 Wei J, DeAngulo G, Sun W, et al. Topotecan enhances immune clearance of gliomas. Cancer Immunol Immunother. 2009;58:259-270.
142 Mizutani Y, Okada Y, Yoshida O, et al. Doxorubicin sensitizes human bladder carcinoma cells to Fas-mediated cytotoxicity. Cancer. 1997;79:1180.
143 Mizutani Y, Yoshida O, Miki T. Adriamycin-mediated potentiation of cytotoxicity against freshly isolated bladder cancer cells by autologous non-activated peripheral blood lymphocytes and tumor infiltrating lymphocytes. J Urol. 1999;162:2170.
144 Uslu R, Borsellino N, Frost P, et al. Chemosensitization of human prostate carcinoma cell lines to anti-Fas-mediated cytotoxicity and apoptosis. Clin Cancer Res. 1997;3:963.
145 Mizutani Y, Wu XX, Yoshida O, et al. Chemoimmunosensitization of the T24 human bladder cancer line to Fas-mediated cytotoxicity and apoptosis by cisplatin and 5-fluorouracil. Oncol Rep. 1999;6:979.
146 Roth W, Wagenknecht B, Grimmel C, et al. Taxol-mediated augmentation of CD95 ligand-induced apoptosis of human malignant glioma cells: association with bcl-2 phosphorylation but neither activation of p53 nor G2/M cell cycle arrest. Br J Cancer. 1998;77:404.
147 Winter S, Roth W, Dichgans J, et al. Synergy of CD95 ligand and teniposide: no role of cleavable complex formation and enhanced CD95 expression. Eur J Pharmacol. 1998;341:323.
148 Chahlavi A, Rayman P, Richmond AL, et al. Glioblastomas induce T-lymphocyte death by two distinct pathways involving gangliosides and CD70. Cancer Res. 2005;65:5428.
149 Friese MA, Platten M, Lutz SZ, et al. MICA/NKG2D-mediated immunogene therapy of experimental gliomas. Cancer Res. 2003;63:8996.
150 Wischhusen J, Friese MA, Mittelbronn M, et al. HLA-E protects glioma cells from NKG2D-mediated immune responses in vitro: implications for immune escape in vivo. J Neuropathol Exp Neurol. 2005;64:523.
150a Heimberger AB, Sampson JH. Immunotherapy coming of age: what will it take to make it standard of care for glioblastoma? Neuro Oncol. 2011;13:3-13.
151 Herrlinger U, Kram CM, Johnson K, et al. Vaccination for experimental gliomas using GM-CSF–transduced glioma cells. Cancer Gene Therapy. 1997;4:345-352.
152 Glick RP, Lichtor T, Mogharbel A, et al. Intracerebral versus subcutaneous immunization with allogenic fibroblasts genetically engineered to secrete IL-2 in the treatment of CNS glioma and melanoma. Neurosurgery. 1997;41:898-907.
153 Glick RP, Lichtor T, de Zoeten E, et al. Prolongation of survival of mice with intracerebral glioma treated with semi-allogeneic fibroblasts secreting IL-2. Neurosurgery. 1999;45:867-874.
154 Fakhrai H, Shawler DL, Gjerset R, et al. Cytokine gene therapy with interleukin-2-transduced fibroblasts: effects of IL-2 dose on anti-tumor immunity. Hum Gene Ther. 1995;6:591.
155 Kim TS, Russell SJ, Collins MK, et al. Immunity to B16 melanoma in mice immunized with IL-2-secreting allogeneic mouse fibroblasts expressing melanoma-associated antigens. Int J Cancer. 1992;51:283.
156 Lichtor T, Glick RP, Kim TS, et al. Prolonged survival of mice with glioma injected intracerebrally with double cytokine-secreting cells. J Neurosurg. 1995;83:1038.
157 Okada H, Villa L, Attanucci J, et al. Cytokine gene therapy of gliomas: effective induction of therapeutic immunity to intracranial tumors by peripheral immunization with interleukin-4 transduced glioma cells. Gene Ther. 2001;8:1157.
158 Tepper RI, Pattengale PK, Leder P. Murine interleukin-4 displays potent anti-tumor activity in vivo. Cell. 1989;57:503.
159 Watanabe Y, Kuribayashi K, Miyatake S, et al. Exogenous expression of mouse interferon gamma cDNA in mouse neuroblastoma C1300 cells results in reduced tumorigenicity by augmented anti-tumor immunity. Proc Natl Acad Sci U S A. 1989;86:9456.
160 Yu JS, Burwick JA, Dranoff G, et al. Gene therapy for metastatic brain tumors by vaccination with granulocyte-macrophage colony-stimulating factor-transduced tumor cells. Hum Gene Ther. 1997;8:1065.
161 Tahara H, Zeh HJ3rd, Storkus WJ, et al. Fibroblasts genetically engineered to secrete interleukin 12 can suppress tumor growth and induce antitumor immunity to a murine melanoma in vivo. Cancer Res. 1994;54:182.
162 Lichtor T, Glick RP, Tarlock K, et al. Application of interleukin-2-secreting syngeneic/allogeneic fibroblasts in the treatment of primary and metastatic brain tumors. Cancer Gene Ther. 2002;9:464.
163 Lichtor T, Glick RP, Lin H, et al. Intratumoral injection of IL-secreting syngeneic/allogeneic fibroblasts transfected with DNA from breast cancer cells prolongs the survival of mice with intracerebral breast cancer. Cancer Gene Ther. 2005;12:708.
164 Lichtor T, Glick RP, O-Sullivan I, et al. DNA-based vaccine for treatment of intracerebral neoplasms. Gene Ther Mol Biol. 2004;8:395.
165 Kim TS, Cohen EP. Interleukin-2-secreting mouse fibroblasts transfected with genomic DNA from murine melanoma cells prolong the survival of mice with melanoma. Cancer Res. 1994;54:2531.
166 Fakhrai H, Mantil J, Gramatikova S, et al. Gene therapy of human gliomas with TGF-β2 antisense gene modified autologous tumor cells. A phase I trial. Proc Am Assoc Cancer Res. 2000;41:543.
167 Borden EC, Sondel PM. Lymphokines and cytokines as cancer treatment. Immunotherapy realized. Cancer. 1990;65:800.
168 Gabrilove JL, Jakubowski A. Hematopoietic growth factors: biology and clinical application. J Natl Cancer Inst Monogr. 1990;10:73-77.
169 Kelso A. Cytokines: structure, function and synthesis. Curr Opin Immunol. 1989;2:215.
170 Lotze MT, Chang AE, Seipp CA, et al. High-dose recombinant interleukin 2 in the treatment of patients with disseminated cancer. Responses, treatment-related morbidity, and histologic findings. JAMA. 1986;256:3117.
171 Pizza G, Viza D, De Vinci C, et al. Intra-lymphatic administration of interleukin-2 (IL-2) in cancer patients: a pilot study. Lymphokine Res. 1988;7:45.
172 Rosenberg SA, Lotze MT, Mule JJ. NIH conference. New approaches to the immunotherapy of cancer using interleukin-2. Ann Intern Med. 1988;108:853.
173 Refaeli Y, Van Parijs L, London CA, et al. Biochemical mechanisms of IL-2-regulated Fas-mediated T cell apoptosis. Immunity. 1998;8:615.
174 Sarna G, Collins J, Figlin R, et al. A pilot study of intralymphatic interleukin-2. II. Clinical and biological effects. J Biol Response Modifiers. 1990;9:81.
175 Bubenik J, Voitenok NN, Kieler J, et al. Local administration of cells containing an inserted IL-2 gene and producing IL-2 inhibits growth of human tumours in nu/nu mice. Immunol Lett. 1988;19:279.
176 Gandolfi L, Solmi L, Pizza GC, et al. Intratumoral echo-guided injection of interleukin-2 and lymphokine-activated killer cells in hepatocellular carcinoma. Hepatogastroenterology. 1989;36:352.
177 Tjuvajev J, Gansbacher B, Desai R, et al. RG-2 glioma growth attenuation and severe brain edema caused by local production of interleukin-2 and interferon-γ. Cancer Res. 1995;55:1902-1910.
178 Okada H, Pollack IF, Lieberman F, et al. Gene therapy of malignant gliomas: a pilot study of vaccination with irradiated autologous glioma and dendritic cells admixed with IL-4 transduced fibroblasts to elicit an immune response. Hum Gene Ther. 2001;12:575.
179 Okada H, Pollack IF, Lotze MT, et al. Gene therapy of malignant gliomas: a phase I study of IL-4-HSV-TK gene-modified autologous tumor to elicit an immune response. Hum Gene Ther. 2000;11:637.
180 Watts RG, Wright JL, Atkinson LL, et al. Histopathological and blood-brain barrier changes in rats induced by an intracerebral injection of human recombinant interleukin 2. Neurosurgery. 1989;25:202.
181 Zhang RD, Price JE, Fujimaki T, et al. Differential permeability of the blood-brain barrier in experimental brain metastases produced by human neoplasms implanted into nude mice. Am J Pathol. 1992;141:1115.
182 Fearon ER, Pardoll DM, Itaya T, et al. Interleukin-2 production by tumor cells bypasses T helper function in the generation of an antitumor response. Cell. 1990;60:397.
183 Gansbacher B, Zier K, Daniels B, et al. Interleukin 2 gene transfer into tumor cells abrogates tumorigenicity and induces protective immunity. J Exp Med. 1990;172:1217.
184 Ehtesham M, Kabos P, Kabosova A, et al. The use of interleukin 12–secreting neural stem cells for the treatment of intracranial glioma. Cancer Res. 2002;62:5657.
185 Glick RP, Lichtor T, Panchal R, et al. Treatment with allogeneic interleukin-2 secreting fibroblasts protects against the development of an intracranial tumor and prolongs survival. J Neurooncol. 2003;64:139-146.
186 Griffitt W, Glick RP, Lichtor T, Cohen EP. Survival and toxicity of an allogeneic cytokine-secreting fibroblast vaccine in the CNS. Neurosurgery. 1998;42:335-340.
187 Giezeman-Smits KM, Okada H, Brissette-Storkus CS, et al. Cytokine gene therapy of gliomas: induction of reactive CD4+ T cells by interleukin-4-transfected 9L gliosarcoma is essential for protective immunity. Cancer Res. 2000;60:2449.
188 Natsume A, Mizuno M, Ryuke Y, et al. Antitumor effect and cellular immunity activation by murine interferon-beta gene transfer against intracerebral glioma in mouse. Gene Ther. 1999;6:1626.
189 Sampson JH, Ashley DM, Archer GE, et al. Characterization of a spontaneous murine astrocytoma and abrogation of its tumorigenicity by cytokine secretion. Neurosurgery. 1997;41:1365.
190 Sobol RE, Fakhrai H, Shawler D, et al. Interleukin-2 gene therapy in a patient with glioblastoma. Gene Ther. 1995;2:164.
191 Birchfield GR, Rodgers GM, Girodias KW, et al. Hypoprothrombinemia associated with interleukin-2 therapy: correction with vitamin K. J Immunother. 1992;11:71.
192 Kim H, Rosenberg SA, Steinberg SM, et al. A randomized double-blinded comparison of the antiemetic efficacy of ondansetron and droperidol in patients receiving high-dose interleukin-2. J Immunother Emphasis Tumor Immunol. 1994;16:60.
193 Ashley DM, Faiola B, Nair S, et al. Bone marrow-generated dendritic cells pulsed with tumor extracts or tumor RNA induce antitumor immunity against central nervous system tumors. J Exp Med. 1997;186:1177.
194 Heimberger AB, Archer GE, Crotty LE, et al. Dendritic cells pulsed with a tumor-specific peptide induce long-lasting immunity and are effective against murine intracerebral melanoma. Neurosurgery. 2002;50:158.
195 Kruse C, Cepeda L, Owens B, et al. Treatment of recurrent glioma with intracavitary alloreactive cytotoxic lymphocytes and interleukin-2. Cancer Immunol Immunother. 1997;45:77-87.
196 Barba D, Saris SC, Holder C, et al. Intratumoral LAK cell and interleukin-2 therapy of human gliomas. J Neurosurg. 1989;70:175-182.
197 Yu J, Wheeler C, Zeltzer P, et al. Vaccination of malignant glioma patients with peptide-pulsed dendritic cells elicits systemic cytotoxicity and intracranial T-cell infiltration. Cancer Res. 2001;61:842.
198 Yu J, Liu G, Ying H, et al. Vaccination with tumor lysate–pulsed dendritic cells elicits antigen-specific, cytotoxic T-cells in patients with malignant glioma. Cancer Res. 2004;64:4973.
199 Sampson JH, Archer GE, Mitchell DA, et al. An epidermal growth factor receptor variant III-targeted vaccine is safe and immunogenic in patients with glioblastoma multiforme. Mol Cancer Ther. 2009;8:2773-2779.
200 Wheeler CJ, Das A, Liu G, et al. Clinical responsiveness of glioblastoma multiforme to chemotherapy after vaccination. Clin Cancer Res. 2004;10:5316.
201 Reardon DA, Akabani G, Coleman RE, et al. Phase II trial of murine131I-labeled antitenascin monoclonal antibody 81C6 administered into surgically created resection cavities of patients with newly diagnosed malignant gliomas. J Clin Oncol. 2002;20:1389.
202 Aldape K, Ballman K, Furth A, et al. Immunohistochemical detection of EGFRvIII in high malignancy grade astrocytomas and evaluation of prognostic significance. J Neuropathol Exp Neurol. 2004;63:700.
203 Hall W. Immunotoxin therapy. Neurosurgery Clin N Am. 1996;7:537-546.
204 Laske D, Youle R, Oldfield E. Tumor regression with regional distribution of the targeted toxin TF-CRM 107 in patients with malignant brain tumors. Nat Med. 1994;3:1362-1368.
205 Sampson JH, Akabani G, Archer GE, et al. Intracerebral infusion of an EGFR-targeted toxin in recurrent malignant brain tumors. Neuro Oncol. 2008;10:320-329.
206 Weber F, Asher A, Bucholz R, et al. Safety, tolerability, and tumor response of IL-4-Pseudomona exotoxin (NBI-3001) in patients with recurrent malignant glioma. J Neurooncol. 2003;64:125-137.
207 Debinski W, Obiri N, Powers S, et al. Human glioma cells overexpress receptors for interleukin 13 and are extremely sensitive to a novel chimeric protein composed of IL-13 and pseudomonas exotoxin. Clin Cancer Res. 1995;1:1253-1258.
208 Kunwar S, Prados MD, Chang SM, et al. Direct intracerebral delivery of cintredekin besudotox (IL13-PE38QQR) in recurrent malignant glioma: a report by the Cintredekin Besudotox Intraparenchymal Study Group. J Clin Oncol. 2007;25:837-844.
209 Bobo R, Laske D, Akbasak A, et al. Convection-enhanced delivery of macromolecules into the brain. Pro Nat Acad Sci U S A. 1994;91:2076-2080.
210 Kleihues P, Burger PC, Plate KH, et al. Astrocytic tumors. In: Kleihues P, Cavenee WJ, editors. Pathology and Genetics: Tumors of the Nervous System, Vol 1. Lyon, France: International Agency for Research on Cancer; 1997.
211 Nestle FO, Alijagic S, Gilliet M, et al. Vaccination of melanoma patients with peptide- or tumor lysate-pulsed dendritic cells. Nat Med. 1998;4:328.
212 Tighe H, Corr M, Roman M, et al. Gene vaccination: plasmid DNA is more than just a blueprint. Immunol Today. 1998;19:89.
213 Kikuchi T, Akasaki Y, Irie M, et al. Results of a phase I clinical trial of vaccination of glioma patients with fusions of dendritic and glioma cells. Cancer Immunol Immunother. 2001;50:337.
214 Yamanaka R, Tsuchiya N, Yajima N, et al. Induction of an antitumor immunological response by an intratumoral injection of dendritic cells pulsed with genetically engineered Semliki Forest virus to produce interleukin-18 combined with the systemic administration of interleukin-12. J Neurosurg. 2003;99:746.
215 Anichini A, Mortarini R, Maccalli C, et al. Cytotoxic T cells directed to tumor antigens not expressed on normal melanocytes dominate HLA-A2.1-restricted immune repertoire to melanoma. J Immunol. 1996;156:208.
216 Heimberger AB, Hlatky R, Suki D, et al. Prognostic effect of epidermal growth factor receptor and EGFRvIII in glioblastoma multiforme patients. Clin Cancer Res. 2005;11:1462.
217 Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987.
217a Parsa, AT. Clinical Trials PDQ®National Cancer Institute GP96 heat shock protein-peptide complex vaccine in treating patients with recurrent or progressive. Unpublished data.
218 Emens LA, Reilly RT, Jaffee EM. Breast cancer vaccines: maximizing cancer treatment by tapping into host immunity. Endocr Relat Cancer. 2005;12:1.
219 Rosenberg SA, Yang JC, Schwartzentruber DJ, et al. Impact of cytokine administration on the generation of antitumor reactivity in patients with metastatic melanoma receiving a peptide vaccine. J Immunol. 1999;163:1690.
220 Rosenberg SA, Yang JC, Schwartzentruber DJ, et al. Prospective randomized trial of the treatment of patients with metastatic melanoma using chemotherapy with cisplatin, dacarbazine, and tamoxifen alone or in combination with interleukin-2 and interferon alfa-2b. J Clin Oncol. 1999;17:968.
221 Ahmadzadeh M, Rosenberg SA. IL-2 administration increases CD4+ CD25(hi) Foxp3+ regulatory T cells in cancer patients. Blood. 2006;107:2409.
222 Liau LM, Prins RM, Kiertscher SM, et al. Dendritic cell vaccination in glioblastoma patients induces systemic and intracranial T-cell responses modulated by the local central nervous system tumor microenvironment. Clin Cancer Res. 2005;11:5515.
223 Heimberger AB, Abou-Ghazal M, Reina-Ortiz C, et al. Incidence and prognostic impact of FoxP3+ regulartory T cells in human gliomas. Clin Cancer Res. 2008;14:5166.
224 Sampson JH, Aldape KD, Gilbert MR, et al. Temozolomide as a vaccine adjuvant in GBM. J Clin Oncol. 2007;25 (suppl):2020.
225 Spagnolo A, Glick RP, Lin H, et al. Prolonged survival of mice with an established intracerebral glioma receiving combined treatment with PPAR-γ thiazolidinedione agonists and IL-2 secreting syngeneic/allogeneic fibroblasts. J Neurosurg. 2007;106:299-305.
[/level-membership-for-neurosurgery-category][not-level-membership-for-neurosurgery-category]
CHAPTER 97 Brain Tumor Immunology and Immunotherapy
An Immune Primer
The function of the immune system is to protect the body.1 Immunotherapy involves the administration of an agent to an individual that stimulates the immune system to react against something foreign or harmful, such as a tumor or an infection. The end result of a vaccine is to develop protective immunity against the foreign agent or tumor. This defensive function is performed by leukocytes (white blood cells) and a number of accessory cells distributed throughout the body. Lymphocytes are the key cells controlling the immune response. They specifically recognize foreign material and distinguish it from the body’s “self” components. Immunity may be cell mediated (T cells, natural killer [NK] cells, and phagocytes) or humoral (B cells, antibody, complement). When stimulated, cytokines are a group of molecules, other than antibodies, produced by lymphocytes that are involved in regulating the immune system. They include the interleukins, the interferons, tumor necrosis factor (TNF), and colony-stimulating factor (CSF). There are two main types of lymphocytes: B cells, which produce antibodies; and T cells, which have a number of functions including helping B cells to make antibodies (CD4 helper T cells, TH2) or helping cytotoxic T-cell responses (CD4 helper T cells, TH1); recognizing and destroying virus-infected cells (CD8 effector T cells); and controlling the level and quality of the immune response (regulatory T cells).
APCs are a group of cells that are capable of taking up antigens, partially degrading them, and presenting them to T cells in a form they can recognize. Although B cells recognize antigen in its native form, T cells only recognize antigenic peptide derivatives of complex antigens that have become associated with major histocompatibility complex (MHC) molecules. Thus, MHC molecules present antigen (i.e., peptides) to T cells. MHC class I molecules are found on all nucleated cells and platelets. MHC class II molecules (Ia antigens) required for helping B cells or making antibodies are expressed on B cells, macrophages, monocytes, APCs, and some T cells. CD8 cells (cytotoxic T lymphocytes [CTLs], killer) are class I restricted, meaning they only recognize antigen presented in the context of MHC class I molecules, whereas CD4 (helper T) cells are MHC class II restricted (Fig. 97-1). Antigens synthesized within a cell, such as viral polypeptides, associate preferentially with MHC class I molecules and present antigen directly to CD8 cells (“direct pathway”). In contrast, antigens that are taken up by an APC and partially degraded (processed) and returned to the cell surface associated with MHC class II molecules are recognized by CD4 cells (“indirect pathway”) (see Fig. 97-1). Syngeneic implies the same MHC as the organism or patient, and allogeneic implies a different MHC; hence the term allograft, which is rejection due to differences in MHC.
T cells in the CNS of healthy humans are a rare finding. However, during inflammatory responses, T cells are evident within the CNS. T cells require activation before entry into the CNS,2 but antigen specificity is not necessary for entry. T-cell infiltrates are commonly identified within human gliomas,3 and multiple studies have attempted to correlate the intensity of infiltration with survival,3–5 but this has not been consistently seen.6 One should bear in mind that these types of immunohistochemical assays do not take into account the functional activity of these cells or the contribution of inhibitor factors such as T regs. Thus, although these T cells have been activated in the systemic circulation, their functional activity has likely become impaired on entry into the tumor microenvironment,7 and it is not surprising that their presence in the tumor is not a definitive prognostic marker.
One concern about the use of immunotherapy in glioma patients is the induction of fatal autoimmunity. However, in previous immunotherapy trials of humans with brain tumors, there were only two possible cases of autoimmunity,8,9 so this remains a consideration but is likely related to the use of strong adjuvants.10 The rationale for selecting a tumor-specific antigen approach in immunotherapy is based on the facts that the targeting antigen preparation does not contain CNS antigens capable of inducing an autoimmune response and that the tumor-specific immune response can be monitored. However, the disadvantage of using a tumor-specific antigen is secondary to the heterogeneity of gliomas—one single antigen is unlikely to produce a durable response because a clonal negative population is likely to arise.11
It is widely recognized that tumors evade the host immune response through a number of mechanisms, including the elaboration of immunosuppressive cytokines, the production of immunosuppressive immune cells (T regs), and signal transduction disruption (Fig. 97-2).12
Mechanisms of Immune Suppression and Therapeutic Agents
Overview: Mechanisms of Immune Evasion
The secretion of immunosuppressive factors by the tumors themselves has recently been identified as a further target for immunotherapy. In particular, interleukin-10 (IL-10) and transforming growth factor-β (TGF-β) are two examples of such immunosuppressive cytokines secreted by tumor cells. Furthermore, there is increased expression of FasL and galectin by glioma that triggers T-cell depletion and T-cell apoptosis.13
Recently, a subset of regulatory T cells (T regs) have been identified in the local tumor environment and in the draining lymph nodes of systemic tumors, and they are elevated in the CD4 compartment of the systemic blood circulation. These cells coexpress CD4 or CD25, or both, and display potent immunosuppressive T-cell activity.14 In vaccines used for tumors outside the CNS, monoclonal antibody inhibition of these T regs has been associated with enhanced antitumor immunogenicity (details provided later). The inhibitory effects of these suppressor cells may also be mediated by cytokines. Interestingly, IL-2–mediated immune regulation involves not only the stimulation of effector cells but also the regulation of T regs.15
Regulatory T Cells
T regs inhibit T-cell activation and effector function by downregulating IL-2 production, inhibiting interferon-γ (IFN- γ) production, and increasing TH2 cytokine production,16 thereby inhibiting the expression of MHC class II molecules, CD40, CD80, and CD86 and suppressing antigen presentation by monocytes and macrophages.17,18 The presence of T reg–mediated suppression has been documented in a variety of human cancers.19 Several studies on immunogenic murine tumors have shown that adoptively transferred T regs are responsible for inhibition of tumor-reactive effector T cells and that the elimination of T regs enhances antitumor immunity.16 Within glioblastoma multiforme (GBM) patients, although reduced in absolute number, T regs comprise a disproportionately large fraction of the patient’s peripheral CD4 T-cell compartment. This increase corresponds with a decrease in T-cell effector functions. Furthermore, in vitro depletion of peripheral blood T reg cells results in successful reversal of effector T-cell function, including increased proliferation and a switch to a TH1 (IL-2–positive, tumor necrosis factor-α [TNF-α]–positive, IFN-γ–positive) cytokine profile. Within murine tumor challenge models of established gliomas, in vivo depletion of CD4+ and CD25+ cells resulted in significant long-term survival.20,21
Within the glioma microenvironment, the host-generated effector T cells can be critically suppressed or overwhelmed by the T reg cells. Tissues from glioma patients obtained after surgical resection have been dissociated and stained for the CD8+ and CD4+ subsets. The tumor-infiltrating CD8+ T cells were phenotypically CD8+ and CD25−, indicating that these cells were not activated or proliferating. CD4+ T cells were more numerous than CD8+ T cells within glioma tissue and the majority of CD4+ T cells were T regs, as evident by positive intracellular staining of FoxP3, a transcription factor that is elevated only in T regs and not in activated effector CD4+ T cells.22 These data indicate that T regs not only can inhibit initial systemic immune activation but also can prevent the effector responses in the tumor microenvironment. Of note, lower grade gliomas have only rare T regs, and oligodendrogliomas none at all. Even within the GBM patients’ specimens, the presence of T reg infiltration is highly variable and not an independent predictor of survival, emphasizing the redundancy of immunosuppressive pathways. Human T regs have been shown to accumulate in tumors and in ascites induced by the tumor and macrophages by the chemokine CCL22, which mediates trafficking of T regs to the tumor.19 Similarly, gliomas elaborate the chemokine CCL2, which induced the intratumoral migration of T regs.23
Regulatory T-Cell Inhibition by Chemotherapeutic Agents
Overcoming T reg immunosuppression is an ongoing immune therapeutic approach that can be achieved through a variety of approaches, including the use of denileukin diftitox (Ontak),24 cyclophosamide,25 or signal transducer and activator of transcription type 3 (STAT-3) blockade agents,26 or by inhibiting the T reg trafficking (i.e., inhibition of CCL2) with temozolomide.27 Inactivation of T regs with an anti-CD25 antibody in murine glioma models has been shown to enhance vaccination-induced antitumor immune responses and to result in the eradication of intracerebral astrocytomas without induction of autoimmunity.28 Similar results were also obtained in this murine model system with systemic cytotoxic T-lymphocyte–associated protein type 4 (CTLA-4) blockade. The CTLA-4 blockade reversed the CD4+ T-cell deficit, similarly seen in malignant glioma patients, and normalized the ratio of T regs in tumor-bearing mice.29 Although eliminating the suppression of endogenous antitumor immune responses through the elimination of regulatory T cells may enhance tumor immune clearance, there is a potential risk for inducing autoimmunity; however, that was not found in the murine model. It is likely that the induction of TH17 responses, and not necessarily the inhibition of T regs, is the primary mechanism of inducing CNS autoimmunity.30,31 In patients with metastatic renal carcinoma, Ontak specifically depleted the T regs without inducing toxicity and significantly improved the stimulation of tumor-specific T-cell responses when those patients were stimulated with RNA-transfected DCs.32 Without vaccination, Ontak has been shown to be efficacious as a single agent in relapsed and refractory B-cell non-Hodgkin’s lymphoma33 and in cutaneous T-cell lymphoma.34 However, Ontak was not particularly effective in clearing the T reg cells in melanoma patients.35 No published studies to date have evaluated this approach in malignant glioma patients. Another set of potential T reg modulation includes agents that block or disrupt Notch signaling. Notch signaling has also been identified as playing a role in regulating the responses of T cells and can affect the differentiation of CD4+ T cells into T regs.36 However, it is unclear which members of the Notch family need to be blocked, and inhibitors are currently under development.
Lymphodepletion to augment immunologic responses has been described in both murine model systems37,38 and human cancer patients.39 These enhanced antitumor responses after lymphodepletion may be secondary to the removal of competition at the surface of APCs,40 enhanced availability of cytokines that augment T-cell activity (e.g., IL-7 and IL-15),41 or depletion of immune inhibitory T regs.42 Cyclophosphamide (CTX), an alkylating agent with a therapeutic effect against tumors at high doses, has preferential effects on inhibiting T regs at lower doses. CTX can abolish the function of CD4+, CD25+, FoxP3+ T cells and enhance cytotoxic T-cell responses.43 Pretreatment with CTX before antitumor vaccination results in the activation of CD8 tumor-specific T cells.44 When CTX is administered at subtumoricidal doses, there is improved immune response and tumor eradication of large established murine tumors (sarcoma) when treated in combination with IL-12. This combination enhances CD4, CD8 T cells and macrophage infiltration within the tumor and skews the immune responses to the TH1 phenotype.45 Similar findings have been made in the murine system treating established melanoma with IL-2, IL-1, and CTX.46 In addition to its induction of immunity to new antigens, CTX can also overcome tolerance. For example, the administration of CTX to mice bearing an established plasmacytoma resulted in the cure of 92% of mice, and further studies demonstrated that the cured mice rejected a subsequent tumor challenge.47 The mechanism appeared to involve both the generation of CD8+ cytotoxic T cells and the upregulation of B7-1 (costimulatory molecule) on tumor cells.48
Multiple clinical trials have demonstrated enhanced immune responses and improved clinical efficacy when CTX was administered before immunotherapy.49,50 Administration of CTX to patients with advanced melanoma and colorectal cancer resulted in enhanced delayed-type hypersensitivity (DTH) reactions and the development of antibodies.51 When CTX was administered to augment autologous melanoma vaccine in patients with metastatic melanoma, the inclusion of CTX resulted in enhanced DTH responses.52 Similar potentiation of immune responses was seen in metastatic breast cancer and renal cell carcinoma. CTX has also been used in a phase I clinical trial of glioma patients and demonstrated some evidence of clinical efficacy. Plautz and colleagues53 treated newly diagnosed glioma patients with a single dose of CTX before the administration of adoptively transferred T cells harvested from the patients’ lymph nodes. This study was conducted before our current understanding of the influence of CTX on T regs, and thus the contribution of the CTX to the clinical efficacy is unknown.
Nevertheless, when combining the different forms of treatment, all aspects of treatment must be taken into account. Different dosing schedules and methods of vaccination can abrogate the desired therapeutic response. The use of low-dose doxorubicin after vaccination with a granulocyte-macrophage colony-stimulating factor (GM-CSF)–secreting cancer cell vaccine in a CT26 colon carcinoma murine model showed a significant increase in CD8+ T-cell response compared with the use of vaccine alone. Yet, when low-dose CTX was administered after or concurrently with a GM-CSF–secreting cancer cell vaccine, the desired immune response was inhibited, and the result was equivalent to that seen in mice treated only with chemotherapy.54 Thus, with CTX administered before vaccination, the reduction of suppressive immunity increased the immunotherapy-mediated tumor clearance. Similar results were also obtained with paclitaxel.55
Another agent that is capable of suppressing T regs is temozolomide (TMZ), the standard of care for GBM. TMZ is an alkylating agent that causes cell death by inducing G2/M-phase cell cycle arrest and likely autophagy without apoptosis.56 TMZ can inhibit the proliferation of lymphocytes, but it also depletes T regs27,57 and the trafficking of T regs into the glioma microenvironment.23 This mechanism may prove beneficial in combination with vaccine therapy and was recently investigated in a phase II clinical trial (ACT II) in combination with PEP-3-KLH vaccination.
In murine models of established intracerebral tumors, the combination of IL-2 immunotherapy and intratumoral administration of bischloroethylnitrosourea (BCNU) polymers enhanced survival.58 The investigators suggested that the cytotoxic agents might increase the number of tumor peptide antigens released or abrogate tumor-derived T-cell suppressor factors, or both. When mice with established tumors (lymphoma) were treated with BCNU, they rejected secondary tumor challenges, indicating the induction of immunologic memory. Furthermore, mice treated with BCNU demonstrated high cytotoxic activity against tumor cells and low T-cell suppressor activity. Thus, although BCNU was directly tumoricidal, it is also possible that the BCNU inhibited the T regs, resulting in antitumor immunity.59 The combination of BCNU with the newer efficacious immunotherapies has not been explored in animal models or clinical trials.
Immuno suppressive Cytokines
Human glioblastoma cells secrete a variety of factors, such as prostaglandin E (PGE), IL-10, and TGF-β2 that suppress multiple immune functions. For example, TGF-β2 inhibits cytotoxic responses of T cells against tumor targets, PGE2 regulates the generation of immune responses, IL-10 downregulates MHC class II expression on monocytes and suppresses T-cell proliferation,60–62 and vascular endothelial growth factor (VEGF) inhibits the maturation of DCs and subsequently downregulates MHC class II molecules. Suppression of TGF-β with an antisense TGF-β has been shown to be efficacious in 9L and C6 glioma intracerebral models.53,63 Other attempts at TGF-β inhibition in murine models have included SD-208, a TGF-βRI kinase inhibitor, which slightly increased median survival,64 and Tranilast, a TGF-β1 antagonist, which resulted in a 50% reduction in intracranial tumor volume.65 A phase I/II clinical trial using an antisense oligodeoxynucleotide (AP 12009) targeted to suppress TGF-β2 in high-grade glioma patients demonstrated an increase in median survival after recurrence that exceeded the current data for chemotherapy.66 The success of this type of approach is dependent on the dependency of the glioma on this particular mechanism of immune suppression and is compounded by the fact that gliomas usually express a variety of immunosuppressive cytokines; the blockade of any one cytokine may not be expected to significantly impact the overall immunosuppressive milieu.
The taxanes, paclitaxel and docetaxel, bind to ß-tubulin, stabilizing microtubules, and resulting in G2/M-phase mitotic arrest. Paclitaxel can diminish the cytotoxicity of NK and T cells by interfering with IL-2–mediated activation. However, the best-known immunomodulatory effect of paclitaxel is its ability to trigger macrophages to secrete a variety of proinflammatory factors, such as IL-1ß, GM-CSF, tumor necrosis factor-α (TNF-α), and IL-12. This lipopolysaccharide-like activity enables paclitaxel, in the presence of a priming signal, to induce the tumoricidal activity of macrophages.67 To date, there have been no reports of combining paclitaxel treatment with cancer immunotherapy.
Clearly, further studies investigating the cytokine profile before chemotherapy are warranted to enhance our understanding of the endogenous immune response. For example, in patients with breast cancer that were evaluated before treatment with chemotherapy, there was a significant decrease in immune cells such as NK and LAK cells and immunostimulatory cytokines such as IL-2, GM-CSF, and interferon compared with healthy individuals. After the administration of docetaxel and paclitaxel, there was a significant increase in the number of immune cells and a decrease in PGE2 and TNF.68 Doxorubicin has also been shown to have immunomodulating effects, including increasing the activity of monocytes and increasing the secretion of IL-1 and IL-2.69
Immune Inhibitory Molecules
To initiate an immune response, T cells must receive two signals: one through the T-cell receptor (TCR), which recognizes antigen presented within the context of MHC; and the second through the costimulatory receptor CD28, which recognizes the costimulatory molecules CD80 and CD86 expressed on the surface of the APC. In response to antigen-specific stimulation, activated T cells produce IL-2 and proliferate. B7-H1 is a negative costimulatory molecule that inhibits CD4+ and CD8+ T-cell activation, can induce T-cell apoptosis,70,71 and is upregulated within the tumor microenvironment72 and specifically in gliomas.70 Blockade of B7-H1 with a monoclonal antibody can activate T cells, which are then able to inhibit tumor.73 This represents a novel mechanism by which glioma cells evade immune recognition and destruction. Blockade of B7-H1 with antibodies in combination with adoptive T-cell transfer has been shown to enhance cancer clearance73 and enhance myeloid DC-mediated immunotherapy72; however, whether sufficient quantities of these types of antibodies can be achieved in the CNS remains to be seen. T-cell anergy can also be induced if the inhibitory molecule CTLA-4 is present on the gliomas.74 CTLA-4 blockade has had some success in promoting antitumor T-cell responses in both preclinical and clinical settings,29,75 but it is not clear whether this approach would be successful against primary brain tumors or CNS metastasis.
HLA-G is a nonclassic MHC molecule with limited tissue distribution that has an immune regulatory function. Soluble HLA-G (sHLA-G) has been identified in the plasma of patients with melanoma, glioma, breast cancer, or ovarian cancer. sHLA-G can inhibit the functions of T and NK cells at high concentration76; thus, at levels within the tumor microenvironment, it would be anticipated that there is reduced immunologic responsiveness. Gene transfer of HLA-G1 or HLA-G5 into HLA-G–negative glioma cells (U87-MG) rendered these cells highly resistant to direct cytolytic clearance, inhibited T-cell proliferation, and prevented efficient priming of cytotoxic T cells. The inhibitory effects of HLA-G were directed against CD8 and CD4 T cells, but appeared to be NK-cell independent.77 Only a few HLA-G–positive cells within the tumor microenvironment are necessary to exert significant immune inhibitory effects77; thus, the inhibition of HLA-G blockade would need to be comprehensive. However, the frequency of expression of HLA-G and B7-H1 has not been assessed across glioma grades or types on tissue arrays; thus, the universality of this type of immune evasion is not currently known.
Phenomenon of Antigen-Loss Variants in Gliomas
Although tumor specificity has the advantage of not inducing potentially fatal autoimmunity, the intrinsic antigenic heterogeneity will prevent clearance of all cancer cells and the development of antigen-loss variants, especially for those antigens whose expression is not required by tumor cells for the maintenance of a transformed phenotype. The propagation of such antigen-loss variants is secondary to epitope immunodominance, that is, the preferential immunodetection of one or a few epitopes among many expressed on a given target.78 The parental tumor cells carrying the immune dominant epitope can be eliminated, and the formerly immune recessive epitopes then become dominant, allowing unrestricted tumor growth. It is well documented that tumor antigen expression is heterogeneous, even within the same tumor,79 and antigen loss has been observed with a variety of immune therapeutic approaches in various cancers.80,81
The epidermal growth factor receptor (EGFR) is often amplified and structurally rearranged in malignant gliomas, with the most common mutation being EGFRvIII. Treatment in a murine model with a vaccine consisting of a peptide encompassing the tumor-specific mutated segment of EGFRvIII (PEP-3) conjugated to keyhole limpet hemocyanin (KLH [PEP-3-KLH]) resulted in the loss of antigen expression. Specifically, EGFRvIII expression in mice that failed to respond to the PEP-3-KLH vaccination was lost in 80% of relapsing tumors, indicating that EGFRvIII-negative escape variants were a potential mechanism of treatment failure in active immunotherapy.11 Although EGFRvIII appears to represent a nearly terminal branch of tumor progression for malignant brain tumors and is expressed clonally within this lineage, antigen-specific tumor targeting might confer a selective growth advantage on neoplastic cells not expressing this epitope. It has been shown previously that treatment failure with antigen-specific passive humoral immunotherapy is not a result of antigen-loss variants,82 suggesting that antigen-negative escape variants are an etiology of treatment failure in active immunotherapy. In two separate phase II clinical trials (ACTIVATE and ACT II) in newly diagnosed EGFRvIII-expressing GBM patients who received the PEP-3-KLH, on tumor progression, the EGFRvIII expression was lost.57,84 To overcome this obstacle, cocktails of different antigens or whole-tumor lysates can be used as immunogens85; however, with nonspecific antigen approaches, there is a risk for autoimmune responses.10 To circumvent this, future approaches may involve screening the patient’s tumor for a panel of antigens86 and targeted immunotherapy selected based on these parameters—similar to selecting antibiotics for a given bacterial infection. Alternatively, upregulating the expression of these antigens with some forms of chemotherapy may restore the ability of the immune system to clear these cancer cells.87
A variety of agents have been shown to be capable of inducing or upregulating antigen expression. For example, gemcitabine, a nucleoside analogue, can increase tumor antigen cross-presentation and T-cell infiltration of the tumor.88 The demethylating agent DAC is capable of inducing the tumor-associated antigen MAGE-1 on melanoma cells with subsequent cytolytic T-cell clearance.89 Irinotecan (CPT-11) has been shown to induce the LYGD/E48 antigen, which resulted in synergistic tumor regression with a cytotoxin-armed antibody in a colorectal xenograft murine model.90 The combination of 5-fluorouracil and cisplatin (CDDP) can upregulate the antigen CEA and MHC class I in human colorectal cancer cell lines91 and was functionally relevant as evidenced by increased CEA-specific, MHC-restricted cytotoxic T-cell killing.87 Thus, many different types of chemotherapy can enhance antigen expression; however, none of these agents has been tested to determine whether it can restore antigen expression after clonal elimination with immunotherapy.
An alternative therapeutic approach for the issue of antigen heterogeneity would be to selectively screen each patient’s tumor for a panel of potential antigens, but this would be somewhat labor intensive and costly at present. Another alternative approach would be to vaccinate cancer patients with a panel of tumor-associated or tumor-specific antigens. Tissue arrays of gliomas from a large population of patients could be used to determine the frequency of the most common tumor antigens. This strategy would be very similar to that used in manufacturing flu vaccines annually, in which the most common virulent strains are incorporated as immunologic targets. Finally, exploration has begun on immunologic approaches for inducing immune responses against immune escape variants based on defects in the transporter associated with antigen processing,92 but these are not yet sufficiently developed for clinical trial application.
STAT-3—A Key Switch Mediating Immuno suppression
The ability of tumor cells to proliferate uncontrollably, resist apoptosis, sustain angiogenesis, and evade immune surveillance is regulated, in part, by the signal transducer and activator of transcription type 3 (STAT-3), which acts as a cytoplasmic signaling protein and nuclear transcription factor. Under normal physiologic conditions, latent STAT-3 activation is dependant on ligand-receptor interaction, primarily under the control of growth factor receptor tyrosine kinases, cytokines, and G-protein receptors.93 For example, EGFR and IL-6 activate Jak2, which then activates STAT-3 by phosphorylation of the tyrosine residue in the transactivation domain of STAT-3.94 In cancer cells, these tyrosine kinases are among the most frequently activated oncogenic proteins. STAT-3 has been shown to be persistently activated in most human cancers, including gliomas.95,96 The phosphorylation of STAT-3 has been attributed to EGFRvIII.97 IL-6 is expressed in the CNS, especially by reactive astrocytes, under a wide variety of conditions, such as hypoxia,98 traumatic and metabolic injury,99 and inflammation,100 which in turn also induces expression of p-STAT-3. The use of decoy antisense STAT-3 oligonucleotides and dominant-negative vectors has provided convincing evidence that STAT-3 is highly relevant to the growth and survival of many tumor types, including gliomas.101–103
STAT-3 has also emerged as a key switch that drives the underlying immuno suppression104 identified in cancer patients by preventing maturation of DCs and inhibiting the proliferation and activation of immune effector populations. STAT-3 has been shown to be a potent regulator of these anti-inflammatory responses by suppressing macrophage activation and limiting inflammatory responses.105,106 It also has been shown to become constitutively active in diverse tumor-infiltrating immune cells.107 For example, STAT-3 activity within NK cells and neutrophils directly reduces their cytotoxicity, whereas STAT-3 activity in DCs reduces the expression of MHC class II, CD80, CD86, and IL-12 in these cells, rendering them unable to stimulate T cells and generate antitumor immunity. Investigators have recently shown that by ablating STAT-3 in only the hematopoietic cells in mice resulted in marked enhancement of activated and functional T cells, NK cells, and DCs in tumor-bearing mice. This ablation of STAT-3 in only the hematopoietic cells also resulted in marked antitumor effects in vivo, indicating that STAT-3 expression in immune cells restrains antitumor immune eradication.107 The tumor microenvironment induces STAT-3 activity in tumor-associated immune cells.104,107,108
The blockade of STAT-3 with small molecular inhibitors in microglia obtained from the CNS of patients with GBM resulted in the upregulation of costimulatory molecules (e.g., CD80, CD86), induced systemic APCs to elaborate proinflammatory cytokines essential for T-cell effector responses, and induced activation and proliferation of T cells, indicating that STAT-3 blockade is a potent approach to modulating both the systemic and local tumor immune microenvironments. Of note, this potent immune activation occurred in immune cells obtained from patients with disease refractory to other conventional immune activators, such as toll-like receptor agonists. When the immune cells from immunosuppressed GBM patients were treated with a STAT-3 inhibitor, Western blot analysis demonstrated phosphorylation of ZAP-70 in T cells, thus bypassing the impairment of signal transduction typically identified in the immune systems of patients with cancer.109 The mechanism of T-cell activation is likely secondary to the inhibition of T regs present in these immune preparations, thus allowing immune activation (i.e., releasing the “brake” on immune cell activation and proliferation). IL-2 has been shown to regulate FoxP3 expression in human CD4+, CD25+ T regs by inducing STAT-3 binding of the first intron of the FoxP3 gene.110 Suppressor of cytokine signaling type 3 has been shown to be an inhibitor of STAT-3 signaling111 and transcriptional activity112,113 but is deficient in T regs.114 STAT-3 inhibitors should have a multiplicity of immune modulatory activities, such as allowance of expression of costimulatory molecules in the tumor microenvironment, inhibition of immune suppressive cytokines, maturation of DCs, and inhibition of T regs, and represents a newly emerging class of drugs that will be used for immune therapeutic purposes.
Immune Modulation by Central Nervous System Antigen-Presenting Cells
T-cell activation requires signals through both the MHC and costimulatory molecules, and the expression of MHC alone results in T-cell anergy.115 Low levels of expression of CD80 provide an immune escape advantage to cancer cells, and CD28-mediated costimulatory signals are essential for differentiation of functional tumor-specific cytotoxic CD8+ effector T cells.116,117 In tumor-bearing animals, repeated stimulation of the T cells is necessary, especially at the effector site, to generate tumor responses,118 indicating that if the APC failed to provide appropriate stimulation, the APC may contribute to immune suppression. Although microglia may be able to act as APCs during autoimmunity (multiple sclerosis) and infection (HIV),119,120 glioma-associated microglia are functionally impaired, are not able to stimulate T-cell responses, and are refractory to activation stimulation (i.e., toll-like receptor agonists).7 Intratumoral DCs may be able to provide these immune-stimulating conditions, but these are rarely present in malignant gliomas.7 Additionally, CD11b+ inhibitory macrophages (iMACs) have been described, which induce apoptotic death in CD8+ T cells. This inhibitory phenotype could be changed into one reflective of highly activated DCs by culturing the iMACs with IL-4 and GM-CSF,121 suggesting that the tumor microenvironment can be manipulated for therapeutic purposes. However, the delivery of cytokines directly to the CNS is problematic. Costimulatory molecules on glioma-associated microglia and macrophages have been shown to be lost or significantly downregulated. In attempts to overcome the failure of APCs, and specifically the lack of sufficient costimulation, vaccines with enhanced costimulatory capacity have been tested and demonstrated antitumor effects that have maintained high avidity effector-memory CD8+ T cells. Other approaches for potentially upregulating the costimulator molecules on APCs include CpG DNA,122 chemotherapy with cytosine arabinoside,123 transfection with poxviruses expressing the costimulatory molecules,124 or transfection of the replication-defective fowlpox recombinant vector rF-TRICOM (TrIad of COstimulatory Molecules).125
An emerging strategy for upregulating costimulatory molecules in the glioma microenvironment is with STAT-3 blockade agents. A small molecular inhibitor (WP1066) of STAT-3 has been shown to upregulate costimulatory molecules and proinflammatory cytokines on microglia obtained from the CNS of patients with GBM that were refractory to modulation by other conventional immune activators, such as toll-like receptor agonists. Furthermore, WP1066 can modulate enhanced T-cell functional activities in physiologic ranges that can be obtained within the CNS.109 These types of agents are anticipated to be in clinical trials of human patients within the next several years.
Immune Modulation by Glioma-Infiltrating Lymphocytes and TH2 Lymphocytes
After activation, CD4+ helper T cells are classified, based on their elaborated cytokine responses, as TH1 (promoting cell-mediated immunity) or TH2 (promoting humoral immunity), which predominantly secrete either interferon-γ (IFN-γ) or IL-4, respectively. A predominance of TH2-type cytokine response has been correlated with enhanced tumor growth.126,127 This TH2 polarization has been demonstrated in gliomas128 and is likely mediated by microglia-secreted macrophage-derived chemokine (MDC)129 or neuropeptides, or both.130 One therapeutic approach altering the TH2 intratumoral skewing would be to limit the chemokines that direct migration of the immunosuppressive immune cells to the tumor microenvironment, such as with a STAT-3 inhibitor.26 Alternatively, preferential TH1 responses could be induced with cyclooxygenase-2 (COX-2) inhibition.131
Impaired Immune Responses
The proliferative response of stimulated T cells, along with the levels of the IL-2 receptor and tyrosine phosphorylation in response to IL-2, from malignant glioma patients are reduced compared with healthy donors.132 These results suggest a defect in the expression of functional IL-2 receptor and T cells isolated directly from human gliomas that are not activated as reflected by expression of the IL-2 receptor CD25.7 Soluble products from tumors may suppress T-cell proliferation through a mechanism that involves downregulation of Jak3 expression and inhibition of IL-2–dependent signaling pathways.133 A potential mechanism for overcoming this is the constitutive activation of the T cells by transfection with CD3 or CD28, or both, for adoptive transfer therapeutic purposes. An antibody, r28M, directed against a melanoma-associated proteoglycan expressed on glioblastoma cells activates T cells through the CD28 molecule without additional stimulus from the TCR-CD3 complex and induces T-cell–mediated killing of glioblastoma cells in vitro and in vivo.134
Doxorubicin, an anthracycline antibiotic, can enhance the cytotoxic T-cell compartment. Pretreatment of mice with doxorubicin produced enhanced cytotoxic T-cell responses and activation of macrophages, as reflected by the secretion of IL-1 and IL-2.51 There was a twofold increase in the number of macrophages that could support the generation of the cytotoxic T cells. Ultimately, the macrophages became tumoricidal, and the effect persisted for 14 to 18 days. Further studies demonstrated that the administration of doxorubicin 5 days after a GM-CSF–secreting vaccine injection potently induced cytotoxic T-cell activity.54 Cancer patients treated with doxorubicin at a dose of 25 mg/m2 developed an increase in the percentage of circulating CD8+ T cells having twofold higher cytotoxic activity.135 Ideally, the chemotherapy selected for use in combination with immunotherapy should have some intrinsic tumoricidal activity. Although doxorubicin has little clinical efficacy against gliomas, this approach could potentially be used to boost the T-cell responses in glioma patients with further clinical development.
T-Cell Apoptosis
T-cell apoptosis has been observed frequently within malignant gliomas.136 The induction of apoptosis involves the activation of caspases, leading to nuclear fragmentation and apoptosis, and is triggered by deprivation of survival stimuli such as costimulators7 or by binding of ligands to death-inducing membrane receptors.137 Both of these mechanisms are operational within gliomas. FasL is predominantly expressed on T cells after activation by antigen and IL-2. When mature T cells are repeatedly stimulated by antigens, they coexpress Fas and FasL.137 Gliomas can evade killing by tumor-infiltrating T cells by overexpressing FasL, rendering the tumor resistant to apoptosis138 while delivering a death signal to Fas-expressing cells, which include the activated cancer-infiltrating T cells. However, high-grade gliomas also express the apoptotic receptors Fas and CD95, rendering them susceptible to antibody-stimulated Fas- and CD95-mediated apoptosis and CD8+ cytotoxic killing.139 Alternatively, agents such as topotecan can be administered to patients to upregulate Fas on gliomas that renders the tumor more susceptible to cytotoxic immune cell clearance.139–141
Topotecan penetrates the blood-brain barrier but has demonstrated only modest clinical efficacy in glioma patients, probably owing to failure to achieve in vivo levels sufficient to induce apoptosis of the tumor. Nevertheless, topotecan has been shown to upregulate Fas and CD95 in high-grade gliomas, rendering them more susceptible to immune clearance.139,141 Thus, although levels achieved are insufficient for direct glioma clearance within the CNS, this agent potentially could be exploited as an immune modulator. Other chemotherapeutics, such as doxorubicin142 (Adriamycin) and its analogues (epirubicin and pirarubicin),143,144 VP-16 (etoposide), cisplatin,144 and the combination of cisplatin and 5-fluorouracil145 can also increase cytotoxicity mediated by T cells and NK cells by upregulating Fas.
A synergistic antitumor effect was observed with paclitaxel and FasL in two human malignant glioma cell lines, T98G and LN-229.146 The synergy of paclitaxel and FasL was achieved independently of either the G2/M-phase cell cycle arrest or p53 activation. At low concentrations of paclitaxel, Bcl-2 phosphorylation was induced in the glioma cells, which in turn interfered with the heterodimerization of Bcl-2 with Bax and the inhibition of Fas-induced apoptosis by Bcl-2. The investigators hypothesized that the synergistic antiproliferative effect of paclitaxel and FasL on malignant glioma cells stemmed from the upregulation of the Bcl-2/Bax rheostat in favor of Bax, thereby sensitizing the tumor cells to apoptosis. Interestingly, this mechanistic synergy was not observed with other agents such as teniposide.147
[/not-level-membership-for-neurosurgery-category]
