30 Blood and Fluid Management during Cardiac Surgery
Transfusion guidelines

Rationale for Guidelines
Clinical guidelines are a ubiquitous part of medicine that are “systematically developed statements to assist practitioner and patient decisions about appropriate healthcare for specific clinical circumstances.”1 The U.S. National Guideline Clearinghouse (http://www.guideline.gov) includes more than 2480 recently generated guidelines. Guidelines are valuable methods for reducing practice variation, errors, and to ensure efficient use of healthcare resources.2 Despite the availability of well-constructed guidelines, clinicians often are reluctant to implement guidelines in daily practice. Several studies have shown that guidelines may do little to change practice behavior.3
Formulation of Guidelines
Groups developing guidelines require support from a national or international society that can endorse and disseminate guidelines. Members of the guideline committee should be recognized experts in the field and be assisted by people with expertise in guideline preparation. American Heart Association guidelines are the model for well-crafted guidelines, developed using well-conducted peer-reviewed studies as their evidence base (Table 30-1).
TABLE 30-1 Requirements for Successful Guideline Development and Implementation
| Guideline Writing Group Formation |
| Support at a societal level |
| Perception of need to define practice standards |
| Senior leadership for guideline preparation |
| Statistical support for examination of evidence |
| Guideline Preparation |
| Sufficient evidence base for guideline preparation |
| Sufficient expertise for guideline preparation |
| Dedicated time and effort for guideline generation |
| Dissemination to content and structure experts for commentary |
| Guideline Dissemination |
| Wide dissemination using an authoritative journal within the field |
| Wide dissemination using traditional and nontraditional mechanisms such as Web sites, conferences, and special interest groups |
| Guideline Conformity at the local institutional level |
| Senior leadership within the institution |
| Incorporation of guidelines into the local context of clinical practice |
| Providing practitioners with the resources to conform to the guidelines |
| Providing practitioners with timely, accurate, and pertinent feedback on measured conformity with guidelines |
Guidelines can be based on a spectrum of evidence from case reports, series without control groups, or randomized, controlled trials. The Society of Thoracic Surgeons/Society of Cardiovascular Anesthesiologists Transfusion Guidelines published in 20074 are a good example of generally well-crafted guidelines, but based on limited available evidence. There were 57 recommendations in the guidelines. For level of evidence, only 13 were level A (best level of evidence), 27 were level B (limited evidence), and 17 were level C (very limited evidence). For class of recommendation, only 7 were class I (benefit strongly outweighs risk), 18 were Class IIa (benefit outweighs risk), 23 were Class IIb (benefit may outweigh risk), and 10 were class III (risk outweighs benefit).5 The relatively low level of evidence for the guidelines likely is reflected by a lack of wide implementation by clinicians.

Implementation of Guidelines
To be effective, guidelines should be widely disseminated using commercial marketing tools. Anesthesia and surgical societies have been comparatively slow in adopting guidelines. Although the recently announced Society of Thoracic Surgeons/Society of Cardiovascular Anesthesiologists Transfusion Guidelines were widely distributed throughout both societies and to the perfusion community, many practitioners were not aware of the content of the guidelines. Several other studies have found similar lack of awareness of other guidelines.6,7
A number of other factors may hinder effective implementation of guidelines by practitioners such as guidelines that are too complicated and difficult to implement or lack rigorous scientific evidence as a basis. In other fields, such as general medicine practices, several studies have demonstrated low rates of change in response to guidelines.8–10 Furthermore, in the surgical environment that relies on multispecialty teams, there is a need for the entire team to understand the rationale for practice guideline implementation. However, it is equally important to realize that “guidelines need to remain flexible enough to permit a degree of patient-specific departures from specified prevention, diagnostic, and treatment protocols.”11
Other key components for successful implementation of guidelines lie in senior institution leadership and endorsement,12 as well as practice-specific feedback of performance. Implementation of guidelines in cardiac surgery previously has been reported to be poor,13,14 but focused implementation of guidelines at single institutions has been reported to be successful.15,16 Quality improvement initiatives used in industry such as Total Quality Management and Six Sigma, which appear applicable to surgical processes, are dependent on collection of verifiable data concerning processes.17,18 The notion that clinicians can improve cardiac surgical care and the performance of individual members of the team without proper timely feedback is not reasonable. In fact, real-time feedback has been demonstrated to be particularly effective.19 In summary, individuals involved in the management of cardiac surgical patients are obligated to provide the best possible clinical care, often guided by guidelines. Societies have a key role in endorsement, dissemination, and use of guidelines to improve care.
Blood groups and transfusion

ABO Blood Groups
ABO and Rh blood groups are the most well-known of more than 30 antigen-based classifications of human blood types. The ABO blood group system is based on identification of A and B antigens present on RBCs and was originally described by Janský and Landsteiner.20 Wide variation exists in the frequency of the ABO groups across different populations, with group B being the most common in Asians but comparatively rare in whites.21
ABO blood grouping is defined by presence or absence of surface antigens on the RBC membrane. The ABO gene is located on chromosome 9q34 and has three principal isoforms (A, B, and O) that are determined by single nucleotide polymorphisms (SNPs) and single-base deletions within the ABO gene (Table 30-2). Four missense SNPs determine the structural and functional differences between the A and B transferases. The different transferases result from four amino acid substitutions at amino acid positions (codons) 176, 235, 266, and 268 and nearly always occur together as a group. Two of these SNPs (L266M and G268A) change the substrate specificity of the ABO enzyme for galactose. The protein produced by the ABO gene is not the antigen. Rather, O, A, and B antigens are formed by the action of three different glycosyltransferases (isoforms) encoded by the ABO gene that modifies a cell membrane glycoprotein (H antigen).22
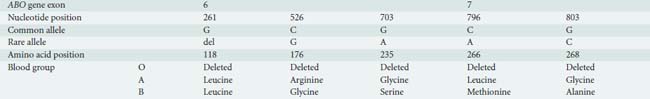
The O blood group is caused by a single base deletion (261delG) in exon 6 of the ABO gene that results in a frameshift mutation and translation of a truncated ABO protein, the O isoform, with no enzymatic activity; therefore, the A and B antigens are not present (the H antigen is unmodified).23
In the A transferase, the amino acids are leucine (L) and glycine (G) at codons 266 and 268, respectively. The A isoform (L266 and G268) encodes a glycosyltransferase (A transferase) that bonds α-N-acetylgalactosamine to the H antigen, producing the A antigen of the ABO blood group system. Individuals who exclusively synthesize A isoforms are blood group A and have the genotype AA (homozygotes: same A allele on both chromosomes) or AO (heterozygotes: A allele only present on one chromosome).23
In the B transferase, the amino acids are methionine (M) and alanine (A) at the same codons 266 and 268, respectively. The B isoform (M266 and A268) encodes a glycosyltransferase (B transferase) that joins α-d-galactose to the H antigen, creating the B antigen. Blood group B individuals have the genotype BB (homozygotes) or BO (heterozygotes). Individuals who express both A and B isoforms of the ABO gene (an A allele on one chromosome 9 and a B allele on the other chromosome 9) are blood group AB.23
Other variations in the ABO gene that create functionally similar antigens also exist. For example, the A(2) isoform, comprising only 20% of group A individuals, is caused by deletion of a protein coding termination point, thus extending the enzyme by 21 extra amino acids and altering its specificity. These structural differences reflect the different catalytic activities of the enzymes encoded by the A(1) and A(2) alleles and result in different antigenic properties of A(1) and A(2) antigens.24 Different molecular mechanisms may be responsible for seemingly identical ABO blood groups. For example, the B3 phenotype may be caused by either a missense mutation (D291N), a splicing mutation (B303), or the combination of a missense mutation and a single nucleotide deletion (V277M and 1060delC).
ABO blood groups also could be measured by genotyping the variants in the ABO gene instead of measuring the presence or absence of the A and B antigens. The ABO antigens are also expressed on the surface of many other cells types, indicating the importance of ABO cross-matching of organ transplants. Several other rare variants of the ABO gene that change activity and/or specificity of the enzyme have been identified and generate several rare blood groups.25
Anti-A and anti-B antibodies (isohemagglutinins) are IgM antibodies that appear in the first years of life. Early in the postnatal period, the immune system generates IgM antibodies against ABO antigen(s) even when they are absent from the individual’s RBCs. The IgM antibodies, if present in the fetus, are too large to cross the placenta, and thus are not a relevant cause of hemolytic disease of the newborn. The antibodies are thought to be produced in response to infantile exposure to influenza virus and gram-negative bacteria. Antibodies usually are not generated against the H antigen. Therefore, an individual with blood group A will make IgM antibodies against the B antigen. An individual with blood group B will make IgM antibodies against the A antigen. An individual with blood group AB will not make IgM antibodies to the A and B antigens. Finally, an individual with blood group O will make IgM antibodies against both the A and B antigens.23
If an individual of blood group A receives group B RBCs, anti-B IgM isohemagglutinins in the recipient plasma bind to the B antigen on donor RBCs, generating a locus for complement-mediated lysis of transfused donor RBCs and generation of a hemolytic transfusion reaction. Similar events occur in group O and B individuals receiving RBCs containing non–self-antigens. However, blood group AB individuals do not generate anti-A and anti-B antibodies. Therefore, they can receive RBCs from all groups and are universal RBC recipients.23

Rhesus Blood Groups
The rhesus (Rh) blood group system consists of ~50 blood group antigens among which five antigens—D, C, c, E, and e—are the most important. The proteins that carry the Rh antigens form a transmembrane transporter complex that resembles NH3 and CO2 transporters of evolutionary origin.26 The proteins are encoded by two adjacent genes on chromosome 1p36.13-p34.3: the RHD gene that encodes the RhD protein with the frequent D antigen and the RHCE gene that encodes the RhCE protein with the C, E, c, and e antigens.27 The term Rh factor refers only to the D antigen that is normally present. Unlike the ABO system, the absence of the normally present D antigen is called the d antigen, but there is no protein corresponding to the d antigen. Lowercase “d” indicates the absence of the D antigen, often as a result of the deletion of the gene or other variants that prevent expression of the antigenic protein on RBCs.27 To be Rh negative, the individual must have the gene absent on both chromosomes.
The frequency of Rh-negative individuals ranges from ~16% in white populations to less than 1% in Asian populations.28 Rh incompatibility of an RhD-negative mother and RhD-positive fetus is the predominant cause of hemolytic disease of the newborn, which occurs when maternal IgG anti-RhD antibodies pass through the placenta into the fetal circulation and cause hemolysis of RhD antigen-positive fetal RBCs.29

Other Blood Groups
At least 30 other antigens are expressed on the RBC surface including the Kell–Cellano, MNS, and Lewis blood groups. These blood groups are capable of causing transfusion reactions, though most are less common and produce less severe transfusion reactions than ABO incompatibility. Several are capable of producing hemolytic disease of the newborn.29
The Kell–Cellano antigens are variants of a transmembrane glycoprotein (ET3) encoded on chromosome 7q33. In contrast with many other blood groups, the function of the enzyme is known and is responsible for producing endothelin-3, a potent bioactive peptide with multiple biologic roles. There are several variants of the gene, of which K1 (Kell) and K2 (Cellano) are the most common and result from an SNP generating a Thr193(K2) from the more frequent Met193(K1) isoform. Kell incompatibility is second to Rh incompatibility for generation of hemolytic disease of the newborn.29
The MNS antigens are variants of two genes, glycophorin A (containing the M and N alleles) and glycophorin B (containing the S and s alleles), adjacent to each other on chromosome 4q28-q31. The glycophorins are the most common sialoglycoproteins present on the RBC membrane, but their function is not clear. Unlike most other blood group systems in which individual SNPs or deletions create the antigens, the MNS antigens are created by complex rearrangements of the protein structure that generate antigens.30
The Lewis blood group antigens are structurally similar to the ABO and the H blood group systems. The antigens are generated by variants in the fucosyl transferase gene (FUT3), residing on chromosome 19p13.3, that acts on the Lewis antigens in a similar fashion to the galactose transferase function of the ABO gene acting on the H antigen. The Lewis antigens are not synthesized in erythrocyte progenitor cells like the ABO antigens, more likely in the gut. The Le-a or Le-b antigens circulate in plasma bound to serum lipoproteins and are adsorbed to circulating erythrocytes, usually only after birth.31

Cross-matching
The use of citrate, refrigeration, and a nascent understanding of blood incompatibility during the First World War enabled the development of blood banking.32 The first U.S. hospital blood bank was created at Cook County Hospital in 1937.32 About 16 million units of blood was transfused in the United States in 2008.33 Cross-matching of donor blood products to an individual recipient is a sequence of procedures performed to prevent transfusion reactions. American Association of Blood Banks’ Standards for Blood Banks and Transfusion Services has defined the procedures performed before blood is transfused to a recipient. The first laboratory component for the recipient blood specimen is a “type and screen,” which consists of two separate tests.34 First, the recipient’s ABO and RhD blood groups are determined (typed) by using commercially produced anti-A and anti-B antibodies that will react with the A or B antigens, if present, on the recipient’s RBCs, causing the RBCs to agglutinate. The RhD antigen is tested in the same manner, with commercially available anti-D antibodies mixed with the recipient’s RBCs.34
Second, antibody screening is to identify whether the recipient has formed antibodies to nonself blood groups, such as Duffy, MNS, Kell, Kidd, and P system antigens, using an updated version of the classic Coombs test, also called the indirect antiglobulin test (Figure 30-1). The recipient’s serum or plasma is mixed separately with three or more commercially available type O washed RBCs that are known to express ~20 of the most “clinically significant” RBC antigens, to detect unexpected RBC antibodies. If transfusion is required, the recipient sample that is already typed and screened is cross-matched with donor units. Provided recipient antibodies were not identified on the type and screen, it is possible to perform a cross-match either serologically, using an immediate spin cross-match, or by an electronic match. If clinically significant antibodies have been found on the type and screen, electronic cross-matching is not sufficient and antiglobulin cross-match must be performed.34 An electronic or computerized cross-match is performed by identifying donor units on hand that have appropriate ABO and RhD blood groups for the recipient.35 Electronic cross-matching only can be used if a patient has a negative antibody screen, which means that they do not have any active RBC atypical antibodies. It is assumed that the proper testing (type and screen) on the recipient and donor blood are sufficient to identify clinically important incompatibility and to identify matching donor blood.36
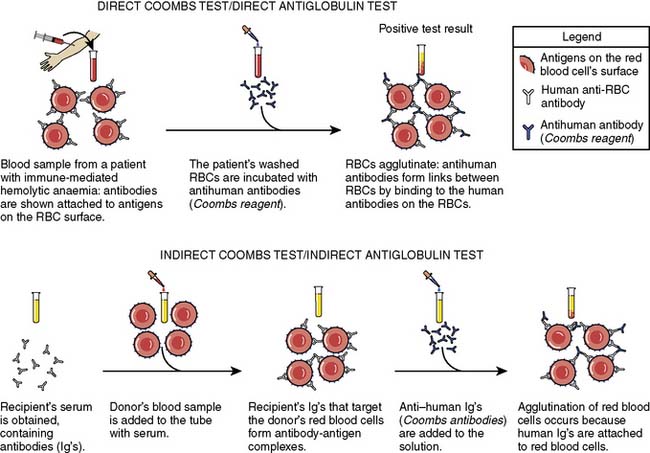
Figure 30-1 The indirect and direct Coombs (antiglobulin) tests.
(Modified from Wikipedia: Coombs test schematic. Available at: http://en.wikipedia.org/wiki/File:Coombs_test_schematic.png.)
When serologic cross-matching (immediate-spin cross-match) is performed, RBCs from an ABO and RhD-compatible RBC unit are mixed with the recipient’s plasma. The mixture is centrifuged and examined for hemolysis or agglutination. Agglutination is considered a positive reaction, indicating that the donor unit is incompatible for that specific patient. If both are absent, ABO compatibility is verified and the RBC unit issued. This procedure is repeated for each donor RBC unit. If agglutination or hemolysis occurs, additional screening of the recipient’s plasma is performed to identify the unexpected antibodies.36 Performing a serologic cross-match, over electronic cross-matching, before transfusing RBCs is preferred by some laboratories because it detects rare ABO errors and also detects most recipient IgM antibodies to antigens on donor RBCs.37 In an emergency, “uncross-matched blood” can be transfused and risk for a serious transfusion reaction minimized by administration of type O and RhD-negative RBCs.38
Components
Because plasma contains the anti-ABO, RhD, and other antibodies of the donor, only ABO and RhD-compatible units are transfused. Thus, the recipient and donor unit must undergo a type and screen; however, cross-matching is not performed. Platelets have ABO antigens on their surface. However, ABO compatibility for platelet transfusion is desirable but not required because of the relatively small volume of plasma present in a bag of platelets. If ABO-incompatible platelets are administered for operational reasons, the recipient subsequently may have a positive direct antiglobulin test result, but significant hemolysis is rare. Furthermore, a donor–recipient ABO mismatch may result in poor function of donor platelets after transfusion.39–41

Immediate Immune-Mediated Complications of Transfusion
Hemolytic transfusion reaction is the result of complement-mediated destruction of transfused RBCs, nearly always because of incompatibility of antigen on the transfused RBCs with antibody in the recipient’s circulation. The most common cause of acute hemolytic reactions is transfusion of ABO-incompatible blood; rarely, undetected serologic incompatibility is a cause of acute hemolysis.42
Transfusion has been associated with greater pulmonary morbidity after cardiac surgery.43 Transfusion-related acute lung injury (TRALI) occurs when increased permeability of the pulmonary endothelium causes pulmonary edema, usually within a few hours of transfusion. Hypotension and fever also may occur.44,45 TRALI constitutes the majority of transfusion-associated mortality in the United States.45,46 The specific cause of TRALI is unknown; however, in many cases, the occurrence of TRALI is associated with the presence of antibodies in donor plasma directed toward the recipient’s leukocyte or neutrophil antigens.45,46 These antibodies are seen more frequently in multiparous women and individuals with prior transfusion.47 Transfusion of blood components, especially products containing high volumes of plasma such as fresh-frozen plasma (FFP), have the greatest risk for TRALI.47 Thus, plasma from women is usually, but not exclusively, used for processed protein fractions rather than transfusion. No routinely available pretransfusion testing is available.
Immune-mediated platelet destruction is the result of recipient antibodies being present to human leukocyte or platelet-specific antigens present on transfused platelets. Most cases of immune-mediated platelet destruction occur in individuals who have had several prior platelet transfusions. In some patients, platelet matching may be required.41
Anaphylactoid reactions rarely occur in transfusion of IgA-containing plasma present in any blood product given to IgA-deficient patients who have anti-IgA antibodies. The reaction is characterized by hypotension, bronchospasm, and laryngeal edema.48
Febrile nonhemolytic reactions are relatively frequent and typically manifested by a temperature increase of 1° C or more occurring during or shortly after transfusion. They may result from antibodies to white blood cells or from the presence of high levels of cytokines in transfused blood products.49

Delayed Immune-Mediated Complications of Transfusion
Delayed hemolytic reactions occur in patients who have had previous exposure to incompatible blood but who do not have circulating antibodies. Re-exposure to the antigen provokes delayed production of antibody that reaches a significant circulating level while the transfused RBCs are still present in the circulation, usually 2 to 14 days after transfusion.50
Graft-versus-host disease (GVHD) is a rare condition that occurs when viable donor T lymphocytes in the transfused blood product successfully engraft in the recipient. Normally, donor T cells are recognized as foreign by the recipient’s immune system. However, in immunocompromised patients, and rarely when the donor is homozygous and the recipient is heterozygous for an HLA haplotype (as can occur in directed donations from first-degree relatives), the recipient’s immune system is not able to destroy the donor T cells. This can result in graft-versus-host disease. Irradiation of the donor unit prevents T-cell proliferation and graft-versus-host disease.51,52
Nonimmune Complications of Transfusion
Transmission of infectious disease may occur with transfusion despite standard blood-banking operational procedures. Routine testing of donor blood is performed for Chagas disease, hepatitis B and C, human immunodeficiency viruses types 1 and 2, human T-lymphotropic virus, syphilis, West Nile virus, and, in some situations, cytomegalovirus.33 Bacterial contamination occurs rarely and is manifested by fever and hemodynamic instability surrounding transfusion of the blood product.53 Circulatory overload, hypothermia, and metabolic and electrolyte derangements are other complications that may occur in high-volume transfusion.54
Transfusion and Morbidity Outcomes
Although transfusion of RBCs is necessary for some patients, its use has been associated with a dose-dependent greater prevalence of morbidity after cardiac surgery.55 Greater rates of postoperative infectious complications, prolonged postoperative ventilatory support, renal injury, and reductions in short- and long-term survival are more common in patients transfused with RBCs.43,55–57 Greater rates of bacteremia, septicemia, and deep and superficial sternal wound infections are thought to be secondary to a downregulation of the immune system.56,58 Transfusion has been related to an increased development of postoperative atrial fibrillation thought to be secondary to the influence of RBC transfusion on inflammation. RBC transfusion results in a direct infusion of inflammatory mediators, as well as augmenting the inflammatory response to cardiopulmonary bypass (CPB) and cardiac surgery.59,60
Storage duration of the RBC product also may be a contributing factor for observed adverse outcomes. The storage lesion consists of a series of structural and functional changes occurring with increasing RBC storage. Some of these changes are reversible, others are not, and together may result in decreased microvascular tissue flow.61–63 A recent investigation reported transfusion of RBCs stored longer than 14 days was associated with a greater risk for death and complications after cardiac surgery.64 A recent laboratory investigation from Sweeney et al65 reported increased thrombin generation for RBC units of increasing storage duration, suggesting a cause for increased complications observed with transfusion of RBCs of increased storage duration.
Genetic causes of hemorrhage
In general, the coagulation system can be thought of as being highly optimized toward rapid cessation of hemorrhage. There likely has been a strong evolutionary pressure toward rapid coagulation and wound healing. Of course, there must be an equally highly developed system that prevents overwhelming coagulation of the entire blood volume in response to a trivial intravascular insult. In contrast, there was likely little or no pressure to avoid a deep venous thrombosis in older age because most individuals did not live beyond 40 years until a few centuries ago. Similarly, there has been no evolutionary pressure to successfully undergo blood exposure to foreign surfaces such as the CPB circuit. Because severe bleeding abnormalities are strongly selected against, in evolutionary terms, they are rare. A sentinel example is the hemophilias, in which a rare variant causes production of a nonfunctional protein with severe consequences. In contrast, trivial abnormalities have not undergone marked evolutionary pressures and are likely to be more common. A good example of this is the wide, but seemingly unimportant, heritable variation seen in the circulating level of many coagulation proteins.66–69
Genetic causes of impaired coagulation can be somewhat simplistically thought of as either a qualitative defect arising from abnormal protein structure because of a coding genetic variant or a quantitative defect arising from abnormal, usually reduced, production of a normal protein because of a noncoding (promoter) genetic variant. This approach is rudimentary but gives the best basic understanding of the genetic mechanisms of coagulation disorders. Like all genetic disorders, the overall effect of a genetic variant on the whole human or surgical population is a product of the frequency of the genetic variant and the biologic effect of the variant.70 If the variant is rare, such as the hemophilias, although the disease is problematic for a single individual, the overall effect on day-to-day practice is low because of its rarity. By contrast, a more common variant may have greater effect. Similarly, for two variants of equal frequency, the one with low biologic effect will have less overall influence than one with a higher biologic effect. To date, no frequent variants with high biologic effect on coagulation have been identified.69

Variation in Coagulation Protein Levels
Several studies have demonstrated the strong heritability of levels of plasma proteins and platelet levels in normal populations.68,71–74 This type of research is undertaken by examining the plasma level of coagulation proteins in multigenerational families and estimating the within-family variation compared with the whole-population variation.75 In accordance with the believed strong evolutionary importance of coagulation function, there is similar strong inheritance within families, with up to 70% of the variation in plasma protein levels determined by genetic heritability. Similar heritability of platelet count and platelet volume has been observed.76,77
Genetic variation associated with the circulating level of a coagulation protein is usually in or around the gene for that coagulation protein. For example, the circulating level of plasminogen activator inhibitor 1 (PAI-1) principally is determined by a common promoter variant that alters the binding of the transcription factor inhibitor.66,67 Similarly, the circulating level of thrombin-activated fibrinolysis inhibitor is determined by two variants,78 and circulating prothrombin levels are determined by a single variant.79 However, some coagulation protein levels are regulated by genetic variation that is not in or around the protein’s gene. Similarly, platelet count and platelet volume are regulated by genes that would not be intuitive choices.76,77

Hemophilias
Hemophilias are the most well-known example of a rare severe genetic disorder of coagulation. Hemophilia A (factor VIII deficiency) is the most common hemophilia, occurring in about 1 in 5000 male births.80 Hemophilia B (factor IX deficiency) occurs in about 1 in 34,000 male births. Both genes lie on the X chromosome, meaning that in male individuals, only one copy of the chromosome is present and a single variant may result in hemophilia (often called a sex-linked or X-linked disease). Accordingly, female hemophiliacs must have two copies of the rare variant—an event that usually occurs only in consanguineous births in which the father has hemophilia. Although most hemophilias are maternally inherited, about 30% are spontaneous mutations not present in the mother. An obvious question is why female individuals do not have twice the factor VIII and IX levels as male individuals. The reason is that in female individuals, one of the X chromosomes is usually “turned off” and does not generate messenger RNA for translation of proteins. However, it is possible for female carriers to have mild hemophilia because of lyonization (inactivation) of the X chromosome that carries the normal gene, leaving the abnormal gene to be the most active. Adult women may experience menorrhagia because of the bleeding tendency. Hemophilia C is an autosomal genetic disorder (not X-linked) involving a lack of functional clotting factor XI. Hemophilia C is not completely recessive because heterozygous individuals also show increased bleeding.81
The gene F8 encodes factor VIII of the intrinsic pathway. Factor VIII is a cofactor for factor IXa, which converts factor X to the activated form Xa. There are almost 700 known coding variations of the F8 gene, many of which have been seen in only one individual or family. More than 170 give severe forms of hemophilia A, and more than 180 produce milder forms.82,83 Some of these mutations change one amino acid; many of these have little effect on protein function because only one amino acid is changed, often to a similar type of amino acid.84 Others cause a frameshift mutation that usually truncates the protein and markedly reduces its function. Another severe mutation is an inversion of a portion of the genome and, therefore, the sequence of a portion of the protein is “back-to-front” with marked reduction in function. Rarely, the entire gene is deleted so that both quantitative and qualitative assays show the absence of factor VIII. The diagnosis of hemophilia A is made by reduced factor VIII activity, prolonged activated partial thromboplastin time except in mild disease, and a normal PT and platelet count.84
The gene F9 encodes the vitamin K–dependent factor IX of the intrinsic pathway. Factor IX is activated by factor XI to factor IXa. Because hemophilia B is rarer than hemophilia A, fewer mutations have been described, but the mechanisms of protein dysfunction from genetic variation are similar. The diagnosis of hemophilia B is made by reduced factor IX activity, prolonged activated partial thromboplastin time except in mild disease, and a normal PT and platelet count.84
Hemophilia is not a contraindication for cardiac surgery, but it is an absolute indication for involvement of a hematologist and blood banker. Most experience has been small case series or single case reports, but all emphasize the need for prolonged factor therapy and subsequent excellent outcomes. For hemophilia A, the factor VIII activity level should be corrected to 100% of normal for cardiac surgery, although some hematologists use 50% to 70% of normal as a goal. One unit of factor VIII is the normal amount of factor VIII in 1 mL plasma, by definition. Because the volume of distribution of factor VIII is that of plasma, the amount of factor VIII in an individual is ~50 mL/kg. The factor VIII dose needed to correct the level is calculated as follows85,86:
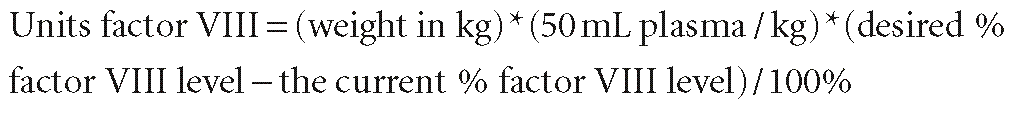
Approximately 30% of people with severe hemophilia A develop antibodies to transfused factor VIII. These antibodies (also called inhibitors) bind to transfused factor VIII and reduce its activity so increased doses of factor VIII are required.87 The next dose should be administered 6 to 12 hours after the initial dose, together with repeated factor VIII activity monitoring with the goal of keeping the trough activity greater than 50%. During hemorrhage, cryoprecipitate and FFP can be given to restore levels of factor VIII and other coagulation factors, as well as blood volume. 1-Deamino-8-d-arginine vasopressin (DDAVP) at a dose of 0.3 μg/kg can be used in mild or moderate hemophilia A, and works by increasing circulating factor VIII and von Willebrand factor (vWF) levels. DDAVP works only when some normal factor VIII activity is present and usually with only modest success.88
For hemophilia B, the factor IX activity level should be corrected to 100% of normal for cardiac surgery, although some hematologists use 50% to 70% of normal as a goal. One unit of factor IX is the normal amount of factor IX in 1 mL plasma, by definition. Unlike factor VIII, the volume of distribution of factor IX in an individual is 100 mL/kg. The factor IX dose needed to correct the level is calculated as follows89:
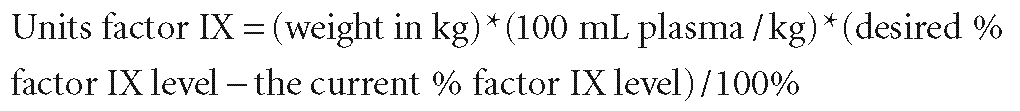
The next dose should be administered 12 to 24 hours after the initial dose, together with repeated factor IX activity monitoring with the goal of keeping the trough activity greater than 50%. During hemorrhage, FFP can be given to restore levels of factor IX and other coagulation factors, as well as blood volume. DDAVP is not effective.88

Von Willebrand Disease
Von Willebrand disease (vWD) is the most common inherited coagulation abnormality in humans, occurring in about 1% of individuals.90 vWD also can be an acquired disease. It arises from quantitative (reduced amounts of usually normal protein) or qualitative (normal amounts of a defective protein) deficits of vWF, which is a multimeric plasma glycoprotein produced by endothelium and platelets. The protein binds factor VIII, the platelet GPIb receptor, the activated form of the GPIIb/IIIa receptor, and collagen.91
Unlike the serine proteases of the intrinsic and extrinsic pathways of the coagulation system, vWF is not an enzyme and serves its functions by binding to other proteins.91 It has an important role in platelet adhesion to exposed subendothelial collagen by binding to exposed collagen and also to the GPIb receptor of circulating platelets, especially in high shear environments such as arterial bleeding. vWF decelerates platelets from rapid flow by uncoiling; thus, it appears to be the critical initiator of platelet adhesion in high-flow environments. Platelet adhesion initiates rapid platelet activation, with switching of the most common platelet receptor, GPIIb/IIIa, from a quiescent protein that poorly binds to fibrinogen and fibrin, to an activated protein that strongly binds fibrinogen and fibrin. Subsequently, vWF also can bind to activated GPIIb/IIIa receptors expressed on activated platelets.
There are four types of hereditary vWD caused by mutations in the gene vWF (Figure 30-2).92,93 Other factors, including having an O blood group, increase the clinical severity of vWD. Type 1 vWD (60% to 80% of all vWD) is a mild quantitative defect of normal vWF, with individuals having about 10% to 50% of normal vWF levels and often having low levels of factor VIII. The disease is inherited in an autosomal dominant fashion, and most individuals are heterozygous (possess one copy) of the abnormal gene with variants principally found between exons 18 and 28 of the gene.94 Rarely, homozygotes (two copies of the abnormal gene) have extremely low levels of vWF. Most heterozygotes have normal or near-normal coagulation and are identified by having abnormal bleeding after tooth extraction or surgery, or having menorrhagia.95
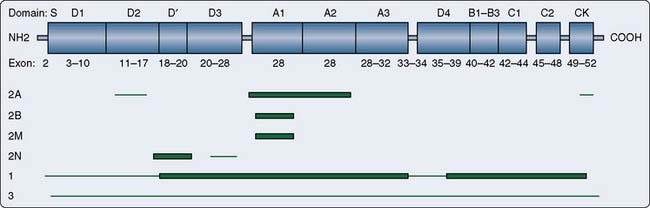
Figure 30-2 Location of von Willebrand factor (vWF) mutations by von Willebrand disease (vWD) type.
(From http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=von-willebrand&rendertype=figure&id=von-willebrand.F1 and http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=von-willebrand. Source: www.genetests.org. Copyright University of Washington, Seattle, WA.)
Type 2 vWD (20% to 30%) is a qualitative defect with four subtypes: 2A, 2B, 2M, and 2N.90 Type 2A vWD is caused by decreased activity, but normal levels, of vWF. The vWF multimers are structurally abnormal and usually small. In contrast with hemophilia, type 2A vWD is inherited in an autosomal dominant fashion and is caused by variants in exons 12 to 16, 28, and 52 of the gene. Type 2B vWF is a rare “gain-of-function” defect leading to spontaneous binding to platelets and subsequent rapid clearance of the platelets and large vWF multimers. Patients with this subtype should not receive desmopressin because it can induce platelet aggregation. Type 2M vWD is characterized by a platelet function defect caused by a decrease in high-molecular-weight multimers. Type 2N vWD is caused by the inability of vWF to bind factor VIII but has an autosomal recessive inheritance and is most often associated with variants in exons 18 to 20. This type gives a normal vWF antigen level and normal functional test results, but has a low factor VIII. This probably has led to some 2N patients being misdiagnosed in the past as having hemophilia A, and should be suspected if the patient has the clinical findings of hemophilia A but a pedigree suggesting autosomal, rather than X-linked, inheritance.90
Type 3 vWD is caused by a quantitative lack of vWF and premature proteolysis of factor VIII, similar to type 2A vWD, but in an autosomal recessive fashion. Type 3 is the most severe form of vWD because individuals are homozygous for the defective gene and protein. They have no detectable vWF antigen and may have sufficiently low factor VIII that they have hemarthroses, similar to hemophilia.95
Acquired vWD can occur in patients with autoantibodies. In this case, the function of vWF is not inhibited, but the vWF-antibody complex is rapidly cleared from the circulation by antibody binding. A form of vWD occurs in patients with aortic valve stenosis, leading to gastrointestinal bleeding (Heyde syndrome).96 This form of acquired vWD may be more prevalent than is currently thought. Acquired vWD also has been described in Wilms tumor and hypothyroidism.
Laboratory diagnosis is made by usually normal hemoglobin, activated partial thromboplastin time, and partial thromboplastin time, but reduced quantity of vWF measured by an antigenic assay, reduced vWF:ristocetin cofactor assay (vWF:RCo), or reduced functional factor VIII assay.92 If abnormalities in these three tests are identified, specialized coagulation studies also may be performed to determine the subtype of vWD. Bleeding can be treated with plasma-derived clotting factor concentrates containing both vWF and FVIII; depending on the vWD type, mild bleeding episodes usually respond to DDAVP.97
Platelet-type or pseudo-vWD is an autosomal dominant type of vWD caused by gain-of-function coding mutations of the vWF receptor (GPIb) on platelets, not of the vWF gene.93 GPIb is a dimeric protein that is part of the larger complex (GPIb/V/IX), which forms the full vWF receptor on platelets. The loss of large vWF multimers is similar to that seen in type 2B vWD, but genetic testing of the vWF gene does not find mutations. The hyper-responsiveness of the platelet receptor results in increased interaction with vWF in response to minimal or no stimulation in vivo. This leads to a decline in plasma vWF and typically to a decreased or low normal platelet count.91 Replacement therapy in the form of VIII/vWF preparations or drugs aimed at increasing the release of endogenous vWF will exacerbate the condition and lead to further reduction of the platelet count. Platelet transfusions are therapeutic.

Factor V
Normal factor Va and its cofactor, factor Xa, are the first members of the final common pathway or thrombin pathway and combine to form the prothrombinase complex.98,99 The prothrombinase complex catalyzes the conversion of prothrombin (factor II) to thrombin (factor IIa). To produce thrombin, the prothrombinase complex cleaves two peptide bonds in prothrombin. The action of factor Va is terminated by cleavage by activated protein C (aPC) (Figure 30-3).
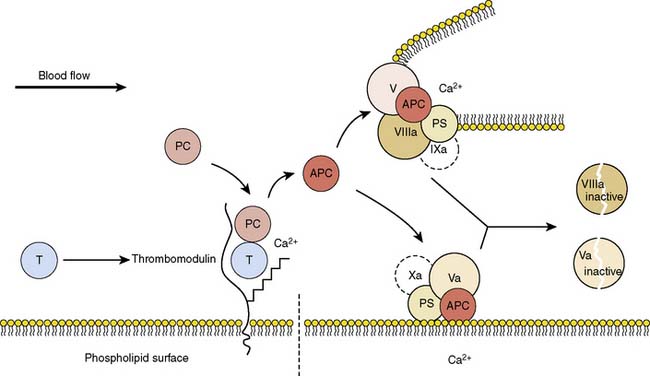
(From Wikipedia: Protein C anticoagulant. Available at: http://en.wikipedia.org/wiki/File:Protein_ C_anticoagulant.jpg)
Factor V Leiden is a common SNP that results in a factor V variant that cannot be as easily degraded by aPC.100 The nucleotide variant (G1691A) results in conversion of an arginine to a glycine (Arg506Gln). This amino acid is normally the cleavage site for aPC, and the protein change markedly reduces the activity of aPC on factor Va.99 When factor Va remains active, it facilitates overproduction of thrombin, leading to excess fibrin generation and excess clotting. In essence, it is creating a gain of function by inhibiting the termination of factor Va. The clotting is almost always venous, resulting in deep vein thrombosis or pulmonary embolus.99
About 5% of whites in North America have factor V Leiden.100 The SNP is less common in Hispanics and African Americans, and is rare in Asians. About 30% of people who have a deep vein thrombosis or pulmonary embolism, especially in younger patients, carry the SNP. Having the SNP and also having other risk factors for deep vein thrombosis including smoking, taking oral contraceptives, pregnancy, and recent surgery, markedly increase risk. Efforts to show that the prothrombotic factor V Leiden variant results in less bleeding have been mixed.101,102

Cold Agglutinins
Cold agglutinin disease is rare (~2/100,000). Causes include a lymphoma-induced monoclonal gammopathy, renal cell carcinoma, or infection; but most commonly, no cause is found.103 Cold agglutinins are usually IgM autoantibodies that react at cold temperatures with RBC polysaccharide antigens. Low titers (1:16) of cold agglutinins often are found in the sera of healthy individuals, but the presence of high titers of cold agglutinins (titers over 1:1000 at 4°C) can lead to hemagglutination and thrombosis at low temperatures, followed by complement activation and subsequent hemolysis on rewarming.104,105 It generally is agreed that screening for cold agglutinins before cardiac surgery is not warranted because of their rarity.104
In contrast with the warm agglutinins, patients with cold agglutinins do not respond to steroids or splenectomy but sometimes respond to rituximab.103 Rituximab destroys both normal and malignant B lymphocytes and is used to treat B-cell lymphomas and rheumatoid arthritis. It specifically targets the CD20 antigen, but the function of CD20 is unknown.103

Variation in Platelet Count and Volume
Mean platelet volume is a measurement of the average size of platelets found in blood and is positively correlated with platelet count and function, as well as with adverse thrombotic outcomes. Genetic variants associated with mean platelet volume and platelet count have been identified in or near the genes WDR66, ARGHEF3, TAOK1, TMCC2, TPM1, PIK3CG, EHD3, ATXN2, PTPN11, and AK3, among others.77 Few of these associations are intuitive or supported by well-understood biologic mechanisms; nevertheless, they indicate the complexity and importance of genetic causes of normal coagulation function.
Bleeding after cardiac surgery
Do genetics affect bleeding after cardiac surgery? For many of the well-identified bleeding diatheses mentioned earlier, the answer is yes. However, clinicians rarely encounter these patients in their practice. More commonly, they see a patient who is bleeding for no apparent reason—without a surgical bleeding site, without a drug cause, and with normal or near-normal coagulation tests. The reason for this occurring in an individual patient is almost never known, and the patient is treated symptomatically until the bleeding stops. It is possible, but unproved, that these patients have a genetically inherited bleeding diathesis. Few studies have examined this question, and their limited findings are unreplicated.102,106–108 (See Chapter 31.)

Reoperation for Bleeding
Re-exploration for bleeding is a serious complication, which significantly impacts a patient’s subsequent postoperative course by increasing both morbidity and mortality. Approximately 2% to 4% of patients undergoing cardiac surgery require reoperation for bleeding with greater reported rates for more complex procedures. A recent investigation evaluating incidence, risk factors, and outcomes for patients requiring reoperation for bleeding in 528,686 CABG patients from the Society of Thoracic Surgeons National Cardiac Database (2004–2007) reported a 2.4% rate of reoperation. Risk factors for reoperation were older age, male sex, comorbidity such as peripheral vascular and cerebrovascular disease, chronic lung disease, renal insufficiency, heart failure, previous interventions, urgent or emergent surgery, preoperative intra-aortic balloon pump, PCI less than 6 hours before CABG, and thienopyridine use less than 24 hours before surgical intervention. Patients requiring reoperation had greater risk for morbidity such as septicemia, stroke, and prolonged ventilatory support after surgery. Risk-adjusted mortality was significantly greater for patients requiring reoperation: 5.9% vs. 1.97%. Of note, risk was not increased in patients receiving aspirin therapy less than 24 hours before surgery.109
Additional studies have characterized risk factors for reoperation and its relation to patient outcome. Moulton et al110 identified a 4.2% reoperation rate and identified increasing age, renal insufficiency, reoperative procedures, and prolonged CPB time as risk factors for this complication. Interestingly, antiplatelet drugs, such as preoperative aspirin, and preoperative use of heparin or thrombolytic agents were not significant predictors. In addition, the authors reported that incidence of death, renal failure, sepsis, and need for prolonged ventilatory support were significantly greater in patients who underwent reoperation for bleeding.110 Choong et al111 reported demographic and comorbidity risk factors previously described, as well as cessation of aspirin within 4 days of surgery, preoperative use of clopidogrel, lack of antifibrinolytic agents during surgery, type of operation, and CPB duration as risk factors for postoperative bleeding necessitating re-exploration.
In a contemporary cohort of patients, Karthik et al112 examined risk factors for reoperation and effect of time delay on morbidity after surgery. The rate of reoperation in their investigation was 3.1%. Factors associated with the need for reoperation included demographics such as increasing age, smaller body mass index, and surgery that was nonelective. They similarly reported that reoperation was associated with increased morbidity, hemodynamic instability, and transfusion of RBC and component therapy. Among the 89 patients requiring reoperation, 31 had greater than 12 hours time delay to re-exploration (4 deaths in delayed reoperation group). The authors concluded that mortality outcomes were worse if the time delay was greater than 12 hours after surgery and recommended an early re-exploration policy for bleeding.112 Ranucci et al113 reported an increased risk for reoperation and noted that much of the morbidity risk was attributable to the amount of RBCs transfused; delay for reoperation was related to risk only if the delay involved excess use of blood products.
Hall et al114 sought to differentiate coagulopathy versus hemorrhage from surgical causes in patients undergoing reoperation for bleeding. Both groups had increased morbidity and mortality compared with those not requiring reoperation. Excess risk was attributed to more hemodynamic instability, transfusions, and inotropic support, which were more common in patients undergoing reoperation. The authors recommended normalization of coagulation profiles within 4 hours of intensive care unit admission, and if significant bleeding persisted with a normal coagulation profile, re-exploration.114

Ratios in Resuscitation: Implications for Massive Transfusion in Cardiac Surgery
Patients requiring massive transfusion in the perioperative period may become coagulopathic secondary to loss, hemodilution, consumption of coagulation factors, and insufficient component replacement. Furthermore, hypothermia is common in the perioperative period and can lead to platelet dysfunction via its effect on platelet activation and adhesion.115 Hypothermia also may contribute to reductions in clotting factor functional activity.116–119 Development of acidosis may act synergistically with hypothermia to further worsen coagulopathy through its impact on pH-sensitive enzyme complexes involved in the clotting cascade.120–122 Furthermore, choice of volume administration (i.e., hetastarch) may further worsen coagulopathy in patients with massive blood loss.120
Borgman et al123 recommended implementation of massive transfusion protocols consisting of greater FFP:RBC (1:1) ratio, noting evidence of improved survival and possibly less use of RBCs primarily in the trauma literature. Holcomb et al124 recommended a massive transfusion protocol that included a 1:1:1 ratio of FFP/platelets/RBC to favorably impact patient outcomes. They analyzed four groups based on high and low plasma and platelet/RBC ratios. The combination of high plasma/RBC and high platelet/RBC ratios was associated with decreased truncal hemorrhage and increased 6-hour, 24-hour, and 30-day survival. Gunter et al125 reported a significant reduction in 30-day mortality for patients who received FFP/RBC of 2:3 or greater compared with those who received less than 2:3. Patients receiving platelets to RBC at a greater ratio (≥ 1:5) also had lower 30-day mortality when compared with patients with less than 1:5 ratio. Similarly, Zink et al126 reported improved survival and decreased overall RBC transfusion in trauma patients receiving early high FFP and platelet-to-RBC transfusion ratios within the first 6 hours of hemorrhage. The authors appropriately addressed the difficulty of basing component therapy on laboratory testing because of time delay for test results.
Others have suggested a U-shaped curve depicting higher mortality at low and higher FFP/RBC ratios, and better outcomes when the ratio was 1:2 or 1:3. A 1:1 ratio of FFP/RBC reduced coagulopathy but failed to improve survival, prompting authors to caution against adopting the 1:1 ratio without further testing.120,127 Interestingly, some have criticized proponents of the greater FFP-to-RBC transfusion protocols, noting that these studies may have a “survival bias” present; that is, the most severely injured patients simply did not survive long enough to receive FFP, which often is not immediately available.120
In a computer simulation model of dilutional coagulopathy, Hirshberg et al128 reported that current massive transfusion protocols in bleeding patients underestimate dilution of clotting factors to correct dilutional coagulopathy. The authors provided a sensitivity analysis of a wide range of replacement protocols by programming their model to administer FFP and platelets after transfusion of a predefined number of RBC units. (Model was calibrated to data from 44 patients.) They determined that the optimal replacement ratios are 2:3 for plasma and 8:10 for platelets with RBC128 (Figures 30-4 and 30-5).
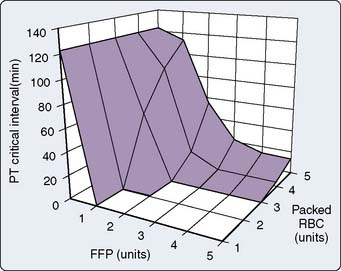
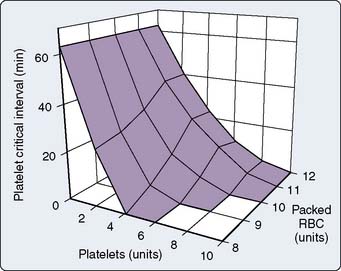
Massive transfusion protocols improve survival in patients with exsanguinating hemorrhage. Early activation of a massive transfusion protocol and achieving predefined ratios have been associated with improved survival.129 Others have reported on benefits of predefined massive transfusion protocols in terms of improved patient outcome in severely injured patients.129 Riskin et al130 reported that it was the implementation of a protocol for massive transfusion that included improved access and availability of blood products that reduced mortality in their patient population rather than use of a greater FFP/RBC ratio. They emphasized the importance of expeditious product availability because survival benefit did not appear to have been related to any alteration in the volume or ratio of blood components used.
Replacement therapy

Factor VIIa
Recombinant factor VIIa (rFVIIa, NovoSeven trademark; Novo Nordisk, Bagsvaerd, Denmark) is approved for the treatment of bleeding in patients with hemophilia A or B with inhibitors against factors VIII and IX. Factor VII acts locally at the site of vessel injury by binding to tissue factor on subendothelial cells and facilitates transformation of factors IX and X to active forms, ultimately resulting in thrombin generation and clot formation.131,132 A laboratory investigation examined the effect of rFVIIa-mediated thrombin generation on platelet adhesion and aggregation at normal and reduced platelet counts. Their results suggested that administration of rFVIIa in patients with and without thrombocytopenia enhanced platelet deposition and aggregation at the site of vascular injury, which may explain, in part, the efficacy of rFVIIa in thrombocytopenic patients.133
In a comprehensive evaluation of off-label use of rFVIIa in Canada, Karkouti et al134 noted rFVIIa was associated with significant reductions in transfusion after administration without increases in mortality or major morbid events. Filsoufi et al131 reported improvement in coagulation variables, blood loss, and reduced need for blood products in patients receiving rFVIIa for management of refractory bleeding after surgery. Similarly, a retrospective investigation from Von Heymann et al135 reported significant reductions in blood loss and transfusion requirements after rFVIIa administration for perioperative bleeding refractory to conventional treatment. Mortality was similar between groups, and no thromboembolic complications were observed in the rFVIIa group. A number of other investigations have reported on use of rFVIIa for treatment of refractory bleeding. Most report that rFVIIa is effective in reducing bleeding and decreasing RBC and component therapy requirements. Clinical use beyond rescue therapy is unclear because of the safety profile of the medication.136–138
A large case series of rFVIIa use from the Australian and New Zealand Hemostasis Registry reported rFVIIa was associated with fewer blood products; however, it was associated with a 7% adverse event rate, 4% of which was related to thromboembolic events.139 In a review of rFVIIa, DiDomenico et al140 noted that 10% of patients had what was believed to be thromboembolic complications after the administration of rFVIIa and addressed the primary concerns with the use of rFVIIa, that is, promoting a hypercoagulable state. Gill et al141 reported that patients who received rFVIIa had fewer transfusions after randomization and fewer reoperations for bleeding; however, they also reported an increase in serious adverse events in patients randomized to rFVIIa compared with placebo; this increase was not significantly different. Similarly, Bowman et al142 reported benefits in controlling hemorrhage and reductions in blood product requirements with rFVIIa; however, patients who received rFVIIa had a greater prevalence of postoperative renal failure, pneumonia, and an 11% incidence rate of thrombosis. Review of the U.S. Food and Drug Administration Adverse Event Reporting System found a total of 431 adverse events related to the use of factor VIIa, among which 185 were thromboembolic events.143

Fibrinogen Concentrates
Human fibrinogen concentrates have been used for substitution therapy in cases of hypofibrinogenemia, dysfibrinogenemia, and afibrinogenemia. Accumulating data suggest that fibrinogen plays a critical role in hemostasis, especially in bleeding patients with an acquired fibrinogen deficiency.144 Clinical use of fibrinogen concentrates is based on the supposition that plasma fibrinogen concentrations may become critically reduced somewhat early in a bleeding patient, and that this may contribute to the coagulopathy associated with hemorrhage. Furthermore, a functional fibrinogen deficiency may develop with excessive hemodilution with colloid plasma expanders. Correction of fibrinogen deficits has included administration of FFP, cryoprecipitate, and plasma-derived fibrinogen concentrates.145
Riastap USA is a commercially available virally inactivated fibrinogen concentrate derived from human plasma. Benefits over FFP and cryoprecipitate include viral inactivation and rapid reconstitution in addition to lower volume of administration. In a pilot study, Rahe-Meyer et al146 reported a reduction in transfusion with administration of fibrinogen concentrate, targeting a greater plasma fibrinogen concentration compared with conventional therapy. Fenger-Eriksen et al145 reported significant reductions in blood loss and RBC, FFP, and platelet transfusion with fibrinogen concentrate without serious adverse events. Others have described benefits of fibrinogen concentrate with outcome measures of fewer transfusions and improved clot firmness.147
Karlsson et al148 hypothesized that preoperative fibrinogen plasma concentrations within the reference range may be a limiting factor for hemostasis. The authors randomized CABG patients with preoperative plasma fibrinogen levels less than 3.8 g/L to an infusion of 2 g fibrinogen concentrate or placebo. End points were vessel occlusion assessed by multislice computed tomography (CT), blood loss, transfusion, and hemoglobin levels 24 hours after surgery. One subclinical vein graft occlusion was reported in the fibrinogen group, with similar global measures of hemostasis assessed by thromboelastography. The fibrinogen group had lower postoperative blood loss and greater hemoglobin concentrations. The authors appropriately addressed the concern of creating a prothrombotic state with increased incidence of graft occlusion. Interesting, they were not able to clarify the mechanism behind the reduced bleeding. Fibrinogen plasma levels were increased in the fibrinogen group immediately after infusion, but there were no differences between the groups 2 hours after surgery, and measures of hemostasis also were similar.148

Prothrombin Complex Concentrates
Prothrombin complex concentrates (PCCs) are prepared from pooled plasma and contain four vitamin K–dependent clotting factors II, VII, IX, and X. PCC can be used for rapid reversal of oral anticoagulants (vitamin K antagonists) in patients with life-threatening bleeding and less commonly for patients with congenital or acquired deficiencies of factor II or X. The U.S. Food and Drug Administration has not approved PCCs for reversal of bleeding associated with warfarin. Although FFP commonly has been used for emergent reversal of vitamin K antagonists, it has a greater volume of administration, it must be patient matched and thawed, and has risks associated with exposure. PCC is virally inactivated and more efficacious in normalizing international normalized ratio in patients treated with vitamin K antagonists. Some have suggested PCC may have a role in the management of massive bleeding; however, side effects include potential for thromboembolic complications.149–152

Volume Replacement: Colloids and Crystalloids
Maintenance of intravascular volume is a common goal for perioperative management in cardiovascular surgery. Merits of specific choices for fluid therapy to replace ongoing fluid loss continue to be debated. Although there may be theoretical advantages of colloid or crystalloid as a choice for volume replacement, clinical trial data do not definitively support one over the other in terms of mortality outcomes. Both have distinct advantages and disadvantages.153 In general, more important than choice of volume replacement is appropriate volume replacement to avoid tissue hypoperfusion.
Volume of distribution for a particular fluid administered will depend on fluid composition. Generally, saline-based crystalloids will result in expansion of plasma volume by approximately 200 to 250 mL for each 1 L administered; glucose-based solutions will expand plasma volume less (approximately 60 to 70 mL/1 L infused); and colloid solutions will result in expansion of plasma volume generally similar to volume infused. Of note, within the semisynthetic colloid solutions there is variability in degree of plasma volume expansion, as well as a differential influence on hemostasis and inflammatory processes.154,155
Broadly, those favoring crystalloids for volume replacement note problems with hemostasis, adverse reactions, and greater risk for volume overload with colloidal fluids. Those favoring colloids highlight larger volumes of crystalloid required to achieve volume resuscitation, tissue edema, and potential for reduction in tissue oxygen delivery.156 It is unclear whether the type of fluid therapy for hypovolemia impacts development of pulmonary edema. Verheij et al157 reported that type of fluid given to patients with pulmonary vascular injury without fluid overloading did not influence pulmonary vascular permeability or pulmonary edema.
A key characteristic of colloid solutions is their persistence within the intravascular space, which is determined by rate of loss from the circulation.155 Albumin is monodisperse (uniformity of particle size molecular weight), whereas semisynthetic colloids are polydisperse (wide distribution of particle molecular weight). The molecular weight of a colloid has pharmacokinetic implications in terms of degree of oncotic effect, viscosity, intravascular persistence, and initial degree of volume expansion. In addition to molecular weight, other properties of colloids such as surface charge impact the degree of loss through capillary endothelium, as well as rate of glomerular filtration.154,155,158
Albumin is derived from pooled human plasma and has minimal side effects or contraindications. Results from the Saline versus Albumin Evaluation (SAFE) trial, which randomized 7000 critically ill medical and surgical patients to 4% albumin or normal saline, provided evidence on the safety of albumin in critically ill patients. The groups had similar mortality outcomes at 28-day follow-up, as well as similar secondary outcomes of length of stay, ventilatory requirements, and renal replacement therapy.159–161
Semisynthetic colloids are dissolved in a crystalloid carrier solvent consisting of either isotonic or hypertonic saline or glucose, or isotonic balanced electrolyte solution. Clinical data provide supportive evidence in terms of patient outcomes for colloids with a balanced solvent solution mirroring composition of plasma.154,155 Semisynthetic colloids have been reported to increase the risk for bleeding largely attributed to hemodilution of clotting factors, reductions in factor VIII/vWF also caused by hemodilution, and/or functional platelet abnormalities.154,155,160,162,163 A retrospective chart review concluded that intraoperative use of hetastarch in cardiac surgery requiring CPB increased bleeding and transfusion requirements.164
A laboratory investigation in a rabbit model examined the effect of fluid resuscitation with three colloids (Hextend, Dextran 70, 5% albumin) on coagulation and uncontrolled bleeding in rabbits subjected to a splenic injury. Although the prothrombin and partial thromboplastin times were prolonged in all rabbits, thromboelastography and thrombin generation assays identified more severe coagulopathy with Hextend and Dextran than with albumin. Their results suggested resuscitation with albumin maintained coagulation function, decreased blood loss, and improved survival time compared with synthetic colloids.165
Lang et al166 examined the impact of volume replacement with 6% hydroxyethyl starch or lactated Ringer solutions on tissue oxygen tension during and after major surgical procedures. Patients who received 6% hydroxyethyl starch had improved tissue oxygenation compared with a crystalloid-based volume replacement strategy. Improvements in tissue oxygen tension in the hydroxyethyl starch–treated group were thought to be due to improved microperfusion and less endothelial swelling.
Jacob et al155 examined the impact of albumin, hydroxyethyl starch, and saline as resuscitation fluids on vascular integrity in an isolated guinea pig perfused heart model. The authors hypothesized that fluid extravasation might lead to myocardial edema and consequent reduction in ventricular function. Albumin more effectively prevented fluid extravasation in the heart than crystalloid or artificial colloid, and this effect was partly independent of colloid osmotic pressure. Others have noted that using albumin for the CPB prime better preserved platelet counts than crystalloid prime and more favorably influenced colloid oncotic pressure, positive fluid balance, and postoperative weight gain.167

Blood Substitutes
Risks associated with RBC transfusion in terms of morbidity, mortality, survival, and availability of supply have been issues that have spurred development of alternatives to blood transfusion for decades. Ideal characteristics of a blood substitute include long shelf-life, no need to cross-match, immediate availability, and lack of toxic side effects.168 Hemoglobin-based oxygen carriers (HBOCs) are engineered human, animal, or recombinant hemoglobin products in a cell-free hemoglobin preparation. There are currently no approved products in the United States.169,170 Perfluorocarbon-based oxygen carriers are aqueous emulsions of perfluorocarbon derivative that dissolve relatively large amounts of oxygen and generally require patients to breathe oxygen-enriched air.169
Failure to bring a product to the clinical arena has been primarily because of toxicity-related issues. First-generation HBOC had issues with the way oxygen was carried and released from RBCs. A number of adverse events have been reported in clinical trials of oxygen carriers that include death, stroke, hypertension, anemia, and abdominal pain.171 Others have reported on adverse effects that included skin rash, diarrhea, hemoglobinuria, elevated lipase, vasoconstrictive effects, and increased hemostatic effect because of reversal of inhibition effect of nitric oxide on platelet aggregation. Free plasma hemoglobin in addition to generating reactive oxygen species is also a potent scavenger of nitric oxide170 (Figure 30-6 and Table 30-3).
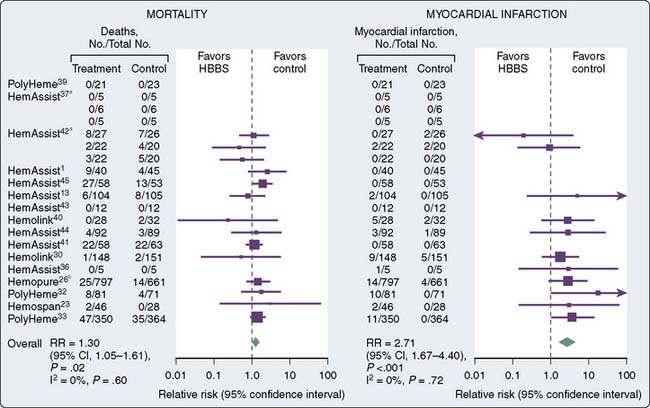
In a review of the current state of oxygen carriers, Winslow171 noted these solutions exhibit side effects of vasoconstriction, which has been one of the limiting factors for clinical use. Vasoconstriction is thought to be due to either scavenging nitric oxide by hemoglobin or an oversupply of oxygen from free hemoglobin via facilitated diffusion. An understanding of the proposed mechanisms of vasoconstriction (i.e., the oversupply theory) led to new product development involving modification of hemoglobin with lower P50 and increased molecule size, thereby reducing release of oxygen in resistance vessels and resultant vasoconstriction.172
Similarly, Yu et al173 noted profound vasoconstrictor side effects limiting clinical utility of HBOC and attributed this side effect to nitric oxide scavenging. The authors noted that by inhaling nitric oxide, changes occur in body stores of nitric oxide metabolites without producing hypotension and may prevent hypertensive side effects of HBOC infusion. Others have reported on nitric oxide scavenging properties of HBOC as a likely mechanism of vasoconstriction associated with infusion and proposed modifications that could potentially ameliorate this side effect.174,175
A basic science working group from the National Heart, Lung and Blood Institute Division of Blood Diseases and Resources summarized and provided recommendations for basic science focus in the area of blood substitutes. The working group highlighted impediments to further HBOC product development secondary to significant side effects of excessive cardiovascular and cerebrovascular events and mortality.176
Lowest hematocrit on cardiopulmonary bypass
Both anemia on CPB and transfusion of RBCs have been associated with adverse perioperative outcomes. Hemodilution secondary to fixed priming volume and pre-CPB fluid administration contribute to anemia and transfusion need.177,178 Minimum hematocrit values less than or equal to 14% on CPB have been associated with postoperative mortality in CABG; and in high-risk patients, values less than 17% increase mortality risk.179 Morbidity risk associated with hemodilutional anemia is thought to be due to inadequate oxygen delivery leading to organ dysfunction.180 Lowest hematocrit on CPB also has been a risk factor for postoperative low-output syndrome and renal failure. Ranucci et al181 reported a cutoff hematocrit value of 23% for renal failure and 24% for development of the low-output syndrome. Transfusion of RBCs also was associated with both renal failure and the low-output syndrome in their investigation. Risk for renal injury further increased when RBC transfusions were associated with a nadir hematocrit on CPB of less than 23%.
Swaminathan et al182 reported more perioperative renal injury with hemodilution, targeting CPB hematocrit levels of 22% to 24%. The authors highlighted renal benefits of hemodilution relating to reductions in blood viscosity and improved regional blood flow; however, they noted that a well-defined cutoff for hemodilution has not been well clarified. They were unable to find an “elbow” for a cutoff between hematocrit and change in creatinine values. The significant association between lowest CPB hematocrit and change in creatinine values was highly influenced by body weight in their investigation.182 Others have reported that hemodilution on CPB to hematocrit values less than 24% and associated renal injury were further exacerbated by longer CPB times and with RBC transfusion.180
Karkouti et al183 suggested a U-shaped relation between nadir hematocrit on CPB and renal failure requiring dialysis. Moderate hemodilution (hematocrit values between 21% and 25%) were associated with lowest risk for acute renal failure; risk increased as nadir hematocrit concentrations decreased to less than 21% or were greater than 25%. In congenital heart surgery, Jonas et al184 reported that lower hematocrit strategies (21% ± 2.9%) versus higher hematocrit strategies (28% ± 3.2%) were associated with higher serum lactate levels 60 minutes after CPB, greater percentage of increase in total body water on postoperative day 1, and at age 1 year, worse scores on the Psychomotor Development Index. The authors concluded that a lower hematocrit strategy was associated with increased risk for developmental impairment.
DeFoe et al185 reported a risk-adjusted increased risk for mortality, need for intra-aortic balloon pump, and return to CPB after initial separation with nadir hematocrit on CPB. Smaller patients and those with lower preoperative hematocrit values were at greater risk for development of lower CPB values. They reported trends toward increasing risk for death in patients with hematocrit values less than 23%; and for those with hematocrit values less than 19%, mortality was almost twice as high as in patients with hematocrit values of 25% or more.185 Others have reported on the association between hemodilution to hematocrit values of 24%, RBC transfusion, and increased risk for renal and splanchnic injury.186
Notably, not all investigations reported adverse consequences with lower nadir hematocrit values on CPB. von Heymann et al187 examined oxygen delivery, oxygen consumption, and outcomes in low-risk CABG patients assigned CPB hematocrit values of 20% or 25%. They reported similar oxygen delivery, oxygen consumption, and blood lactate levels between the two groups, as well as clinical outcome measures. The authors concluded that CPB hematocrit values of 20% were adequate to maintain calculated whole-body oxygen delivery above critical levels. Similarly, Berger et al randomized patients to profound hemodilution with CPB hematocrit values between 19% and 21% versus standard values of 24% to 26%. They reported similar changes in intestinal permeability and cytokine release between the two groups and concluded that CPB hematocrit values between 19% and 21% did not adversely impact outcomes.188
Orlov et al189 reported on the clinical utility of using oxygen extraction ratio as an adjunct to hemoglobin concentration for guiding cardiac surgical RBC transfusion decisions. The authors suggested that a normal oxygen extraction ratio in patients with anemia with no evidence of organ dysfunction indicated adequate tissue oxygen delivery, and by incorporating this into the transfusion decisions, RBC transfusions could be reduced.
1 Field M., Lohr K., editors. Clinical Practice Guidelines: Directions for a New Program. Washington, DC: National Academy Press, Institute of Medicine, 1990. Committee to Advise the Public Health Service on Clinical Practice Guidelines
2 DioDato C.P., Likosky D.S., DeFoe G.R., et al. Cardiopulmonary bypass recommendations in adults: The northern New England experience. J Extra Corpor Technol. 2008;40:16-20.
3 Woolf S.H., Grol R., Hutchinson A., et al. Clinical guidelines: Potential benefits, limitations, and harms of clinical guidelines. BMJ. 1999;318:527-530.
4 Ferraris V.A., Ferraris S.P., Saha S.P., et al. Perioperative blood transfusion and blood conservation in cardiac surgery: The Society of Thoracic Surgeons and the Society of Cardiovascular Anesthesiologists clinical practice guideline. Ann Thorac Surg. 2007;83(Suppl 5):S27-S86.
5 Guidelines MaPftAATFoP, Methodology Manual for ACCF/AHA Guideline Writing Committees; January 1, 2010; Available at: http://www.americanheart.org/downloadable/heart/12604770597301209Methodology_Manual_for_ACC_AHA_Writing_Committees.pdf Accessed February 2, 2010
6 Arroll B., Jenkins S., North D., et al. Management of hypertension and the core services guidelines: Results from interviews with 100 Auckland general practitioners. N Z Med J. 1995;108:55-57.
7 Christakis D.A., Rivara F.P. Pediatricians’ awareness of and attitudes about four clinical practice guidelines. Pediatrics. 1998;101:825-830.
8 Grilli R., Lomas J. Evaluating the message: The relationship between compliance rate and the subject of a practice guideline. Med Care. 1994;32:202-213.
9 Rhew D.C., Riedinger M.S., Sandhu M., et al. A prospective, multicenter study of a pneumonia practice guideline. Chest. 1998;114:115-119.
10 Grol R., Dalhuijsen J., Thomas S., et al. Attributes of clinical guidelines that influence use of guidelines in general practice: Observational study. BMJ. 1998;317:858-861.
11 Merritt T.A., Palmer D., Bergman D.A., et al. Clinical practice guidelines in pediatric and newborn medicine: Implications for their use in practice. Pediatrics. 1997;99:100-114.
12 Fleming M., Wentzell N. Patient safety culture improvement tool: Development and guidelines for use. Healthc Q. 2008;11(3 spec no.):10-15.
13 Hiratzka L.F., Eagle K.A., Liang L., et al. Atherosclerosis secondary prevention performance measures after coronary bypass graft surgery compared with percutaneous catheter intervention and nonintervention patients in the Get With the Guidelines database. Circulation. 2007;116(Suppl 11):I207-I212.
14 Steinberg B.A., Steg P.G., Bhatt D.L., et al. Comparisons of guideline-recommended therapies in patients with documented coronary artery disease having percutaneous coronary intervention versus coronary artery bypass grafting versus medical therapy only (from the REACH International Registry). Am J Cardiol. 2007;99:1212-1215.
15 Yam F.K., Akers W.S., Ferraris V.A., et al. Interventions to improve guideline compliance following coronary artery bypass grafting. Surgery. 2006;;140:541-547. discussion 547–552
16 Berry S.A., Doll M.C., McKinley K.E., et al. ProvenCare: Quality improvement model for designing highly reliable care in cardiac surgery. Qual Saf Health Care. 2009;18:360-368.
17 Grimshaw J.M., Thomson M.A. What have new efforts to change professional practice achieved? Cochrane Effective Practice and Organization of Care Group. J R Soc Med. 1998;91(Suppl 35):20-25.
18 Mowatt G., Grimshaw J.M., Davis D.A., et al. Getting evidence into practice: The work of the Cochrane Effective Practice and Organization of Care Group (EPOC). J Contin Educ Health Prof. 2001;21:55-60.
19 Vasaiwala S., Nolan E., Ramanath V.S., et al. A quality guarantee in acute coronary syndromes: The American College of Cardiology’s Guidelines Applied in Practice program taken real-time. Am Heart J. 2007;153:16-21.
20 Bayne-Jones S. Dr. Karl Landsteiner Nobel Prize Laureate in Medicine, 1930. Science. 1931;73:599-604.
21 , Racial and Ethnic Distribution of ABO blood types; July 13, 2008; Available at: http://www.bloodbook.com/world-abo.html Accessed February 2, 2010
22 Yamamoto F., Clausen H., White T., et al. Molecular genetic basis of the histo-blood group ABO system. Nature. 1990;345:229-233.
23 Reid M., Lomas-Francis C. The Blood Group Antigen Facts Book, ed 2. New York: Elsevier Academic Press, 2004.
24 Yamamoto F., McNeill P.D., Hakomori S. Human histo-blood group A2 transferase coded by A2 allele, one of the A subtypes, is characterized by a single base deletion in the coding sequence, which results in an additional domain at the carboxyl terminal. Biochem Biophys Res Commun. 1992;187:366-374.
25 Yamamoto F., Molecular Genetic Basis of the Blood Group ABO System; 2008; Available at: http://sites.google.com/site/abobloodgroup/17.aboalleles2008 Accessed February 2, 2010
26 Kustu S., Inwood W. Biological gas channels for NH3 and CO2: Evidence that Rh (rhesus) proteins are CO2 channels. Transfus Clin Biol. 2006;13:103-110.
27 Avent N., Reid M. The Rh blood group system: A review. Blood. 2002;95:375-387.
28 Mwangi J. Blood group distribution in an urban population of patient targeted blood donors. East Afr Med J. 1999;76:615-618.
29 Urbaniak S., Greiss M. RhD haemolytic disease of the fetus and the newborn. Blood Rev. 2000;14:44-61.
30 National Center for Biotechnology Information, MNS (MNS) Blood Group System; December 11, 2009; Available at: http://www.ncbi.nlm.nih.gov/gv/mhc/xslcgi.cgi?cmd=bgmut/systems_info& system=mns Accessed February 2, 2010
31 National Center for Biotechnology Information, Lewis (LE) Blood Group System; January 29 2009; Available at: http://www.ncbi.nlm.nih.gov/projects/gv/mhc/xslcgi.cgi?cmd=bgmut/systems_info& system=lewis Accessed February 2, 2010
32 Blood Bank, January 28, 2010. Available at: http://en.wikipedia.org/wiki/Blood_bank Accessed February 2, 2010
33 U.S. Food and Drug Administration, Fatalities Reported to FDA Following Blood Collection and Transfusion: Annual Summary for Fiscal Year 2008; February 19, 2010; Available at: http://www.fda.gov/BiologicsBloodVaccines/SafetyAvailability/ReportaProblem/TransfusionDonationFatalities/ucm113649.htm Accessed February 2, 2010
34 Napier J.A. The crossmatch. Br J Haematol. 1991;78:1-4.
35 Chapman J.F., Milkins C., Voak D. The computer crossmatch: A safe alternative to the serological crossmatch. Transfus Med. 2000;10:251-256.
36 Roback J., Combs M., Grossman B., et al. AABB Technical Manual, ed 16. Bethesda, MD: American Association of Blood Banks, 2008.
37 Judd W.J. Requirements for the electronic crossmatch. Vox Sang. 1998;74(Suppl 2):409-417.
38 Inaba K., Teixeira P.G., Shulman I., et al. The impact of uncross-matched blood transfusion on the need for massive transfusion and mortality: Analysis of 5,166 uncross-matched units. J Trauma. 2008;65:1222-1226.
39 Lee E.J., Schiffer C.A. ABO compatibility can influence the results of platelet transfusion. Results of a randomized trial. Transfusion. 1989;29:384-389.
40 Julmy F., Ammann R.A., Taleghani B.M., et al. Transfusion efficacy of ABO major-mismatched platelets (PLTs) in children is inferior to that of ABO-identical PLTs. Transfusion. 2009;49:21-33.
41 Kanda J., Ichinohe T., Matsuo K., et al. Impact of ABO mismatching on the outcomes of allogeneic related and unrelated blood and marrow stem cell transplantations for hematologic malignancies: IPD-based meta-analysis of cohort studies. Transfusion. 2009;49:624-635.
42 Leo A., Pedal I. Diagnostic approaches to acute transfusion reactions. Forens Sci Med Pathol. 2010;6:135-145.
43 Koch C., Li L., Figueroa P., et al. Transfusion and pulmonary morbidity after cardiac surgery. Ann Thorac Surg. 2009;88:1410-1418.
44 Cherry T., Steciuk M., Reddy V.V., et al. Transfusion-related acute lung injury: Past, present, and future. Am J Clin Pathol. 2008;129:287-297.
45 Curtis B.R., McFarland J.G. Mechanisms of transfusion-related acute lung injury (TRALI): Anti-leukocyte antibodies. Crit Care Med. 2006;34(Suppl 5):S118-123.
46 Popovsky M.A. Transfusion and lung injury. Transfus Clin Biol. 2001;8:272-277.
47 Holness L., Knippen M.A., Simmons L., et al. Fatalities caused by TRALI. Transfus Med Rev. 2004;18:184-188.
48 Sandler S.G. How I manage patients suspected of having had an IgA anaphylactic transfusion reaction. Transfusion. 2006;46:10-13.
49 Hirayama F. Recent advances in laboratory assays for nonhemolytic transfusion reactions. Transfusion. 2010;50:252-263.
50 Davenport R.D. Pathophysiology of hemolytic transfusion reactions. Semin Hematol. 2005;42:165-168.
51 Hendrickson J.E., Hillyer C.D. Noninfectious serious hazards of transfusion. Anesth Analg. 2009;108:759-769.
52 Ruhl H., Bein G., Sachs U.J. Transfusion-associated graft-versus-host disease. Transfus Med Rev. 2009;23:62-71.
53 Hsueh J.C., Ho C.F., Chang S.H., et al. Blood surveillance and detection on platelet bacterial contamination associated with septic events. Transfus Med. 2009;19:350-356.
54 Dzik W.H., Kirkley S.A. Citrate toxicity during massive blood transfusion. Transfus Med Rev. 1988;2:76-94.
55 Koch C.G., Li L., Duncan A.I., et al. Transfusion in coronary artery bypass grafting is associated with reduced long-term survival. Ann Thorac Surg. 2006;81:1650-1657.
56 Banbury M.K., Brizzio M.E., Rajeswaran J., et al. Transfusion increases the risk of postoperative infection after cardiovascular surgery. J Am Coll Surg. 2006;202:131-138.
57 Koch C.G., Li L., Duncan A.I., et al. Morbidity and mortality risk associated with red blood cell and blood-component transfusion in isolated coronary artery bypass grafting. Crit Care Med. 2006;34:1608-1616.
58 Blajchman M.A. Immunomodulation and blood transfusion. Am J Ther. 2002;9:389-395.
59 Koch C.G., Li L., Van Wagoner D.R., et al. Red cell transfusion is associated with an increased risk for postoperative atrial fibrillation. Ann Thorac Surg. 2006;82:1747-1756.
60 Fransen E., Maessen J., Dentener M., et al. Systemic inflammation present in patients undergoing CABG without extracorporeal circulation. Chest. 1998;113:1290-1295.
61 Bennett-Guerrero E., Veldman T.H., Doctor A., et al. Evolution of adverse changes in stored RBCs. Proc Natl Acad Sci U S A. 2007;104:17063-17068.
62 Relevy H., Koshkaryev A., Manny N., et al. Blood banking-induced alteration of red blood cell flow properties. Transfusion. 2008;48:136-146.
63 Rigamonti A., McLaren A.T., Mazer C.D., et al. Storage of strain-specific rat blood limits cerebral tissue oxygen delivery during acute fluid resuscitation. Br J Anaesth. 2008;100:357-364.
64 Koch C.G., Li L., Sessler D.I., et al. Duration of red-cell storage and complications after cardiac surgery. N Engl J Med. 2008;358:1229-1239.
65 Sweeney J., Kouttab N., Kurtis J., et al. Stored red blood cell supernatant facilitates thrombin generation. Transfusion. 2009;49:1569-1579.
66 Burzotta F., Iacoviello L., Di Castelnuovo A., et al. 4G/5G PAI-1 promoter polymorphism and acute-phase levels of PAI-1 following coronary bypass surgery: A prospective study. J Thromb Thrombolysis. 2003;16:149-154.
67 de Lange M., Snieder H., Ariens R.A., et al. The genetics of haemostasis: A twin study. Lancet. 2001;357:101-105.
68 Freeman M.S., Mansfield M.W., Barrett J.H., et al. Genetic contribution to circulating levels of hemostatic factors in healthy families with effects of known genetic polymorphisms on heritability. Arterioscler Thromb Vasc Biol. 2002;22:506-510.
69 Muehlschlegel J.D., Body S.C. Impact of genetic variation on perioperative bleeding. Am J Hematol. 2008;83:732-737.
70 Souto J.C., Almasy L., Borrell M., et al. Genetic susceptibility to thrombosis and its relationship to physiological risk factors: The GAIT study. Genetic Analysis of Idiopathic Thrombophilia. Am J Hum Genet. 2000;67:1452-1459.
71 de Lange M., de Geus E.J., Kluft C., et al. Genetic influences on fibrinogen, tissue plasminogen activator-antigen and von Willebrand factor in males and females. Thromb Haemost. 2006;95(3):414-419.
72 Dunn E.J., Ariens R.A., de Lange M., et al. Genetics of fibrin clot structure: A twin study. Blood. 2004;103:1735-1740.
73 Miller C.H., Haff E., Platt S.J., et al. Measurement of von Willebrand factor activity: Relative effects of ABO blood type and race. J Thromb Haemost. 2003;1:2191-2197.
74 Eriksson-Berg M., Deguchi H., Hawe E., et al. Influence of factor VII gene polymorphisms and environmental factors on plasma coagulation factor VII concentrations in middle-aged women with and without manifest coronary heart disease. Thromb Haemost. 2005;93:351-358.
75 Eyre-Walker A. Evolution in health and medicine Sackler colloquium: Genetic architecture of a complex trait and its implications for fitness and genome-wide association studies. Proc Natl Acad Sci U S A. 2010;107(Suppl 1):1752-1756.
76 Ganesh S.K., Zakai N.A., van Rooij F.J., et al. Multiple loci influence erythrocyte phenotypes in the CHARGE Consortium. Nat Genet. 2009;41:1191-1198.
77 Soranzo N., Spector T.D., Mangino M., et al. A genome-wide meta-analysis identifies 22 loci associated with eight hematological parameters in the HaemGen consortium. Nat Genet. 2009;41:1182-1190.
78 Miah M.F., Boffa M.B. Functional analysis of mutant variants of thrombin-activatable fibrinolysis inhibitor resistant to activation by thrombin or plasmin. J Thromb Haemost. 2009;7:665-672.
79 Peyvandi F., Jayandharan G., Chandy M., et al. Genetic diagnosis of haemophilia and other inherited bleeding disorders. Haemophilia. 2006;12(Suppl 3):82-89.
80 Coppola A., Franchini M., Tagliaferri A. Prophylaxis in people with haemophilia. Thromb Haemost. 2009;101:674-681.
81 Seligsohn U. Factor XI deficiency in humans. J Thromb Haemost. 2009;7(Suppl 1):84-87.
82 National Institutes of Health, Genetic Home Reference; February 21, 2010; Available at: http://ghr.nlm.nih.gov/condition=hemophilia#genes Accessed February 25, 2010
83 James P., Lillicrap D. The role of molecular genetics in diagnosing von Willebrand disease. Semin Thromb Hemost. 2008;34:502-508.
84 Wagenman B.L., Townsend K.T., Mathew P., et al. The laboratory approach to inherited and acquired coagulation factor deficiencies. Clin Lab Med. 2009;29:229-252.
85 Brettler D., Levine P., Clinical manifestations and therapy of inherited coagulation factor deficiencies, Colman R., Hirsh J., Marder V., Salzman E., editors, Hemostasis and Thrombosis: Basic Principles and Clinical Practice, ed 3, 1993,:169-183
86 ClinLab Navigator, Factor VIII concentrate; 2010; Available at: http://www.clinlabnavigator.com/transfusion/factorviiiconcentrate.html Accessed February 2, 2010
87 ClinLab Navigator, Factor VIII inhibitors, Available at: http://www.clinlabnavigator.com/transfusion/factorviiiinhibitors.html Accessed February 2, 2010
88 Levy J.H. Pharmacologic methods to reduce perioperative bleeding. Transfusion. 2008;48(Suppl 1):31S-38S.
89 ClinLab Navigator. Factor IX complex & concentrates. http://www.clinlabnavigator.com/transfusion/factorixcomplex.html. Available at:Accessed February 2, 2010
90 Michiels J.J., Berneman Z., Gadisseur A., et al. Classification and characterization of hereditary types 2A, 2B, 2C, 2D, 2E, 2M, 2N, and 2U (unclassifiable) von Willebrand disease. Clin Appl Thromb Hemost. 2006;12:397-420.
91 Federici A.B. Classification of inherited von Willebrand disease and implications in clinical practice. Thromb Res. 2009;124(Suppl 1):S2-S6.
92 Favaloro E.J. Laboratory identification of von Willebrand disease: Technical and scientific perspectives. Semin Thromb Hemost. 2006;32:456-471.
93 Lillicrap D. Von Willebrand disease—phenotype versus genotype: Deficiency versus disease. Thromb Res. 2007;120(Suppl 1):S11-16.
94 Lillicrap D. Genotype/phenotype association in von Willebrand disease: Is the glass half full or empty? J Thromb Haemost. 2009;7(Suppl 1):65-70.
95 Budde U. Diagnosis of von Willebrand disease subtypes: Implications for treatment. Haemophilia. 2008;14(Suppl 5):27-38.
96 Pate G.E., Chandavimol M., Naiman S.C., et al. Heyde’s syndrome: A review. J Heart Valve Dis. 2004;13:701-712.
97 Cattaneo M. The use of desmopressin in open-heart surgery. Haemophilia. 2008;14(Suppl 1):40-47.
98 Nicolaes G.A., Dahlback B. Factor V and thrombotic disease: Description of a janus-faced protein. Arterioscler Thromb Vasc Biol. 2002;22:530-538.
99 Segers K., Dahlback B., Nicolaes G.A. Coagulation factor V and thrombophilia: Background and mechanisms. Thromb Haemost. 2007;98:530-542.
100 Andreassi M.G., Botto N., Maffei S. Factor V Leiden, prothrombin G20210A substitution and hormone therapy: Indications for molecular screening. Clin Chem Lab Med. 2006;44:514-521.
101 Boehm J., Grammer J.B., Lehnert F., et al. Factor V Leiden does not affect bleeding in aprotinin recipients after cardiopulmonary bypass. Anesthesiology. 2007;106:681-686.
102 Donahue B.S., Gailani D., Higgins M.S., et al. Factor V Leiden protects against blood loss and transfusion after cardiac surgery. Circulation. 2003;107:1003-1008.
103 Berentsen S., Beiske K., Tjonnfjord G.E. Primary chronic cold agglutinin disease: An update on pathogenesis, clinical features and therapy. Hematology. 2007;12:361-370.
104 Agarwal S.K., Ghosh P.K., Gupta D. Cardiac surgery and cold-reactive proteins. Ann Thorac Surg. 1995;60:1143-1150.
105 Fischer G.D., Claypoole V., Collard C.D. Increased pressures in the retrograde blood cardioplegia line: An unusual presentation of cold agglutinins during cardiopulmonary bypass. Anesth Analg. 1997;84:454-456.
106 Welsby I.J., Jones R., Pylman J., et al. ABO blood group and bleeding after coronary artery bypass graft surgery. Blood Coagul Fibrinolysis. 2007;18:781-785.
107 Welsby I.J., Podgoreanu M.V., Phillips-Bute B., et al. Association of the 98G/T ELAM-1 polymorphism with increased bleeding and transfusion after cardiac surgery. Anesth Analg. 2007;104:SCA39.
108 Welsby I.J., Podgoreanu M.V., Phillips-Bute B., et al. Genetic factors contribute to bleeding after cardiac surgery. J Thromb Haemost. 2005;3:1206-1212.
109 Mehta R.H., Sheng S., O’Brien S.M., et al. Reoperation for bleeding in patients undergoing coronary artery bypass surgery: Incidence, risk factors, time trends, and outcomes. Circulation. 2009;2:583-590.
110 Moulton M.J., Creswell L.L., Mackey M.E., et al. Reexploration for bleeding is a risk factor for adverse outcomes after cardiac operations. J Thorac Cardiovasc Surg. 1996;111:1037-1046.
111 Choong C.K., Gerrard C., Goldsmith K.A., et al. Delayed re-exploration for bleeding after coronary artery bypass surgery results in adverse outcomes. Eur J Cardiothorac Surg. 2007;31:834-838.
112 Karthik S., Grayson A.D., McCarron E.E., et al. Reexploration for bleeding after coronary artery bypass surgery: Risk factors, outcomes, and the effect of time delay. Ann Thorac Surg. 2004;78:527-534.
113 Ranucci M., Bozzetti G., Ditta A., et al. Surgical reexploration after cardiac operations: Why a worse outcome? Ann Thorac Surg. 2008;86:1557-1562.
114 Hall T.S., Brevetti G.R., Skoultchi A.J., et al. Re-exploration for hemorrhage following open heart surgery differentiation on the causes of bleeding and the impact on patient outcomes. Ann Thorac Cardiovasc Surg. 2001;7:352-357.
115 Kermode J.C., Zheng Q., Milner E.P. Marked temperature dependence of the platelet calcium signal induced by human von Willebrand factor. Blood. 1999;94:199-207.
116 Johnston T.D., Chen Y., Reed R.L.2nd. Functional equivalence of hypothermia to specific clotting factor deficiencies. J Trauma. 1994;37:413-417.
117 Wolberg A.S., Meng Z.H., Monroe D.M.3rd, et al. A systematic evaluation of the effect of temperature on coagulation enzyme activity and platelet function. J Trauma. 2004;56:1221-1228.
118 Meng Z.H., Wolberg A.S., Monroe D.M.3rd, et al. The effect of temperature and pH on the activity of factor VIIa: Implications for the efficacy of high-dose factor VIIa in hypothermic and acidotic patients. J Trauma. 2003;55:886-891.
119 Martini W.Z., Pusateri A.E., Uscilowicz J.M., et al. Independent contributions of hypothermia and acidosis to coagulopathy in swine. J Trauma. 2005;58:1002-1009. discussion 1009–1010
120 Alam H.B., Bice L.M., Butt M.U., et al. Testing of blood products in a polytrauma model: Results of a multi-institutional randomized preclinical trial. J Trauma. 2009;67:856-864.
121 Dirkmann D., Hanke A.A., Gorlinger K., et al. Hypothermia and acidosis synergistically impair coagulation in human whole blood. Anesth Analg. 2008;106:1627-1632.
122 Maani C.V., DeSocio P.A., Holcomb J.B. Coagulopathy in trauma patients: What are the main influence factors? Curr Opin Anaesthesiol. 2009;22:255-260.
123 Borgman M.A., Spinella P.C., Perkins J.G., et al. The ratio of blood products transfused affects mortality in patients receiving massive transfusions at a combat support hospital. J Trauma. 2007;63:805-813.
124 Holcomb J.B., Wade C.E., Michalek J.E., et al. Increased plasma and platelet to red blood cell ratios improves outcome in 466 massively transfused civilian trauma patients. Ann Surg. 2008;248:447-458.
125 Gunter O.L.Jr, Au B.K., Isbell J.M., et al. Optimizing outcomes in damage control resuscitation: Identifying blood product ratios associated with improved survival. J Trauma. 2008;65:527-534.
126 Zink K.A., Sambasivan C.N., Holcomb J.B., et al. A high ratio of plasma and platelets to packed red blood cells in the first 6 hours of massive transfusion improves outcomes in a large multicenter study. Am J Surg. 2009;197:565-570.
127 Kashuk J.L., Moore E.E., Johnson J.L., et al. Postinjury life threatening coagulopathy: Is 1:1 fresh frozen plasma:packed red blood cells the answer? J Trauma. 2008;;65:261-270. discussion 270–271
128 Hirshberg A., Dugas M., Banez E.I., et al. Minimizing dilutional coagulopathy in exsanguinating hemorrhage: A computer simulation. J Trauma. 2003;54:454-463.
129 Cotton B.A., Au B.K., Nunez T.C., et al. Predefined massive transfusion protocols are associated with a reduction in organ failure and postinjury complications. J Trauma. 2009;66:41-48. discussion 48–49
130 Riskin D.J., Tsai T.C., Riskin L., et al. Massive transfusion protocols: The role of aggressive resuscitation versus product ratio in mortality reduction. J Am Coll Surg. 2009;209:198-205.
131 Filsoufi F., Castillo J.G., Rahmanian P.B., et al. Effective management of refractory postcardiotomy bleeding with the use of recombinant activated factor VII. Ann Thorac Surg. 2006;82:1779-1783.
132 Hoffman M., Monroe D.M.3rd. A cell-based model of hemostasis. Thromb Haemost. 2001;85:958-965.
133 Lisman T., Adelmeijer J., Cauwenberghs S., et al. Recombinant factor VIIa enhances platelet adhesion and activation under flow conditions at normal and reduced platelet count. J Thromb Haemost. 2005;3:742-751.
134 Karkouti K., Beattie W.S., Arellano R., et al. Comprehensive Canadian review of the off-label use of recombinant activated factor VII in cardiac surgery. Circulation. 2008;118:331-338.
135 von Heymann C., Redlich U., Jain U., et al. Recombinant activated factor VII for refractory bleeding after cardiac surgery—a retrospective analysis of safety and efficacy. Crit Care Med. 2005;33:2241-2246.
136 McCall P., Story D.A., Karalapillai D. Audit of factor VIIa for bleeding resistant to conventional therapy following complex cardiac surgery. Can J Anaesth. 2006;53:926-933.
137 Tatoulis J., Theodore S., Meswani M., et al. Safe use of recombinant activated factor VIIa for recalcitrant postoperative haemorrhage in cardiac surgery. Interact Cardiovasc Thorac Surg. 2009;9:459-462.
138 Bishop C.V., Renwick W.E., Hogan C., et al. Recombinant activated factor VII: Treating postoperative hemorrhage in cardiac surgery. Ann Thorac Surg. 2006;81:875-879.
139 Dunkley S., Phillips L., McCall P., et al. Recombinant activated factor VII in cardiac surgery: Experience from the Australian and New Zealand Haemostasis Registry. Ann Thorac Surg. 2008;85:836-844.
140 DiDomenico R.J., Massad M.G., Kpodonu J., et al. Use of recombinant activated factor VII for bleeding following operations requiring cardiopulmonary bypass. Chest. 2005;127:1828-1835.
141 Gill R., Herbertson M., Vuylsteke A., et al. Safety and efficacy of recombinant activated factor VII: A randomized placebo-controlled trial in the setting of bleeding after cardiac surgery. Circulation. 2009;120:21-27.
142 Bowman L.J., Uber W.E., Stroud M.R., et al. Use of recombinant activated factor VII concentrate to control postoperative hemorrhage in complex cardiovascular surgery. Ann Thorac Surg. 2008;;85:1669-1676. discussion 1676–1667
143 O’Connell K.A., Wood J.J., Wise R.P., et al. Thromboembolic adverse events after use of recombinant human coagulation factor VIIa. JAMA. 2006;295:293-298.
144 Fenger-Eriksen C., Ingerslev J., Sorensen B. Fibrinogen concentrate—a potential universal hemostatic agent. Expert Opin Biol Ther. 2009;9:1325-1333.
145 Fenger-Eriksen C., Lindberg-Larsen M., Christensen A.Q., et al. Fibrinogen concentrate substitution therapy in patients with massive haemorrhage and low plasma fibrinogen concentrations. Br J Anaesth. 2008;101:769-773.
146 Rahe-Meyer N., Pichlmaier M., Haverich A., et al. Bleeding management with fibrinogen concentrate targeting a high-normal plasma fibrinogen level: A pilot study. Br J Anaesth. 2009;102:785-792.
147 Fenger-Eriksen C., Jensen T.M., Kristensen B.S., et al. Fibrinogen substitution improves whole blood clot firmness after dilution with hydroxyethyl starch in bleeding patients undergoing radical cystectomy: A randomized, placebo-controlled clinical trial. J Thromb Haemost. 2009;7:795-802.
148 Karlsson M., Ternstrom L., Hyllner M., et al. Prophylactic fibrinogen infusion reduces bleeding after coronary artery bypass surgery. A prospective randomised pilot study. Thromb Haemost. 2009;102:137-144.
149 Samama C.M. Prothrombin complex concentrates: A brief review. Eur J Anaesthesiol. 2008;25:784-789.
150 Riess H.B., Meier-Hellmann A., Motsch J., et al. Prothrombin complex concentrate (Octaplex) in patients requiring immediate reversal of oral anticoagulation. Thromb Res. 2007;121:9-16.
151 Leissinger C.A., Blatt P.M., Hoots W.K., et al. Role of prothrombin complex concentrates in reversing warfarin anticoagulation: A review of the literature. Am J Hematol. 2008;83:137-143.
152 Warren O., Simon B. Massive, fatal, intracardiac thrombosis associated with prothrombin complex concentrate. Ann Emerg Med. 2009;53:758-761.
153 Vincent J.L. Fluid resuscitation: Colloids vs crystalloids. Acta Clin Belg Suppl. 2007:408-411.
154 Sakka S.G. Resuscitation of hemorrhagic shock with normal saline versus lactated Ringer’s: Effects on oxygenation, extravascular lung water, and hemodynamics. Crit Care. 2009;13:128.
155 Jacob M., Bruegger D., Rehm M., et al. Contrasting effects of colloid and crystalloid resuscitation fluids on cardiac vascular permeability. Anesthesiology. 2006;104:1223-1231.
156 Grocott M.P., Hamilton M.A. Resuscitation fluids. Vox Sang. 2002;82:1-8.
157 Verheij J., van Lingen A., Raijmakers P.G., et al. Effect of fluid loading with saline or colloids on pulmonary permeability, oedema and lung injury score after cardiac and major vascular surgery. Br J Anaesth. 2006;96:21-30.
158 Dieterich H.J. Recent developments in European colloid solutions. J Trauma. 2003;54(Suppl 5):S26-S30.
159 Finfer S., Bellomo R., Boyce N., et al. A comparison of albumin and saline for fluid resuscitation in the intensive care unit. N Engl J Med. 2004;350:2247-2256.
160 Boldt J., Suttner S. Plasma substitutes. Minerva Anestesiol. 2005;71:741-758.
161 Fan E., Stewart T.E. Albumin in critical care: SAFE, but worth its salt? Crit Care. 2004;8:297-299.
162 Vercueil A., Grocott M.P., Mythen M.G. Physiology, pharmacology, and rationale for colloid administration for the maintenance of effective hemodynamic stability in critically ill patients. Transfus Med Rev. 2005;19:93-109.
163 Roberts J.S., Bratton S.L. Colloid volume expanders. Problems, pitfalls and possibilities. Drugs. 1998;55:621-630.
164 Knutson J.E., Deering J.A., Hall F.W., et al. Does intraoperative hetastarch administration increase blood loss and transfusion requirements after cardiac surgery? Anesth Analg. 2000;90:801-807.
165 Kheirabadi B.S., Crissey J.M., Deguzman R., et al. Effects of synthetic versus natural colloid resuscitation on inducing dilutional coagulopathy and increasing hemorrhage in rabbits. J Trauma. 2008;;64:1218-1228. discussion 1228–1229
166 Lang K., Boldt J., Suttner S., et al. Colloids versus crystalloids and tissue oxygen tension in patients undergoing major abdominal surgery. Anesth Analg. 2001;93:405-409. 403rd contents page,
167 Russell J.A., Navickis R.J., Wilkes M.M. Albumin versus crystalloid for pump priming in cardiac surgery: Meta-analysis of controlled trials. J Cardiothorac Vasc Anesth. 2004;18:429-437.
168 Jahr J.S., Walker V., Manoochehri K. Blood substitutes as pharmacotherapies in clinical practice. Curr Opin Anaesthesiol. 2007;20:325-330.
169 Kim H.W., Greenburg A.G. Artificial oxygen carriers as red blood cell substitutes: A selected review and current status. Artif Organs. 2004;28:813-828.
170 Napolitano L.M. Hemoglobin-based oxygen carriers: First, second or third generation? Human or bovine? Where are we now? Crit Care Clin. 2009;25:279-301. Table of Contents
171 Winslow R.M. Current status of oxygen carriers (‘blood substitutes’): 2006. Vox Sang. 2006;91:102-110.
172 Winslow R.M. Cell-free oxygen carriers: Scientific foundations, clinical development, and new directions. Biochim Biophys Acta. 2008;1784:1382-1386.
173 Yu B., Bloch K.D., Zapol W.M. Hemoglobin-based red blood cell substitutes and nitric oxide. Trends Cardiovasc Med. 2009;19:103-107.
174 Raat N.J., Liu J.F., Doyle M.P., et al. Effects of recombinant-hemoglobin solutions rHb2.0 and rHb1.1 on blood pressure, intestinal blood flow, and gut oxygenation in a rat model of hemorrhagic shock. J Lab Clin Med. 2005;145:21-32.
175 Hermann J., Corso C., Messmer K.F. Resuscitation with recombinant hemoglobin rHb2.0 in a rodent model of hemorrhagic shock. Anesthesiology. 2007;107:273-280.
176 Estep T., Bucci E., Farmer M., et al. Basic science focus on blood substitutes: A summary of the NHLBI Division of Blood Diseases and Resources Working Group Workshop, March 1, 2006. Transfusion. 2008;48:776-782.
177 Campbell J.A., Holt D.W., Shostrom V.K., et al. Influence of intraoperative fluid volume on cardiopulmonary bypass hematocrit and blood transfusions in coronary artery bypass surgery. J Extra Corpor Technol. 2008;40:99-108.
178 Pappalardo F., Corno C., Franco A., et al. Reduction of hemodilution in small adults undergoing open heart surgery: A prospective, randomized trial. Perfusion. 2007;22:317-322.
179 Fang W.C., Helm R.E., Krieger K.H., et al. Impact of minimum hematocrit during cardiopulmonary bypass on mortality in patients undergoing coronary artery surgery. Circulation. 1997;96(Suppl 9):II-194-II-199.
180 Habib R.H., Zacharias A., Schwann T.A., et al. Role of hemodilutional anemia and transfusion during cardiopulmonary bypass in renal injury after coronary revascularization: Implications on operative outcome. Crit Care Med. 2005;33:1749-1756.
181 Ranucci M., Biagioli B., Scolletta S., et al. Lowest hematocrit on cardiopulmonary bypass impairs the outcome in coronary surgery: An Italian Multicenter Study from the National Cardioanesthesia Database. Tex Heart Inst J. 2006;33:300-305.
182 Swaminathan M., Phillips-Bute B.G., Conlon P.J., et al. The association of lowest hematocrit during cardiopulmonary bypass with acute renal injury after coronary artery bypass surgery. Ann Thorac Surg. 2003;;76:784-791. discussion 792
183 Karkouti K., Beattie W.S., Wijeysundera D.N., et al. Hemodilution during cardiopulmonary bypass is an independent risk factor for acute renal failure in adult cardiac surgery. J Thorac Cardiovasc Surg. 2005;129:391-400.
184 Jonas R.A., Wypij D., Roth S.J., et al. The influence of hemodilution on outcome after hypothermic cardiopulmonary bypass: Results of a randomized trial in infants. J Thorac Cardiovasc Surg. 2003;126:1765-1774.
185 DeFoe G.R., Ross C.S., Olmstead E.M., et al. Lowest hematocrit on bypass and adverse outcomes associated with coronary artery bypass grafting. Northern New England Cardiovascular Disease Study Group. Ann Thorac Surg. 2001;71:769-776.
186 Huybregts R.A., de Vroege R., Jansen E.K., et al. The association of hemodilution and transfusion of red blood cells with biochemical markers of splanchnic and renal injury during cardiopulmonary bypass. Anesth Analg. 2009;109:331-339.
187 von Heymann C., Sander M., Foer A., et al. The impact of an hematocrit of 20% during normothermic cardiopulmonary bypass for elective low risk coronary artery bypass graft surgery on oxygen delivery and clinical outcome—a randomized controlled study [ISRCTN35655335]. Crit Care. 2006;10:R58.
188 Berger K., Sander M., Spies C.D., et al. Profound haemodilution during normothermic cardiopulmonary bypass influences neither gastrointestinal permeability nor cytokine release in coronary artery bypass graft surgery. Br J Anaesth. 2009;103:511-517.
189 Orlov D., O’Farrell R., McCluskey S.A., et al. The clinical utility of an index of global oxygenation for guiding red blood cell transfusion in cardiac surgery. Transfusion. 2009;49:682-688.


{kind=link}
{kind=link}