[level-membership-for-neurology-category]
Chapter 59 Brain Edema and Disorders of Cerebrospinal Fluid Circulation
Brain tissue, which is composed of 80% water, is separated from the systemic circulation by a complex series of interfaces. The major site is the endothelial cells that are a component of the neurovascular unit (Iadecola and Nedergaard, 2007). Cells that form these interfaces have specialized proteins that form tight junctions; some have carrier proteins that shuttle essential molecules, and multiple electrolyte pumps on cell membranes. Cellular membranes preserve the compartmental structure with water in extracellular and intracellular spaces. When shifts in water from one compartment to another occur under pathological conditions, swelling in the various compartments leads to increased intracranial pressure (ICP). If the increased water is within the ventricles, hydrocephalus results. Hydrocephalus leads to transependymal flow of water into the periventricular white matter, resulting in interstitial edema. Cytotoxic edema is cell swelling with reduction in the extracellular space; this occurs in ischemic or traumatic injury to the brain, with the loss of nutrients that form the energy stores (Marmarou, 2007). The loss of adenosine triphosphate (ATP) that drives chemiosmotic work through ATPase-mediated membrane pumps causes the cellular swelling. A disturbance in the cerebral blood vessels, which produces leakage of serum proteins across the damaged vessels, leads to vasogenic edema. Each of these shifts in water from its normal compartment into another alters ICP.
In the past several years, new information emerged describing the molecular events underlying the changes in water and proteins within the fluid spaces of the brain. A pore-forming molecule, aquaporin, was discovered; it facilitates movement of water along osmotic gradients (Agre and Kozono, 2003; Verkman, 2009). Novel molecules, such as hypoxia inducible factors (HIF), have been discovered that contribute to inflammation secondary to hypoxic/ischemic insults and brain trauma (Semenza, 2007). Cytokines and free radicals amplify the tissue damage. Advances in imaging modalities, including computed tomography (CT) and magnetic resonance imaging (MRI), have improved the diagnosis of cerebrospinal fluid (CSF) disorders and brain edema. Although we understand the underlying molecular processes involved in edema formation and have better ways of observing its evolution, we remain behind in our attempts to treat brain edema, which remains a major challenge.
Examination of the CSF by lumbar puncture (LP) can provide unique information, aiding diagnosis and patient management. Increased ICP can only be determined by measurements made during removal of CSF; this information is critical in the diagnosis of raised CSF pressure in idiopathic intracranial hypertension. Studies of cells and proteins in the CSF provide information about infection and inflammation. Cancer cells can be detected and antibodies to infectious agents identified. When the BBB is disrupted, increased proteins, mainly albumin, appear in the CSF. Diagnosis of multiple sclerosis (MS) is strengthened by the detection of myelin basic protein and immunoglobulin (Ig)G endogenous production. Alzheimer disease leads to alterations in the amyloid protein, Aβ1-42, and tau proteins that can aid in early diagnosis (Mattsson et al., 2009). Thus, LP to obtain CSF is one of the most cost-effective procedures in daily clinical practice, and when done correctly, it can obtain important information only available from CSF.
The recognition that the total volume of fluid and tissue contained within the skull is constant is called the Monro-Kellie doctrine, named after the two early anatomists. Changes in volume of blood, CSF, or brain compartments produce compensatory changes in the others, with a resultant increase in CSF pressure. When CSF outflow pathways are blocked, enlargement of the ventricles or hydrocephalus follow, resulting in a buildup of pressure in the ventricles that forces the CSF to move transependymally into the periventricular white matter. Masses enlarge the tissue space and compress CSF and blood spaces. When the compensatory mechanisms are overwhelmed, ICP increases. Disruption of the blood vessels leads to vasogenic edema that moves through the more compliant extracellular space of the white matter. On the contrary, damaged swollen cells lead to cytotoxic edema, with narrowing of the extracellular space. Finally, an increase in blood volume, as seen in hypercapnia and hypoxia, increases the ICP (Table 59.1).
Table 59.1 Causes of Increased Intracranial Pressure
| Site of Increased Intracranial Pressure | Diseases |
|---|---|
| Increased tissue volume | Tumor, abscess |
| Increased blood volume | Hypercapnia, hypoxia, venous sinus occlusion |
| Cytotoxic edema | Ischemia, trauma, toxins, metabolic diseases |
| Vasogenic edema | Infections, brain tumors, hyperosmolar states, inflammation |
| Interstitial edema | Hydrocephalus with transependymal flow |
Blood-Brain Interfaces
Cerebral Blood Vessels and the Neurovascular Unit
The total surface of the capillary endothelial cells forms the major interface between the blood and brain. Other, less extensive, interface surfaces include choroid plexuses and arachnoid granulations (Table 59.2). At each of the BBB interfaces, high-resistance junctions between cells, which make the surface into an epithelial-like structure, restrict bulk transport. The epithelial sheets impede non–lipid-soluble substances, charged substances, or large molecules, whereas lipid-soluble substances, such as anesthetic gases, pass easily through the cells. Water has an anomalous structure that allows it to pass rapidly through endothelial cells but with slight restrictions.
Table 59.2 Characteristic Features of the Blood-Brain Interfaces
| Interface | Tight-Junction Location | Functional Aspects |
|---|---|---|
| Blood-CSF | Choroid plexus cell | Active secretion of CSF via ATPase and carbonic anhydrase |
| CSF-blood | Arachnoid membrane | Arachnoid granulations absorb CSF by one-way valve mechanism |
| Blood-brain | Capillary endothelial cell | Active transport of ISF via ATPase; increased mitochondria and glucose transporters in capillary endothelial cells |
ATPase, Adenosine triphosphatase; CSF, cerebrospinal fluid; ISF, interstitial fluid.
In 1925, Cushing and Weed described the pathways involved in the movement of CSF and ISF through the ventricles and around the brain, recognizing that the CSF/ISF acted as a lymph-like fluid in brain. They compared that circulation to those of the blood and lymph. The importance of this circulation has grown with the discovery that ISF is formed by cerebral blood vessels, which have electrolyte pumps that make fluid in a fashion similar to that of the epithelial cells. Flowing around cells, ISF brings nutrients such as glucose and oxygen to neurons and astrocytes and removes the products of metabolism. ISF is absorbed either into the blood via terminal capillaries and venules or into CSF for eventual absorption through the arachnoid granulations (Fig. 59.1).
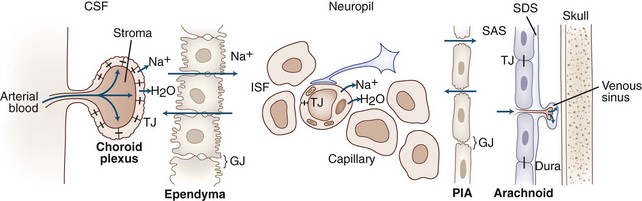
Brain extracellular space comprises 15% to 20% of the total brain volume. Complex carbohydrates are found in the extracellular space, including hyaluronic acid, chondroitin sulfate, and heparan sulfate. Hyaluronic acid forms large water domains. These large extracellular matrix glycoproteins impede cell movement. After an injury, astrocytes secrete an extracellular molecule, hyaluron, which impedes movement of fluids in the extracellular space, slowing tissue repair. Treatment with hyaluronidase reduces hyaluron and improves regrowth of injured fibers (Back et al., 2005).
Proteases are secreted during development, angiogenesis, and neurogenesis to clear a path for the growing cells, similar to the secretion of proteases by spreading cancer cells (Candelario-Jalil et al., 2009; Yong, 2005). An important concept that has emerged over the past few years is that of the neurovascular (or gliovascular) unit. Neurons, astrocytes, and pericytes comprise the neurovascular unit (Neuwelt et al., 2008). On the abluminal surface of the endothelial cells is a thin layer of basal lamina composed of type IV collagen, fibronectin, heparan sulfate, laminin, and entactin. Entactin connects type IV collagen and laminin to add a structural element to the capillary. Fibronectin from the cells joins the basal lamina to the endothelium. Basal lamina provides structure through type IV collagen, charge barriers by heparan sulfate, and binding sites on the laminin and fibronectin molecules. Within the basal lamina reside the pericytes, which are a combination of smooth muscle and macrophage. Astrocyte foot processes surround the basal lamina. Glia limitans is found at the pial surface and at the interface between astrocytes and blood vessels (Owens et al., 2008). Neurons complete the group of cells that comprise the neurovascular unit (Girouard and Iadecola, 2006) (Fig. 59.2).
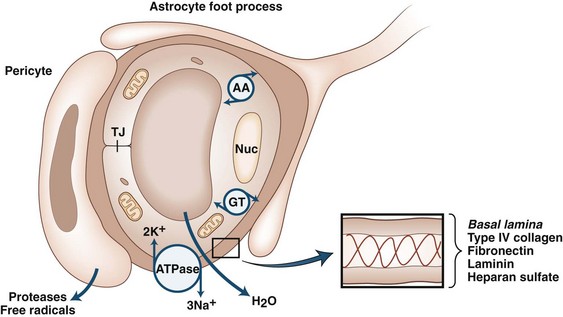
Cerebral blood vessels have very low permeability and high electrical resistance, making them more similar to epithelial cells than systemic capillaries, which are passive structures with low electrical resistance and fenestrations that permit passage of large protein molecules. In addition, cerebral blood vessels have highly selective molecular transport properties. During development, cerebral blood vessels acquire the characteristics that distinguish them from systemic capillaries. Astrocytes are critical in this differentiation process, which involves interactions between blood vessels and astrocytes. The critical nature of the astrocytes in this process was shown in transplantation studies involving chicken and quail cells, which can be separated histologically. Quail brain grafts from 3-day-old quails transplanted into the coelomic cavity of chick embryos become vascularized by chick endothelial cells and form a competent BBB. On the other hand, when avascular embryonic quail coelomic grafts are transplanted into embryonic chick brain, chick endothelial cells form leaky capillaries and venules (Stewart and Wiley, 1981). Subsequent studies showed that astrocytes induced BBB properties in non-neural endothelial cells in vivo (Janzer and Raff, 1987).
Electron microscopic studies with electron-dense tracers identified a major site of the BBB as the endothelial tight junctions (Reese and Karnovsky, 1967). Isolation of tight-junction proteins and cloning of the molecules permitted production of antibodies that identified their location. Zona occludins tether the tight-junction proteins to actin within the endothelial cells (Hawkins and Davis, 2005). Occludin and claudin form the actual tight junctions within the endothelial clefts (Furuse et al., 1993). Occludin attaches to the zona occludins, while claudins attach to occludin and protrude into the clefts between cells. The extracellular tails of claudins from adjacent cells self-assemble to form the tight junctions that are “zip-locked” together (Nitta et al., 2003).
Tight junctions between the endothelial cells create the unique membrane properties of the cerebral capillaries by greatly increasing the electrical resistance, which blocks transport of non–lipid-soluble substances (Box 59.1). Brain tissue has a very high demand for glucose and essential amino acids, which can be met by specialized molecules that transport glucose and amino acids across the BBB. Glucose transporters are densely distributed in the capillaries. At low levels of blood glucose, the carriers function at full capacity to meet metabolic needs, but at higher levels of blood glucose, the carriers are saturated and transport is dominated by diffusion rather than active transport. Several isoforms of the glucose transporter molecule have been isolated and cloned (Vannucci et al., 1997). High concentrations of one isoform, GLUT1, are found on cerebral blood vessels. GLUT3 is found on neurons and GLUT5 in microglia. GLUT2 is found predominantly in the liver, intestine, kidney, and pancreas. Impairment of glucose transport due to lack of transporters has been described in patients with Alzheimer disease and in other neurological disorders (Kalaria and Harik, 1989). Amino acid transporters carry essential amino acids into the brain. Competition for the amino acid transporters can lead to a deficiency state; serotonin uptake is decreased in patients with phenylketonuria, which competes for the transporter.
Box 59.1 Unique Features of Cerebral Capillaries
Tight junctions create high electrical resistance
Adenosine triphosphatase pumps on abluminal surfaces form interstitial fluid
Increased numbers of mitochondria for high-energy needs
Glucose transporters and amino acid carriers
Basal lamina contributes to the barrier
Steady-state levels of brain electrolytes are preserved by transport mechanisms at the BBB. Potassium is maintained at a constant level in the CSF and brain by the BBB (Katzman and Pappius, 1973). This prevents fluctuations of electrolyte levels in the blood from influencing brain levels. Calcium is similarly regulated. Glutamate, which is an excitotoxin, is excluded from the brain. Highly lipid-soluble gases such as carbon dioxide and oxygen are rapidly exchanged across the capillary. Anesthetic gases are effective because they readily cross the BBB and enter the brain.
Different rates for equilibration of various substances between blood and brain can cause paradoxical clinical situations. For example, to compensate for a metabolic acidosis, bicarbonate levels fall in both the blood and the brain. Metabolic acidosis is balanced by a respiratory alkalosis due to lowering of carbon dioxide by hyperventilation, which compensates for the acidosis. Carbon dioxide is reduced in both the blood and CSF compartments, since it readily crosses the BBB, while bicarbonate is much more slowly exchanged between the two compartments. This adjustment results in a stable, albeit pathological, situation. However, when the metabolic acidosis is corrected by intravenous infusion of bicarbonate, there is a rapid adjustment of Pco2 as the hyperventilation stops and CO2 builds up. Bicarbonate adjusts very slowly because of the limited transport across the BBB, and the CO2 entering the brain causes a further fall in brain pH. This dangerous situation continues until the bicarbonate levels in the brain rise. Although treatment is necessary to correct the metabolic acidosis, patients may become worse due to brain acidosis if treatment is too rapid (Posner and Plum, 1967).
Production of Cerebrospinal Fluid and Interstitial Fluid
While choroid plexuses are the major source of CSF, capillaries contribute ISF to varying degrees, depending on the species. Production of CSF is constant across species when the volume of fluid formed is divided by the weight of the choroid plexus (Cserr, 1971). In humans, the volume of CSF in the ventricles is 140 mL, with a rate of CSF production of 0.35 mL/min or about 500 mL/day. Obstructive hydrocephalus can rapidly cause life-threatening symptoms when the rate of production remains constant but there is no absorption. On the other hand, removal of 20 mL of CSF at the time of LP for the treatment of idiopathic intracranial hypertension does not make physiological sense (although it seems to help at times, which may be due to the hole placed in the dura, with a slow leak of CSF).
CSF production occurs at both choroidal and extrachoroidal sites, and estimates of the proportion of CSF from each site vary, depending on the species and the method of measurement. Extrachoroidal production accounts for 30% of total CSF production in the cat (Rosenberg et al., 1980) and approximately 60% in the nonhuman primate (Milhorat, 1969). No measurements of the relative proportion of CSF produced at each source have been made in humans.
Water Molecules: Basis for Magnetic Resonance Imaging
Resonance signals detected by MRI are from water molecule protons. Since water is the most abundant source of protons in the brain, water protons dominate the signals. New rapid-acquisition pulse sequences are fast enough to show diffusion of water. Cytotoxic edema shrinks the extracellular space and restricts the diffusion of water (Moseley et al., 1990). The ability to monitor water diffusion by MRI has greatly improved our ability to diagnose an acute ischemic event. Water diffusion between cells in the extracellular space occurs normally. When there is cellular swelling and the extracellular space shrinks, the diffusion of water slows, and the apparent diffusion coefficient (ADC) shows a loss of signal, which appears black on the image. The diffusion-weighted image (DWI) has a bright signal. Because the DWI may show T2 shine-through that will be misinterpreted as reduced diffusion, both a darkened ADC and a bright DWI should be seen in the region of the infarct. In cerebral ischemia, the DWI is abnormal within minutes after the onset of the ischemia, making this an excellent diagnostic test for the presence of cerebral ischemia (Adami et al., 2002).
Diffusion tensor imaging (DTI) reveals the patterns of white matter tracts in three dimensions. Taking advantage of the directional flow of water protons along white matter, diffusion is measured in three planes, and the separate pathways for water movement between the fibers are traced. In patients with white-matter pathology, such as in vascular cognitive impairment and MS, injury patterns in the white matter can be revealed by DTI (Nitkunan et al., 2008).
Anatomical Sites of Central Nervous System Infection
The terminology used to describe various types of central nervous system (CNS) infections is anatomically based (Table 59.3). An infection limited to the subarachnoid space, with inflammation of the meninges, is called meningitis. Meningeal signs of headache, stiff neck, and photophobia are present without focal findings that would indicate spread into the parenchyma. When the infection spreads contiguously from the subarachnoid space through the pial surface or along Virchow-Robin spaces, crossing the gap-junctioned linings, the brain parenchyma is infected, and the term meningoencephalitis is used. In addition to meningeal signs, there are focal findings and possibly impaired consciousness and seizures. An infection in the brain tissue that is most likely spread via blood begins as a loose collection of invading cells referred to as a cerebritis; walling off of the infected brain tissue leads to an abscess. Finally, the term encephalitis is used to describe a more diffuse brain infection in both the gray and white matter, which is usually indicative of a viral infection. Occasionally the infection spreads in a potential space beneath the dura but outside the arachnoid; subdural empyema describes a life-threatening collection of pus over the brain surface that has often spread from an infected sinus through the venous plexus of the ethmoid or sphenoid sinuses into the subdural space. The presence of a subdural empyema should be suspected in a patient with sinus infection, fever, seizures, focal findings, and altered consciousness. Diagnosis of meningitis can be done by examination of CSF for signs of infection such as increased white blood cells or protein. Infections that invade the brain are best diagnosed with MRI, which can readily demonstrate a meningoencephalitis, cerebritis, abscess, or encephalitis. Use of contrast agents increases the potential of reaching a correct diagnosis based on site of infection. Subdural empyema is the most difficult condition to diagnose because it may only be a thin layer of pus on the surface of the brain and be obscured by the skull. Diagnosis can be missed on LP or CT.
Arachnoid Granulations and Absorption of Cerebrospinal Fluid
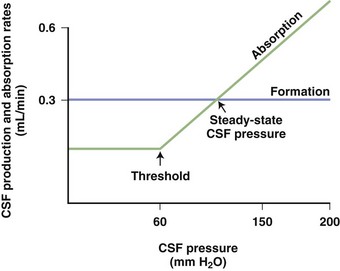
Arachnoid granulations (pacchionian granulations) are the major sites for the drainage of CSF into the blood. They protrude through the dura into the superior sagittal sinus and act as one-way valves. As CSF pressure increases, more fluid is absorbed. When CSF pressure falls below a threshold value, the absorption of CSF ceases (Fig. 59.3). In this way, CSF pressure is maintained at a constant level, with the rate of CSF production as one determining factor.
Cerebrospinal Fluid Pressure
The opening CSF pressure is measured with a manometer attached to the needle. Normal CSF pressure ranges from 80 to 180 mm H2O but may go as high as 200 mm H2O in obese patients or those who are not relaxed. Three components contribute to the measured pressure: volume of blood within the cranial cavity, amount of CSF, and the brain tissue. The CSF pressure recorded by the manometer represents the venous pressure transmitted from the right side of the heart through the venous sinuses. Small fluctuations from the cardiac systolic pulse and larger fluctuations from respirations can be seen in the column of fluid in the manometer. Pulsations in the manometer represent the fluctuations in the thin-walled veins (Davson, 1967). Arteries have thick elastic walls that dampen the pulsations from arteries. Deep respirations cause wide fluctuations in the CSF pressure, whereas changes in arterial pressure are barely visible. As ICP rises, tissue compliance falls and reserve capacity of the intracranial contents is lost. When tissue compliance is lost, small changes in fluid volume may lead to large increases in ICP.
In addition to complications of elevated CSF pressure, there are circumstances that lead to low pressure. Some patients have low pressure after LP owing to a persistent tear in the dura. Headaches occur with standing up and are often relieved with bedrest. Low-pressure headaches after LP generally resolve spontaneously. However, rarely they persist and require placement of a epidural blood-patch, which is accomplished by injecting the patient’s own blood to close the hole in the dura. Postsurgical and posttraumatic leakage of CSF can cause low-pressure headaches, and occasionally a spontaneous tear occurs. Diagnosis of a spontaneous leak can at times be difficult and may require injection of radiolabelled substances or contrast agents into the CSF (Schievink et al., 2008). Enhancement of the meninges can be seen on MRI in patients with low CSF pressure, but the cause of this abnormality is uncertain.
Composition of the Cerebrospinal Fluid
Diagnosis of MS is aided by obtaining CSF for a demyelinating test profile. Acute MS attacks cause an increase in myelin basic protein, which represents breakdown of myelin; oligoclonal bands suggest a longer disease course (Noseworthy et al., 2000). The ratio of IgG to albumin in both the blood and brain is calculated according to the formula: (CSF IgG × serum albumin) / (serum IgG × CSF albumin). Dividing the ratio in the brain by that in the blood indicates whether the IgG comes from the blood across a leaky BBB, in which case the ratio is low, or whether the source of IgG is the brain, in which case the IgG index is elevated. An IgG index above 0.7 indicates intrathecal IgG synthesis.
Brain Edema
Molecular Cascade in Injury
Cerebral edema is the end result of many neurological diseases. Excess fluid can accumulate in the intracellular or extracellular spaces. A convenient (though simplified) classification separates brain edema into cytotoxic or cellular swelling, and vasogenic or vascular leakage (Klatzo, 1967). Another proposed category is interstitial edema, which represents the accumulation of fluid in interstitial spaces in hydrocephalus (Fishman, 1975). Separation into distinct categories, while useful, is often difficult because of the overlap between the various types of edema.
Cellular and blood vessel damage follows activation of an injury cascade (Dirnagl et al., 1999). The cascade begins with depletion of energy and glutamate release into the extracellular space (Fig. 59.4). This occurs during a hypoxic, ischemic, or traumatic injury and causes cytotoxic damage. Release into the extracellular space of excessive amounts of the excitatory neurotransmitter, glutamate, opens calcium channels on cell membranes, allowing extracellular calcium to enter the brain. Because one calcium ion is exchanged for three sodium ions, the removal of excess calcium from the cell, which requires an intact cellular membrane, causes a buildup of sodium within the cell, creating an osmotic gradient that pulls water into the cell. While the cell membrane is intact, the increase in water causes dysfunction but not necessarily permanent damage.
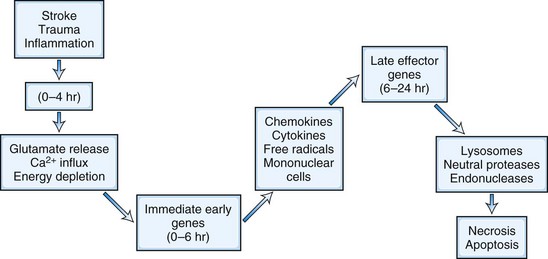
Accumulation of calcium ions within the cell activates intracellular cytotoxic processes, leading to cell death. An inflammatory response is initiated by the formation of immediate early genes (e.g., c-fos and c-jun) and cytokines, chemokines, and other intermediary substances. Microglial cells are activated and release free radicals and proteases, which contribute to the attack on cell membranes and capillaries. Irreversible damage to the cell occurs when the integrity of the membrane is lost. Free radicals are pluripotential substances produced in the ischemic brain and after traumatic injury. The arachidonic acid cascade produces reactive oxygen species such as superoxide ion, hydrogen peroxide, and hydroxyl ion. Release of fatty acids (e.g., arachidonic acid) provides a supply of damaging molecules. Superoxide dismutase-1 and catalase are the major enzymes that catalyze the breakdown of reactive oxygen species. Other defenses include glutathione, ascorbic acid, vitamin E, and iron chelators such as the 21-amino steroids. The role of oxygen radicals has been extensively studied. Transgenic mice that overexpress the superoxide dismutase-1 gene have smaller ischemic lesions than controls (Jung et al., 2009).
Nitric oxide (NO) is another source of free radicals, which have both positive and negative effects. NO synthetase (NOS) has three forms: neuronal NOS (nNOS), endothelial NOS (eNOS), and inducible or immunological NOS (iNOS). Macrophages and activated microglial cells form NO through the action of iNOS in response to ischemia, injury, and inflammatory stimuli. NO acts both as a normal vasodilator of blood vessels, by release of cyclic guanosine monophosphate in smooth muscle, and as a toxic compound in pathological conditions through the action of peroxynitrite anions (ONOO−), which are formed from the reaction of NO with superoxide anions (Beckman et al., 1990; Endres et al., 2004).
Manipulation of the NOS gene has helped reveal the action of the enzyme. Neuronal NOS produces toxic free radicals early in ischemic injury. Deletion of the nNOS gene in transgenic mice results in smaller infarcts from middle cerebral artery occlusion (Huang et al., 1994; Iadecola et al., 1994). On the other hand, eNOS causes vasodilatation and increases cerebral blood flow. Removing the eNOS genes leads to increased infarct size. Inflammation induces iNOS, which enhances injury and reaches a maximum at 24 hours (Huang and Lo, 1998).
Neuroinflammation and Vasogenic Edema
Vasogenic edema occurs when there is damage to the capillary and subsequent disruption of the BBB. Protein and blood products enter brain tissue, increasing the oncotic pressure in the brain and exposing brain cells to toxic products from the blood. Opening of the BBB could occur by loosening of tight junctions, development of pinocytotic vesicles in the endothelial cell, or an alteration in the basal lamina surrounding the capillaries. Tight junctions in the endothelial cells are the first line of protection. Proteases and free radicals are the major substances that attack the capillaries. The layer of basal lamina around the capillary, containing type IV collagen, fibronectin, and laminin, is degraded by proteases. The proteases involved include the serine proteases, plasminogen activators/plasmin system, and matrix metalloproteinases (MMPs) (Cunningham et al., 2005). Free radicals activate the proteases and attack the membranes directly (Jian and Rosenberg, 2005). Brain cells and infiltrating leukocytes are the sources of proteases and free radicals. Neutrophils contain prepackaged gelatinase B (MMP-9), which is released at the injury site and activated.
Extracellular matrix undergoes remodeling by the action of MMPs during development and repair (Yong, 2005). The MMPs are a gene family of over 24 enzymes that are expressed constitutively during normal remodeling but are induced in an injury. MMPs are expressed in a latent form that requires activation. Constitutively expressed MMP-2 is normally expressed by astrocytic foot processes around cerebral blood vessels, where it modulates the permeability of the BBB. Membrane-type MMP (MT-MMP) is membrane bound and forms a trimolecular complex with tissue inhibitor to metalloproteinases 2 (TIMP-2) to activate MMP-2. This configuration keeps the action of MMP-2 close to the membrane where it can gradually remodel the extracellular matrix around the blood vessel (Candelario-Jalil et al., 2009).
Bacterial meningitis initiates an inflammatory response in the meninges caused by the invading organisms and by the secondary release of cytokines and chemokines. The secondary inflammatory response may aggravate the infection. Cytokines, including tumor necrosis factor (TNF)-α and interleukin (IL)-6, are elevated in the CSF of patients with bacterial meningitis and contribute to the secondary tissue damage. MMPs are increased in bacterial meningitis, and MMP inhibitors (e.g., doxycycline) block the damage secondary to infection (Leib et al., 2001). Steroids suppress the expression of MMPs and other inflammatory mediators. In children, treatment of bacterial meningitis with steroids along with the antibiotic reduces secondary injury. Use of steroids in adults with bacterial meningitis is more controversial. Doxycycline, a tetracycline derivative, suppresses MMP-9 expression and has a beneficial effect in reducing inflammation in meningitis when combined with another antibiotic (Meli et al., 2006).
Cytotoxic Brain Edema
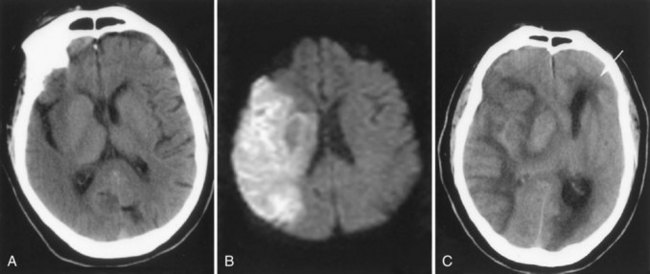
Stroke, trauma, and toxins induce cytotoxic edema. After a stroke, brain water increases rapidly owing to energy failure and loss of ATP. Cytotoxic edema is seen between 24 and 72 hours after the stroke, when the danger of brain herniation is greatest (Fig. 59.5). Damage to the blood vessels, resulting in vasogenic edema, occurs at multiple times after the insult. In brain trauma, there is an early opening of the BBB along with extensive damage to the brain tissue, and a mixture of cytotoxic and vasogenic edema leads to severe brain edema in the early stages after injury. Permanent occlusion of a blood vessel decreases blood flow to the vessel territory, and unless collateral vessels take over, there is infarction of the ischemic tissue. Greater damage occurs in transient ischemia, because the restoration of blood flow returns oxygen and white blood cells to the region, enhancing the damage. Reperfusion injury particularly damages the capillary, with disruption of the BBB seen in two phases: an early opening after several hours and a more disruptive secondary opening after several days. Emboli are more likely to lead to reperfusion injury than thrombosis because the breaking up of the clot can restore blood flow to a previously ischemic region. When that occurs, the risk of hemorrhage is increased (Fig. 59.6). In animal studies of reperfusion injury after stroke, there is a biphasic opening of the BBB, with the first opening within several hours after reperfusion (Rosenberg et al., 1998). The initial opening, which is transient, is related to the activation of MMP-2, which is constitutively expressed and normally found in the latent form. Opening of the tight junctions is seen transiently after the onset of reperfusion, where disruption of tight junction proteins is observed (Yang et al., 2007). A second, more disruptive, phase of injury to the capillary begins around 24 to 48 hours after the onset of reperfusion. This is related to activation of MMP-3 and MMP-9, along with cycloxygenase-2, which are induced from several cell types including microglia/macrophages during the amplification phase of the secondary inflammatory response.
Effect of Blood Pressure and Osmolality Changes on Brain Edema
Rapid elevation of blood pressure causes hypertensive encephalopathy. In experimental animals, hyperemia is present, suggesting that the blood vessels are dilated and have increased permeability. Confusion, focal findings, seizures with papilledema, and increased CSF protein are present in some patients with hypertensive encephalopathy. MRI shows vasogenic edema, primarily in the posterior white matter of the brain (Fig. 59.7), a condition referred to by some as reversible posterior leukoencephalopathy syndrome (Hinchey et al., 1996).
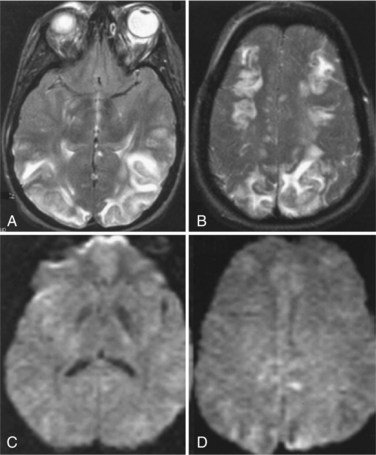
Common causes of rapid elevations of blood pressure are kidney disease, particularly in children with lupus erythematosus or pyelonephritis, and in the pregnancy-induced syndrome of eclampsia. Changes may be transient, and complete recovery is possible if treatment is instituted before hemorrhage or infarction occurs. A characteristic pattern of vasogenic edema without cytotoxic edema is present on MRI: there is extensive edema seen in the white matter, generally in the posterior regions, but spread in frontal regions can be seen, and an absence of DWI lesions indicating this is only vasogenic edema. Absence of signs of ischemia, such as a normal DWI in the face of marked white matter edema, supports a good prognosis for recovery (Covarrubias et al., 2002). Rapid reduction in blood pressure is necessary. The reason for involvement of the posterior circulation is uncertain. Eclamptic patients have visual disturbances due to involvement of the occipital lobes; on postmortem examination, petechial hemorrhages may be seen in the occipital lobes, explaining the visual symptoms.
Another cause of cerebral edema is a rapid change in serum osmolality. For example, rapid reduction of plasma glucose and sodium puts patients treated for diabetic ketoacidosis at risk for edema secondary to water shifts into the brain (Bohn and Daneman, 2002). Long-standing hyperosmolality leads to solute accumulation in the brain to compensate for hyperosmolar plasma levels. These idiogenic osmoles are thought to include taurine and other amino acids. During treatment of the diabetic ketoacidosis, blood osmolality is reduced, and water moves into brain along the osmotic gradient, resulting in cerebral edema. Rapid reduction of serum hyperosmolality, as in diabetic ketoacidosis, should be avoided to prevent brain edema due to the residual idiogenic osmoles (Edge et al., 2001). Dialysis disequilibrium may also be due to an osmotic imbalance that results from urea buildup in brain tissue.
Rapid correction of chronic serum hyponatremia can cause central pontine myelinolysis (Murase et al., 2006). In this syndrome, patients have very low sodium, usually less than 120 mEq/L, secondary to a variety of causes including inappropriate secretion of antidiuretic hormone (ADH), excessive water drinking, anorexia nervosa, alcohol withdrawal, meningitis, and subarachnoid hemorrhage. When there is inappropriate secretion of ADH, serum osmolality is low in the face of high urine osmolality. Treatment involves water restriction. In other patients, there is a salt-wasting syndrome that is treated by careful salt replacement. Low serum sodium can develop over an extended time period and be remarkably well tolerated. Shifts of water during treatment can result in central pontine myelinolysis due to damage to the myelinated tracts, particularly in the brainstem, but extrapontine myelinolysis may also be present.
Cerebral edema is a complication of acute mountain sickness, which in rare circumstances may be life threatening (Wilson et al., 2009). Cerebral symptoms are prominent, and there is an increase in cerebral blood volume related to the hypoxia. Raised ICP causes headaches, ataxia, and confusion. Papilledema has been seen in people with high-altitude cerebral edema. MRI shows changes in the white matter, particularly the corpus callosum, with involvement of the splenium, which may accompany high-altitude pulmonary edema (Yarnell et al., 2000). At high altitude, hypoxia occurs along with extreme exertion; hyperventilation may lead to a drastic reduction in carbon dioxide, resulting in vasoconstriction and cerebral ischemia (Hackett and Roach, 2001). Climbers develop headaches and impaired thinking. Paradoxically, re-breathing carbon dioxide was shown to improve symptoms by reducing vasoconstriction and restoring cerebral blood flow (Harvey et al., 1988).
Edema in Venous Occlusion and Intracerebral Hemorrhage
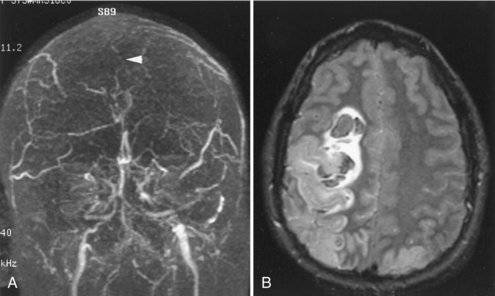
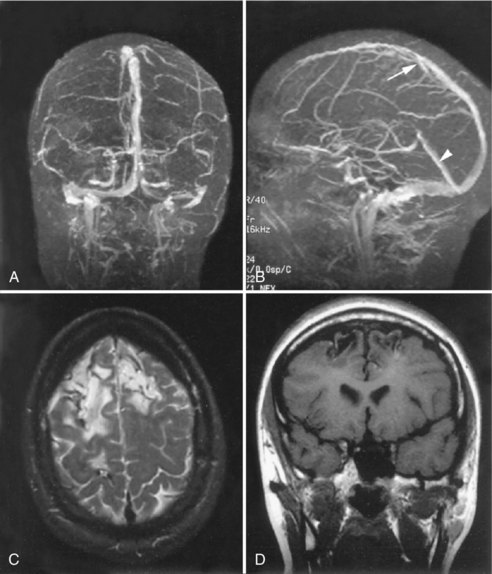
Occlusion of the venous sinuses draining the brain can cause increased ICP and venous hemorrhagic infarction. When the superior sagittal sinus is involved, there may be hemorrhagic infarction in both hemispheres (Fig. 59.8). Dehydration and hypercoagulable states are often found in such patients. Early symptoms may be subtle, with headache due to vessel occlusion or increased ICP. As infarction develops, however, other symptoms such as seizures develop, leading to hemorrhagic conversion of the infarction, herniation, and death. CT scan is usually unhelpful, and MRI may have subtle findings. Definitive diagnosis can be made with an MR venogram showing the occluded veins. Partial occlusions resulting in increased ICP are underdiagnosed. Patients may recanalize the thrombosed superior sagittal sinus and have an excellent outcome (Fig. 59.9). Although still controversial, most studies suggest that anticoagulation of the patient with sagittal sinus thrombosis is indicated even when there is hemorrhage into the brain.
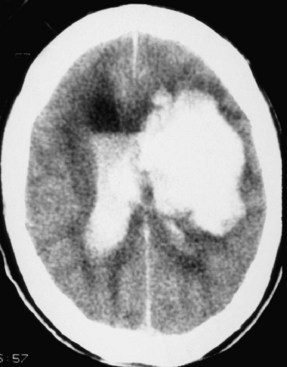
Intracerebral hemorrhage (ICH) causes brain edema around the hemorrhagic mass (Qureshi et al., 2001). This edema is both cytotoxic (direct damage to cells) and vasogenic (inflammatory response induced by toxic blood products). Growth of hematoma was observed after 24 hours in 38% of patients who were imaged within 3 hours of hemorrhage onset and again within 24 hours (Brott et al., 1997). The origin of the intracranial bleeding is obscured by the tissue destruction following the bleed and cellular necrosis. In primary ICH, a vessel ruptures, releasing blood into the brain. Secondary hemorrhage occurs in an area of infarction, particularly when the ischemic region is large. Generally, the hemorrhagic transformation is found 24 to 72 hours after the insult, but occasionally it can be seen relatively soon after the infarct and appear as a primary intracerebral hemorrhage. Since the tissue is massively destroyed, the origin of the blood is difficult to determine.
Primary ICH most commonly occurs in the region of the basal ganglia, where the lenticulostriate arteries are subjected to hypertensive changes. The pons and cerebellum are less common sites (Fig. 59.10). Accumulation of blood causes both mass effect on the surrounding tissues and release of toxic blood products into adjacent tissues. Mass effect can lead to herniation. Several blood products have been shown to cause a secondary inflammatory response that leads to BBB damage and cytotoxic edema. Blood contains coagulation cascade enzymes such as thrombin and plasmin which are pluripotential molecules that can damage cells both directly by their toxic effects and indirectly by activation of other proteases. In experimental animals, injection of thrombin into the brain produces a focal increase in brain water content (Lee et al., 1996). Thrombin stimulates production of hypoxia-inducing factor (HIF)-1α and induces the tumor suppressor gene, which promotes apoptosis (Xi et al., 2006). In addition to proteases, free radicals are thought to be involved in hemorrhagic injury, but evidence of free radical involvement is indirect and comes from studies showing that free radical scavengers and spin trap agents reduce bleeding and improve function in experimental models of ICH (Peeling et al., 1998).
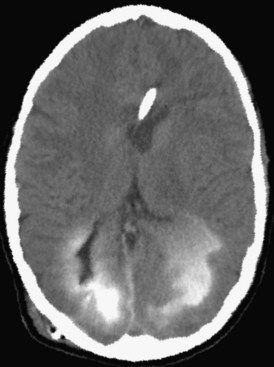
Another cause of intracerebral bleeding is the breakdown of tissue and blood vessels after ischemia. Emboli from extracranial sources produce a region of ischemic injury. When the clot dissolves and blood returns to the damaged areas, there is a high risk of bleeding. This results in a hemorrhagic transformation. If the process is rapid, it can produce a large tissue mass similar to a primary ICH. A recent study in humans showed that treatment with recombinant factor VII reduced the growth of the hemorrhage (Mayer et al., 2005), but initial enthusiasm was dampened when a second study could not confirm the positive results of the first, owing to thrombotic side effects (Diringer et al., 2008). Treatment of ICH is conservative, with control of cerebral edema being most important. In spite of many well-controlled studies of surgical treatment of ICH, no benefit can be found (Qureshi et al., 2009).
Treatment of Brain Edema
Treatment of brain edema has lagged behind the advances in understanding the mechanisms producing the edema (Rabinstein, 2006). Reduction of volume in one of the three compartments may be helpful. Blood volume can be reduced with hyperventilation, which lowers carbon dioxide. However, excessive hyperventilation can cause vasoconstriction and ischemia. Reduction of CSF volume can be done mechanically by placing a drainage catheter into one of the ventricles. This can be difficult when cerebral edema has compressed the ventricular system. Intraventricular drainage is mainly used in patients with head injuries or acute hydrocephalus, or is done postsurgically. Agents that reduce the production of CSF (e.g., acetazolamide, diuretics) may be used but are of marginal benefit.
Idiopathic Intracranial Hypertension
Before the advent of CT or MRI scanners, the complaint of headache and the finding of papilledema raised the suspicion of hydrocephalus or tumor. When tests were negative for either of these conditions, confusing names for the syndrome were invented, which have lead to the use of inappropriate terms for this syndrome. It was first noted that otitis media was at times associated with papilledema that was suspected to be due to hydrocephalus, leading to the pre-imaging term, otitic hydrocephalus (Symonds, 1931). During the era of pneumoencephalography, which was done to show distortion of the ventricles to diagnose hydrocephalus or tumors, the term pseudotumor cerebri was invented to describe patients with papilledema who had neither. More recently, the syndrome has been called benign intracranial hypertension, but when blindness occurs it cannot be considered benign. None of these terms is satisfactory, and the descriptive term idiopathic intracranial hypertension (IIH) is preferred, although through common usage, pseudotumor cerebri has persisted in the literature.
Clinical Features
Patients with IIH have a constellation of symptoms that includes headaches, transient visual obscurations, pulsatile tinnitus, diplopia, and sustained visual loss. Headache is the most frequent symptom; it is the presenting symptom in most patients and is an important reason for searching for papilledema in all headache patients. The pain characteristically wakes the patient from sleep in the early morning hours. Sudden movements such as coughing aggravate the headache. Headaches may be present for months before a diagnosis is made. Some patients complain of dizziness. Transient obscuration of vision occurs when changing position from sitting to standing. Visual fields show an enlarged blind spot due to the encroachment of the swollen optic nerve head. Prolonged papilledema may lead to sector scotomas and, rarely, vision loss when the swollen disc encroaches on the region of the macula. It is important to differentiate papillitis due to inflammation from papilledema due to increased CSF pressure. In the former, vision loss is prominent early in the course and the papillary response is abnormal, whereas with papilledema, the vision is preserved until the late stages when the swollen disc encroaches on the macula. Dysfunction of one or both sixth cranial nerves may occur as an effect of shifts of cerebral tissue. Because the sixth cranial nerve is remote from the site of the process producing intracranial hypertension, the cranial neuropathy is a false localizing sign. The sixth nerve has a long course as it travels to the eye. Before entering the eye socket, it makes a ninety degree turn and goes through the canal of Dorello at the tip of the temporal bone. It is possibly at this site where compression of the abducens nerve could occur (Nathan et al., 1974).
Obesity is often found in women with IIH. Endocrine abnormalities have been extensively investigated in both obese and nonobese subjects, but none has been identified. Drugs associated with the syndrome include tetracycline-type antibiotics, nalidixic acid, nitrofurantoin, sulfonamides, and trimethoprim-sulfamethoxazole (Box 59.2). Paradoxically, the withdrawal of corticosteroids used to treat increased ICP can cause an increase in ICP. Large doses of vitamin A, which are used in the treatment of various skin conditions, may cause the syndrome. Hypercapnia leads to retention of carbon dioxide and increase in blood volume. Sleep apnea and lung diseases may cause headaches and papilledema due to this mechanism. Less frequent causes include Guillain-Barré syndrome, in which increased CSF protein clogs the arachnoid villa, leading to an increase in ICP. Similarly, a cellular response in meningitis may increase CSF pressure by blocking outflow pathways. Uremic patients have an increased incidence of papilledema with IIH. Renal failure patients have increased levels of vitamin A, use corticosteroids, and take cyclosporine, which have all been linked to IIH.
Table 59.3 Terms Used to Describe Different Sites of Inflammation in the CNS
| Infection | Symptoms | Site of Inflammation |
|---|---|---|
| Meningitis | Fever, stiffness, photophobia, headache | Cells confined to subarachnoid space (SAS) |
| Meningoencephalitis | Meningeal symptoms with focal findings | SAS and brain inflammation |
| Encephalitis | Headache, seizures, altered mental state | Multiple sites of cellular response in brain tissue |
| Cerebritis/abscess | Fever, seizures, focal findings | Cerebritis, early collection of inflammatory cells around vessels; abscess is the walled-off stage |
| Subdural empyema | Fever, seizures, coma | Diffuse collection of pus over the surface of the brain between the dura and arachnoid |
Other less well-substantiated causes of elevated CSF pressure include obstruction to venous outflow. Venous pressure measurement has shown high pressure in the superior sagittal sinus and proximal transverse sinuses, with a drop in venous pressure distal to the transverse sinus (King et al., 1995). Angiography does not show this well. In patients without a documented structural defect in the venous sinuses, increased right atrial filling pressure that was transmitted to the venous sinuses has been shown (Karahalios et al., 1996). Whether the high venous pressure and imaging evidence of venous narrowing is the cause or the result of the increased ICP is controversial. Several patients with venous sinus occlusion as the cause of increased ICP have had intravascular stents placed to improve flow. In a series of 12 patients with refractory IIH who had venous pressure gradients, after stenting 7 were improved, with 5 of these becoming asymptomatic and 5 patients being unimproved (Higgins et al., 2002). There are no controlled studies of the efficacy and long-term consequences of placing venous stents in this population of younger patients, and since the normal course is resolution with time, this invasive procedure should be considered experimental until such studies are done.
Treatment
Patients with fulminant IIH are rare but require urgent treatment with acetazolamide, high-dose steroids, and optic nerve fenestration or ventriculoperitoneal shunting. In one study from two institutions, a total of 16 patients were studied, all of whom were women between the ages of 14 and 39 years. All were obese with mean CSF pressures of 541 mm H2O. All had surgical treatment, which reduced headaches and vomiting, but 50% remained legally blind, showing the serious nature of this form of the illness (Thambisetty et al., 2007).
Brain Edema in Idiopathic Intracranial Hypertension
Several studies have suggested the presence of brain edema in patients with IIH. A biopsy showed brain edema in one patient who was subjected to a temporal decompression, a procedure that is no longer done (Sahs and Joynt, 1956). Two recent MRI studies showed edema in the white matter in patients with IIH; there was an increase in white-matter water signal of a heavily T2-weighted imaging sequence obtained at 1.5 T (Gideon et al., 1995). Another study compared diffusion maps of the apparent diffusion coefficient (ADC) in 12 patients fulfilling conventional diagnostic criteria for IIH and in 12 healthy volunteers. They reported a significantly larger ADC within subcortical white matter in the patient group than in the control group, without significant differences within cortical gray matter, the basal nuclei, the internal capsule, or the corpus callosum. In addition, 4 of 7 patients with increased ADC in subcortical white matter also had increased ADC within gray matter (Moser et al., 1988). Another group measured mean diffusivity of water and the proton longitudinal relaxation time in 10 patients with IIH and 10 age-, sex-, and weight-matched controls. They failed to find significant differences in DWI and T1 values between patient and control groups in any of the brain regions investigated, concluding that IIH is not associated with abnormalities of convective transependymal water flow leading to diffuse brain edema (Bastin et al., 2003). Thus, based on the results of MRI studies, there is no consensus as to the presence of brain edema.
Hydrocephalus
Hydrocephalus in Children
Long-standing hydrocephalus may cause atrophy in the white matter surrounding the ventricles but rarely affects the gray matter. When the rate of ventricular enlargement stabilizes in patients with incomplete ventricular obstruction, CSF production is balanced by transependymal absorption (Fig. 59.11). Occasionally a patient escapes detection of hydrocephalus in early life, and an enlarged head is the only sign of an underlying problem. Many years may elapse before the hydrocephalus manifests symptoms, and they may decompensate after many years of stability.
Premature infants weighing less than 1500 g at birth have a high risk of intraventricular hemorrhage, and approximately 25% of these infants develop progressive ventricular enlargement, as shown by CT or ultrasound (Papile et al., 1978). Ventricular size in the neonate may be followed at the bedside with B-mode ultrasound through the open fontanelle. Long-term follow-up studies of children with intraventricular hemorrhage due to prematurity show that 5% require shunting for hydrocephalus. The survivors of a large germinal plate hemorrhage often have multiple disabilities. Angiogenic factors play a role in the development of the hemorrhages (Ballabh et al., 2007).
Adult-Onset Hydrocephalus
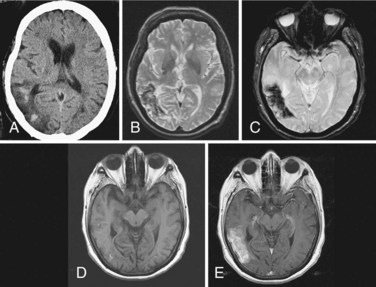
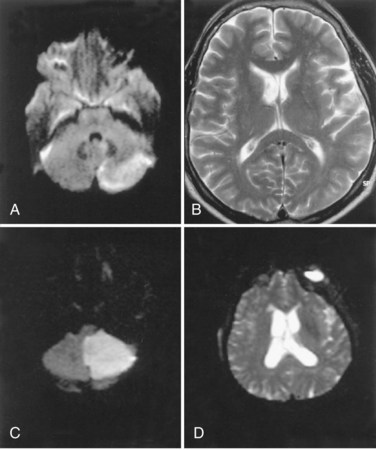
Cerebellar hemorrhage and cerebellar infarction with edema cause an acute hydrocephalus by compression of the brainstem, occluding the cerebral aqueduct and fourth ventricle outflow pathways and causing noncommunicating hydrocephalus and acute elevation in intraventricular pressure. Patients with cerebellar hemorrhage usually have a history of hypertension. Increasing drowsiness and difficulty walking often follow the acute onset of headache. Hemiparesis and brainstem findings evolve after the ataxia, providing a clue that the origin of the problem is in the posterior fossa. The expanding hemorrhagic mass in the posterior fossa, if it is encroaching on the brainstem, requires urgent neurosurgical attention, with placement of a ventricular catheter to decompress the lateral and third ventricles, followed by posterior fossa craniectomy to remove the mass and reduce pressure on the brainstem (Fisher et al., 1965; Ott et al., 1974). In patients with cerebellar infarction, the progression is generally slower, since the maximum swelling takes place in 24 to 48 hours, but the consequences of the enlarging posterior fossa mass are the same as with hemorrhage, and surgery may be necessary to remove the necrotic tissues and restore normal CSF flow. CT is helpful to show enlargement of the ventricle, but MRI is better for imaging the cerebellar infarction (Fig. 59.12).
Adult-onset hydrocephalus that is communicating may be due to a tumor in the basal cisterns, subarachnoid bleeding, or infection or inflammation of the meninges. In the preantibiotic era, syphilis, tuberculosis, and fungal infections were a common cause of hydrocephalus due to chronic obstruction of subarachnoid pathways (McHugh, 1964). CSF cultures are indicated in the elderly patient with enlarged ventricles, and searching for other sources of infection in lungs and other organs may be helpful in establishing the type of infection.
Normal-Pressure Hydrocephalus
Chronic hydrocephalus in the adult can produce symptoms of gait disturbance, incontinence, and memory loss, with or without symptoms and signs of raised ICP including headache, papilledema, and false localizing signs (Adams et al., 1965). Causes of chronic hydrocephalus include post subarachnoid hemorrhage, chronic meningeal infections (e.g., fungal, tuberculosis, syphilis), and slow-growing tumors blocking the CSF pathways.
Normal-pressure hydrocephalus (NPH) is a term commonly used to describe chronic communicating adult-onset hydrocephalus (Marmarou et al., 2005). Typically, patients with NPH have the triad of mental impairment, gait disturbance, and incontinence. NPH can develop secondary to trauma, infection, or subarachnoid hemorrhage, but in about one-third of patients, no etiology is found. Enlarged ventricles are seen on CT, and MRI shows both the enlarged ventricles and transependymal CSF absorption. By definition, LP generally reveals a normal or minimally elevated CSF pressure. Normal pressure is an unfortunate term, because patients who have undergone long-term monitoring with this syndrome have intermittently elevated pressures, often during the night.
Diagnosis of adult-onset hydrocephalus and selection of patients for placement of a ventriculoperitoneal shunt has been difficult. Many of these patients have hypertensive vascular disease with lacunar infarcts. Features of Parkinson disease were noted in earlier reports of the syndrome, and it is now recommended that all patients with Parkinson disease have scans to rule out hydrocephalus. CT and MRI have aided in separating Parkinson disease, lacunar state, and NPH, although NPH may occasionally coexist with these diseases. Patients diagnosed with vascular diseases such as lacunar state or subcortical arteriosclerotic encephalopathy (Binswanger disease) along with the hydrocephalus respond poorly to shunting, and if there is a positive response, it may be transient as the underlying disease progresses (Boon et al., 1999; Tullberg et al., 2002). Selection of patients for shunting requires a combination of clinical findings and diagnostic test results, because no test can totally predict whether a patient will likely benefit from an operation.
There may be a correlation between improvement in gait after a large volume of CSF is removed, which suggests that the patient would benefit from a shunt. Cisternography has been used in diagnosis; it is a procedure that involves injecting a radiolabeled tracer into the CSF, then monitoring its absorption for 3 days. Normally the radiolabeled material fails to enter the ventricles, moving over the convexity of the brain and leaving the CSF space within 12 to 24 hours. In patients with large ventricles due to atrophy, there may be a delay in circulation time, with some isotope being seen in the ventricles during the first 24 hours (Benson et al., 1970). Communicating hydrocephalus with abnormal CSF circulation shows persistent ventricular filling for more than 48 hours. In patients with NPH, there is reflux of the tracer into the cerebral ventricles by 24 hours and retention in the ventricles for 48 to 72 hours. This suggests that transependymal absorption is occurring and that periventricular white matter has become an alternate route of CSF absorption. A positive cisternogram is seen in some patients with hypertensive cerebrovascular disease and Binswanger encephalopathy because of the overlap in the three syndromes.
Infusion of artificial CSF into the lumbar sac at a constant rate shows increased resistance to absorption of CSF, presumably across the arachnoid granulations, in patients with NPH (Katzman and Hussey, 1970). Variations on the infusion test are used, and some investigators report usefulness of the test in making treatment decisions. There is a revival of interest in determining both the resistance to absorption and the compliance or elasticity of the ventricles as an aid in selecting shunt patients.
A large number of patients underwent placement of ventriculoperitoneal shunts after the initial report of NPH and the subsequent enthusiasm that this would cure dementia (Adams et al., 1965). As the number of patients that showed no improvement with shunts grew and the complication rates of placing a shunt in an elderly patient became evident, the number of patients undergoing shunt operations at most centers declined. This trend may be reversing with the improvements in patient selection. However, none of the currently available tests by themselves identifies the patients who will benefit from shunting. Most helpful is a combination of clinical signs and judiciously chosen laboratory tests.
Various success rates for shunt placement have been reported; some reports describe improvement in approximately 80% of treated patients, while others report lower rates. In the early days of treatment of NPH patients with shunts, a high rate of shunt failure occurred, with complications of shunting being a major problem (Katzman and Wells, 1977). Serious complications occurred in as many as one-fourth of the patients, including infection and subdural hematomas. More recently, the rates of correct diagnosis and complication-free treatments have improved, but the definitive diagnostic test and complication-free treatment remain elusive goals (Marmarou et al., 2005). Clearly, more information is needed to aid in the diagnosis and management of patients with this potentially treatable syndrome.
Adami A., Thijs V., Tong D.C., et al. Use of diffusion weighted MRI to predict the occurrence and severity of hemorrhagic transformation in a rabbit model of embolic stroke. Brain Res. 2002;944:32-39.
Adams R.D., Fisher C.M., Hakim S. Symptomatic occult hydrocephalus with “normal” cerebrospinal fluid pressure: a treatable syndrome. N Engl J Med. 1965;273:117-126.
Agre P., Kozono D. Aquaporin water channels: molecular mechanisms for human diseases. FEBS Lett. 2003;555:72-78.
Back S.A., Tuohy T.M., Chen H., et al. Hyaluronan accumulates in demyelinated lesions and inhibits oligodendrocyte progenitor maturation. Nat Med. 2005;11:966-972.
Ballabh P., Xu H., Hu F., et al. Angiogenic inhibition reduces germinal matrix hemorrhage. Nat Med. 2007;13:477-485.
Bastin M.E., Sinha S., Farrall A.J., et al. Diffuse brain oedema in idiopathic intracranial hypertension: a quantitative magnetic resonance imaging study. J Neurol Neurosurg Psychiatry. 2003;74:1693-1696.
Beckman J.S., Beckman T.W., Chen J., et al. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci U S A. 1990;87:1620-1624.
Benson D.F., LeMay M., Patten D.H., et al. Diagnosis of normal-pressure hydrocephalus. N Engl J Med. 1970;283:609-615.
Bohn D., Daneman D. Diabetic ketoacidosis and cerebral edema. [Review] [43 refs]. Curr Opin Pediatr. 2002;14:287-291.
Boon A.J., Tans J.T., Delwel E.J., et al. Dutch Normal-Pressure Hydrocephalus Study: the role of cerebrovascular disease. J Neurosurg. 1999;90:221-226.
Brott T., Broderick J., Kothari R., et al. Early hemorrhage growth in patients with intracerebral hemorrhage. Stroke. 1997;28:1-5.
Candelario-Jalil E., Yang Y., Rosenberg G.A. Diverse roles of matrix metalloproteinases and tissue inhibitors of metalloproteinases in neuroinflammation and cerebral ischemia. Neuroscience. 2009;158:983-994.
Covarrubias D.J., Luetmer P.H., Campeau N.G. Posterior reversible encephalopathy syndrome: prognostic utility of quantitative diffusion-weighted MR images. AJNR Am J Neuroradiol. 2002;23:1038-1048.
Cserr H.F. Physiology of the choroid plexus. Physiol Rev. 1971;51:273-311.
Cunningham L.A., Wetzel M., Rosenberg G.A. Multiple roles for MMPs and TIMPs in cerebral ischemia. Glia. 2005;50:329-339.
Davson H. Physiology of the Cerebrospinal Fluid. London: J. & A. Churchill; 1967.
Diringer M.N., Skolnick B.E., Mayer S.A., et al. Risk of thromboembolic events in controlled trials of rFVIIa in spontaneous intracerebral hemorrhage. Stroke. 2008;39:850-856.
Dirnagl U., Iadecola C., Moskowitz M.A. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22:391-397.
Edge J.A., Hawkins M.M., Winter D.L., et al. The risk and outcome of cerebral oedema developing during diabetic ketoacidosis. Arch Dis Child. 2001;85:16-22.
Endres M., Laufs U., Liao J.K., et al. Targeting eNOS for stroke protection. Trends Neurosci. 2004;27:283-289.
Fisher C.M., Picard E.H., Polak A., et al. Acute hypertensive cerebellar hemorrhage: diagnosis and surgical treatment. J Nerv Ment Dis. 1965;140:38-57.
Fishman R.A. Brain edema. N Engl J Med. 1975;293:706-711.
Furuse M., Hirase T., Itoh M., et al. Occludin: a novel integral membrane protein localizing at tight junctions. J Cell Biol. 1993;123:1777-1788.
Gideon P., Sorensen P.S., Thomsen C., et al. Increased brain water self-diffusion in patients with idiopathic intracranial hypertension. AJNR Am J Neuroradiol. 1995;16:381-387.
Girouard H., Iadecola C. Neurovascular coupling in the normal brain and in hypertension, stroke, and Alzheimer disease. J Appl Physiol. 2006;100:328-335.
Hackett P.H., Roach R.C. High-altitude illness. [Comments.]. N Engl J Med. 2001;345:107-114.
Harvey T.C., Raichle M.E., Winterborn M.H., et al. Effect of carbon dioxide in acute mountain sickness: a rediscovery. Lancet. 1988;2:639-641.
Hawkins B.T., Davis T.P. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev. 2005;57:173-185.
Higgins J.N., Owler B.K., Cousins C., et al. Venous sinus stenting for refractory benign intracranial hypertension. Lancet. 2002;359:228-230.
Hinchey J., Chaves C., Appignani B., et al. A reversible posterior leukoencephalopathy syndrome [see comments]. N Engl J Med. 1996;334:494-500.
Huang P.L., Lo E.H. Genetic analysis of NOS isoforms using nNOS and eNOS knockout animals. [Review] [88 refs]. Prog Brain Res. 1998;118:13-25.
Huang Z., Huang P.L., Panahian N., et al. Effects of cerebral ischemia in mice deficient in neuronal nitric oxide synthase. Science. 1994;265:1883-1885.
Iadecola C., Nedergaard M. Glial regulation of the cerebral microvasculature. Nat Neurosci. 2007;10:1369-1376.
Iadecola C., Pelligrino D.A., Moskowitz M.A., et al. Nitric oxide synthase inhibition and cerebrovascular regulation. J Cereb Blood Flow Metab. 1994;14:175-192.
Janzer R.C., Raff M.C. Astrocytes induce blood-brain barrier properties in endothelial cells. Nature. 1987;325:253-257.
Jian L.K., Rosenberg G.A. Matrix metalloproteinases and free radicals in cerebral ischemia. Free Radic Biol Med. 2005;39:71-80.
Jung J.E., Kim G.S., Narasimhan P., Song Y.S., et al. Regulation of Mn-superoxide dismutase activity and neuroprotection by STAT3 in mice after cerebral ischemia. J Neurosci. 2009;29:7003-7014.
Kalaria R.N., Harik S.I. Reduced glucose transporter at the blood-brain barrier and in cerebral cortex in Alzheimer disease. J Neurochem. 1989;53:1083-1088.
Karahalios D.G., Rekate H.L., Khayata M.H., et al. Elevated intracranial venous pressure as a universal mechanism in pseudotumor cerebri of varying etiologies. Neurology. 1996;46:198-202.
Katzman R., Hussey F. A simple constant-infusion manometric test for measurement of CSF absorption. I. Rationale and method. Neurology. 1970;20:534-544.
Katzman R., Pappius H.M. Brain Electrolytes and Fluid Metabolism. Baltimore: Williams & Wilkins Co; 1973.
Katzman R., Wells C.E. Normal pressure hydrocephalus. In: Dementia. Philadelphia: FA Davis; 1977:69-92.
King J.O., Mitchell P.J., Thomson K.R., et al. Cerebral venography and manometry in idiopathic intracranial hypertension. Neurology. 1995;45:2224-2228.
Klatzo I. Presidential address. Neuropathological aspects of brain edema. J Neuropathol Exp Neurol. 1967;26:1-14.
Lee K.R., Colon G.P., Betz A.L., et al. Edema from intracerebral hemorrhage: the role of thrombin. J Neurosurg. 1996;84:91-96.
Leib S.L., Clements J.M., Lindberg R.L., et al. Inhibition of matrix metalloproteinases and tumour necrosis factor alpha converting enzyme as adjuvant therapy in pneumococcal meningitis. Brain. 2001;124:1734-1742.
Marmarou A. A review of progress in understanding the pathophysiology and treatment of brain edema. Neurosurg Focus. 2007;22:E1.
Marmarou A., Young H.F., Aygok G.A., et al. Diagnosis and management of idiopathic normal-pressure hydrocephalus: a prospective study in 151 patients. J Neurosurg. 2005;102:987-997.
Mattsson N., et al. CSF biomarkers and incipient Alzheimer disease in patients with mild cognitive impairment. JAMA. 2009;302:385-393.
Mayer S.A., Brun N.C., Begtrup K., et al. Recombinant activated factor VII for acute intracerebral hemorrhage. N Engl J Med. 2005;352:777-785.
McHugh P.R. Occult hydrocephalus. QJM. 1964;33:297-308.
Meli D.N., Coimbra R.S., Erhart D.G., et al. Doxycycline reduces mortality and injury to the brain and cochlea in experimental pneumococcal meningitis. Infect Immun. 2006;74:3890-3896.
Milhorat T.H. Choroid plexus and cerebrospinal fluid production. Science. 1969;166:1514.
Moseley M.E., Cohen Y., Mintorovitch J., et al. Early detection of regional cerebral ischemia in cats: comparison of diffusion- and T2-weighted MRI and spectroscopy. Magn Reson Med. 1990;14:330-346.
Moser F.G., Hilal S.K., Abrams G., et al. MR imaging of pseudotumor cerebri. AJR Am J Roentgenol. 1988;150:903-909.
Murase T., Sugimura Y., Takefuji S., et al. Mechanisms and therapy of osmotic demyelination. Am J Med. 2006;119:S69-S73.
Nathan H., Ouaknine G., Kosary I.Z. The abducens nerve. Anatomical variations in its course. J Neurosurg. 1974;41:561-566.
Neuwelt E., Abbott N.J., Abrey L., et al. Strategies to advance translational research into brain barriers. Lancet Neurol. 2008;7:84-96.
Nitkunan A., Charlton R.A., McIntyre D.J., et al. Diffusion tensor imaging and MR spectroscopy in hypertension and presumed cerebral small vessel disease. Magn Reson Med. 2008;59:528-534.
Nitta T., Hata M., Gotoh S., Seo, Y., et al. Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J Cell Biol. 2003;161:653-660.
Noseworthy J.H., Lucchinetti C., Rodriguez M., et al. Multiple sclerosis. N Engl J Med. 2000;343:938-952.
Ott K.H., Kase C.S., Ojemann R.G., et al. Cerebellar hemorrhage: diagnosis and treatment. A review of 56 cases. Arch Neurol. 1974;31:160-167.
Owens T., Bechmann I., Engelhardt B. Perivascular spaces and the two steps to neuroinflammation. J Neuropathol Exp Neurol. 2008;67:1113-1121.
Papile L.A., Burstein J., Burstein R., et al. Incidence and evolution of subependymal and intraventricular hemorrhage: a study of infants with birth weights less than 1,500 gm. J Pediatr. 1978;92:529-534.
Peeling J., Yan H.J., Chen S.G., et al. Protective effects of free radical inhibitors in intracerebral hemorrhage in rat. Brain Res. 1998;795:63-70.
Posner J.B., Plum F. Spinal-fluid pH and neurologic symptoms in systemic acidosis. N Engl J Med. 1967;277:605-613.
Qureshi A.I., Mendelow A.D., Hanley D.F. Intracerebral haemorrhage. Lancet. 2009;373:1632-1644.
Qureshi A.I., Tuhrim S., Broderick J.P., et al. Spontaneous intracerebral hemorrhage. N Engl J Med. 2001;344:1450-1460.
Rabinstein A.A. Treatment of cerebral edema. Neurologist. 2006;12:59-73.
Reese T.S., Karnovsky M.J. Fine structural localization of a blood-brain barrier to exogenous peroxidase. J Cell Biol. 1967;34:207-217.
Rosenberg G.A., Kyner W.T., Estrada E. Bulk flow of brain interstitial fluid under normal and hyperosmolar conditions. Am J Physiol. 1980;238:F42-F49.
Rosenberg G.A., Estrada E.Y., Dencoff J.E. Matrix metalloproteinases and TIMPs are associated with blood-brain barrier opening after reperfusion in rat brain. Stroke. 1998;29:2189-2195.
Sahs A.L., Joynt R.J. Brain swelling of unknown cause. Neurology. 1956;6:791-803.
Schievink W.I., Maya M.M., Louy C., et al. Diagnostic criteria for spontaneous spinal CSF leaks and intracranial hypotension. AJNR Am J Neuroradiol. 2008;29:853-856.
Semenza G.L. Life with oxygen. Science. 2007;318:62-64.
Stewart P.A., Wiley M.J. Developing nervous tissue induces formation of blood-brain barrier characteristics in invading endothelial cells: a study using quail-chick transplantation chimeras. Dev Biol. 1981;84:183-192.
Symonds C.P. Otitic hydrocephalus. Brain. 1931;54:55.
Thambisetty M., Lavin P.J., Newman N.J., et al. Fulminant idiopathic intracranial hypertension. Neurology. 2007;68:229-232.
Tullberg M., Hultin L., Ekholm S., et al. White matter changes in normal pressure hydrocephalus and Binswanger disease: specificity, predictive value and correlations to axonal degeneration and demyelination. Acta Neurol Scand. 2002;105:417-426.
Vannucci S.J., Maher F., Simpson I.A. Glucose transporter proteins in brain: delivery of glucose to neurons and glia. Glia. 1997;21:2-21.
Verkman A.S. Aquaporins: translating bench research to human disease. J Exp Biol. 2009;212:1707-1715.
Wilson M.H., Newman S., Imray C.H. The cerebral effects of ascent to high altitudes. Lancet Neurol. 2009;8:175-191.
Xi G., Keep R.F., Hoff J.T. Mechanisms of brain injury after intracerebral haemorrhage. Lancet Neurol. 2006;5:53-63.
Yang Y., Estrada E.Y., Thompson J.F., et al. Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J Cereb Blood Flow Metab. 2007;27:697-709.
Yarnell P.R., Heit J., Hackett P.H. High-altitude cerebral edema (HACE): the Denver/Front Range experience. Semin Neurol. 2000;20:209-217.
Yong V.W. Metalloproteinases: mediators of pathology and regeneration in the CNS. Nat Rev Neurosci. 2005;6:931-944.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Chapter 59 Brain Edema and Disorders of Cerebrospinal Fluid Circulation
Brain tissue, which is composed of 80% water, is separated from the systemic circulation by a complex series of interfaces. The major site is the endothelial cells that are a component of the neurovascular unit (Iadecola and Nedergaard, 2007). Cells that form these interfaces have specialized proteins that form tight junctions; some have carrier proteins that shuttle essential molecules, and multiple electrolyte pumps on cell membranes. Cellular membranes preserve the compartmental structure with water in extracellular and intracellular spaces. When shifts in water from one compartment to another occur under pathological conditions, swelling in the various compartments leads to increased intracranial pressure (ICP). If the increased water is within the ventricles, hydrocephalus results. Hydrocephalus leads to transependymal flow of water into the periventricular white matter, resulting in interstitial edema. Cytotoxic edema is cell swelling with reduction in the extracellular space; this occurs in ischemic or traumatic injury to the brain, with the loss of nutrients that form the energy stores (Marmarou, 2007). The loss of adenosine triphosphate (ATP) that drives chemiosmotic work through ATPase-mediated membrane pumps causes the cellular swelling. A disturbance in the cerebral blood vessels, which produces leakage of serum proteins across the damaged vessels, leads to vasogenic edema. Each of these shifts in water from its normal compartment into another alters ICP.
In the past several years, new information emerged describing the molecular events underlying the changes in water and proteins within the fluid spaces of the brain. A pore-forming molecule, aquaporin, was discovered; it facilitates movement of water along osmotic gradients (Agre and Kozono, 2003; Verkman, 2009). Novel molecules, such as hypoxia inducible factors (HIF), have been discovered that contribute to inflammation secondary to hypoxic/ischemic insults and brain trauma (Semenza, 2007). Cytokines and free radicals amplify the tissue damage. Advances in imaging modalities, including computed tomography (CT) and magnetic resonance imaging (MRI), have improved the diagnosis of cerebrospinal fluid (CSF) disorders and brain edema. Although we understand the underlying molecular processes involved in edema formation and have better ways of observing its evolution, we remain behind in our attempts to treat brain edema, which remains a major challenge.
Examination of the CSF by lumbar puncture (LP) can provide unique information, aiding diagnosis and patient management. Increased ICP can only be determined by measurements made during removal of CSF; this information is critical in the diagnosis of raised CSF pressure in idiopathic intracranial hypertension. Studies of cells and proteins in the CSF provide information about infection and inflammation. Cancer cells can be detected and antibodies to infectious agents identified. When the BBB is disrupted, increased proteins, mainly albumin, appear in the CSF. Diagnosis of multiple sclerosis (MS) is strengthened by the detection of myelin basic protein and immunoglobulin (Ig)G endogenous production. Alzheimer disease leads to alterations in the amyloid protein, Aβ1-42, and tau proteins that can aid in early diagnosis (Mattsson et al., 2009). Thus, LP to obtain CSF is one of the most cost-effective procedures in daily clinical practice, and when done correctly, it can obtain important information only available from CSF.
The recognition that the total volume of fluid and tissue contained within the skull is constant is called the Monro-Kellie doctrine, named after the two early anatomists. Changes in volume of blood, CSF, or brain compartments produce compensatory changes in the others, with a resultant increase in CSF pressure. When CSF outflow pathways are blocked, enlargement of the ventricles or hydrocephalus follow, resulting in a buildup of pressure in the ventricles that forces the CSF to move transependymally into the periventricular white matter. Masses enlarge the tissue space and compress CSF and blood spaces. When the compensatory mechanisms are overwhelmed, ICP increases. Disruption of the blood vessels leads to vasogenic edema that moves through the more compliant extracellular space of the white matter. On the contrary, damaged swollen cells lead to cytotoxic edema, with narrowing of the extracellular space. Finally, an increase in blood volume, as seen in hypercapnia and hypoxia, increases the ICP (Table 59.1).
Table 59.1 Causes of Increased Intracranial Pressure
| Site of Increased Intracranial Pressure | Diseases |
|---|---|
| Increased tissue volume | Tumor, abscess |
| Increased blood volume | Hypercapnia, hypoxia, venous sinus occlusion |
| Cytotoxic edema | Ischemia, trauma, toxins, metabolic diseases |
| Vasogenic edema | Infections, brain tumors, hyperosmolar states, inflammation |
| Interstitial edema | Hydrocephalus with transependymal flow |
Blood-Brain Interfaces
Cerebral Blood Vessels and the Neurovascular Unit
The total surface of the capillary endothelial cells forms the major interface between the blood and brain. Other, less extensive, interface surfaces include choroid plexuses and arachnoid granulations (Table 59.2). At each of the BBB interfaces, high-resistance junctions between cells, which make the surface into an epithelial-like structure, restrict bulk transport. The epithelial sheets impede non–lipid-soluble substances, charged substances, or large molecules, whereas lipid-soluble substances, such as anesthetic gases, pass easily through the cells. Water has an anomalous structure that allows it to pass rapidly through endothelial cells but with slight restrictions.
Table 59.2 Characteristic Features of the Blood-Brain Interfaces
| Interface | Tight-Junction Location | Functional Aspects |
|---|---|---|
| Blood-CSF | Choroid plexus cell | Active secretion of CSF via ATPase and carbonic anhydrase |
| CSF-blood | Arachnoid membrane | Arachnoid granulations absorb CSF by one-way valve mechanism |
| Blood-brain | Capillary endothelial cell | Active transport of ISF via ATPase; increased mitochondria and glucose transporters in capillary endothelial cells |
ATPase, Adenosine triphosphatase; CSF, cerebrospinal fluid; ISF, interstitial fluid.
In 1925, Cushing and Weed described the pathways involved in the movement of CSF and ISF through the ventricles and around the brain, recognizing that the CSF/ISF acted as a lymph-like fluid in brain. They compared that circulation to those of the blood and lymph. The importance of this circulation has grown with the discovery that ISF is formed by cerebral blood vessels, which have electrolyte pumps that make fluid in a fashion similar to that of the epithelial cells. Flowing around cells, ISF brings nutrients such as glucose and oxygen to neurons and astrocytes and removes the products of metabolism. ISF is absorbed either into the blood via terminal capillaries and venules or into CSF for eventual absorption through the arachnoid granulations (Fig. 59.1).
Brain extracellular space comprises 15% to 20% of the total brain volume. Complex carbohydrates are found in the extracellular space, including hyaluronic acid, chondroitin sulfate, and heparan sulfate. Hyaluronic acid forms large water domains. These large extracellular matrix glycoproteins impede cell movement. After an injury, astrocytes secrete an extracellular molecule, hyaluron, which impedes movement of fluids in the extracellular space, slowing tissue repair. Treatment with hyaluronidase reduces hyaluron and improves regrowth of injured fibers (Back et al., 2005).
Proteases are secreted during development, angiogenesis, and neurogenesis to clear a path for the growing cells, similar to the secretion of proteases by spreading cancer cells (Candelario-Jalil et al., 2009; Yong, 2005). An important concept that has emerged over the past few years is that of the neurovascular (or gliovascular) unit. Neurons, astrocytes, and pericytes comprise the neurovascular unit (Neuwelt et al., 2008). On the abluminal surface of the endothelial cells is a thin layer of basal lamina composed of type IV collagen, fibronectin, heparan sulfate, laminin, and entactin. Entactin connects type IV collagen and laminin to add a structural element to the capillary. Fibronectin from the cells joins the basal lamina to the endothelium. Basal lamina provides structure through type IV collagen, charge barriers by heparan sulfate, and binding sites on the laminin and fibronectin molecules. Within the basal lamina reside the pericytes, which are a combination of smooth muscle and macrophage. Astrocyte foot processes surround the basal lamina. Glia limitans is found at the pial surface and at the interface between astrocytes and blood vessels (Owens et al., 2008). Neurons complete the group of cells that comprise the neurovascular unit (Girouard and Iadecola, 2006) (Fig. 59.2).
Cerebral blood vessels have very low permeability and high electrical resistance, making them more similar to epithelial cells than systemic capillaries, which are passive structures with low electrical resistance and fenestrations that permit passage of large protein molecules. In addition, cerebral blood vessels have highly selective molecular transport properties. During development, cerebral blood vessels acquire the characteristics that distinguish them from systemic capillaries. Astrocytes are critical in this differentiation process, which involves interactions between blood vessels and astrocytes. The critical nature of the astrocytes in this process was shown in transplantation studies involving chicken and quail cells, which can be separated histologically. Quail brain grafts from 3-day-old quails transplanted into the coelomic cavity of chick embryos become vascularized by chick endothelial cells and form a competent BBB. On the other hand, when avascular embryonic quail coelomic grafts are transplanted into embryonic chick brain, chick endothelial cells form leaky capillaries and venules (Stewart and Wiley, 1981). Subsequent studies showed that astrocytes induced BBB properties in non-neural endothelial cells in vivo (Janzer and Raff, 1987).
Electron microscopic studies with electron-dense tracers identified a major site of the BBB as the endothelial tight junctions (Reese and Karnovsky, 1967). Isolation of tight-junction proteins and cloning of the molecules permitted production of antibodies that identified their location. Zona occludins tether the tight-junction proteins to actin within the endothelial cells (Hawkins and Davis, 2005). Occludin and claudin form the actual tight junctions within the endothelial clefts (Furuse et al., 1993). Occludin attaches to the zona occludins, while claudins attach to occludin and protrude into the clefts between cells. The extracellular tails of claudins from adjacent cells self-assemble to form the tight junctions that are “zip-locked” together (Nitta et al., 2003).
Tight junctions between the endothelial cells create the unique membrane properties of the cerebral capillaries by greatly increasing the electrical resistance, which blocks transport of non–lipid-soluble substances (Box 59.1). Brain tissue has a very high demand for glucose and essential amino acids, which can be met by specialized molecules that transport glucose and amino acids across the BBB. Glucose transporters are densely distributed in the capillaries. At low levels of blood glucose, the carriers function at full capacity to meet metabolic needs, but at higher levels of blood glucose, the carriers are saturated and transport is dominated by diffusion rather than active transport. Several isoforms of the glucose transporter molecule have been isolated and cloned (Vannucci et al., 1997). High concentrations of one isoform, GLUT1, are found on cerebral blood vessels. GLUT3 is found on neurons and GLUT5 in microglia. GLUT2 is found predominantly in the liver, intestine, kidney, and pancreas. Impairment of glucose transport due to lack of transporters has been described in patients with Alzheimer disease and in other neurological disorders (Kalaria and Harik, 1989). Amino acid transporters carry essential amino acids into the brain. Competition for the amino acid transporters can lead to a deficiency state; serotonin uptake is decreased in patients with phenylketonuria, which competes for the transporter.
Box 59.1 Unique Features of Cerebral Capillaries
Tight junctions create high electrical resistance
Adenosine triphosphatase pumps on abluminal surfaces form interstitial fluid
Increased numbers of mitochondria for high-energy needs
Glucose transporters and amino acid carriers
Basal lamina contributes to the barrier
Steady-state levels of brain electrolytes are preserved by transport mechanisms at the BBB. Potassium is maintained at a constant level in the CSF and brain by the BBB (Katzman and Pappius, 1973). This prevents fluctuations of electrolyte levels in the blood from influencing brain levels. Calcium is similarly regulated. Glutamate, which is an excitotoxin, is excluded from the brain. Highly lipid-soluble gases such as carbon dioxide and oxygen are rapidly exchanged across the capillary. Anesthetic gases are effective because they readily cross the BBB and enter the brain.
Different rates for equilibration of various substances between blood and brain can cause paradoxical clinical situations. For example, to compensate for a metabolic acidosis, bicarbonate levels fall in both the blood and the brain. Metabolic acidosis is balanced by a respiratory alkalosis due to lowering of carbon dioxide by hyperventilation, which compensates for the acidosis. Carbon dioxide is reduced in both the blood and CSF compartments, since it readily crosses the BBB, while bicarbonate is much more slowly exchanged between the two compartments. This adjustment results in a stable, albeit pathological, situation. However, when the metabolic acidosis is corrected by intravenous infusion of bicarbonate, there is a rapid adjustment of Pco2 as the hyperventilation stops and CO2 builds up. Bicarbonate adjusts very slowly because of the limited transport across the BBB, and the CO2 entering the brain causes a further fall in brain pH. This dangerous situation continues until the bicarbonate levels in the brain rise. Although treatment is necessary to correct the metabolic acidosis, patients may become worse due to brain acidosis if treatment is too rapid (Posner and Plum, 1967).
Production of Cerebrospinal Fluid and Interstitial Fluid
While choroid plexuses are the major source of CSF, capillaries contribute ISF to varying degrees, depending on the species. Production of CSF is constant across species when the volume of fluid formed is divided by the weight of the choroid plexus (Cserr, 1971). In humans, the volume of CSF in the ventricles is 140 mL, with a rate of CSF production of 0.35 mL/min or about 500 mL/day. Obstructive hydrocephalus can rapidly cause life-threatening symptoms when the rate of production remains constant but there is no absorption. On the other hand, removal of 20 mL of CSF at the time of LP for the treatment of idiopathic intracranial hypertension does not make physiological sense (although it seems to help at times, which may be due to the hole placed in the dura, with a slow leak of CSF).
CSF production occurs at both choroidal and extrachoroidal sites, and estimates of the proportion of CSF from each site vary, depending on the species and the method of measurement. Extrachoroidal production accounts for 30% of total CSF production in the cat (Rosenberg et al., 1980) and approximately 60% in the nonhuman primate (Milhorat, 1969). No measurements of the relative proportion of CSF produced at each source have been made in humans.
Water Molecules: Basis for Magnetic Resonance Imaging
Resonance signals detected by MRI are from water molecule protons. Since water is the most abundant source of protons in the brain, water protons dominate the signals. New rapid-acquisition pulse sequences are fast enough to show diffusion of water. Cytotoxic edema shrinks the extracellular space and restricts the diffusion of water (Moseley et al., 1990). The ability to monitor water diffusion by MRI has greatly improved our ability to diagnose an acute ischemic event. Water diffusion between cells in the extracellular space occurs normally. When there is cellular swelling and the extracellular space shrinks, the diffusion of water slows, and the apparent diffusion coefficient (ADC) shows a loss of signal, which appears black on the image. The diffusion-weighted image (DWI) has a bright signal. Because the DWI may show T2 shine-through that will be misinterpreted as reduced diffusion, both a darkened ADC and a bright DWI should be seen in the region of the infarct. In cerebral ischemia, the DWI is abnormal within minutes after the onset of the ischemia, making this an excellent diagnostic test for the presence of cerebral ischemia (Adami et al., 2002).
Diffusion tensor imaging (DTI) reveals the patterns of white matter tracts in three dimensions. Taking advantage of the directional flow of water protons along white matter, diffusion is measured in three planes, and the separate pathways for water movement between the fibers are traced. In patients with white-matter pathology, such as in vascular cognitive impairment and MS, injury patterns in the white matter can be revealed by DTI (Nitkunan et al., 2008).
Anatomical Sites of Central Nervous System Infection
The terminology used to describe various types of central nervous system (CNS) infections is anatomically based (Table 59.3). An infection limited to the subarachnoid space, with inflammation of the meninges, is called meningitis. Meningeal signs of headache, stiff neck, and photophobia are present without focal findings that would indicate spread into the parenchyma. When the infection spreads contiguously from the subarachnoid space through the pial surface or along Virchow-Robin spaces, crossing the gap-junctioned linings, the brain parenchyma is infected, and the term meningoencephalitis is used. In addition to meningeal signs, there are focal findings and possibly impaired consciousness and seizures. An infection in the brain tissue that is most likely spread via blood begins as a loose collection of invading cells referred to as a cerebritis; walling off of the infected brain tissue leads to an abscess. Finally, the term encephalitis is used to describe a more diffuse brain infection in both the gray and white matter, which is usually indicative of a viral infection. Occasionally the infection spreads in a potential space beneath the dura but outside the arachnoid; subdural empyema describes a life-threatening collection of pus over the brain surface that has often spread from an infected sinus through the venous plexus of the ethmoid or sphenoid sinuses into the subdural space. The presence of a subdural empyema should be suspected in a patient with sinus infection, fever, seizures, focal findings, and altered consciousness. Diagnosis of meningitis can be done by examination of CSF for signs of infection such as increased white blood cells or protein. Infections that invade the brain are best diagnosed with MRI, which can readily demonstrate a meningoencephalitis, cerebritis, abscess, or encephalitis. Use of contrast agents increases the potential of reaching a correct diagnosis based on site of infection. Subdural empyema is the most difficult condition to diagnose because it may only be a thin layer of pus on the surface of the brain and be obscured by the skull. Diagnosis can be missed on LP or CT.
Arachnoid Granulations and Absorption of Cerebrospinal Fluid
Arachnoid granulations (pacchionian granulations) are the major sites for the drainage of CSF into the blood. They protrude through the dura into the superior sagittal sinus and act as one-way valves. As CSF pressure increases, more fluid is absorbed. When CSF pressure falls below a threshold value, the absorption of CSF ceases (Fig. 59.3). In this way, CSF pressure is maintained at a constant level, with the rate of CSF production as one determining factor.
Cerebrospinal Fluid Pressure
The opening CSF pressure is measured with a manometer attached to the needle. Normal CSF pressure ranges from 80 to 180 mm H2O but may go as high as 200 mm H2O in obese patients or those who are not relaxed. Three components contribute to the measured pressure: volume of blood within the cranial cavity, amount of CSF, and the brain tissue. The CSF pressure recorded by the manometer represents the venous pressure transmitted from the right side of the heart through the venous sinuses. Small fluctuations from the cardiac systolic pulse and larger fluctuations from respirations can be seen in the column of fluid in the manometer. Pulsations in the manometer represent the fluctuations in the thin-walled veins (Davson, 1967). Arteries have thick elastic walls that dampen the pulsations from arteries. Deep respirations cause wide fluctuations in the CSF pressure, whereas changes in arterial pressure are barely visible. As ICP rises, tissue compliance falls and reserve capacity of the intracranial contents is lost. When tissue compliance is lost, small changes in fluid volume may lead to large increases in ICP.
In addition to complications of elevated CSF pressure, there are circumstances that lead to low pressure. Some patients have low pressure after LP owing to a persistent tear in the dura. Headaches occur with standing up and are often relieved with bedrest. Low-pressure headaches after LP generally resolve spontaneously. However, rarely they persist and require placement of a epidural blood-patch, which is accomplished by injecting the patient’s own blood to close the hole in the dura. Postsurgical and posttraumatic leakage of CSF can cause low-pressure headaches, and occasionally a spontaneous tear occurs. Diagnosis of a spontaneous leak can at times be difficult and may require injection of radiolabelled substances or contrast agents into the CSF (Schievink et al., 2008). Enhancement of the meninges can be seen on MRI in patients with low CSF pressure, but the cause of this abnormality is uncertain.
