CHAPTER 8 Blood-Brain Barrier
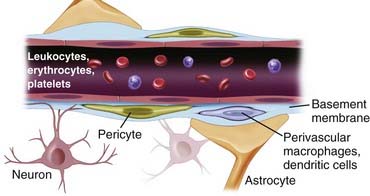
The blood-brain barrier (BBB) is a “neurovascular unit” composed of microvascular endothelium, basement membrane, neurons, and neuroglial structures: astrocytes, pericytes, and microglia. More recently, it has become apparent that in human brain pathology, the BBB also interacts substantially with intravascular signals and circulating blood cells. In this respect, it becomes clear that despite its location at the blood-brain interface, the potential impact and topography of BBB cells are more widespread than initially believed. For example, the interaction of circulating white blood cells infected by human immunodeficiency virus (HIV) has been shown to have an impact on BBB function; in contrast, the BBB in patients with acquired immunodeficiency syndrome seems to act as a reservoir for the virus, thus further extending the reach of this cellular interface.1,2 The BBB is an active and dynamic organ that ensures adequate concentration of essential compounds such as oxygen and glucose and at the same time protects the brain from deleterious substances in the peripheral circulation. The BBB selectively prevents transportation of substances into brain via tight junctions (TJs), enzymatic reactions, and neurotransmitter signaling and selectively transports small and large molecules by passive and facilitated diffusion and active transport. The synergistic integration of all molecular and structural components gives rise to this functional complex called the BBB. Disruption of the BBB is seen in numerous pathologic processes. However, the discriminatory nature of the neurovascular unit also prevents the delivery of therapies to the brain, including chemotherapy agents, antiviral drugs, and beneficial neuromodulators. There are novel methods of circumventing the BBB that may provide novel therapies to treat a variety of neurological disorders. Scientific investigation of the BBB continues to provide insight into this complex and dynamic system and may generate much needed therapies to treat numerous neurological diseases.
History
Scientific investigation in identifying the BBB dates back to the 19th century. In 1885, Paul Ehrlich, a bacteriologist, observed that aniline dyes intravenously injected into animals colored all organs with the exception of the brain and spinal cord.3 His interpretation at the time was that there was poor uptake of the dye by the brain. Later, one of Ehrlich’s protégés, Edwin Goldmann, injected trypan blue intravenously and was able to visualize the dye in the choroid plexus and meninges but not the brain itself.4 However, when he injected trypan blue into cerebrospinal fluid (CSF), the dye was present throughout the brain, although it was absent in the rest of the body.5
In the 1920s, experiments performed by Stern and Gautier led to greater understanding of the blood-CSF barrier. They studied the transport of substances from blood into CSF. Chemicals such as bromide, bile salts, and morphine injected into the bloodstream were found in CSF, whereas fluorescein and epinephrine were absent, even though they were administered in the same fashion.6 Moreover, substances that entered the brain affected its activity and substances that were unable to penetrate the brain had no functional consequence.7 They coined this semipermeable protection of agents entering the brain “barrière hématoencéphalique.”
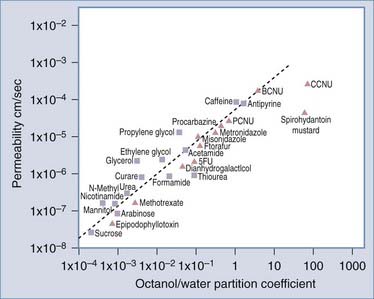
A series of investigations in the 1960s led to the identification of properties of molecular compounds that facilitated transport across the BBB, such as lipid solubility.8 Moreover, a gradient existed between extracellular fluid in the brain and CSF. This allows substances to be filtered out of the brain through CSF, termed the sink effect.9 More recently, it was shown that molecular transport of substances across the BBB could be determined from their log octanol-water partition coefficients (Fig. 8-1).10 A plot of log BBB permeability (cm/sec) versus log octanol-water partition coefficient shows that increased lipophilicity directly correlates with increased membrane permeability in that the more lipid soluble a molecule is, the more readily it moves from the aqueous environment of blood across the lipid environment of the endothelial cell (EC) membrane and enters the brain. Compounds subject to active transport will exceed their predicted permeability based on membrane lipophilicity. Counteracting influences that may slow diffusion across the BBB include pH, temperature, and retention in blood because of protein binding. As a general rule, lipid-soluble molecules with a molecular size less than 400 daltons can cross the BBB. Unfortunately, few central nervous system (CNS) diseases respond to small-molecule drugs.
Finally, between 1965 and 1967, a number of scientists identified the structure of the BBB as consisting of a network of capillaries and ECs known as TJs.11–13
Anatomy of the Blood-Brain Barrier
The anatomic BBB is formed by a monolayer of microvascular ECs that line the intraluminal space of brain capillaries. The BBB consists of ECs packed close together and forming TJs. The EC layer has a luminal (inside) and abluminal (outside) compartment separated by 300 to 500 nm of cytoplasm between the blood and brain. The EC layer is composed of TJs, which consist of occludin and claudin; adherent junctions, including cadherin, catenins, vinculin, and actinin; and junctional adhesion molecules (Fig. 8-2).
In addition to the structural integrity of the BBB, there exists an enzymatic surveillance system that metabolizes drugs and other compounds bypassing the structural barrier. Three main catalytic agents regulate transportation across the BBB: γ-glutamyl transpeptidase (γ-GTP), alkaline phosphatase, and aromatic acid decarboxylase. All are highly concentrated in cerebral vessels.14
There is charge polarity between the abluminal and luminal surface of ECs. This polarity influences permeability of the barrier. Alkaline phosphatase and γ-GTP are concentrated on the luminal compartment, whereas sodium-potassium adenosine triphosphatase (Na+,K+-ATPase) and other transporters are clustered on the abluminal side. Other shuttling proteins that contribute to transport polarity include glucose transporter-1 (GLUT-1), which is concentrated at the abluminal membrane,15 and the drug transporter P-glycoprotein (Pgp), which is concentrated at the luminal membrane.16
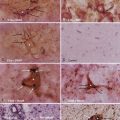
The tissue microenvironment is necessary for continued regulation of barrier function. The BBB, also known as the neurovascular unit, consists of astrocytes, pericytes, microglia, neurons, and the extracellular matrix (ECM), all of which play a supportive role in maintaining integrity of the BBB.17,18 Astrocytes have end-feet that border the basement membrane of vessels of the parenchyma. More than 90% of astrocyte foot processes surround ECs.19 They are associated with adrenergic and cholinergic nerve terminals, as well as those that respond to peptides (Fig. 8-3).
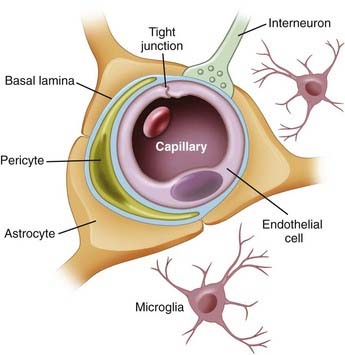
Astrocytes also densely surround TJs and augment ECs by reducing the size of the gap of TJs.20 In vitro experiments imply that without astrocytes, the integrity of the BBB is significantly compromised.20,21 In contrast, other studies indicate that BBB integrity is retained amid degradation of astrocytes,22 thus suggesting that astrocytes regulate BBB activity indirectly rather than through physical obstruction. Astrocytes are considered to be inducers of both the barrier and permeability properties of the endothelium. Stewart and Wiley in 1981 first demonstrated that newly formed vessels originating from the coelomic cavity display BBB characteristics when placed in contact with grafts of neural tissue.23 Later, Janzer and Raff first demonstrated that a functional BBB was induced in nonbrain ECs in the anterior chamber of the eye in the presence of astrocytic aggregates.24
Pericytes are undifferentiated contractile connective tissue cells that localize to capillary walls and share a common basement membrane with brain ECs. They may not be involved in vessel contraction because they lack a contractile actin subtype.25 In vitro studies have revealed communication between ECs and pericytes. The proposed mechanism of this communication is through cellular projections, which penetrate the basal lamina and cover 20% to 30% of the microvascular circumference.25 Pericytes express macrophage functions and are actively involved in the immune response, where they operate as a second line of defense at the BBB. Pericytes are the most abundant on venules, for which they provide mechanical support and also synthesize ECM proteins such as laminin and fibronectin. Platelet-derived growth factor receptor (PDGFR) is a tyrosine kinase receptor expressed on the surface of pericytes that has been targeted for the treatment of malignant brain tumors. Clinical trials were conducted with imatinib, a PDGFR inhibitor, on patients with glioblastoma multiforme (GBM) who were refractory to chemotherapy and radiation therapy. Patients treated with imatinib and hydroxyurea had a 20% response rate, and the drug combination was reasonably well tolerated in phase II studies.26,27 Pathologic conditions that increase BBB permeability, such as trauma or hypoxia, result in a significantly decreased pericyte concentration as they migrate away from the site of injury.28
Neurons are the building blocks of the CNS. The role of neuronal modulation at the BBB is principally enzymatic (Table 8-1). Functional brain imaging studies, such as positron emission tomography (PET) and functional magnetic resonance imaging (MRI), are based on regional increases in cerebral blood flow and glucose and oxygen consumption, which are associated with regional increases in neuronal activity.29 Neurons upregulate catalytic factors specific to ECs.30 Astrocytes and their associated ECs are innervated by noradrenergic,31 serotoninergic,32 cholinergic,33 and GABAergic (transmitting or secreting γ-aminobutyric acid [GABA]) neurons.34 Lesions of the norepinephrine-producing locus caeruleus sensitize the BBB to hypertension. In Alzheimer’s disease, cholinergic inhibition impairs cerebrovascular blood flow.33,35
|
Decreased Blood-Brain Barrier Permeability |
Adapted from Abbott NJ. Dynamics of CNS barriers: Evolution, differentiation, and modulation. Cell Mol Neurobiol. 2005;25:5-23.
Microglia serve as surveillance cells for the BBB. They identify foreign compounds that have bypassed the BBB and act as antigen-presenting cells by engulfing these substances and presenting them to activated T cells for destruction. Microglia also secrete cytokines, or proinflammatory molecules, and rapidly proliferate to contain the offending agent.15
The ECM provides physical stability to the BBB. It is a critical anchoring site that mediates polarity at the EC-astrocyte interface. Disruption of the ECM predictably impairs the structural integrity of the BBB, which in turn compromises its activity. Structural integrity of the BBB is achieved through interaction with several structural proteins, including laminin, collagen type IV, and integrins.36 Matrix proteins also upregulate TJ protein expression.37
The permeability of the BBB to macromolecules is determined by both TJ-controlled paracellular permeability (through cell-cell junctions) and caveolae-mediated transcellular permeability. Caveolae are sites of endothelial transcytosis, endocytosis, and signal transduction. The relationship between paracellular and transcellular permeability is of crucial importance for the regulation of transendothelial permeability. Using an electron microscope, Majno and colleagues found that carbon particles injected into blood entered the parenchyma after brain tissue had been exposed to histamine.38 In addition, these authors were able to see gaps between ECs. The concept of osmotic control of the BBB was also based on electron microscope studies, in which it was shown that the nuclei of ECs seemed to have a contracted, raisin-like appearance after exposure to histamine.39 This method of osmotic regulation of the BBB has since been further described.40
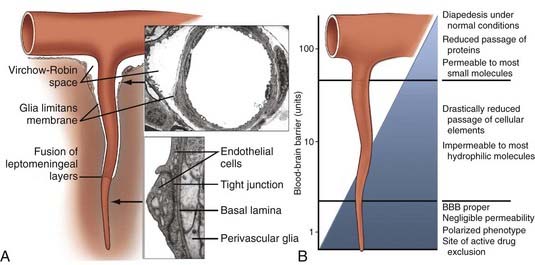
The precise vascular localization of the functional term BBB might be extended beyond the capillary segments to CNS microvessels (Fig. 8-4). The average total surface area of the brain microvasculature is 20 m2, whereas the surface area of cerebral capillary endothelium is 100 cm2/g tissue.41 The total length is 650 km, the inner capillary lumen is 6 µm, and capillaries are 20 µm apart from one another. The BBB occupies more than 99% of brain capillaries, with the exception of the circumventricular organs, which have a blood-CSF barrier. Circumventricular organs include the median eminence, pituitary gland, choroid plexus, subfornical organ, lamina terminalis, and area postrema. Although not as stringent as the BBB, the blood-CSF barrier prevents blood-borne substances from entering the brain. Other mechanisms of controlling traffic across the BBB besides the structural support include ion channels and transport carriers, which control the traffic of hydrophilic nutrients, metabolites, vitamins and hormones, and ions across the BBB.
Transport Across the Blood-Brain Barrier
The biochemical BBB is established by transport systems of the BBB, which can be grouped into four types (Fig. 8-5):
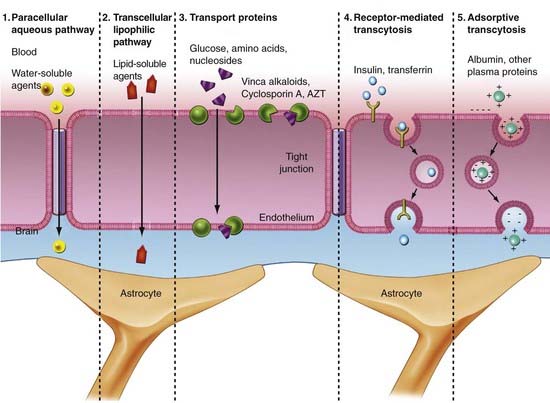
Glucose
The glucose transporter at the BBB is of special importance because glucose is the primary source of energy for the brain and is required for normal brain activity and function. Transportation across the BBB occurs via a glucose transporter. There are five members of the sodium-independent glucose transporters in the brain, including GLUT-1 (ECs), GLUT-3 (neurons),43 and GLUT-5 (microglia).44 Each transports 2-deoxyglucose, 3-O-methylglucose, mannose, galactose, and glucose across the membrane.44 GLUT-1 is a 45- to 55-kD protein, depending on its glycosylation state. It is present in high concentration in the ECs of arterioles, venules, and capillaries and facilitates movement of d-glucose enantiomers from the peripheral circulation to the brain.45 Although the BBB is small, glucose transport is the rate-limiting step of primary energy acquisition. GLUT-1 expression is three to four times higher on the abluminal membrane and is altered with processes such as diabetes, epilepsy, trauma, and tumors.
Amino Acids
Delivery of amino acids across the BBB is achieved by carrier-mediated transport across the abluminal and luminal membranes in both a concentration-dependent and stereospecific manner.46 Factors affecting amino acid uptake include plasma concentration, affinity of the transport system for a particular amino acid, and competition among amino acids for a particular transport system. Amino acid transport is classified into four systems based on seminal experiments conducted on the BBB and amino acid transport.47 The first group is the large, neutral-charged amino acid transporter (L type), which has a preference for leucine residues. These transporters are sodium independent, saturable, and stereospecific. The rate of transfer across the BBB is high because of the high requirement for this subtype of amino acids by the brain. The second group of transporters consists of small, neutral-charged amino acids (A type). These transporters are dependent on sodium for movement across the BBB. Independent transporters for acidic and basic amino acids make up the third transport system. The rate of transfer of basic amino acids is high because of a high requirement for this subtype by the brain. Finally, alanine, serine, cysteine, and threonine residues use a sodium-dependent transport system that shuttles small neutral amino acids across the BBB. Investigation of this transport system has suggested that its primary role is to transport amino acids out of the brain.48
Glutamate is the most abundant amino acid in the CNS and is stored intracellularly against its concentration gradient. It is an excitatory neurotransmitter that serves a number of functions in the brain. It is involved in energy metabolism, molecular synthesis of glutathione and GABA, and breakdown and removal of ammonia. In addition to transport systems, the BBB also plays a role in glutamate metabolism.49
Ions and Water
Nitric oxide (NO) is well known for its properties as a vasodilatory agent and also plays an important role in BBB signal regulation and autocrine activity.50,51 It mediates the transport of ions and nutrients essential for brain function and is regulated by cyclic guanosine monophosphate and cyclic adenosine monophosphate.
All movement of ions across the BBB is also associated with the movement of water. Water passes through the plasma membranes via facilitated diffusion through water channels called aquaporins, by cotransport with organic or inorganic ions, and by diffusion across the lipid bilayer. The water channel expressed most in the CNS is aquaporin-4, principally by astrocytes. Immunolocalization studies with double staining for aquaporin-4 and glial fibrillary acidic protein (GFAP), which stains astrocytes, show strong colocalization at the level of the BBB.52,53 Involvement of aquaporin-4 in brain edema and water homeostasis has been well established.
Lipoproteins
The mechanism underlying lipoprotein transport across the BBB has garnered considerable interest among researchers for potential exploitation of this channel for delivering drug therapy. Low-density lipoprotein (LDL) receptors are expressed on the BBB, and LDL is transported across the BBB by endocytosis. Scientists are now attempting to use this receptor as a vehicle for delivering therapeutic drugs into the brain.54
Multidrug Resistance
Multidrug resistance (MDR) protein has also been intensely studied as a possible vehicle for drug delivery. Pgp is an efflux transporter protein found in ECs, astrocytes, and microglia.55 It is expressed on the luminal surface of the endothelial membrane and glia55 and prevents toxins from entering the brain.56 Many drugs are substrates for Pgp, which limits their accumulation in the brain. Vinca alkaloids, anthracyclines, and taxanes are among the anticancer agents known to be transported by Pgp. Preclinical models have revealed that patients with deletion of Pgp have 100-fold increased sensitivity to chemotherapy agents and antiviral compounds in comparison to control subjects.57–60 Pgp is also protective in disease states such as Alzheimer’s, in which there is decreased deposition of β amyloid.61 However, overexpression of Pgp is also found in patients with epilepsy, although it is unclear whether upregulation of the transporter is a pathologic process of epilepsy or secondary to resistance to antiepileptic drugs (AEDs).62 In vivo studies have shown depletion of Pgp in patients with Parkinson’s disease.63
Leukocytes
It was an early notion that leukocytes are rare within the brain and that the architecture of brain microvessels maintains its immune-privileged status. There is now evidence that leukocytes traverse microvessels via a transcellular route. In addition, activated T lymphocytes can cross the endothelial wall in the normal state. Immunologic response to foreign pathogens, surveillance, and inflammation are regulated by leukocyte transportation across the BBB. Transportation occurs by means of integrins, intercellular adhesion molecules (ICAMs), and direct intercellular activity.64,65 In HIV encephalitis, actin cytoskeletal proteins such as Rho guanosine triphosphatase (GTPase) facilitate leukocyte migration, and blocking these compounds maintains the integrity of the BBB against leukocyte transport.66
Tight Junctions
The most prominent feature of the BBB is the presence of complex TJs between CNS ECs, which establish high transendothelial electrical resistance and determine the permeability of the BBB to hydrophilic molecules. TJs form a protective layer around ECs and are composed of various adhesive molecules linked to cytoskeletal signaling proteins that reside intracellularly. The proteins that make up the cellular architecture of the TJ and secure its connection to the actin cytoskeleton provide the structural integrity for the junctional complex to regulate transportation of solutes into the brain (Fig. 8-6).
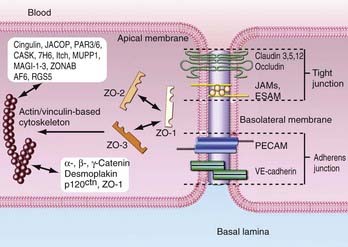
Membrane-Associated Guanylate Kinase Homologue
This class of proteins forms multimers that anchor TJs to the cell membrane. There are three main adhesion proteins: zona occludin-1 (ZO-1), ZO-2, and ZO-3. The first TJ-associated protein identified was ZO-1, a 220-kD membrane-associated protein expressed on both EC and epithelial cell surfaces.67 It serves as a scaffold for TJ formation by binding to the C-terminal of the cytoplasmic tail of occludin and the cytoskeletal protein spectrin.68 The importance of ZO-1 in TJ stability is illustrated by research showing that barrier stringency is compromised when ZO-1 expression is reduced or dissociated from membrane proteins.69 ZO-2 is a 160-kD protein with homologous regions to ZO-1 and has been identified as a ZO-1–associated protein.70 It binds to a transcription factor and transmembrane domains of the TJ. During proliferation of the TJ, ZO-2 migrates to the nucleus.71,72 Although ZO-1 and ZO-2 are seen in both brain and peripheral ECs, ZO-1 is continuous in brain but discontinuous in peripheral ECs.73 ZO-3 is seen solely in epithelial cells, unlike ZO-1 and ZO-2, which are intricate components of ECs.74
Occludin
Occludin is a 65-kD transmembrane protein located at TJ margins19,75 and is associated with cytoskeletal signaling proteins, including ZO-1, and ZO-2.70 Antibodies against occludin cluster around cell-cell contacts in brain ECs.76 Occludin is composed of four transmembrane domains. The terminal ends of the protein face the cytoplasm, and two extracellular loops span the intracellular cleft.77 The concentration of occludin in peripheral ECs is much lower than in brain ECs. Occludin is thought to be transcriptionally regulated, as measured by significant differences in mRNA levels between EC types.73 Occludin contains numerous phosphorylation sites, which are directly related to substance permeability. The cytoplasmic C-terminal provides association among occludin, ZO-1, and ZO-2. Occludin dysfunction is seen in numerous diseases such as HIV.66,78 Furthermore, downregulation of 55-kD occludin expression is seen in high-grade (III and IV) brain tumors.79
Claudins
Claudins are a third group of membrane proteins. These proteins function to selectively permit entry of cations through TJs.80 There is sequence conservation in the first and fourth transmembrane domains in the approximately 24 claudins identified in mammals.81 Both homophilic and heterophilic binding of extracellular domains facilitates close interaction of cell layers.82 In elucidating the roles of claudins and occludin in TJ formation, overexpression of claudin generated cell layers resembling TJs, which was not the case with occludin.83 Based on staining experiments, claudins are arranged continuously along ECs, in contrast to occludins.83 These results imply that claudins form the fundamental scaffold of TJs and occludins secure TJs. ECs express at least four claudin subtypes, claudin-1, claudin-3, claudin-5, and claudin-12. In vitro studies have revealed that claudin expression is significantly downregulated in anaplastic astrocytoma and GBM cell cultures.84,85
Junctional Adhesion Molecules
Members of the immunoglobulin supergene family are localized within TJs. Junctional adhesion molecule-1 (JAM-1) is a 40-kD IgG protein that regulates cell membrane attachment between large-domain and smaller chains.86,87 In animal models, JAMs regulate transendothelial cell migration, but their role in the BBB in vivo remains unclear.86
Endothelial TJs can rapidly and reversibly alter their conformation to permit plasma constituents to pass. The mechanism of control of this system is the degree of tyrosine phosphorylation of ZO-1 or other contact proteins; with increased phosphorylation, there is decreased electrical resistance and TJ permeability increases.88 Other mechanisms of control of cell-cell junctions are regulated by factors secreted by astroglial cells. These factors alter the microenvironment of the junction, thereby modifying permeability of the TJ.
Tight Junction Regulation
Ion Signaling
Phosphorylation indirectly controls TJ permeability by acting on associated proteins.89 Various pathologic states affect the function of phosphorylation on TJs. For example, there is significant phosphorylation of TJs in the presence of vascular endothelial growth factor (VEGF), which compromises the integrity of the BBB.90 In contrast, in bacterial infections there is significant dephosphorylation of TJs, which increases BBB permeability.91,92
Calcium
Calcium (Ca2+) acts as second messenger for regulation of BBB activity. Low levels of extracellular Ca2+ decrease TJ barrier stability,93 possibly dispersing ZO-1 and occludin away from TJ sites.94 Intracellular Ca2+ levels also affect BBB integrity by activating signaling cascades, regulating TJ transcription, and modulating the distribution of TJs.95 Release of intracellular Ca2+ compromises the BBB by inducing phosphorylation of TJs.96
G Protein
G proteins regulate scaffold proteins responsible for the cytoarchitecture of the TJ and membrane-associated proteins regulating TJ activity.97 They also facilitate leukocyte transport into brain via Rho GTPases.66 Protein kinase C (PKC) regulation also modulates BBB function. Control of PKC-α and PKC-ζ influences BBB permeability.98 In HIV disease, the cell surface protein gp120 activates PKC. Inhibitors of gp120 activity block PKC activation and prevent breakdown of the BBB.99 AF-6 is a Ras-binding protein that also binds to ZO-1 to stabilize the BBB. Overexpression of Ras results in disruption of ZO-1 and increased permeability of the BBB.100
Pathologic Changes in the Blood-Brain Barrier
Many of the same factors that regulate permeability under normal conditions are altered during pathologic conditions and result in enhanced vascular permeability and edema formation. Many neurologic conditions, including trauma, inflammatory and autoimmune disease, infection, cerebrovascular disease, neurodegenerative disease, epilepsy, and neoplasia, result in disruption of the BBB. Head trauma alters transporter activity, which may inhibit essential compounds from crossing the BBB while permitting pathogens and toxins to do so. Lymphoid surveillance of the CNS occurs via lymphatic vessels such as olfactory nerves or arachnoid granulations. BBB permeability is increased in autoimmune inflammatory diseases such as multiple sclerosis. Studies have also revealed that extravasation of lymphoid cells mediated by the vascular and intracellular adhesion molecules VCAM-1 and ICAM-1 is a seminal event in the pathophysiology of multiple sclerosis.101 Extravasation of leukocytes is achieved by three fundamental processes:
In addition, leukocytes secrete matrix metalloproteinases (MMPs), which degrade the ECM.
Infectious agents cross the BBB by transcytosis. HIV-1 enters the brain by adsorptive endocytosis.66,102 Because brain ECs lack CD4 and galactosylceramide receptors, they are protected from direct infection. HIV enters the CNS via infected white blood cells. The virus continues to proliferate in glial cells and is protected from therapy by the BBB.2
Clostridium perfringens has high affinity for TJ claudin proteins, which when bound, increases BBB permeability.103 Pneumococci bind to ECs through receptors for platelet-activating factor, thereby resulting in increased paracellular transport.104 Subsequent degradation of TJs results in the release of cytokines, including interleukin-1 (IL-1), tumor necrosis factor (TNF), and MMPs.105 In contrast, Neisseria meningitidis adheres to ECs, induces phosphorylation of EC binding proteins, inhibits leukocyte transportation, and in turn, inhibits inflammation.106 In patients with bacterial meningitis, steroids are given in conjunction with antibiotics to minimize inflammation in the brain. Dexamethasone, a synthetic glucocorticoid, may impede antibiotic permeability by tightening the BBB in the setting of meningitis. Herpes enters the CNS through olfactory nerves, whereas rabies enters through spinal nerves. In general, viruses perturb the BBB less than bacterial infection does.
Cerebrovascular disease and ischemia have deleterious effects on the BBB by depleting the brain of nutrients, inducing inflammation, and activating the cytokine cascade. This results in release of MMP, which causes vasogenic edema and degradation of the ECM. Like the edema seen in trauma, no significant improvement is provided with steroids. Stroke causes disruption of TJs and destruction of basal lamina proteins, including collagen type IV, laminin, and fibronectin.107 The severity of infarction is correlated with the degree of BBB dysfunction,10 which may place the patient at greater risk for hemorrhagic conversion after reperfusion. Klatzo first characterized brain edema as a cytotoxic versus a vasogenic process.108 In cytotoxic edema, the brain cells swell at the expense of the extracellular space, whereas the BBB remains intact. In vasogenic edema, permeability is increased because of disruption of the BBB, which allows an influx of plasma constituents and expansion of the extracellular space. During stroke, features of both cytotoxic edema and vasogenic edema occur simultaneously.
Breakdown of the BBB is implicated in the pathophysiology of Alzheimer’s disease, amyotrophic lateral sclerosis, and Parkinson’s disease. In Alzheimer’s disease, Aβ amyloid deposition is a histopathologic hallmark of the disease. Amyloid is transported across the BBB by the receptor for advanced glycation end products (RAGE). This receptor transports Aβ amyloid into the brain, whereas LDL receptor–related protein-1 transports Aβ amyloid out.109 Alzheimer’s patients have changes in RAGE and LDL receptor–related protein-1 concentrations on the hippocampus and cerebral cortex, thus indicating that Aβ amyloid deposition secondary to BBB dysfunction is involved in the early pathogenesis of the disease.110 Anti-RAGE therapies are currently being developed to test this hypothesis.
Seizures induce extravasation of intravascular markers, which results in a transient increase in BBB permeability. The BBB characteristics seen in epilepsy include downregulation of GLUT-1, inflammation, and MDR to AEDs, findings implicating both MDR receptors and efflux transporters.111
Brain tumors represent a disease process in which the dynamic characteristics of the BBB present significant challenges for therapy. ECs in tumor vessels are characterized by frequent membrane fenestrations, prominent pinocytotic vesicles, and lack of perivascular glial end-feet, and they display abnormal TJ morphology. The blood-tumor barrier is characterized by higher permeability to small molecules, although a large majority of drug therapies are still unable to penetrate the CNS in adequate concentration because of their large size and transport mechanisms, such as Pgp, that shuttle drug away from the brain.112 The center of a tumor’s BBB is often vastly compromised, thus making it ideal for drug delivery. However, surrounding normal parenchyma has an intact BBB, which prevents clinically significant doses of drugs from reaching the lesion. Corticosteroid therapy after brain tumor surgery has reduced mortality because it helps reestablish BBB integrity.113
Central Nervous System Drug Administration and the Blood-Brain Barrier
Some speculate that strong selective pressure must have existed to allow such a complex structure as the BBB to evolve.114,115 The CNS has no lymphatic system or other method of parenchymal drainage and is enclosed within the cranium, a rigid, nonexpandable structure. A net influx of molecules into the CNS would increase osmolarity and allow water from the vasculature to enter the brain, thereby leading to an increase in intracranial pressure. The evolution of the BBB fortunately makes large increases in intracranial pressure rare occurrences.116,117 Additionally, the BBB serves to prevent potentially harmful toxins from reaching the brain.
Despite these important functional roles, the BBB’s unique selectivity has created a strong challenge for medicine by hindering the ability of CNS medications to pass through it. Many drugs that have potentially useful action in vitro are found on in vivo evaluation to be unable to enter the CNS. New techniques to make the BBB more permeable would allow a number of potentially useful drugs currently unable to traverse the barrier to reach the CNS. Considerable research is currently under way to accomplish this task via a variety of approaches. The difficulty in administering drugs for the CNS can be seen in surveys of currently available medications. One study analyzing 6304 medications, excluding diagnostic dyes, revealed that only 6% of the drugs were used for treatment of the CNS118 despite heavy research in CNS pharmaceutical development. Other groups are focusing on strengthening the barrier, which would help prevent damage from exposure to neurotoxins and limit CNS side effects caused by drugs acting on other organ systems. In a related area, studies are searching to find more effective methods of analyzing BBB permeability, which is often disrupted in many disease states. All these new areas of research have developed rapidly in recent years, and some interesting and unique strategies to both “read” and “write” the BBB have been formulated.
Importance of Adequate Central Nervous System Drug Penetration
Improved regulation of BBB permeability would result in increased efficacy in treating the vast majority of CNS disorders. Nevertheless, most research in this area has focused on a handful of specific diseases, including brain tumors, HIV disease, epilepsy, Parkinson’s disease, Alzheimer’s disease, and infections. Both primary and metastatic CNS neoplasms, the most common of which in adults are astrocytomas, oligodendrogliomas, and mixed oligoastrocytomas,119 are difficult to treat because most chemotherapeutic agents are unable to adequately traverse the BBB as a result of their low lipophilicity.40,119 Partly as a consequence of poor drug delivery to the CNS, many tumors are currently treated by surgery and radiation therapy. However, difficulties often arise in surgery because precise boundaries of some tumors are difficult to locate. Other tumors may lie in inoperable locations or have already metastasized to multiple sites. Although radiotherapy is often beneficial as well, it may lead to secondary, more aggressive tumors.120,121 Chemotherapy is the best option in these cases; however, the drugs must be able to enter the CNS to be effective.
AEDs are often excluded from the CNS by the BBB and are therefore clinically unusable despite demonstrating potent and selective in vitro action.122 Pgp has been implicated as an important transport protein for some of these AEDs,123 including phenytoin124 and carbamazepine.125 However, other reports question the role of Pgp in AED extrusion.126,127 Approximately 30% of epileptic patients do not respond to the common pharmaceutical treatments, thereby resulting in increased morbidity and mortality in these resistant patients.128 In an immunohistochemical study, Volk and Loscher129 demonstrated that rats resistant to AED therapy had higher levels of Pgp expression in brain capillary ECs than did control animals. Similar variations in Pgp in human epileptics would help explain the existence of patients with medically intractable epilepsy who do not respond to AEDs.
In the treatment of Parkinson’s disease, it is estimated that only 5% of orally administered levodopa reaches the circulation after first pass through the liver. Less than 1% of the oral dose enters the brain because of blockage at the BBB.130 Recent studies are beginning to elucidate the potential mechanisms for exclusion of levodopa from the CNS. Levodopa enters the brain via the luminal neutral amino acid transporter L1 on ECs, but it may be excreted by the sodium-dependent large neutral amino acid (LNAA) transporter on the abluminal side of the membrane.131,132 Other molecules of levodopa are degraded by the endothelial enzyme monoamine oxidase B in the periphery,133 although drugs such as carbidopa are now available to inhibit this enzyme.
Medications for Alzheimer’s disease are excluded by the BBB as well.134 Given the rising proportion of elderly individuals in whom this disease is diagnosed in the United States, effective treatment of Alzheimer’s disease will be critical. Certain modifications of the BBB may eventually be used to provide better treatment of this debilitating illness.
Infectious disease could also be treated more successfully by opening the BBB, which would allow CNS penetration of more effective antibacterial, antifungal, and antiviral agents for the treatment of meningitis, encephalitis, abscesses, and other infectious diseases of the CNS. Once bacteria are killed, the BBB must be permeable to the remnants of their cell walls, which could otherwise irritate sensitive nervous tissue.135
HIV may harbor itself in the brain where antiviral drugs cannot penetrate effectively and then re-emerge later.136,137 Several studies have shown that zidovudine (azidothymidine [AZT]) does not effectively penetrate the BBB in rodents,138–140 although this does not appear to be the result of Pgp export.141 Strazielle and colleagues142 suggested that the organic anion transporters, a different class of protein pumps at the BBB, may be responsible for transport of AZT. Other anti-HIV medications are exported by similar means. The nucleoside reverse transcriptase inhibitor (NRTI) stavudine (d4T) may be removed from the BBB by the same transporter as AZT.143 Another NRTI, lamivudine (3TC), does not significantly penetrate the CNS either.140 Some protease inhibitors used for the treatment of HIV also appear to be susceptible to BBB exporters, primarily Pgp.144 However, there are some anti-HIV drugs that do enter the CNS in significant amounts. Some non-NRTIs, including nevirapine, appear to cross the BBB,145 whereas other anti-HIV drugs are believed to cross the BBB as a result of their CNS side effects.146
Drug Modifications
Expanding on the initial experiments of Brodie and associates8 in the 1960s, recent studies have shown that the permeability of most molecules can be predicted by determining their octanol-water coefficients based on their respective nonpolar and polar solubilites.147,148 Specifically, substances with the greatest ability to pass through the BBB generally have a log octanol-water coefficient between −0.5 and 6.0149 and a molecular mass of less than 400 to 500 daltons and do not form hydrogen bonds with water.150 With this understanding, many pharmaceutical researchers have conjugated their bioactive compounds to lipophilic moieties in the hope that they will become sufficiently lipid soluble to passively move through the BBB. Others have masked the hydrophilic groups of the compounds in an effort to increase lipophilicity.42 Conjugation by esters and disulfide bonds allows enzymes to cleave the lipids from the drug once it has passively entered the CNS, thus making the drug polar and trapping it inside. An example is heroin, an opiate that passes through the BBB 100 times more easily than morphine and is subsequently converted to morphine in the CNS.141,151 Such prodrugs have proved to be useful, although more research is needed to evaluate their efficacy and safety.152 Additionally, cleavage to form the active drug may not occur at a sufficient rate and with the necessary accuracy to produce localized therapeutic concentrations of the drug.
Lipophilic conjugation has been used successfully for introduction of the chemotherapeutic agent chlorambucil into the CNS.153,154 Kitagawa and coworkers155 evaluated the conjugative properties of adamantine, a compound related to the drug memantine used for the treatment of Parkinson’s disease. By conjugating adamantine to [D-Ala2] leu-enkephalin, the opioid gained the ability to pass through the BBB into the CNS. In another experiment, Prokai-Tatrai and colleagues156 successfully conjugated a different leu-enkephalin analogue to a lipophilic moiety for CNS administration. However, conjugation of drugs may not always be necessary. The simple reduction of hydrogen bonding potential by altering polar side groups has successfully increased permeability of the BBB to some small peptides.157
Despite these successes, many difficulties have arisen in the search for successful methods to increase the lipophilicity of pharmacologic compounds. Modification often increases the mass of the drug, and even lipophilic drugs do not cross the BBB effectively when their mass has increased to greater than the 400- to 500-dalton threshold.42 Increased size of the drugs can affect transport as well. The limit for molecular area appears to be around 80 Å2, and increases in size to greater than this seem to decrease BBB permeability dramatically.158 In addition to the physical constraints on lipophilic conjugation, chemical constraints have been demonstrated as well. Conjugation or masking of hydrophilic side groups may make the drug biologically inactive.152 An increase in lipophilicity can also make the drug susceptible to transport by Pgp and other export proteins despite having little if any susceptibility before the modification. Alternatively, conjugation of some drugs that are already substrates for Pgp can have an added beneficial effect by successfully preventing their export through hindrance of their ability to attach to the Pgp binding site. A derivative of the chemotherapeutic agent paclitaxel has been successfully modified in this fashion while still maintaining cytotoxic action against cells of the breast cancer lineage.159 Drug modification can alter pharmacokinetic parameters as well. Conjugation may decrease the solubility, plasma protein binding, and liver and reticuloendothelial uptake, thus altering the bioavailability of the drug.152 New side effects may also be due to increases in drug uptake into other organs as a result of lipophilic modification and potentially damage the more sensitive organs.
Another strategy of drug modification for bypassing the BBB is conjugation to bioactive molecules, either those that are normally transported into the CNS by specific transport proteins or some that enter the CNS via receptor-mediated endocytosis by cerebrovascular endothelium. Friden and associates160 successfully conjugated nerve growth factor to an antibody for the transferrin receptor in the rat. Binding of the antibody to the receptor stimulates receptor-mediated endocytosis and provides transcellular passage through the endothelium. Other researchers have successfully conjugated drugs to insulin fragments or antibodies to insulin factor to permit transfer through the BBB.46,161 Even large molecules, such as the enzyme β-galactosidase, have been successfully transported into the CNS via similar methods of bioactive conjugation.162
Exporter Protein Modulation
The most well characterized export protein of the BBB, Pgp, was first described in hamsters in 1976 by Juliano and Ling.163 As a member of the APT-binding cassette family of transport proteins, Pgp serves to protect the CNS by pumping xenobiotic compounds out of the brain and spinal cord into the vasculature.164 In general, substrates for Pgp are lipophilic, planar molecules that are either neutral or cationic.165 Pgp may have developed to remove hydrophobic substances that partitioned into the lipid core of the plasma membrane166,167 and therefore may be activated by sensing disruptions of the lipid bilayer.165 Unfortunately, the broad specificity of Pgp, although beneficial in preventing penetration of neurotoxins into the brain and spinal cord, also hinders therapeutic drugs from reaching their targets, thereby creating great difficulty for those researching CNS pharmaceutical design.
The specific location of Pgp in the BBB has recently been a source of debate. Studies have shown that Pgp is expressed in both cerebrovascular ECs and astrocytes in the human brain.168,169 Many studies suggest that the primary localization of Pgp is on the luminal EC membrane, where it serves to pump compounds directly into the lumen of the microvasculature.23,170–173 However, some evidence points to localization of Pgp on astrocyte foot processes as well.168,169 Although Pgp is important in the case of toxin exposure, animal experiments suggest that it may not have any necessary function during normal homeostasis. Mice deficient in the export protein have no changes relative to the wild type unless exposed to drugs that are normally pumped out of the cerebrovascular endothelium by Pgp.164 In a related experiment, dogs with increased susceptibility to neurotoxicity with the administration of ivermectin were found to be deficient in Pgp as a result of a deletion mutation in the mdr1 gene.174
Despite its high lipophilicity, cyclosporine was found to ineffectively penetrate the CNS as a result of interactions with Pgp. Studies have shown that cyclosporine inhibits Pgp, but it is not generally used for this purpose because of its immunosuppressive effects.175–177 The cyclosporine analogue PSC 833 was developed later and maintains the Pgp inhibitory action of its parent drug without the resulting immunosuppression.178,179 An array of other Pgp inhibitors have since been developed180–182 and used successfully with a variety of medications.165 For example, Pgp inhibitors have been shown to enhance delivery of the chemotherapeutic drugs paclitaxel183,184 and docetaxel to the CNS in animals.184 Recently, some natural products have been demonstrated to have Pgp inhibitory characteristics as well, including psoralen found in grapefruit,185,186 ginsenoside Rg3 from red ginseng,187 and piperine from black pepper.188 Clearly a variety of compounds have effects on Pgp, and as knowledge of these substances increases, researchers should be able to develop more effective compounds.
The major drawback of Pgp inhibitors is that historically, many have been toxic in concentrations sufficient for inhibition. However, the toxicologic profiles of the inhibitors have improved in recent years. First-generation inhibitors such as cyclosporine and verapamil were very toxic, whereas second-generation inhibitors such as valspodar and biricodar had improved tolerability.189 Second-generation inhibitors were later found to have unpredictable interactions when coadministered with chemotherapeutic agents. The recently developed third-generation inhibitors, including tariquidar, zosuquidar, and laniquidar, are very specific for Pgp and have less interaction with coadministered drugs. These new Pgp inhibitors are currently undergoing clinical trials.190
Opening of Endothelial Tight Junctions—Osmotic Disruption
Although most vascular ECs contain no TJs, epithelial barriers throughout the body possess these intercellular structures to ensure that the luminal contents remain separate from underlying tissue. TJs between ECs of the cerebral microvasculature seem to be able to separate, effectively open up the BBB, and allow the paracellular passage of water and other molecules. Although the actual mechanism for this type of nonspecific BBB opening remains to be elucidated, a variety of substances appear to alter the permeability of the BBB in this manner. The most common of these techniques used today is osmotic disruption, which involves the injection of hypertonic nonmetabolizable solutes such as mannitol directly into either the internal carotid arteries or the vertebral arteries. The location of injection is important because the permeability of the BBB will be affected only in the vasculature distal to the site of solute injection.191 CNS drug delivery via this method of BBB opening has been shown to be increased by up to 100-fold.10
The predominant hypothesis for the mechanism of osmotic BBB disruption is that shrinking of ECs occurs as a result of hyperosmolarity of the vasculature. This reduction in cell volume causes the TJs to separate and thereby open up the paracellular space for molecular movement.192,193 However, newer studies suggest that this model may be an oversimplification. Farkas and coworkers194 have provided evidence of phosphorylation of the multifunctional protein β-catenin during osmotic BBB opening, thus suggesting a more active cell response than passive shrinkage. A calcium-mediated contraction mechanism involving circumferential bundles of actin that interact with a variety of proteins in ECs has been proposed as well.195,196 Exposure to hypertonic substances appears to increase intracellular calcium in cultured cerebrovascular ECs, which may trigger pathways leading to cell shrinkage itself.197
Several studies have demonstrated the benefits of osmotic BBB disruption for the delivery of chemotherapeutic agents to the CNS to treat both primary and metastatic malignancies. Neoplasia represents a unique challenge for administration of pharmaceuticals. The BBB is often more permeable at the core of a tumor, but the brain tissue adjacent to the tumor maintains a high level of BBB integrity.10,198 Consequently, significantly less drug reaches the periphery and most goes to the center of the tumor.199 This “sink effect” causes very low levels of drug in the brain tumor periphery, in essence sparing tumor cells and contributing to neoplastic recurrence.10 Additionally, the surrounding normal brain tissue may contain metastatic seeds that are hidden behind a relatively normal BBB, thus further confounding effective treatment.133 Evidence also suggests that although the BBB is leaky in the center of tumors, it does not disappear completely in either primary or metastatic lesions.200,201 In one experiment in mice, Pgp still played a role in the microvasculature of brain tumors despite their increased permeability in comparison to normal brain tissue.202 The use of osmotic BBB disruption in conjunction with chemotherapy helps maintain a relatively constant level of chemotherapeutic agents throughout the entire region around the tumor, thereby preventing the “sink effect” and exposing any smaller metastatic lesions that would otherwise be inaccessible to the drugs. Osmotic BBB disruption also allows prolonged exposure of tumor to higher localized concentrations of drugs.203,204
The first phase I trial of osmotic BBB disruption began in 1979 and used mannitol (25%, 1.37 mol/L) infused via a catheter into the internal carotid artery at 4 to 8 mL/sec for 30 seconds. A transient rise in intracranial pressure occurred in this study, but no clinical sequelae were observed. In a study by Dahlborg and colleagues,205 645 osmotic BBB disruptions in conjunction with chemotherapy were performed on 34 patients, and more than 80% showed a partial response and 62% showed a complete response. Kraemer and coworkers206 found increased survival rates in patients with primary CNS lymphomas who underwent osmotic BBB disruption for delivery of chemotherapeutic drugs. They also identified a positive correlation between patient survival and the number of BBB disruptions. In another study, the estimated 5-year survival rate for patients with non–AIDS-related primary CNS lymphoma treated by osmotic BBB disruption and chemotherapy was 42% with a median survival time of 40.7 months.207 Such techniques have also been used successfully in pediatric patients.208
Despite its increased use in recent years, osmotic BBB disruption is far from perfect. Early hints of potential difficulties with the procedure came from studies in rats in which seizure-like events occurred during BBB modification.209 Transient increases in intracranial pressure are common during osmotic disruption. In the aforementioned study by Dahlborg and associates,205 which included 34 patients, one episode of tonsillar herniation occurred with no neurological sequelae, seizures developed in 4% of patients, and sepsis or granulocytopenic fever developed in 3%. In a study by McAllister and coworkers207 on primary CNS lymphoma, seizures occurred in 6% to 8% of those who underwent osmotic BBB opening. Four of the 74 patients in this study died within 30 days of the procedure, all attributed to infection, with three deaths occurring before the administration of granulocyte colony-stimulating factor. In one case study, expressive aphasia developed in a patient after osmotic BBB disruption but later resolved.210 Vasospasm is another problem associated with osmotic BBB disruption211 that can lead to ischemic stroke. Higher relative permeability of the BBB to viruses and proteins also occurs during osmotic BBB disruption.212,213 Exposure of nervous tissue to protein, specifically serum albumin, may cause astrocyte activation and seizures,214 although others argue that osmotic BBB disruption does not allow extravasation of serum albumin.215 Finally, osmotic BBB disruption is a costly and invasive procedure that requires highly trained practitioners to coordinate the treatment effectively.
Opening of Endothelial Tight Junctions—Parasympathetic Stimulation
Although the sympathetic division of the autonomic nervous system is responsible for maintaining and altering vasomotor tone in the body and thus peripheral blood flow, it has little if any effect on cerebral blood flow.216 This discovery prompted researchers to evaluate alternative pathways for cerebrovascular control. Interestingly, sensory innervation to the circle of Willis travels via the nasociliary nerve, a branch of the ophthalmic nerve, after leaving the cranial cavity through the ethmoidal foramen.217 Previous studies have shown that substance P–containing pain fibers may also dilate blood vessels via separate branches ending in motor terminals.218
Parasympathetic input to the cerebral vasculature was evaluated by Suzuki and associates.219,220 In this study, the nasociliary nerve was cut to interrupt sensory stimulation, and then the parasympathetic fibers entering the ethmoidal foramen were stimulated, which resulted in a 17% increase in cerebral blood flow. Sectioning the fibers did not affect blood flow, so parasympathetic stimulation also did not contribute to resting levels of vasomotor tone. Moreover, anticholinergic agents such as atropine and scopolamine did not attenuate the increase in cerebral blood flow, thus suggesting that the active neurotransmitter was not acetylcholine. The neurotransmitter mediating this effect was later shown to be NO.221–223 In another study, stimulation of the rat sphenopalatine ganglion (SPG) increased cerebral blood flow by up to 50% in the ipsilateral parietal cortex.224–227
Studies by Mayhan228,229 showed that both NO donors and histamine increase permeability of the BBB. Yarnitsky and coworkers230 subsequently hypothesized that stimulation of parasympathetic fibers may increase the permeability of the BBB because parasympathetic fibers release NO. They evaluated the permeability of the BBB by using fluorescein isothiocyanate–labeled dextran in rat parietal cortex exposed by craniotomy. Evans blue–labeled albumin and two chemotherapeutic agents, anti-HER2 monoclonal antibody used for the treatment of breast cancer and etoposide, were used in closed-cranium experiments to evaluate permeability of the BBB as well. All these agents entered through the BBB in significant amounts throughout the ipsilateral brain after stimulation of the postganglionic parasympathetic fibers in the SPG. Some of the contralateral brain was affected as well. Smaller molecules penetrated the BBB more easily than larger ones, thus suggesting that the mechanism of this effect works by opening TJs rather than vesicular transport. No injurious effect on the brain, as measured by nicotinamide adenine dinucleotide (NAD)/reduced NAD (NADH) balance, was found, and no significant brain edema occurred. In another experiment by Yarnitsky and colleagues,231 SPG stimulation in dogs showed similar effects on the BBB and was also observed to increase BBB permeability of the optic nerve, an extension of the CNS. Unlike the experiments in rodents, however, the effect in dogs and domestic pigs was entirely ipsilateral and only the anterior circulation was affected because the posterior vasculature is innervated by branches from the otic ganglion in these animals. Such findings suggest that SPG stimulation in humans would have similar effects on the ipsilateral anterior cerebral circulation.
Convection-Enhanced Delivery
Chemotherapy is infused into the brain tumor under constant pressure to deliver drug by bulk flow through catheters placed into the tumor bed.232 Image guidance is used to optimize the accuracy of drug delivery. There are numerous variables that determine whether effective doses of drugs have been delivered to the tumor: the rate of infusion and total volume infused, the molecular weight of the drug agent, the affinity for and concentration in and around the target receptor, and finally, the density of brain tissue.233 Technical challenges of convection-enhanced delivery include injury to the brain during insertion of the catheter, backflow of infusate, drug infused into the EC space of the CNS, drug efflux by transport mechanisms related to the BBB, premature metabolism of the infusate, and finally, interstitial pressure gradients preventing adequate concentration of drug to remain in the tumor bed.234
Local Delivery of Polymer-Infused Chemotherapy
Incorporation of chemotherapy into biodegradable polymers was first developed in 1991235 and approved for used in 1996 in patients with malignant gliomas. A craniotomy must be performed to implant the drug wafers into the tumor resection cavity. There is variability in the amount of total drug released into the resection cavity, as well as nonuniform release of carmustine into a tumor. This approach relies on passive diffusion of drugs into the brain, unlike convection-enhanced delivery, which forces fluid drugs through the brain.
Targeted Toxin Therapy
This drug delivery system relies on a fusion protein consisting of a protein with affinity for the IL-4 receptor, which is upregulated on tumor cells, and a bacterial toxin that causes apoptosis by inhibiting translation.236
Trojan Horse Liposome
An appropriately named therapy, immunoliposomes carry a gene for protein correction gene therapy or small hairpin RNA expressed in plasma to inhibit protein synthesis by RNA interference (RNAi). The molecular Trojan Horse liposome is conjugated to a polyethylene glycol moiety, which increases the circulating half-life of the fusion construct. Polyethylene glycol is a peptidomimetic that binds to BBB transport receptors such as insulin or transferrin to enter the brain parenchyma. Preclinical studies have shown this technique to be a means of gene therapy for treating Parkinson’s disease.237 The most widely characterized receptor-mediated transcytosis system for targeting of drugs to the brain is the transferrin receptor, which mediates cellular uptake of iron bound to transferrin. Drugs targeting the transferrin receptor can be developed either by using an endogenous ligand, transferrin, or by using an antibody directed against the transferrin receptor. The insulin receptor has also been used for targeted delivery of drugs to the brain.
Focused Ultrasound Disruption of the Blood-Brain Barrier
This method uses ultrasound to create lesions in the BBB to increase its permeability. It is monitored closely by MRI thermometry.238 Vascular penetration was generated without measurable tissue injury by using intravenous contrast dye to visualize disruption of the BBB.239 Imaging revealed expanded TJs and increased migration across ECs.240 Preclinical studies with doxorubicin showed increased drug in sites targeted by ultrasound and retained binding of antibodies attached to a chemotherapeutic agent for their ligand.241
Imaging the Blood-Brain Barrier
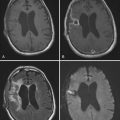
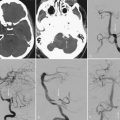
Neuroimaging technology has advanced to provide insight into the effect of brain tumors on the BBB by enabling clinicians to view the invasiveness of the disease and the effect of the tumor on BBB permeability, as well as to evaluate the efficacy of treatment strategies. Disruption of the BBB by disease processes such as brain tumors can be seen on contrast-enhanced computed tomography (CT) and MRI.242 Brain tumor lesions have a higher relative cerebral blood volume than normal tissue does, largely because of secretion of VEGF by brain tumors. To assess the relationship between BBB penetration and tumor perfusion, patients with high-grade gliomas underwent MRI screening with a T1-weighted fast spoiled gradient echo technique for measurement of BBB permeability, followed by dynamic susceptibility contrast-enhanced (DSC) imaging to measure regional cerebral blood volume. There was a correlation between T1-weighted and DSC images, which implies a direct relationship between BBB permeability and regional cerebral blood volume (Fig. 8-7).242
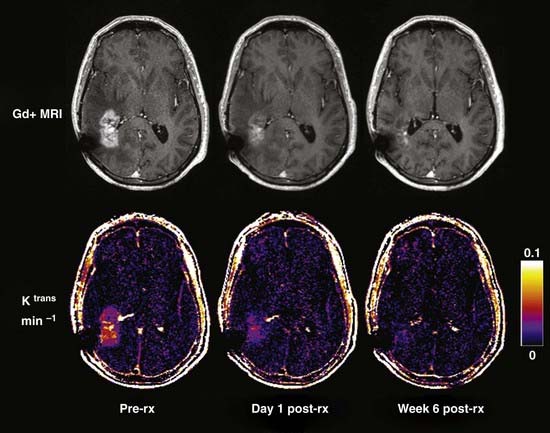
Brain MRI has been used to monitor drug delivery and evaluate the success of treatment of CNS tumors.241 Patients with intracranial mass lesions underwent MRI before and after corticosteroid therapy. In comparing normal and pathologic brain tissue, there was a significant difference in T1-weighted contrast enhancement, as well as a difference between the two groups on T2-weighted contrast-enhanced images.243
More recently, iron oxide nanoparticles have been used for tumor imaging. When compared with conventional gadolinium contrast, nanoparticles exhibit superior intravascular retention and may more accurately delineate brain tumor perfusion.244 Advances in PET may help distinguish tumor regrowth from radiation necrosis and even help stage a tumor.245
S-100β: A Peripheral Marker of Blood-Brain Barrier Damage
Opening of the BBB provides molecules normally present in blood open passage into the CNS. However, this opening, unless involving specific transporters, does not work in just one direction. Proteins normally present in blood are free to diffuse into the CNS, and in turn, proteins normally present in high concentration in the CNS are free to diffuse down concentration gradients into blood. These peripheral BBB markers can be detected in blood to evaluate the permeability characteristics of the BBB at any given time. In a recent review article, Marchi and associates246 discussed the ideal properties of a peripheral marker of BBB disruption. Such proteins should have low or undetectable plasma levels in normal subjects, be normally present in CSF, and have a higher normal concentration in CSF than in plasma. Additionally, the CSF concentration of the protein should increase in response to insults. The protein should be normally blocked by the BBB and exhibit flux across the BBB during barrier disruption. Several proteins, including S-100β, neuron-specific enolase, and GFAP, have been evaluated for this purpose, but only S-100β meets the characteristics of having very low plasma levels with a concentration less than that found in CSF in normal subjects.
Moore discovered the S-100 protein family in 1965 by isolating a fraction of subcellular material containing proteins from bovine brain.247 Moore named the fraction S-100 because its contents were soluble in 100% saturated ammonium sulfate at neutral pH. The S-100 family contains more than 15 different calcium-binding proteins, including S-100β. All of them contain EF-hand calcium-binding domains and are approximately 10,000 daltons in size,247 significantly larger than the 400- to 500-dalton limit for passage through the BBB. Of the proteins in this family, S-100β is unique in its predominant location in the CNS248–251 and is specifically located primarily in both astrocyte end-foot processes and Schwann cells.252–254 Although S-100β is found in several other body tissues, its concentrations in these peripheral locations are significantly lower than in the CNS.248,249 The primary role of S-100β remains to be elucidated, but it interacts with a number of cytoplasmic proteins with calcium-dependent actions and thus exerts a variety of influences on cells. It may exert its effects via a cyclic adenosine monophosphate–related mechanism.255
Plasma levels of S-100β are normally a third of those found in CSF and are nearly undetectable.256 Several diseases cause an elevation in plasma levels of S-100β, which can be detected and used for both diagnostic and prognostic purposes, as well as for evaluation of disease progression. Plasma S-100β levels increase with cerebral ischemia, with peak levels occurring approximately 3 days after infarction.257–260 These levels have served as a useful marker of both infarct size and long-term clinical outcome.257–259,261,262 Traumatic brain injury has also been shown to increase S-100β levels in plasma,263,264 with a positive correlation between the extent of damage after head injury and elevation in plasma S-100β.265 Ingebrigtsen and coworkers266,267 found a negative predictive value of 0.99 for detecting intracranial pathology via serum S-100β levels versus CT studies. The highest S-100β levels in one study of traumatic brain injury were observed in samples taken approximately 2.5 hours after trauma, which is a considerably shorter period than that required for the maximal peak in plasma concentrations during ischemic stroke.268 A positive correlation was also found between early rises in blood plasma levels, up to 5 hours after trauma, and unfavorable outcome.262,265,267–270
Plasma levels of S-100β have also been elevated in patients with hemorrhagic shock,263 aneurysmal subarachnoid hemorrhage,271 hypoxia secondary to cardiac arrest,272 and brain damage subsequent to attempted cardiopulmonary resuscitation.273 In all these cases, the elevation in S-100β plasma concentration is thought to result from damage to nervous tissue, including both neurons and glial cells. Additionally, depleted ATP levels in the brain during ischemic, hypoxic, and traumatic injury lead to increased adenosine levels,274–276 which may activate A1 adenosine receptors and cause release of S-100β from astrocytes. In experiments on cultured astrocytes, Ciccarelli and colleagues277 observed that adenosine receptor agonists cause the released S-100β to be nearly 160% that of controls. Plasma S-100β has also been elevated during cardiothoracic surgery,278–280 although these findings remain debatable.281 Currently, plasma levels of S-100β are predominantly used clinically for the monitoring of melanoma because these malignant cells express the protein. High plasma levels of S-100β correlate with disease progression.282
With the exception of melanoma, all the aforementioned disease processes involve some degree of brain damage. Diseases of the CNS that are associated with opening of the BBB do not necessarily cause brain damage or increased adenosine levels, so S-100β plasma levels must rise in response to BBB disruption without coexisting nervous tissue damage. To evaluate this, plasma S-100β levels were determined in patients with primary CNS lymphoma treated by chemotherapy, with intra-arterial infusion of mannitol used for osmotic BBB disruption. Plasma S-100β concentrations increased with mannitol administration. Intra-arterial administration of methotrexate once again caused plasma levels to rise, although patients receiving methotrexate without BBB disruption had no change in plasma S-100β. This elevation in peripheral S-100β levels occurred nearly immediately after BBB disruption, which excludes protein synthesis as a source for the increased S-100β.283 Intra-arterial infusion of methotrexate with BBB disruption does not appear to damage nervous tissue,284 thus suggesting that the elevated plasma concentrations reflect release of baseline levels of S-100β protein in the CNS and not release as a result of damaged brain tissue. Further studies have also shown that peripheral S-100β levels rise even in the absence of brain damage.246,285
Three hypotheses have been proposed for how the rises in peripheral S-100β occur. CNS concentrations of the protein may increase first because of neuronal damage, with plasma levels rising after subsequent opening of the BBB. Alternatively, the BBB may open first, with subsequent neuronal damage elevating plasma levels. Finally, the rise in peripheral S-100β levels may be due to release of the normal amount of the protein in the CNS after opening of the BBB. This last hypothesis of plasma S-100β elevation suggests that it is a useful specific marker of BBB permeability. To evaluate the diagnostic utility of S-100β levels, Kanner and associates285 conducted a prospective study by determining S-100β levels in 51 patients undergoing MRI with and without gadolinium contrast enhancement for diagnostic and volumetric purposes. Normal MRI findings were seen in six patients from this group, and their plasma S-100β levels were found to be close to normal as well. In two patients treated for trigeminal neuralgia, one had a normal MRI study, whereas the other showed lacunar infarcts and related white matter changes. Plasma S-100β levels were elevated in this second patient, probably because of ischemia from the infarcts. All the remaining patients had MRI studies that demonstrated gadolinium enhancement and had significantly elevated basal S-100β levels. No significant differences in S-100β levels were found in patients with metastatic tumors of various origins. Moreover, there was no correlation between tumor size and rise in plasma S-100β concentrations. Primary CNS lymphoma was the only tumor evaluated that did not cause an elevation in S-100β. Immunocytochemical studies showed that this tumor had a uniform cytology devoid of normal astrocytic markers, including GFAP and S-100β, thus explaining the absence of plasma S-100β elevations in these patients.
Based on these studies on the use of peripheral detection of S-100β, Janigro and Marchi published methods to determine plasma levels of S-100β for use in the diagnosis of new conditions, determining the prognosis and progression of various disorders, and providing insight into the permeability characteristics of the BBB for proper drug administration.286 Research is currently under way to ensure that peripheral S-100β levels correlate well with gadolinium enhancement on MRI. If so, plasma S-100β could be used to screen patients for MRI for the diagnosis of brain tumors. Because BBB disruption can occur transiently for a variety of reasons, elevated plasma S-100β would not necessarily mean that a lesion was present. However, a normal plasma level would suggest that MRI would show no gadolinium enhancement, thus making MRI unnecessary. A simple and inexpensive blood test could be used regularly in place of expensive and time-consuming MRI studies.
Blood-Brain Barrier and Neurological Disorders: Epilepsy
Patients with epilepsy have seizures intermittently, and depending on the underlying cause, many patients are completely seizure free for months or even years. This sporadic appearance of seizures implies that precipitating factors induce seizures in patients with epilepsy. Numerous groups have described a number of vascular/blood-related factors that may be tipping the fragile epileptic brain toward seizures.214,287–294 Seizures are a result of a shift in the normal balance of excitation and inhibition within the CNS. Given the numerous properties that control neuronal activity, it is not surprising that there are many different ways to perturb homeostasis and therefore precipitate seizures. One of the main determinants of neuronal firing rate and synchronicity is the extracellular potassium level. Potassium controls glial and neuronal resting potential, repolarization, ion channel conductance, cerebral blood flow, and Na+/K+ pump activity. The complexity of CNS potassium homeostasis underscores its importance in the mammalian brain and also the role of the BBB. The process involves different cell types (neurons, glia, and ECs), several extracellular mechanisms (spatial buffering, cerebral blood flow), and strictly controlled segregation of potassium concentrations between blood (4.0 to 5.0 mM) and brain parenchyma (2.5 to 3.0 mM). Other molecular elements that may either participate in seizure onset or decrease the seizure threshold are brain levels of albumin, antibodies, or drugs.
Seizures and epilepsy are commonly observed in conjunction with stroke, traumatic brain injury, and CNS infections—all conditions known to compromise BBB function. It remains debatable whether the compromised integrity of the BBB is a factor involved in the etiology of epilepsy or secondary to such pathologies. The etiologic role that the BBB plays in seizures is supported by the fact that BBB disruption after acute head trauma is a well-known pathologic finding in both animal and human studies of S-100β.294–297 BBB disruption may persist for weeks to years after the injury and may colocalize with abnormal electroencephalographic (EEG) activity. The increased interest in osmotic opening of the BBB as a viable mechanism of increasing drug delivery to the brain provides an opportunity to explore the connection between BBB disruption and seizures in a controlled clinical environment. The marked increase in BBB permeability to intravascular substances (10- to 100-fold for small molecules) after this osmotic disruption procedure is due to both increased diffusion and bulk fluid flow across the TJs. The permeability effect is largely reversed within minutes.298 In rodents, loss of BBB integrity by intra-arterial administration of hyperosmotic mannitol has been shown to rapidly lead to EEG changes consistent with epileptic seizures (spike/wave complexes interspersed with decreased EEG voltage that persist for several hours after the BBB disruption event).290,299
Given these findings, it is not surprising that seizures are the primary complication of osmotic BBB disruption. Indeed, seizures occur in a relatively large number of these patients (13% to 55%). This high incidence was initially attributed to meglumine iothalamate, a known epileptogenic agent used as a contrast agent for CT.300 However, seizures associated with BBB disruption continued to occur (albeit with decreased frequency) when the disruption was monitored by radionuclide scanning rather than CT. Current research is focused on attempting to establish a correlation between the level of BBB disruption and the probability of a seizure occurring.290,301
Abbott NJ. Dynamics of CNS barriers: evolution, differentiation, and modulation. Cell Mol Neurobiol. 2005;25:5-23.
Agre P, Nielsen S, Ottersen OP. Towards a molecular understanding of water homeostasis in the brain. Neuroscience. 2004;129:849-850.
Begley DJ. Delivery of therapeutic agents to the central nervous system: the problems and the possibilities. Pharmacol Ther. 2004;104:29-45.
Betz AL, Goldstein GW. Polarity of the blood-brain barrier: neutral amino acid transport into isolated brain capillaries. Science. 1978;202:225-227.
Brightman MW. The distribution within the brain of ferritin injected into cerebrospinal fluid compartments. II. Parenchymal distribution. Am J Anat. 1965;117:193-219.
Cairncross JG, Macdonald DR, Pexman JH, et al. Steroid-induced CT changes in patients with recurrent malignant glioma. Neurology. 1988;38:724-726.
Cervos-Navarro J, Kannuki S, Nakagawa Y. Blood-brain barrier (BBB). Review from morphological aspect. Histol Histopathol. 1988;3:203-213.
Davson H. Review lecture. The blood-brain barrier. J Physiol. 1976:1-28.
Demeule M, Regina A, Annabi B, et al. Brain endothelial cells as pharmacological targets in brain tumors. Mol Neurobiol. 2004;30:157-183.
Dore-Duffy P, Owen C, Balabanov R, et al. Pericyte migration from the vascular wall in response to traumatic brain injury. Microvasc Res. 2000;60:55-69.
Fricker G, Miller DS. Modulation of drug transporters at the blood-brain barrier. Pharmacology. 2004;70:169-176.
Hanin I. The Gulf War, stress and a leaky blood-brain barrier. Nat Med. 1996;2:1307-1308.
Hirase T, Staddon JM, Saitou M, et al. Occludin as a possible determinant of tight junction permeability in endothelial cells. J Cell Sci. 1997;110:1603-1613.
Janzer RC, Raff MC. Astrocytes induce blood-brain barrier properties in endothelial cells. Nature. 1987;325:253-257.
Kapural M, Krizanac-Bengez L, Barnett G, et al. Serum S-100beta as a possible marker of blood-brain barrier disruption. Brain Res. 2002;940:102-104.
Karnovsky MJ. The ultrastructural basis of capillary permeability studied with peroxidase as a tracer. J Cell Biol. 1967;35:213-236.
Kroll RA, Neuwelt EA. Outwitting the blood-brain barrier for therapeutic purposes: osmotic opening and other means. Neurosurgery. 1998;42:1083-1099.
Loscher W, Potschka H. Role of drug efflux transporters in the brain for drug disposition and treatment of brain diseases. Prog Neurobiol. 2005;76:22-76.
Neuwelt EA. Mechanisms of disease: the blood-brain barrier. Neurosurgery. 2004;54:131-140.
Oby E, Janigro D. The blood-brain barrier and epilepsy. Epilepsia. 2006;47:1761-1774.
Pardridge WM. Molecular biology of the blood-brain barrier. Mol Biotechnol. 2005;30:57-70.
Pardridge WM. Blood-brain barrier drug targeting: the future of brain drug development. Mol Interv. 2003;3:90-105.
Provenzale JM, Mukundan S, Dewhirst M. The role of blood-brain barrier permeability in brain tumor imaging and therapeutics. AJR Am J Roentgenol. 2005;185:763-767.
Reese TS, Karnovsky MJ. Fine structural localization of a blood-brain barrier to exogenous peroxidase. J Cell Biol. 1967;34:207-217.
Schinkel AH, Smit JJ, van Tellingen O, et al. Disruption of the mouse mdr1a P-glycoprotein gene leads to a deficiency in the blood-brain barrier and to increased sensitivity to drugs. Cell. 1994;77:491-502.
Smith QR. A review of blood-brain barrier transport techniques. Methods Mol Med. 2003;89:193-208.
Stewart PA, Wiley MJ. Developing nervous tissue induces formation of blood-brain barrier characteristics in invading endothelial cells: a study using quail-chick transplantation chimeras. Dev Biol. 1981;84:183-192.
Tao-Cheng JH, Brightman MW. Development of membrane interactions between brain endothelial cells and astrocytes in vitro. Int J Dev Neurosci. 1988;6:25-37.
Thomas H, Coley HM. Overcoming multidrug resistance in cancer: an update on the clinical strategy of inhibiting p-glycoprotein. Cancer Control. 2003;10:159-165.
Tontsch U, Bauer HC. Glial cells and neurons induce blood-brain barrier related enzymes in cultured cerebral endothelial cells. Brain Res. 1991;539:247-253.
Volk H, Potschka H, Loscher W. Immunohistochemical localization of P-glycoprotein in rat brain and detection of its increased expression by seizures are sensitive to fixation and staining variables. J Histochem Cytochem. 2005;53:517-531.
Volk HA, Loscher W. Multidrug resistance in epilepsy: rats with drug-resistant seizures exhibit enhanced brain expression of P-glycoprotein compared with rats with drug-responsive seizures. Brain. 2005;128:1358-1368.
1 Strelow L, Janigro D, Nelson JA. Persistent SIV infection of a blood-brain barrier model. J Neurovirol. 2002;8:270-280.
2 Strelow LI, Janigro D, Nelson JA. The blood-brain barrier and AIDS. Adv Virus Res. 2001;56:355-388.
3 Ehrlich P. Ueber die Beziehungen von chemischer Constitution, Verteilung, und pharmacologische Wirkung. New York: John Wiley; 1906.
4 Goldmann E. Die äussere und innere Sekretion des Genden und gekranken Organismus im Licht der vitalen Färbung. Beitr Klin Chir. 1909;64:192-265.
5 Goldmann E. Vitalfärbung am Zentrainervensystem. Abh Preuss Akad Wiss Phys-Math. 1913;1:1-60.
6 Stern L, Gautier R. Repports entre le liquide céphalo-rachidien et la circulation sanquine. Arch Int Physiol. 1921;17:138-192.
7 Stern L, Gautier R. Les repports entre le liquide céphalorachidien et les éléments nerveux de l’axe cérébrospinal. Arch Int Physiol. 1922;17:391-448.
8 Brodie BB, Kurz H, Schanker LS. The importance of dissociation constant and lipid-solubility in influencing the passage of drugs into the cerebrospinal fluid. J Pharmacol Exp Ther. 1960;130:20-25.
9 Davson H. Review lecture. The blood-brain barrier. J Physiol. 1976;255:1-28.
10 Neuwelt EA. Mechanisms of disease: the blood-brain barrier. Neurosurgery. 2004;54:131-140.
11 Brightman MW. The distribution within the brain of ferritin injected into cerebrospinal fluid compartments. II. Parenchymal distribution. Am J Anat. 1965;117:193-219.
12 Karnovsky MJ. The ultrastructural basis of capillary permeability studied with peroxidase as a tracer. J Cell Biol. 1967;35:213-236.
13 Reese TS, Karnovsky MJ. Fine structural localization of a blood-brain barrier to exogenous peroxidase. J Cell Biol. 1967;34:207-217.
14 Pardridge WM. Molecular biology of the blood-brain barrier. Mol Biotechnol. 2005;30:57-70.
15 Abbott NJ. Dynamics of CNS barriers: evolution, differentiation, and modulation. Cell Mol Neurobiol. 2005;25:5-23.
16 Loscher W, Potschka H. Role of drug efflux transporters in the brain for drug disposition and treatment of brain diseases. Prog Neurobiol. 2005;76:22-76.
17 Ikeda J, Nagashima G, Saito N, et al. Putative neuroexcitation in cerebral ischemia and brain injury. Stroke. 1990;21(suppl 11):III65-III70.
18 Migeon MB, Street VA, Demas VP, et al. Cloning, sequence and chromosomal localization of MK6, a murine potassium channel gene. Epilepsy Res Suppl. 1992;9:173-180.
19 Hawkins BT, Davis TP. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev. 2005;57:173-185.
20 Tao-Cheng JH, Brightman MW. Development of membrane interactions between brain endothelial cells and astrocytes in vitro. Int J Dev Neurosci. 1988;6:25-37.
21 Tao-Cheng JH, Nagy Z, Brightman MW. Tight junctions of brain endothelium in vitro are enhanced by astroglia. J Neurosci. 1987;7:3293-3299.
22 Willis CL, Nolan CC, Reith SN, et al. Focal astrocyte loss is followed by microvascular damage, with subsequent repair of the blood-brain barrier in the apparent absence of direct astrocytic contact. Glia. 2004;45:325-337.
23 Stewart PA, Wiley MJ. Developing nervous tissue induces formation of blood-brain barrier characteristics in invading endothelial cells: a study using quail-chick transplantation chimeras. Dev Biol. 1981;84:183-192.
24 Janzer RC, Raff MC. Astrocytes induce blood-brain barrier properties in endothelial cells. Nature. 1987;325:253-257.
25 Lai CH, Kuo KH. The critical component to establish in vitro BBB model: pericyte. Brain Res Brain Res Rev. 2005;50:258-265.
26 Dresemann G. Imatinib and hydroxyurea in pretreated progressive glioblastoma multiforme: a patient series. Ann Oncol. 2005;16:1702-1708.
27 Reardon DA, Egorin MJ, Quinn JA, et al. Phase II study of imatinib mesylate plus hydroxyurea in adults with recurrent glioblastoma multiforme. J Clin Oncol. 2005;23:9359-9368.
28 Dore-Duffy P, Owen C, Balabanov R, et al. Pericyte migration from the vascular wall in response to traumatic brain injury. Microvasc Res. 2000;60:55-69.
29 Paemeleire K. The cellular basis of neurovascular metabolic coupling. Acta Neurol Belg. 2002;102:153-157.
30 Tontsch U, Bauer HC. Glial cells and neurons induce blood-brain barrier related enzymes in cultured cerebral endothelial cells. Brain Res. 1991;539:247-253.
31 Cohen RM, Andreason PJ, Doudet DJ, et al. Opiate receptor avidity and cerebral blood flow in Alzheimer’s disease. J Neurol Sci. 1997;148:171-180.
32 Cohen Z, Molinatti G, Hamel E. Astroglial and vascular interactions of noradrenaline terminals in the rat cerebral cortex. J Cereb Blood Flow Metab. 1997;17:894-904.
33 Tong XK, Hamel E. Regional cholinergic denervation of cortical microvessels and nitric oxide synthase–containing neurons in Alzheimer’s disease. Neuroscience. 1999;92:163-175.
34 Vaucher E, Tong XK, Cholet N, et al. GABA neurons provide a rich input to microvessels but not nitric oxide neurons in the rat cerebral cortex: a means for direct regulation of local cerebral blood flow. J Comp Neurol. 2000;421:161-171.
35 Ben-Menachem E, Johansson BB, Svensson TH. Increased vulnerability of the blood-brain barrier to acute hypertension following depletion of brain noradrenaline. J Neural Transm. 1982;53:159-167.
36 del Zoppo GJ, Hallenbeck JM. Advances in the vascular pathophysiology of ischemic stroke. Thromb Res. 2000;98:73-81.
37 Tilling T, Engelbertz C, Decker S, et al. Expression and adhesive properties of basement membrane proteins in cerebral capillary endothelial cell cultures. Cell Tissue Res. 2002;310:19-29.
38 Majno G, Palade GE, Schoefl GI. Studies on inflammation. II. The site of action of histamine and serotonin along the vascular tree: a topographic study. J Biophys Biochem Cytol. 1961;11:607-626.
39 Majno G, Shea SM, Leventhal M. Endothelial contraction induced by histamine-type mediators: an electron microscopic study. J Cell Biol. 1969;42:647-672.
40 Kroll RA, Neuwelt EA. Outwitting the blood-brain barrier for therapeutic purposes: osmotic opening and other means. Neurosurgery. 1998;42:1083-1099.
41 Pardridge WM. Blood-brain barrier drug targeting: the future of brain drug development. Mol Interv. 2003;3:90-105.
42 Cervos-Navarro J, Kannuki S, Nakagawa Y. Blood-brain barrier (BBB). Review from morphological aspect. Histol Histopathol. 1988;3:203-213.
43 Nagamatsu S, Kornhauser JM, Burant CF, et al. Glucose transporter expression in brain. cDNA sequence of mouse GLUT3, the brain facilitative glucose transporter isoform, and identification of sites of expression by in situ hybridization. J Biol Chem. 1992;267:467-472.
44 Maher F, Vannucci SJ, Simpson IA. Glucose transporter proteins in brain. FASEB J. 1994;8:1003-1011.
45 Mueckler M, Caruso C, Baldwin SA, et al. Sequence and structure of a human glucose transporter. Science. 1985;229:941-945.
46 Pardridge WM. Blood-brain barrier carrier-mediated transport and brain metabolism of amino acids. Neurochem Res. 1998;23:635-644.
47 Oldendorf WH, Szabo J. Amino acid assignment to one of three blood-brain barrier amino acid carriers. Am J Physiol. 1976;230:94-98.
48 Betz AL, Goldstein GW. Polarity of the blood-brain barrier: neutral amino acid transport into isolated brain capillaries. Science. 1978;202:225-227.
49 Benrabh H, Lefauconnier JM. Glutamate is transported across the rat blood-brain barrier by a sodium-independent system. Neurosci Lett. 1996;210:9-12.
50 Utepbergenov DI, Mertsch K, Sporbert A, et al. Nitric oxide protects blood-brain barrier in vitro from hypoxia/reoxygenation-mediated injury. FEBS Lett. 1998;424:197-201.
51 Janigro D, West GA, Nguyen TS, et al. Regulation of blood-brain barrier endothelial cells by nitric oxide. Circ Res. 1994;75:528-538.
52 Agre P, Nielsen S, Ottersen OP. Towards a molecular understanding of water homeostasis in the brain. Neuroscience. 2004;129:849-850.
53 Papadopoulos MC, Manley GT, Krishna S, et al. Aquaporin-4 facilitates reabsorption of excess fluid in vasogenic brain edema. FASEB J. 2004;18:1291-1293.
54 Mousazadeh M, Palizban A, Salehi R, et al. Gene delivery to brain cells with apoprotein E derived peptide conjugated to polylysine (apoEdp-PLL). J Drug Target. 2007;15:226-230.
55 Lee G, Bendayan R. Functional expression and localization of P-glycoprotein in the central nervous system: relevance to the pathogenesis and treatment of neurological disorders. Pharm Res. 2004;21:1313-1330.
56 Fricker G, Miller DS. Modulation of drug transporters at the blood-brain barrier. Pharmacology. 2004;70:169-176.
57 Schinkel AH, Smit JJ, van Tellingen O, et al. Disruption of the mouse mdr1a P-glycoprotein gene leads to a deficiency in the blood-brain barrier and to increased sensitivity to drugs. Cell. 1994;77:491-502.
58 Hayashi K, Pu H, Andras IE, et al. HIV-TAT protein upregulates expression of multidrug resistance protein 1 in the blood-brain barrier. J Cereb Blood Flow Metab. 2006;26:1052-1065.
59 Hayashi K, Pu H, Tian J, et al. HIV-Tat protein induces P-glycoprotein expression in brain microvascular endothelial cells. J Neurochem. 2005;93:1231-1241.
60 Graff CL, Pollack GM. Drug transport at the blood-brain barrier and the choroid plexus. Curr Drug Metab. 2004;5:95-108.
61 Cirrito JR, Deane R, Fagan AM, et al. P-glycoprotein deficiency at the blood-brain barrier increases amyloid-beta deposition in an Alzheimer disease mouse model. J Clin Invest. 2005;115:3285-3290.
62 Volk H, Potschka H, Loscher W. Immunohistochemical localization of P-glycoprotein in rat brain and detection of its increased expression by seizures are sensitive to fixation and staining variables. J Histochem Cytochem. 2005;53:517-531.
63 Kortekaas R, Leenders KL, van Oostrom JC, et al. Blood-brain barrier dysfunction in parkinsonian midbrain in vivo. Ann Neurol. 2005;57:176-179.
64 Yang L, Froio RM, Sciuto TE, et al. ICAM-1 regulates neutrophil adhesion and transcellular migration of TNF-alpha–activated vascular endothelium under flow. Blood. 2005;106:584-592.
65 Cullere X, Shaw SK, Andersson L, et al. Regulation of vascular endothelial barrier function by Epac, a cAMP-activated exchange factor for Rap GTPase. Blood. 2005;105:1950-1955.
66 Persidsky Y, Heilman D, Haorah J, et al. Rho-mediated regulation of tight junctions during monocyte migration across the blood-brain barrier in HIV-1 encephalitis (HIVE). Blood. 2006;107:4770-4780.
67 Stevenson BR, Siliciano JD, Mooseker MS, et al. Identification of ZO-1: a high molecular weight polypeptide associated with the tight junction (zonula occludens) in a variety of epithelia. J Cell Biol. 1986;103:755-766.
68 Hanin I. The Gulf War, stress and a leaky blood-brain barrier. Nat Med. 1996;2:1307-1308.
69 Mark KS, Davis TP. Cerebral microvascular changes in permeability and tight junctions induced by hypoxia-reoxygenation. Am J Physiol Heart Circ Physiol. 2002;282:H1485-H1494.
70 Zahraoui A, Joberty G, Arpin M, et al. A small rab GTPase is distributed in cytoplasmic vesicles in non polarized cells but colocalizes with the tight junction marker ZO-1 in polarized epithelial cells. J Cell Biol. 1994;124:101-115.
71 Islas S, Vega J, Ponce L, et al. Nuclear localization of the tight junction protein ZO-2 in epithelial cells. Exp Cell Res. 2002;274:138-148.
72 Traweger A, Fuchs R, Krizbai IA, et al. The tight junction protein ZO-2 localizes to the nucleus and interacts with the heterogeneous nuclear ribonucleoprotein scaffold attachment factor-B. J Biol Chem. 2003;278:2692-2700.
73 Hirase T, Staddon JM, Saitou M, et al. Occludin as a possible determinant of tight junction permeability in endothelial cells. J Cell Sci. 1997;110:1603-1613.
74 Inoko A, Itoh M, Tamura A, et al. Expression and distribution of ZO-3, a tight junction MAGUK protein, in mouse tissues. Genes Cells. 2003;8:837-845.
75 Wolburg H, Lippoldt A. Tight junctions of the blood-brain barrier: development, composition and regulation. Vascul Pharmacol. 2002;38:323-337.
76 Tsukita S, Furuse M, Itoh M. Structural and signalling molecules come together at tight junctions. Curr Opin Cell Biol. 1999;11:628-633.
77 Furuse M, Hirase T, Itoh M, et al. Occludin: a novel integral membrane protein localizing at tight junctions. J Cell Biol. 1993;123:1777-1788.
78 Dallasta LM, Pisarov LA, Esplen JE, et al. Blood-brain barrier tight junction disruption in human immunodeficiency virus-1 encephalitis. Am J Pathol. 1999;155:1915-1927.
79 Papadopoulos MC, Saadoun S, Woodrow CJ, et al. Occludin expression in microvessels of neoplastic and non-neoplastic human brain. Neuropathol Appl Neurobiol. 2001;27:384-395.
80 Hou J, Gomes AS, Paul DL, et al. Study of claudin function by RNA interference. J Biol Chem. 2006;281:36117-36123.
81 Morita K, Sasaki H, Furuse M, et al. Endothelial claudin: claudin-5/TMVCF constitutes tight junction strands in endothelial cells. J Cell Biol. 1999;147:185-194.
82 Morita K, Sasaki H, Furuse K, et al. Expression of claudin-5 in dermal vascular endothelia. Exp Dermatol. 2003;12:289-295.
83 Kubota K, Furuse M, Sasaki H, et al. Ca(2+)-independent cell-adhesion activity of claudins, a family of integral membrane proteins localized at tight junctions. Curr Biol. 1999;9:1035-1038.
84 Ishihara H, Kubota H, Lindberg RL, et al. Endothelial cell barrier impairment induced by glioblastomas and transforming growth factor beta2 involves matrix metalloproteinases and tight junction proteins. J Neuropathol Exp Neurol. 2008;67:435-448.
85 Liebner S, Fischmann A, Rascher G, et al. Claudin-1 and claudin-5 expression and tight junction morphology are altered in blood vessels of human glioblastoma multiforme. Acta Neuropathol. 2000;100:323-331.
86 Del Maschio A, De Luigi A, Martin-Padura I, et al. Leukocyte recruitment in the cerebrospinal fluid of mice with experimental meningitis is inhibited by an antibody to junctional adhesion molecule (JAM). J Exp Med. 1999;190:1351-1356.
87 Martin-Padura I, Lostaglio S, Schneemann M, et al. Junctional adhesion molecule, a novel member of the immunoglobulin superfamily that distributes at intercellular junctions and modulates monocyte transmigration. J Cell Biol. 1998;142:117-127.
88 Maher PA, Pasquale EB. Tyrosine phosphorylated proteins in different tissues during chick embryo development. J Cell Biol. 1988;106:1747-1755.
89 Sakakibara A, Furuse M, Saitou M, et al. Possible involvement of phosphorylation of occludin in tight junction formation. J Cell Biol. 1997;137:1393-1401.
90 Antonetti DA, Wolpert EB, DeMaio L, et al. Hydrocortisone decreases retinal endothelial cell water and solute flux coincident with increased content and decreased phosphorylation of occludin. J Neurochem. 2002;80:667-677.
91 Denker BM, Nigam SK. Molecular structure and assembly of the tight junction. Am J Physiol. 1998;274:F1-F9.
92 Tsukamoto T, Nigam SK. Role of tyrosine phosphorylation in the reassembly of occludin and other tight junction proteins. Am J Physiol. 1999;276:F737-F750.
93 Said HM, Ma TY. Mechanism of riboflavine uptake by Caco-2 human intestinal epithelial cells. Am J Physiol. 1994;266:G15-G21.
94 Ma TY, Tran D, Hoa N, et al. Mechanism of extracellular calcium regulation of intestinal epithelial tight junction permeability: role of cytoskeletal involvement. Microsc Res Tech. 2000;51:156-168.
95 Brown RC, Davis TP. Calcium modulation of adherens and tight junction function: a potential mechanism for blood-brain barrier disruption after stroke. Stroke. 2002;33:1706-1711.
96 Haorah J, Knipe B, Leibhart J, et al. Alcohol-induced oxidative stress in brain endothelial cells causes blood-brain barrier dysfunction. J Leukoc Biol. 2005;78:1223-1232.
97 Hopkins AM, Li D, Mrsny RJ, et al. Modulation of tight junction function by G protein–coupled events. Adv Drug Deliv Rev. 2000;30(41):329-340.
98 Stamatovic SM, Dimitrijevic OB, Keep RF, et al. Protein kinase Calpha-RhoA cross-talk in CCL2-induced alterations in brain endothelial permeability. J Biol Chem. 2006;281:8379-8388.
99 Kanmogne GD, Schall K, Leibhart J, et al. HIV-1 gp120 compromises blood-brain barrier integrity and enhances monocyte migration across blood-brain barrier: implication for viral neuropathogenesis. J Cereb Blood Flow Metab. 2007;27:123-134.
100 Yamamoto T, Harada N, Kawano Y, et al. In vivo interaction of AF-6 with activated Ras and ZO-1. Biochem Biophys Res Commun. 1999;259:103-107.
101 Prat A, Biernacki K, Lavoie JF, et al. Migration of multiple sclerosis lymphocytes through brain endothelium. Arch Neurol. 2002;59:391-397.
102 Banks WA, Ercal N, Price TO. The blood-brain barrier in neuroAIDS. Curr HIV Res. 2006;4:259-266.
103 Ebihara C, Kondoh M, Harada M, et al. Role of Tyr306 in the C-terminal fragment of Clostridium perfringens enterotoxin for modulation of tight junction. Biochem Pharmacol. 2007;73:824-830.
104 Ring A, Weiser JN, Tuomanen EI. Pneumococcal trafficking across the blood-brain barrier. Molecular analysis of a novel bidirectional pathway. J Clin Invest. 1998;102:347-360.
105 Paul R, Lorenzl S, Koedel U, et al. Matrix metalloproteinases contribute to the blood-brain barrier disruption during bacterial meningitis. Ann Neurol. 1998;44:592-600.
106 Doulet N, Donnadieu E, Laran-Chich MP, et al. Neisseria meningitidis infection of human endothelial cells interferes with leukocyte transmigration by preventing the formation of endothelial docking structures. J Cell Biol. 2006;173:627-637.
107 Cunningham LA, Wetzel M, Rosenberg GA. Multiple roles for MMPs and TIMPs in cerebral ischemia. Glia. 2005;50:329-339.
108 Klatzo I. Presidental address. Neuropathological aspects of brain edema. J Neuropathol Exp Neurol. 1967;26:1-14.
109 Deane R, Wu Z, Zlokovic BV. RAGE (yin) versus LRP (yang) balance regulates Alzheimer amyloid beta-peptide clearance through transport across the blood-brain barrier. Stroke. 2004;35(suppl 1):2628-2631.
110 Donahue JE, Flaherty SL, Johanson CE, et al. RAGE, LRP-1, and amyloid-beta protein in Alzheimer’s disease. Acta Neuropathol. 2006;112:405-415.
111 Oby E, Janigro D. The blood-brain barrier and epilepsy. Epilepsia. 2006;47:1761-1774.
112 Smith QR. A review of blood-brain barrier transport techniques. Methods Mol Med. 2003;89:193-208.
113 Cairncross JG, Macdonald DR, Pexman JH, et al. Steroid-induced CT changes in patients with recurrent malignant glioma. Neurology. 1988;38:724-726.
114 Abbott NJ, Pichon Y. The glial blood-brain barrier of crustacea and cephalopods: a review. J Physiol (Paris). 1987;82:304-313.
115 Butt AM, Jones HC, Abbott NJ. Electrical resistance across the blood-brain barrier in anaesthetized rats: a developmental study. J Physiol. 1990;429:47-62.
116 Olesen SP. Rapid increase in blood-brain barrier permeability during severe hypoxia and metabolic inhibition. Brain Res. 1986;368:24-29.
117 Schilling L, Wahl M. Brain edema: pathogenesis and therapy. Kidney Int Suppl. 1997;59:S69-S75.
118 Ghose AK, Viswanadhan VN, Wendoloski JJ. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J Comb Chem. 1999;1:55-68.
119 Behin A, Hoang-Xuan K, Carpentier AF, et al. Primary brain tumours in adults. Lancet. 2003;361:323-331.
120 Wick W, Wick A, Schulz JB, et al. Prevention of irradiation-induced glioma cell invasion by temozolomide involves caspase 3 activity and cleavage of focal adhesion kinase. Cancer Res. 2002;62:1915-1919.
121 Wild-Bode C, Weller M, Rimner A, et al. Sublethal irradiation promotes migration and invasiveness of glioma cells: implications for radiotherapy of human glioblastoma. Cancer Res. 2001;61:2744-2750.
122 Janigro D. Blood-brain barrier, ion homeostasis and epilepsy: possible implications towards the understanding of ketogenic diet mechanisms. Epilepsy Res. 1999;37:223-232.
123 Sills GJ, Kwan P, Butler E, et al. P-glycoprotein–mediated efflux of antiepileptic drugs: preliminary studies in mdr1a knockout mice. Epilepsy Behav. 2002;3:427-432.
124 Potschka H, Loscher W. In vivo evidence for P-glycoprotein–mediated transport of phenytoin at the blood-brain barrier of rats. Epilepsia. 2001;42:1231-1240.
125 Potschka H, Fedrowitz M, Loscher W. P-glycoprotein and multidrug resistance–associated protein are involved in the regulation of extracellular levels of the major antiepileptic drug carbamazepine in the brain. Neuroreport. 2001;12:3557-3560.
126 Awasthi S, Hallene KL, Fazio V, et al. RLIP76, a non-ABC transporter, and drug resistance in epilepsy. BMC Neurosci. 2005;6:61.
127 Marchi N, Hallene KL, Kight KM, et al. Significance of MDR1 and multiple drug resistance in refractory human epileptic brain. BMC Med. 2004;2:37.
128 Regesta G, Tanganelli P. Clinical aspects and biological bases of drug-resistant epilepsies. Epilepsy Res. 1999;34:109-122.
129 Volk HA, Loscher W. Multidrug resistance in epilepsy: rats with drug-resistant seizures exhibit enhanced brain expression of P-glycoprotein compared with rats with drug-responsive seizures. Brain. 2005;128:1358-1368.
130 Hardman J, Limbird L, Gilman A. Goodman and Gilman’s the Pharmacological Basis of Therapeutics. New York: McGraw-Hill; 1991.
131 Hawkins RA, Mokashi A, Simpson IA. An active transport system in the blood-brain barrier may reduce levodopa availability. Exp Neurol. 2005;195:267-271.
132 O’Kane RL, Hawkins RA. Na+-dependent transport of large neutral amino acids occurs at the abluminal membrane of the blood-brain barrier. Am J Physiol Endocrinol Metab. 2003;285:E1167-E1173.
133 Calne DB Parkinsonism. Clinical and neuropharmacologic aspects. Postgrad Med. 1978;64:82-88.
134 Parnetti L. Clinical pharmacokinetics of drugs for Alzheimer’s disease. Clin Pharmacokinet. 1995;29:110-129.
135 Chaudhuri JD. Blood brain barrier and infection. Med Sci Monit. 2000;6:1213-1222.
136 Cashion MF, Banks WA, Bost KL, et al. Transmission routes of HIV-1 gp120 from brain to lymphoid tissues. Brain Res. 1999;822:26-33.
137 Schrager LK, D’Souza MP. Cellular and anatomical reservoirs of HIV-1 in patients receiving potent antiretroviral combination therapy. JAMA. 1998;280:67-71.
138 Ahmed AE, Jacob S, Loh JP, et al. Comparative disposition and whole-body autoradiographic distribution of [2-14C]azidothymidine and [2-14C]thymidine in mice. J Pharmacol Exp Ther. 1991;257:479-486.
139 de Miranda P, Burnette TC, Good SS. Tissue distribution and metabolic disposition of zidovudine in rats. Drug Metab Dispos. 1990;18:315-320.
140 Wu D, Clement JG, Pardridge WM. Low blood-brain barrier permeability to azidothymidine (AZT), 3TC, and thymidine in the rat. Brain Res. 1998;27(791):313-316.
141 Lucia MB, Cauda R, Landay AL, et al. Transmembrane P-glycoprotein (P-gp/P-170) in HIV infection: analysis of lymphocyte surface expression and drug-unrelated function. AIDS Res Hum Retroviruses. 1995;11:893-901.
142 Strazielle N, Belin MF, Ghersi-Egea JF. Choroid plexus controls brain availability of anti-HIV nucleoside analogs via pharmacologically inhibitable organic anion transporters. AIDS. 2003;17:1473-1485.
143 Yang Z, Brundage RC, Barbhaiya RH, et al. Microdialysis studies of the distribution of stavudine into the central nervous system in the freely-moving rat. Pharm Res. 1997;14:865-872.
144 Huisman MT, Smit JW, Wiltshire HR, et al. P-glycoprotein limits oral availability, brain, and fetal penetration of saquinavir even with high doses of ritonavir. Mol Pharmacol. 2001;59:806-813.
145 Glynn SL, Yazdanian M. In vitro blood-brain barrier permeability of nevirapine compared to other HIV antiretroviral agents. J Pharm Sci. 1998;87:306-310.
146 Thomas SA. Anti-HIV drug distribution to the central nervous system. Curr Pharm Des. 2004;10:1313-1324.
147 Buchwald P, Bodor N. Octanol-water partition: searching for predictive models. Curr Med Chem. 1998;5:353-380.
148 Kay GG. The effects of antihistamines on cognition and performance. J Allergy Clin Immunol. 2000;105:S622-S627.
149 Bodor N, Buchwald P. Brain-targeted drug delivery: experiences to date. Am J Drug Deliv. 2003;1:13-26.
150 Pardridge WM. Brain Drug Targeting: The Future of Brain Drug Development. Cambridge, UK: Cambridge University Press; 2001.
151 Bodor N, Buchwald P. Barriers to remember: brain-targeting chemical delivery systems and Alzheimer’s disease. Drug Discov Today. 2002;7:766-774.
152 Begley DJ. Delivery of therapeutic agents to the central nervous system: the problems and the possibilities. Pharmacol Ther. 2004;104:29-45.
153 Genka S, Deutsch J, Shetty UH, et al. Development of lipophilic anticancer agents for the treatment of brain tumors by the esterification of water-soluble chlorambucil. Clin Exp Metastasis. 1993;11:131-140.
154 Greig NH, Genka S, Daly EM, et al. Physicochemical and pharmacokinetic parameters of seven lipophilic chlorambucil esters designed for brain penetration. Cancer Chemother Pharmacol. 1990;25:311-319.
155 Kitagawa K, Mizobuchi N, Hama T, et al. Synthesis and antinociceptive activity of [D-Ala2]Leu-enkephalin derivatives conjugated with the adamantane moiety. Chem Pharm Bull (Tokyo). 1997;45:1782-1787.
156 Prokai-Tatrai K, Prokai L, Bodor N. Brain-targeted delivery of a leucine-enkephalin analogue by retrometabolic design. J Med Chem. 1996;39:4775-4782.
157 Chikhale EG, Ng KY, Burton PS, et al. Hydrogen bonding potential as a determinant of the in vitro and in situ blood-brain barrier permeability of peptides. Pharm Res. 1994;11:412-419.
158 Fischer H, Gottschlich R, Seelig A. Blood-brain barrier permeation: molecular parameters governing passive diffusion. J Membr Biol. 1998;165:201-211.
159 Rice A, Liu Y, Michaelis ML, et al. Chemical modification of paclitaxel (Taxol) reduces P-glycoprotein interactions and increases permeation across the blood-brain barrier in vitro and in situ. J Med Chem. 2005;48:832-838.
160 Friden PM, Walus LR, Watson P, et al. Blood-brain barrier penetration and in vivo activity of an NGF conjugate. Science. 1993;259:373-377.
161 Shi N, Pardridge WM. Noninvasive gene targeting to the brain. Proc Natl Acad Sci U S A. 2000;97:7567-7572.
162 Schwarze SR, Ho A, Vocero-Akbani A, et al. In vivo protein transduction: delivery of a biologically active protein into the mouse. Science. 1999;285:1569-1572.
163 Juliano RL, Ling V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim Biophys Acta. 1976;455:152-162.
164 Schinkel AH, Wagenaar E, Mol CA, et al. P-glycoprotein in the blood-brain barrier of mice influences the brain penetration and pharmacological activity of many drugs. J Clin Invest. 1996;97:2517-2524.
165 Begley DJ. ABC transporters and the blood-brain barrier. Curr Pharm Des. 2004;10:1295-1312.
166 Gottesman MM, Pastan I. Biochemistry of multidrug resistance mediated by the multidrug transporter. Annu Rev Biochem. 1993;62:385-427.
167 van Veen HW, Konings WN. Multidrug transporters from bacteria to man: similarities in structure and function. Semin Cancer Biol. 1997;8:183-191.
168 Golden PL, Pardridge WM. P-Glycoprotein on astrocyte foot processes of unfixed isolated human brain capillaries. Brain Res. 1999;819:143-146.
169 Pardridge WM, Golden PL, Kang YS, et al. Brain microvascular and astrocyte localization of P-glycoprotein. J Neurochem. 1997;68:1278-1285.
170 Cordon-Cardo C, O’Brien JP, Casals D, et al. Multidrug-resistance gene (P-glycoprotein) is expressed by endothelial cells at blood-brain barrier sites. Proc Natl Acad Sci U S A. 1989;86:695-698.
171 Demeule M, Regina A, Jodoin J, et al. Drug transport to the brain: key roles for the efflux pump P-glycoprotein in the blood-brain barrier. Vascul Pharmacol. 2002;38:339-348.
172 Miller DS, Nobmann SN, Gutmann H, et al. Xenobiotic transport across isolated brain microvessels studied by confocal microscopy. Mol Pharmacol. 2000;58:1357-1367.
173 Virgintino D, Robertson D, Errede M, et al. Expression of P-glycoprotein in human cerebral cortex microvessels. J Histochem Cytochem. 2002;50:1671-1676.
174 Mealey KL, Bentjen SA, Gay JM, et al. Ivermectin sensitivity in collies is associated with a deletion mutation of the mdr1 gene. Pharmacogenetics. 2001;11:727-733.
175 Ford JM, Hait WN. Pharmacology of drugs that alter multidrug resistance in cancer. Pharmacol Rev. 1990;42:155-199.
176 Tsuji A, Tamai I, Sakata A, et al. Restricted transport of cyclosporin A across the blood-brain barrier by a multidrug transporter, P-glycoprotein. Biochem Pharmacol. 1993;46:1096-1099.
177 Twentyman PR, Fox NE, White DJ. Cyclosporin A and its analogues as modifiers of Adriamycin and vincristine resistance in a multi-drug resistant human lung cancer cell line. Br J Cancer. 1987;56:55-57.
178 Friche E, Jensen PB, Nissen NI. Comparison of cyclosporin A and SDZ PSC833 as multidrug-resistance modulators in a daunorubicin-resistant Ehrlich ascites tumor. Cancer Chemother Pharmacol. 1992;30:235-237.
179 Twentyman PR, Bleehen NM. Resistance modification by PSC-833, a novel non-immunosuppressive cyclosporin [corrected]. Eur J Cancer. 1991;27:1639-1642.
180 Evers R, Kool M, Smith AJ, et al. Inhibitory effect of the reversal agents V-104, GF120918 and Pluronic L61 on MDR1 Pgp-, MRP1- and MRP2-mediated transport. Br J Cancer. 2000;83:366-374.
181 Hyafil F, Vergely C, Du Vignaud P, et al. In vitro and in vivo reversal of multidrug resistance by GF120918, an acridonecarboxamide derivative. Cancer Res. 1993;53:4595-4602.
182 Shepard RL, Cao J, Starling JJ, et al. Modulation of P-glycoprotein but not MRP1- or BCRP-mediated drug resistance by LY335979. Int J Cancer. 2003;103:121-125.
183 Fellner S, Bauer B, Miller DS, et al. Transport of paclitaxel (Taxol) across the blood-brain barrier in vitro and in vivo. J Clin Invest. 2002;110:1309-1318.
184 Kemper EM, Verheij M, Boogerd W, et al. Improved penetration of docetaxel into the brain by co-administration of inhibitors of P-glycoprotein. Eur J Cancer. 2004;40:1269-1274.
185 Spahn-Langguth H, Langguth P. Grapefruit juice enhances intestinal absorption of the P-glycoprotein substrate talinolol. Eur J Pharm Sci. 2001;12:361-367.
186 Wang EJ, Casciano CN, Clement RP, et al. Inhibition of P-glycoprotein transport function by grapefruit juice psoralen. Pharm Res. 2001;18:432-438.
187 Kim SW, Kwon HY, Chi DW, et al. Reversal of P-glycoprotein–mediated multidrug resistance by ginsenoside Rg(3). Biochem Pharmacol. 2003;65:75-82.
188 Bhardwaj RK, Glaeser H, Becquemont L, et al. Piperine, a major constituent of black pepper, inhibits human P-glycoprotein and CYP3A4. J Pharmacol Exp Ther. 2002;302:645-650.
189 Thomas H, Coley HM. Overcoming multidrug resistance in cancer: an update on the clinical strategy of inhibiting p-glycoprotein. Cancer Control. 2003;10:159-165.
190 Demeule M, Regina A, Annabi B, et al. Brain endothelial cells as pharmacological targets in brain tumors. Mol Neurobiol. 2004;30:157-183.
191 Doolittle ND, Miner ME, Hall WA, et al. Safety and efficacy of a multicenter study using intraarterial chemotherapy in conjunction with osmotic opening of the blood-brain barrier for the treatment of patients with malignant brain tumors. Cancer. 2000;88:637-647.
192 Brightman MW, Hori M, Rapoport SI, et al. Osmotic opening of tight junctions in cerebral endothelium. J Comp Neurol. 1973;152:317-325.
193 Rapoport SI. Effect of concentrated solutions on blood-brain barrier. Am J Physiol. 1970;219:270-274.
194 Farkas A, Szatmari E, Orbok A, et al. Hyperosmotic mannitol induces Src kinase–dependent phosphorylation of beta-catenin in cerebral endothelial cells. J Neurosci Res. 2005;80:855-861.
195 Nag S. Role of the endothelial cytoskeleton in blood-brain-barrier permeability to protein. Acta Neuropathol. 1995;90:454-460.
196 Wong MK, Gotlieb AI. Endothelial cell monolayer integrity. I. Characterization of dense peripheral band of microfilaments. Arteriosclerosis. 1986;6:212-219.
197 Nagashima T, Shijing W, Mizoguchi A, et al. A possible role of calcium ion in osmotic opening of blood-brain barrier. J Auton Nerv Syst. 1994;49(suppl):S145-S149.
198 Siegal T, Zylber-Katz E. Strategies for increasing drug delivery to the brain: focus on brain lymphoma. Clin Pharmacokinet. 2002;41:171-186.
199 Walker MD, Weiss HD. Chemotherapy in the treatment of malignant brain tumors. Adv Neurol. 1975;13:149-191.
200 Sawada T, Kato Y, Kobayashi M, et al. Immunohistochemical study of tight junction–related protein in neovasculature in astrocytic tumor. Brain Tumor Pathol. 2000;17:1-6.
201 Yuan F, Salehi HA, Boucher Y, et al. Vascular permeability and microcirculation of gliomas and mammary carcinomas transplanted in rat and mouse cranial windows. Cancer Res. 1994;54:4564-4568.
202 Gallo JM, Li S, Guo P, et al. The effect of P-glycoprotein on paclitaxel brain and brain tumor distribution in mice. Cancer Res. 2003;63:5114-5117.
203 Markowsky SJ, Zimmerman CL, Tholl D, et al. Methotrexate disposition following disruption of the blood-brain barrier. Ther Drug Monit. 1991;13:24-31.
204 Robinson PJ, Rapoport SI. Model for drug uptake by brain tumors: effects of osmotic treatment and of diffusion in brain. J Cereb Blood Flow Metab. 1990;10:153-161.
205 Dahlborg SA, Petrillo A, Crossen JR, et al. The potential for complete and durable response in nonglial primary brain tumors in children and young adults with enhanced chemotherapy delivery. Cancer J Sci Am. 1998;4:110-124.
206 Kraemer DF, Fortin D, Doolittle ND, et al. Association of total dose intensity of chemotherapy in primary central nervous system lymphoma (human non-acquired immunodeficiency syndrome) and survival. Neurosurgery. 2001;48:1033-1040.
207 McAllister LD, Doolittle ND, Guastadisegni PE, et al. Cognitive outcomes and long-term follow-up results after enhanced chemotherapy delivery for primary central nervous system lymphoma. Neurosurgery. 2000;46:51-60.
208 Neuwelt EA, Wiliams PC, Mickey BE, et al. Therapeutic dilemma of disseminated CNS germinoma and the potential of increased platinum-based chemotherapy delivery with osmotic blood-brain barrier disruption. Pediatr Neurosurg. 1994;21:16-22.
209 Somjen GG, Segal MB, Herreras O. Osmotic-hypertensive opening of the blood-brain barrier in rats does not necessarily provide access for potassium to cerebral interstitial fluid. Exp Physiol. 1991;76:507-514.
210 Bell S. Breaking down barriers to treat a patient with a germinoma: a case study. J Neurosci Nurs. 2004;36:195-199.
211 Fortin D, Desjardins A, Benko A, et al. Enhanced chemotherapy delivery by intraarterial infusion and blood-brain barrier disruption in malignant brain tumors: the Sherbrooke experience. Cancer. 2005;103:2606-2615.
212 Doolittle ND, Abrey LE, Ferrari N, et al. Targeted delivery in primary and metastatic brain tumors: summary report of the seventh annual meeting of the Blood-Brain Barrier Disruption Consortium. Clin Cancer Res. 2002;8:1702-1709.
213 Kraemer DF, Fortin D, Neuwelt EA. Chemotherapeutic dose intensification for treatment of malignant brain tumors: recent developments and future directions. Curr Neurol Neurosci Rep. 2002;2:216-224.
214 Seiffert E, Dreier JP, Ivens S, et al. Lasting blood-brain barrier disruption induces epileptic focus in the rat somatosensory cortex. J Neurosci. 2004;24:7829-7836.
215 Brown RC, Egleton RD, Davis TP. Mannitol opening of the blood-brain barrier: regional variation in the permeability of sucrose, but not 86Rb+ or albumin. Brain Res. 2004;1014:221-227.
216 Heistad DD, Marcus ML, Gross PM. Effects of sympathetic nerves on cerebral vessels in dog, cat, and monkey. Am J Physiol. 1978;235:H544-H552.
217 Suzuki N, Hardebo JE, Owman C. Origins and pathways of cerebrovascular nerves storing substance P and calcitonin gene–related peptide in rat. Neuroscience. 1989;31:427-438.
218 Liu-Chen LY, Liszczak TM, King JC, et al. Immunoelectron microscopic study of substance P–containing fibers in feline cerebral arteries. Brain Res. 1986;369:12-20.
219 Suzuki N, Hardebo JE, Kahrstrom J, et al. Effect on cortical blood flow of electrical stimulation of trigeminal cerebrovascular nerve fibres in the rat. Acta Physiol Scand. 1990;138:307-316.
220 Suzuki N, Hardebo JE, Kahrstrom J, et al. Selective electrical stimulation of postganglionic cerebrovascular parasympathetic nerve fibers originating from the sphenopalatine ganglion enhances cortical blood flow in the rat. J Cereb Blood Flow Metab. 1990;10:383-391.
221 Matthew JD, Wadsworth RM. The role of nitric oxide in inhibitory neurotransmission in the middle cerebral artery of the sheep. Gen Pharmacol. 1997;28:393-397.
222 Toda N. Mediation by nitric oxide of neurally-induced human cerebral artery relaxation. Experientia. 1993;49:51-53.
223 Toda N, Ayajiki K, Tanaka T, et al. Preganglionic and postganglionic neurons responsible for cerebral vasodilation mediated by nitric oxide in anesthetized dogs. J Cereb Blood Flow Metab. 2000;20:700-708.
224 Seylaz J, Hara H, Pinard E, et al. Effect of stimulation of the sphenopalatine ganglion on cortical blood flow in the rat. J Cereb Blood Flow Metab. 1988;8:875-878.
225 Suzuki N, Hardebo JE, Owman C. Origins and pathways of cerebrovascular vasoactive intestinal polypeptide–positive nerves in rat. J Cereb Blood Flow Metab. 1988;8:697-712.
226 Hara H, Zhang QJ, Kuroyanagi T, et al. Parasympathetic cerebrovascular innervation: an anterograde tracing from the sphenopalatine ganglion in the rat. Neurosurgery. 1993;32:822-827.
227 Suzuki N, Hardebo JE, Skagerberg G, et al. Central origins of preganglionic fibers to the sphenopalatine ganglion in the rat. A fluorescent retrograde tracer study with special reference to its relation to central catecholaminergic systems. J Auton Nerv Syst. 1990;30:101-109.
228 Mayhan WG. Role of nitric oxide in histamine-induced increases in permeability of the blood-brain barrier. Brain Res. 1996;743:70-76.
229 Mayhan WG. Nitric oxide donor–induced increase in permeability of the blood-brain barrier. Brain Res. 2000;866:101-108.
230 Yarnitsky D, Gross Y, Lorian A, et al. Blood-brain barrier opened by stimulation of the parasympathetic sphenopalatine ganglion: a new method for macromolecule delivery to the brain. J Neurosurg. 2004;101:303-309.
231 Yarnitsky D, Gross Y, Lorian A, et al. Increased BBB permeability by parasympathetic sphenopalatine ganglion stimulation in dogs. Brain Res. 2004;1018:236-240.
232 Bobo RH, Laske DW, Akbasak A, et al. Convection-enhanced delivery of macromolecules in the brain. Proc Natl Acad Sci U S A. 1994;91:2076-2080.
233 Laske DW, Youle RJ, Oldfield EH. Tumor regression with regional distribution of the targeted toxin TF-CRM107 in patients with malignant brain tumors. Nat Med. 1997;3:1362-1368.
234 Ali MJ, Navalitloha Y, Vavra MW, et al. Isolation of drug delivery from drug effect: problems of optimizing drug delivery parameters. Neuro Oncol. 2006;8:109-118.
235 Brem H, Mahaley MSJr, Vick NA, et al. Interstitial chemotherapy with drug polymer implants for the treatment of recurrent gliomas. J Neurosurg. 1991;74:441-446.
236 Hall WA. Targeted toxin therapy for malignant astrocytoma. Neurosurgery. 2000;46:544-551.
237 Xia CF, Boado RJ, Zhang Y, et al. Intravenous glial-derived neurotrophic factor gene therapy of experimental Parkinson’s disease with Trojan horse liposomes and a tyrosine hydroxylase promoter. J Gene Med. 2008;10:306-315.
238 Hynynen K, Clement GT, McDannold N, et al. 500-element ultrasound phased array system for noninvasive focal surgery of the brain: a preliminary rabbit study with ex vivo human skulls. Magn Reson Med. 2004;52:100-107.
239 Hynynen K, McDannold N, Vykhodtseva N, et al. Noninvasive MR imaging–guided focal opening of the blood-brain barrier in rabbits. Radiology. 2001;220:640-646.
240 Sheikov N, McDannold N, Vykhodtseva N, et al. Cellular mechanisms of the blood-brain barrier opening induced by ultrasound in presence of microbubbles. Ultrasound Med Biol. 2004;30:979-989.
241 Muldoon LL, Soussain C, Jahnke K, et al. Chemotherapy delivery issues in central nervous system malignancy: a reality check. J Clin Oncol. 2007;25:2295-2305.
242 Provenzale JM, Mukundan S, Dewhirst M. The role of blood-brain barrier permeability in brain tumor imaging and therapeutics. AJR Am J Roentgenol. 2005;185:763-767.
243 Wilkinson ID, Jellineck DA, Levy D, et al. Dexamethasone and enhancing solitary cerebral mass lesions: alterations in perfusion and blood-tumor barrier kinetics shown by magnetic resonance imaging. Neurosurgery. 2006;58:640-646.
244 Neuwelt EA, Varallyay CG, Manninger S, et al. The potential of ferumoxytol nanoparticle magnetic resonance imaging, perfusion, and angiography in central nervous system malignancy: a pilot study. Neurosurgery. 2007;60:601-611.
245 Becherer A, Karanikas G, Szabo M, et al. Brain tumour imaging with PET: a comparison between [18F]fluorodopa and [11C]methionine. Eur J Nucl Med Mol Imaging. 2003;30:1561-1567.
246 Marchi N, Rasmussen P, Kapural M, et al. Peripheral markers of brain damage and blood-brain barrier dysfunction. Restor Neurol Neurosci. 2003;21:109-121.
247 Zimmer DB, Cornwall EH, Landar A, et al. The S-100 protein family: history, function, and expression. Brain Res Bull. 1995;37:417-429.
248 Haimoto H, Hosoda S, Kato K. Differential distribution of immunoreactive S-100-alpha and S-100-beta proteins in normal nonnervous human tissues. Lab Invest. 1987;57:489-498.
249 Kato K, Kimura S. S-100ao (alpha alpha) protein is mainly located in the heart and striated muscles. Biochim Biophys Acta. 1985;842:146-150.
250 Kato K, Suzuki F, Morishita R, et al. Selective increase in S-100 beta protein by aging in rat cerebral cortex. J Neurochem. 1990;54:1269-1274.
251 Reiber H. Cerebrospinal fluid—physiology, analysis and interpretation of protein patterns for diagnosis of neurological diseases. Mult Scler. 1998;4(3):99-107.
252 Dyck RH, Van Eldik LJ, Cynader MS. Immunohistochemical localization of the S-100 beta protein in postnatal cat visual cortex: spatial and temporal patterns of expression in cortical and subcortical glia. Brain Res Dev Brain Res. 1993;72:181-192.
253 Ludwin SK, Kosek JC, Eng LF. The topographical distribution of S-100 and GFA proteins in the adult rat brain: an immunohistochemical study using horseradish peroxidase–labelled antibodies. J Comp Neurol. 1976;165:197-207.
254 Mercier F, Hatton GI. Immunocytochemical basis for a meningeo-glial network. J Comp Neurol. 2000;420:445-465.
255 Rebaudo R, Melani R, Balestrino M, et al. Antiserum against S-100 protein prevents long term potentiation through a cAMP-related mechanism. Neurochem Res. 2000;25:541-545.
256 Grocott H, Laskowitz D, Newman M. The Brain and Cardiac Surgery. Amsterdam: Harwood Academic Publishers; 2000.
257 Buttner T, Weyers S, Postert T, et al. S-100 protein: serum marker of focal brain damage after ischemic territorial MCA infarction. Stroke. 1997;28:1961-1965.
258 Fassbender K, Schmidt R, Schreiner A, et al. Leakage of brain-originated proteins in peripheral blood: temporal profile and diagnostic value in early ischemic stroke. J Neurol Sci. 1997;148:101-105.
259 Missler U, Wiesmann M, Friedrich C, et al. S-100 protein and neuron-specific enolase concentrations in blood as indicators of infarction volume and prognosis in acute ischemic stroke. Stroke. 1997;28:1956-1960.
260 Persson L, Hardemark HG, Gustafsson J, et al. S-100 protein and neuron-specific enolase in cerebrospinal fluid and serum: markers of cell damage in human central nervous system. Stroke. 1987;18:911-918.
261 Abraha HD, Butterworth RJ, Bath PM, et al. Serum S-100 protein, relationship to clinical outcome in acute stroke. Ann Clin Biochem. 1997;34:366-370.
262 Elting JW, de Jager AE, Teelken AW, et al. Comparison of serum S-100 protein levels following stroke and traumatic brain injury. J Neurol Sci. 2000;181:104-110.
263 Pelinka LE, Toegel E, Mauritz W, et al. Serum S 100 B: a marker of brain damage in traumatic brain injury with and without multiple trauma. Shock. 2003;19:195-200.
264 Slemmer JE, Weber JT, De Zeeuw CI. Cell death, glial protein alterations and elevated S-100 beta release in cerebellar cell cultures following mechanically induced trauma. Neurobiol Dis. 2004;15:563-572.
265 Rothoerl RD, Woertgen C, Brawanski A. S-100 serum levels and outcome after severe head injury. Acta Neurochir Suppl. 2000;76:97-100.
266 Ingebrigtsen T, Romner B, Marup-Jensen S, et al. The clinical value of serum S-100 protein measurements in minor head injury: a Scandinavian multicentre study. Brain Inj. 2000;14:1047-1055.
267 Ingebrigtsen T, Waterloo K, Jacobsen EA, et al. Traumatic brain damage in minor head injury: relation of serum S-100 protein measurements to magnetic resonance imaging and neurobehavioral outcome. Neurosurgery. 1999;45:468-475.
268 Woertgen C, Rothoerl RD, Holzschuh M, et al. Comparison of serial S-100 and NSE serum measurements after severe head injury. Acta Neurochir (Wien). 1997;139:1161-1164.
269 Regner A, Kaufman M, Friedman G, et al. Increased serum S-100beta protein concentrations following severe head injury in humans: a biochemical marker of brain death? Neuroreport. 2001;12:691-694.
270 Ucar T, Baykal A, Akyuz M, et al. Comparison of serum and cerebrospinal fluid protein S-100b levels after severe head injury and their prognostic importance. J Trauma. 2004;57:95-98.
271 Wiesmann M, Missler U, Hagenstrom H, et al. S-100 protein plasma levels after aneurysmal subarachnoid haemorrhage. Acta Neurochir (Wien). 1997;139:1155-1160.
272 Rosen H, Rosengren L, Herlitz J, et al. Increased serum levels of the S-100 protein are associated with hypoxic brain damage after cardiac arrest. Stroke. 1998;29:473-477.
273 Bottiger BW, Mobes S, Glatzer R, et al. Astroglial protein S-100 is an early and sensitive marker of hypoxic brain damage and outcome after cardiac arrest in humans. Circulation. 2001;103:2694-2698.
274 Headrick JP, Bendall MR, Faden AI, et al. Dissociation of adenosine levels from bioenergetic state in experimental brain trauma: potential role in secondary injury. J Cereb Blood Flow Metab. 1994;14:853-861.
275 Nilsson P, Hillered L, Ponten U, et al. Changes in cortical extracellular levels of energy-related metabolites and amino acids following concussive brain injury in rats. J Cereb Blood Flow Metab. 1990;10:631-637.
276 Rudolphi KA, Schubert P, Parkinson FE, et al. Adenosine and brain ischemia. Cerebrovasc Brain Metab Rev. 1992;4:346-369.
277 Ciccarelli R, Di Iorio P, Bruno V, et al. Activation of A(1) adenosine or mGlu3 metabotropic glutamate receptors enhances the release of nerve growth factor and S-100beta protein from cultured astrocytes. Glia. 1999;27:275-281.
278 Anderson RE, Hansson LO, Nilsson O, et al. Increase in serum S-100A1-B and S-100BB during cardiac surgery arises from extracerebral sources. Ann Thorac Surg. 2001;71:1512-1517.
279 Blomquist S, Johnsson P, Luhrs C, et al. The appearance of S-100 protein in serum during and immediately after cardiopulmonary bypass surgery: a possible marker for cerebral injury. J Cardiothorac Vasc Anesth. 1997;11:699-703.
280 Grocott HP, Croughwell ND, Amory DW, et al. Cerebral emboli and serum S-100beta during cardiac operations. Ann Thorac Surg. 1998;65:1645-1649.
281 Fazio V, Bhudia SK, Marchi N, et al. Peripheral detection of S-100beta during cardiothoracic surgery: what are we really measuring? Ann Thorac Surg. 2004;78:46-52.
282 Brochez L, Naeyaert JM. Serological markers for melanoma. Br J Dermatol. 2000;143:256-268.
283 Kapural M, Krizanac-Bengez L, Barnett G, et al. Serum S-100beta as a possible marker of blood-brain barrier disruption. Brain Res. 2002;940:102-104.
284 Roman-Goldstein S, Mitchell P, Crossen JR, et al. MR and cognitive testing of patients undergoing osmotic blood-brain barrier disruption with intraarterial chemotherapy. AJNR Am J Neuroradiol. 1995;16:543-553.
285 Kanner AA, Marchi N, Fazio V, et al. Serum S-100beta: a noninvasive marker of blood-brain barrier function and brain lesions. Cancer. 2003;97:2806-2813.
286 Janigro D, Marchi N. Markers of Blood Barrier Permeability and Methods of Using Same. United States inventors. 2004.
287 Fabene PF, Marzola P, Sbarbati A, et al. Magnetic resonance imaging of changes elicited by status epilepticus in the rat brain: diffusion-weighted and T2-weighted images, regional blood volume maps, and direct correlation with tissue and cell damage. Neuroimage. 2003;18:375-389.
288 Fabene PF, Merigo F, Galie M, et al. Pilocarpine-induced status epilepticus in rats involves ischemic and excitotoxic mechanisms. PLoS One. 2007;2(10):e1105.
289 Ivens S, Kaufer D, Flores LP, et al. TGF-beta receptor–mediated albumin uptake into astrocytes is involved in neocortical epileptogenesis. Brain. 2007;130:535-547.
290 Marchi N, Angelov L, Masaryk T, et al. Seizure-promoting effect of blood-brain barrier disruption. Epilepsia. 2007;48:732-742.
291 Marchi N, Fan Q, Ghosh C, et al. Antagonism of peripheral inflammation reduces the severity of status epilepticus. Neurobiol Dis. 2009;33:171-181.
292 Marchi N, Oby E, Batra A, et al. In vivo and in vitro effects of pilocarpine: relevance to ictogenesis. Epilepsia. 2007;48:1934-1946.
293 Tomkins O, Friedman O, Ivens S, et al. Blood-brain barrier disruption results in delayed functional and structural alterations in the rat neocortex. Neurobiol Dis. 2007;25:367-377.
294 Tomkins O, Shelef I, Kaizerman I, et al. Blood-brain barrier disruption in post-traumatic epilepsy. J Neurol Neurosurg Psychiatry. 2008;79:774-777.
295 Pavlovsky L, Seiffert E, Heinemann U, et al. Persistent BBB disruption may underlie alpha interferon–induced seizures. J Neurol. 2005;252:42-46.
296 D’Ambrosio R, Fairbanks JP, Fender JS, et al. Post-traumatic epilepsy following fluid percussion injury in the rat. Brain. 2004;127:304-314.
297 Korn A, Golan H, Melamed I, et al. Focal cortical dysfunction and blood-brain barrier disruption in patients with Postconcussion syndrome. J Clin Neurophysiol. 2005;22:1-9.
298 Zylber-Katz E, Gomori JM, Schwartz A, et al. Pharmacokinetics of methotrexate in cerebrospinal fluid and serum after osmotic blood-brain barrier disruption in patients with brain lymphoma. Clin Pharmacol Ther. 2000;67:631-641.
299 Fieschi C, Lenzi GL, Zanette E, et al. Effects on EEG of the osmotic opening of the blood-brain barrier in rats. Life Sci. 1980;27:239-243.
300 Roman-Goldstein SM, Barnett PA, McCormick CI, et al. Effects of gadopentetate dimeglumine administration after osmotic blood-brain barrier disruption: toxicity and MR imaging findings. AJNR Am J Neuroradiol. 1991;12:885-890.
301 Elkassabany NM, Bhatia J, Deogaonkar A, et al. Perioperative complications of blood brain barrier disruption under general anesthesia: a retrospective review. J Neurosurg Anesthesiol. 2008;20:45-48.