206 Bleeding Disorders
 Key Points
Key Points• Bleeding disorders are common and can be classified as congenital or acquired.
• A thorough history, including medication history, can help screen for many potential bleeding disorders.
• The prothrombin time is a measurement of abnormalities in the tissue factor pathway, whereas the activated partial thromboplastic time is a measurement of abnormalities in the contact activation pathway.
• Hemophilia A and B are both X-linked conditions, and factor replacement may be needed as prophylaxis or treatment for bleeding. Recombinant factor VIIa can be used to treat patients with inhibitors.
• von Willebrand disease is the most common congenital bleeding disorder. Treatment options vary, depending on the severity of the condition and bleeding, but they include desmopressin, aminocaproic acid, tranexamic acid, and factor replacement.
• Common causes of acquired coagulopathies include trauma, massive transfusion, disseminated intravascular coagulation, drugs, and toxins.
• The definitive treatment of disseminated intravascular coagulation is reversal or treatment of the underlying cause.
Epidemiology
Under normal physiologic conditions, a constant balance exists between clot formation and clot breakdown, with a preference favoring anticoagulation.1 Hemostasis results from a complex interaction among the vascular endothelium, platelets, the coagulation cascade, and the fibrinolytic system. Any disorder that affects this interplay can alter the equilibrium, thereby producing either excessive thrombosis or excessive hemorrhage.
Broadly speaking, bleeding disorders can be either inherited or acquired. Of the inherited disorders, the von Willebrand syndromes and hemophilia A are the most common, with a prevalence between 1 per 100 individuals and 1 per 100,000 individuals. Less common inherited disorders include deficiencies of factors II, V, VII, X, XIII, and fibrinogen, with a prevalence between 1 and 2 per 100,000 individuals.2 The most common acquired bleeding disorders are drug induced, from therapeutic administration of agents expressly intended to decrease the risk of thrombosis. Oral anticoagulants are taken by up to 2% of the population and up to 8% of persons more than 65 years old, and the use of antiplatelet agents is vastly more common than that.3 Patients with inherited bleeding disorders will likely present to the emergency department (ED) for treatment of abnormal bleeding at some point during their lifetime,4 and ED visits for bleeding in patients who are taking antithrombotic agents are not uncommon.5 The clinician must be familiar with these disease states to provide prompt care because treatment delays can increase morbidity and mortality.4,6
Physiology and Biology of Hemostasis
Coagulation Cascade
The coagulation cascade (Fig. 206.1) is typically thought of as two pathways working separately. In reality, however, the tissue factor pathway (formerly called the extrinsic system) and the contact activation pathway (formerly called the intrinsic system) function intimately with each other to produce thrombin, a critical enzyme in the coagulation system.1,7
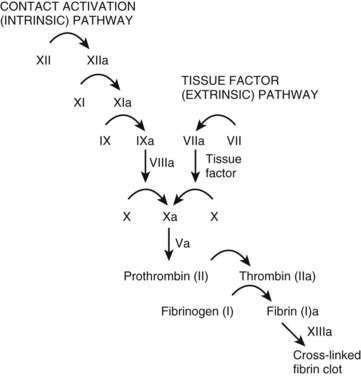
Tissue factor typically resides on smooth muscle cells, fibroblasts, and pericytes.8–10 Binding of tissue factor to factor VII results in an activated factor VII (factor VIIa). Factor VIIa subsequently catalyzes the conversion of factors IX and X to their active forms (factors IXa and Xa, respectively).
The contact activation pathway begins with the activation of factor XII by the high-molecular-weight enzymes kininogen and prekallikrein.1 This activation subsequently triggers a series of reactions leading to the conversion of X to Xa, the first step of the common pathway. The exact relevance of the contact activation pathway is debatable, however, because its activation is not needed in trauma-initiated coagulopathy.1 Furthermore, with the exception of a deficiency of factor XI, deficiencies in enzymes involved in the contact activation pathway generally do not produce substantial coagulopathies.1
Platelet Function
Following injury to the vascular endothelium, endothelin is released, resulting in vasoconstriction. In addition, injury to the vascular endothelium results in tissue factor exposure, with subsequent binding of platelets to the endothelium.8,10 Platelet adhesion primarily involves the binding of platelets to damaged endothelium by von Willebrand factor (vWF).11,12 Additional platelet activation occurs through the release of numerous mediators including adenosine diphosphate (ADP), epinephrine, thromboxane A2, and thrombin.11 Ultimately, platelet activation and aggregation require glycoprotein IIb/IIIa.11 Thrombus stability, however, requires fibrin, which is formed from the coagulation cascade on the surface of platelets.11
Differential Diagnosis and Medical Decision Making
The initial assessment of a patient with a known or suspected bleeding disorder begins with a thorough history and physical examination. Laboratory studies can be used to assess the severity of the disorder and to support a diagnosis. The initial laboratory studies that should be obtained when evaluating such patients include a complete blood count, prothrombin time (PT; a measure of the tissue factor pathway), and an activated partial thromboplastin time (aPTT; a measure of the contact activation pathway). A fibrinogen count can also assist in the initial management. These laboratory tests can help screen for clinically significant factor deficiencies, such as hemophilia A or B, as well as hypofibrinogenemic states. Additional tests may be indicated, depending on the condition of interest. For example, the concentration of D-dimers may be helpful in establishing the diagnosis of disseminated coagulation (DIC), whereas factor concentrations may be useful in assessing the severity of hemophilia. Patients with acute fulminant hepatic failure may demonstrate abnormalities similar to those seen in DIC. In such cases, factor V, VII, and VIII levels can be obtained. Although factor V levels are likely to be decreased in both DIC and liver failure, factor VIII levels are normal or elevated in liver failure–induced coagulopathy, yet markedly decreased in DIC-induced coagulopathy. Factor VII levels are also reduced in coagulopathy resulting from hepatic failure. Mixing studies can be performed to determine whether a factor deficiency or inhibitor is present in patients with prolonged aPTT.13 Examination of the peripheral blood film can be very helpful in patients with unexplained bleeding disorders.
Coagulation Cascade Defects
Platelet-related defects are discussed in Chapter 205.
Hereditary Defects
Hemophilia A and B
Hemophilia A and hemophilia B are X-linked recessive disorders involving factors VIII and IX, respectively. Clinically, hemophilia B is indistinguishable from hemophilia A. However, nearly half of all cases of hemophilia A, the most common form of hemophilia, represent de novo mutations, in which boys are born to parents without the disease.1 These disorders can be classified as mild, moderate, or severe, corresponding to a plasma coagulation factor concentration of 6% to 30%, 2% to 5%, or 1% or lower, respectively.14 Those patients with a mild form of the disease generally have bleeding only after trauma or surgery. In contrast, patients with the severe form of the disease have an average of 20 to 30 bleeding episodes annually, and bleeding occurs spontaneously or after minor trauma.14 Hemarthrosis is one of the most common forms of bleeding in severe forms of hemophilia.1 Historically, treatment involved transfusions of plasma concentrates of coagulation factors.14 However, such an approach was associated with the acquisition of numerous infectious diseases, including human immunodeficiency virus infection or hepatitis, as well as the development of inhibitors to the clotting factors.14,15 Now, both plasma and recombinant-derived factor VIII and IX exist. Treatment can be administered as an on-demand regimen, in which therapy is given based on bleeding, or in a prophylactic regimen. Because the plasma half-life of factor IX is longer than of factor VIII (18 to 24 hours versus 8 to 12 hours), dosing of factor replacement is less frequent for hemophilia B than for hemophilia A.16 Table 206.1 shows a common treatment regimen. Minor bleeding requires factor replacement to achieve factor concentrations of 30% of normal (typically 30 units/kg). In contrast, major hemorrhagic events, including hemarthrosis and large muscle bleeding, requires factor replacement to achieve factor concentrations of at least 50% (typically 50 units/kg). Life-threatening hemorrhage requires factor replacement to achieve factor concentrations of at least 80% (typically 80 units/kg).16 In general, each unit per kilogram of body weight of factor VIII increases plasma factor VIII concentrations by 2%, whereas each unit per kilogram of body weight of factor IX increases plasma factor IX concentrations by 1%.16 In patients with life-threatening bleeding, however, in vivo factor concentrations should be followed to ensure adequate replacement. When patients with hemophilia A have developed inhibitors to factor VIII, recombinant factor VIIa (rFVIIa) (90 mcg/kg) has been used.4
| TREATMENT OPTIONS | NOTES | |
|---|---|---|
| Disease-Induced Coagulopathy | ||
| Factor V deficiency | FFP | |
| Factor VIII deficiency | Recombinant factor VIIIa | 1 U/kg IV increases plasma factor VIII concentrations by 2%. A typical dose will be 40 U/kg for severe bleeding. |
| Recombinant factor VIIa can be used for those with inhibitors to factor VIII | 90 mcg/kg IV | |
| Factor IX deficiency | Recombinant factor IXa | 1 U/kg IV increases plasma factor IX concentrations by 1%. A typical dose will be 80 U/kg for severe bleeding. |
| Factor X deficiency | FFP or PCC | |
| Factor XI deficiency | FFP | |
| von Willebrand disease | DDAVP Aminocaproic acid Tranexamic acid |
0.3 mcg/kg IV 50-60 mg/kg IV q4-6h 10-15 mg/kg IV q8-12h |
| Congenital fibrinogen disorders | FFP Cryoprecipitate Fibrinogen concentrates† |
|
| Coagulopathy of trauma | FFP Platelets |
|
| Disseminated intravascular coagulation | Cryoprecipitate Platelets |
Primary treatment is to treat the underlying cause |
| Transfusion-related coagulopathy | FFP Platelets |
|
| Drug/toxin–Induced Coagulopathy | ||
| Heparin | Protamine | 1 Unit reverses 100 U UFH |
| Factor Xa inhibitors | rFVIIa FFP |
90 mcg/kg IV |
| Warfarin | Vitamin K, FFP, PCC | |
| Crotalid envenomation | Crotalidae polyvalent immune Fav (ovine); CroFab | |
DDAVP, Desmopressin; FFP, fresh frozen plasma; PCC, pool complex concentrates.
* Many etiologies of coagulopathy do not require factor replacement or transfusion. If these products are to be administered, the specific agent is listed in the table. However, for many conditions, no blood product should be transfused. The text can help guide when specific therapies should be administered.
von Willebrand Disease
von Willebrand disease (VWD) is the most common hereditary bleeding disorder. Although laboratory evidence of this disorder is present in up to 2% of the population, clinically relevant bleeding occurs much less commonly.17 vWF has two roles in maintaining hemostasis. First, vWF forms a link between platelets and the vascular endothelium of injured blood vessels. Second, vWF serves as a plasma carrier for factor VIII.18 The primary defect in VWD is a quantitative deficiency (types I, III) or a qualitative defect (type II) in vWF.1,17 However, because vWF serves as a plasma carrier and stabilizer for factor VIII, VWD is also associated with a secondary decrease in factor VIII.17
Mucocutaneous bleeding is the most common clinical manifestation of VWD. Epistaxis, hematomas, and menorrhagia are relatively common. Patients with type III disease, the most severe form, develop spontaneous hemarthrosis resembling that seen in hemophilia.18 The diagnosis can be confirmed by obtaining a vWF antigen, vWF ristocetin cofactor assay, and factor VIII activity. Minor bleeding, including mucosal bleeding, usually does not warrant specific treatment. For more severe bleeding, however, antifibrinolytic agents and desmopressin should be administered (see Table 206.1) Desmopressin produces a transient increase in factor VIII and vWF three to five times higher than baseline within 1 hour of intravenous administration, and it should be administered at a dose of 0.3 mcg/kg intravenously. Antifibrinolytic agents include aminocaproic acid (50 to 60 mg/kg every 4 to 6 hours) and tranexamic acid (10 to 15 mg/kg every 8 to 12 hours).17 For those patients with life-threatening bleeding, factor VIII and vWF concentrates may be needed.4
Miscellaneous Factor Deficiencies
Numerous other congenital coagulation factor deficiencies are recognized. Defects in factor V can result in either hemorrhagic or thrombotic complications.19 Factor V deficiencies can be classified as either type I (low or unmeasurable antigen level) or type II (normal or mildly low antigen level). Parahemophilia, a severe form of the type I variety, is generally associated with mild bleeding.19,20 No factor V–specific concentrate is available, so bleeding, if it occurs, should be treated with fresh frozen plasma (FFP) (see Table 206.1).20
Factor X, a vitamin K–dependent clotting factor, is the first enzyme in the common pathway in the coagulation cascade. Factor X deficiency is one of the rarest inherited coagulation disorders.21 Patients with severe deficiency usually develop hemorrhage early in life, including umbilical stump bleeding. Patients with less severe deficiency, in contrast, typically develop bleeding only following trauma or menorrhagia, for example.21,22 In addition to congenital deficiencies, several states are associated with acquired factor X deficiency, including liver disease, vitamin K deficiency, myeloma, and various malignant diseases.21 In addition, the AL, but not the AA, form of amyloidosis has been associated with acquired factor X deficiency.21 Sodium valproate use is associated with a transient form of factor X deficiency.23 If bleeding occurs, the patient should receive FFP or prothrombin complex concentrates (PCC) (see Table 206.1).
A deficiency of either factor XI or factor XII (Hageman trait) is rarely associated with clinically relevant bleeding.24–26 Severe forms of factor XI deficiency can be clinically relevant, however, and bleeding is best treated with the administration of FFP (see Table 206.1). Factor XI is more common among individuals of Ashkenazi heritage.
Congenital Fibrinogen Disorders
Congenital fibrinogen disorders include hypofibrinogenemia and afibrinogenemia. Patients frequently present at birth with umbilical stump bleeding.27 In older individuals, life-threatening bleeding is rare, but it can include gastrointestinal hemorrhages and central nervous system bleeding.27 In contrast to patients with various factor deficiencies, patients with fibrinogen defects typically do not have significant spontaneous bleeding, and when bleeding does occur, it is typically less severe and primarily associated with trauma or surgery.2 Fibrinogen deficiency can be treated with FFP, cryoprecipitate, or (where licensed) fibrinogen concentrates (see Table 206.1).
Acquired Defects
Drugs
Numerous pharmaceutical agents can produce coagulopathy by interfering with the coagulation cascade at various levels. Furthermore, salicylates, ADP antagonists (e.g., ticlopidine, clopidogrel, prasugrel, and ticagrelor), and glycoprotein IIb/IIIa inhibitors (abciximab, eptifibatide, and tirofiban) can result in hemorrhage, but they induce platelet dysfunction, rather than disorders in fibrin formation. In addition, innumerable drugs can cause thrombocytopenia with prolonged use. Drugs that alter platelet function are discussed elsewhere (see Chapter 205).
Warfarin is a vitamin K antagonist, which inhibits the cyclic conversion of vitamin K 2,3-epoxide to vitamin K quinone, as well as the conversion of vitamin K quinone to the active form of vitamin K, vitamin K quinol. The inhibition of these enzymes prevents the γ-carboxylation of the vitamin K–dependent clotting factors, namely, factors II, VII, IX, and X, along with protein C and protein S.28 Patients taking warfarin frequently present to the ED either because of a supratherapeutic international normalized ratio (INR), which was checked during an outpatient visit, or because of hemorrhage. The treatment of an elevated INR depends on the clinical circumstances.29,30 For example, if a patient is asymptomatic but has an incidental INR of 4, simply withholding a dose of warfarin may be appropriate, whereas a patient with an intracranial hemorrhage with an INR of 2 requires aggressive reversal. Table 206.2 outlines the recommended guidelines for warfarin reversal. In general, if vitamin K is to be administered, oral and intravenous routes are preferred because subcutaneous and intramuscular administration leads to poor and erratic absorption.31
| INR | CHEST GUIDELINES | AUSTRALASIAN SOCIETY OF THROMBOSIS AND HAEMOSTASIS |
|---|---|---|
| 2-5; no bleeding | Lower or omit dose | Lower or omit dose |
| 5-9; no bleeding | Hold 1-2 doses or 1-2.5 mg PO vitamin K* |
Hold warfarin; if bleeding risk high, 1-2 mg PO or 0.5-1 mg IV vitamin K |
| ≥9; no bleeding | Hold warfarin; give 5 mg PO vitamin K | Hold warfarin; if bleeding risk low, give 2.5-5 mg PO vitamin K or 1 mg IV vitamin K If high risk, give 1 mg IV vitamin K, consider PCC, FFP |
| Serious bleeding | Hold warfarin; give 10 mg IV vitamin K, supplemented by FFP, PCC, or rVIIa | Hold warfarin; give 5-10 mg IV vitamin K, PCC, and FFP |
| Life-threatening bleeding | Hold warfarin; give FFP, PCC, or rVIIa, supplemented by 10 mg IV vitamin K |
FFP, Fresh frozen plasma; INR, international normalized ratio; IV, intravenous; PCC, prothrombin complex concentrates; PO, oral; rVIIa, recombinant factor VIIa.
Date from Ansell J, Hirsh J, Hylek E, et al. Pharmacology and management of the vitamin K antagonists: American College of Chest Physicians evidence-based clinical practice guidelines. Chest 2008;133:160S–98S; and Baker RL, Coughlin PB, Gallus AS, et al. Warfarin-reversal: consensus guidelines, on behalf of the Australasian Society of Thrombosis and Haemostasis. Med J Aust 2004;181:492–7.)
Heparin products can be classified as either unfractionated heparin (UFH) or low-molecular-weight heparin (LMWH). Commonly used LMWHs include dalteparin, enoxaparin, tinzaparin, and ardeparin. Traditionally, UFH was the primary xenobiotic used for both prophylaxis and treatment of venothromboembolic disease.32 However, because of increased ease of administration, a lack of need for laboratory monitoring, and a more predictable dose-response curve, the use of LMWH has substantially increased.32 UFH produces anticoagulation by binding to antithrombin III, thereby producing a conformational change. The modified antithrombin III–heparin complex is a potent inhibitor of factors IIa and Xa. UFH also has some activity against factors IXa, XIIa, and kallikrein.33,34 The LMWHs, in contrast, also bind to antithrombin III, but they have greater effect on factor Xa. Comparing the LMWHs with UFH, the LMWHs have a higher ratio of antifactor Xa to IIa than does UFH.32,34
The anticoagulant effects of heparin are measured by following the aPTT. Monitoring is typically not needed for the LMWHs. However, if monitoring is desired, an antifactor Xa level should be obtained 4 hours after subcutaneous injection.35 Heparin reversal is accomplished by the intravenous administration of protamine sulfate (see Table 206.1). Each unit of protamine sulfate reverses 100 units of UFH. Because protamine sulfate primarily reverses factor IIa, and not Xa, its use in patients with LMWH produces only a partial reversal.36
Factor Xa inhibitors can inhibit factor Xa either indirectly (idraparinux or fondaparinux) or directly (rivaroxaban). Unlike the LMWH agents, which inhibit both factors IIa and Xa, the factor Xa inhibitors produce no inhibition of factor IIa. Rivaroxaban, which is available orally, is considered a direct factor Xa inhibitor because it binds directly to factor X, unlike idraparinux and fondaparinux, which bind to antithrombin III.32 In the event of life-threatening hemorrhage in patients taking a factor Xa inhibitor, no consensus on therapy exists, although some investigators have recommended rFVIIa for this purpose.37 Outcome data are insufficient to recommend the routine administration of rFVIIa for the reversal of factor Xa inhibitors; the use of rFVIIa for this purpose must be weighed against the potential for thromboembolic events.
Coagulopathy of Trauma
Coagulopathy is common among trauma patients. Nearly 25% of critically ill trauma patients are found to have some laboratory or clinical evidence of coagulopathy on admission. This coagulopathy is related to injury severity but not to fluid resuscitation.38 Coagulopathy, along with hypothermia and acidosis, is part of the “triad of death” because its development produces synergistic effects that can ultimately culminate in mortality.39
The mechanism by which this process occurs is unclear. First, local tissue damage after trauma is thought to result in exposure to tissue factor from the damaged endothelium, thereby activating the coagulation cascade. In addition, traumatic brain injury results in the release of brain-specific thromboplastins, leading to consumption of clotting factors.40 Finally, severely injured trauma patients can present in shock. This leads to diffuse endothelial disruption and early production of factors II, V, VIII, and X. Furthermore, shock results in decreased clearance of thrombin, thereby leading to activation of protein C with resultant inactivation of factors Va, VIIIa, and plasminogen activator inhibitor-1.40,41 Acidemia alters coagulation protease function, increases fibrinogen degradation, and further contributing to coagulopathy. Hypothermia, which is common in trauma patients, results in coagulopathy secondary to decreased activity of factor VIIa, inhibition of coagulation protease activity, and impaired platelet function.39,40
Treatment of patients with trauma-induced coagulopathy begins with basic resuscitation, including the removal of wet clothes, as well as the application of warmed blankets, warmed fluids, and maintenance of high ambient heat. Coagulopathy can be treated with FFP (see Table 206.1). In the setting of acute blood loss, platelet transfusions can be given to maintain a platelet count of at least 50 × 109/L.42 rFVIIa has been used on an off-label basis in numerous studies as a treatment of trauma-induced coagulopathy. Although the use of rFVIIa may be associated with reduced transfusion of blood products, no effect on outcome has yet been demonstrated.41 Its use must be weighed against its associated cost, lack of clear proven benefit, and potential for thromboembolic complications.
Transfusion-Related Coagulopathy
The administration of large volumes of fluids to actively bleeding patients can lead to a dilution of clotting factors and platelets that contributes to coagulopathy.43 Furthermore, the aggressive use of colloids (e.g., hydroxyethyl starch) is associated with migration of plasma proteins to the interstitial space, with a resulting reduction of plasma concentrations of factors VII and vWF, as well as impaired platelet function.43 The liberal use of crystalloid fluids, especially lactated Ringer solution with racemic lactate, is associated with a significant increase in immune mediators.44
The term massive transfusion commonly refers to the replacement of one’s blood volume within 24 hours, or the replacement of 50% of the total blood volume within 3 hours.45 The transfusion of large volumes of packed red blood cells (PRBCs), as occurs during a massive transfusion, is associated with numerous complications, including coagulopathy.46 In the military setting, the use of massive transfusion protocols has been associated with improved survival. In the civilian trauma setting, however, mortality benefit from such protocols is conflicting. An example of such a protocol includes a set transfusion ratio of FFP to PRBCs of 1 : 2 or 1 : 1.41,47,48 Many protocols also include platelets, which are transfused at a ratio of FFP to PRBCs to platelets of 1 : 1 : 1.
Toxin
Pit vipers include rattlesnakes, copperheads, and cottonmouths. Pit viper envenomations, especially those from rattlesnakes, can lead to local pain and edema, myotoxicity, and hematotoxicity. Initial evaluation of these patients should include a complete blood count, fibrinogen, and prothrombin time. Thrombocytopenia, hypofibrinogenemia, and coagulopathy can develop quickly after an envenomation. Because the venom does not result in thrombin production, fibrin is not cross-linked. This lack of thrombin production and fibrin cross-linkage separates crotalid-induced coagulopathy from true DIC.49 Afibrinogenemia, which can occur following envenomation, is not a result of a consumptive coagulopathy, but rather of primary fibrinogenolysis.50 Furthermore, despite profoundly abnormal laboratory findings, because the patient is able to produce endogenous thrombin, hemostasis remains intact, and gross hemorrhage remains rare.49
Treatment of pit viper–induced coagulopathy and thrombocytopenia is with antivenom. Crotalidae polyvalent immune Fab (ovine) (CroFab) is the only available antivenom currently approved for the management of pit viper envenomation in the United States. A full review of pit viper envenomations is beyond the scope of this chapter, but in general, stable patients should receive an initial dose of four to six vials intravenously over 1 hour.51 In the absence of life-threatening hemorrhage, blood products are not indicated, despite the presence of critically abnormal platelets, PT, or fibrinogen.52
Following therapy with CroFab for rattlesnake envenomation, late hematotoxicity can occur. Patients can develop recurrence, in which severe thrombocytopenia or coagulopathy can occur days after therapy, even if these laboratory abnormalities were not initially present. Thus, these patients should have a fibrinogen, PT, and complete blood count assessed 2 to 4 days and again 5 to 7 days after envenomation.51,53
Disseminated Intravascular Coagulation
In the setting of severe systemic illness or injury, the body’s coagulation processes can become activated, thereby resulting in widespread, excessive microvascular thrombosis. This thrombosis can result in impaired end-organ perfusion that contributes to multisystem organ failure. Because of this extreme, disproportionate activation of the coagulation system, consumptive coagulopathy can occur, in which platelets, clotting factors, and protease inhibitors are consumed. Intravascular fibrin strand formation results in the shearing of red blood cells, with subsequent development of microangiopathic hemolytic anemia.54 Thus, this condition, DIC, results in excessive clot formation, bleeding, or both simultaneously.55 DIC does not occur as a primary process but is always secondary to some underlying disorder, including sepsis, trauma, malignant disease (particularly metastatic adenocarcinoma, acute promyelocytic leukemia), tumor lysis syndrome, and obstetric conditions (e.g., abruptio placentae, placenta previa, amniotic fluid embolism, HELLP [hemolysis, elevated liver enzymes, low platelets] syndrome, uterine atony), among others.54,56
DIC can be considered either acute or chronic.54 The diagnosis of DIC can be confirmed by the finding of several laboratory abnormalities, including the presence of thrombocytopenia, prolongation of the PT and aPTT, hypofibrinogenemia, elevated D-dimer, and the presence of schistocytes on peripheral smear. Not all these laboratory abnormalities need to be present to make the diagnosis of DIC, and no single laboratory test can confirm or exclude the diagnosis.54,56 The primary treatment of DIC is to treat the underlying inciting cause.54,55 Platelet transfusion is indicated only in those patients with active bleeding or in those who are at risk for significant bleeding. Such patients include those with platelet counts lower than 50 × 109/L who need an invasive procedure or those with platelet counts lower than 10 to 20 × 109/L.55 Cryoprecipitate can be used to correct severe hypofibrinogenemia (<50 mg/dL) in patients with active bleeding.54 No role exists for routine transfusion of cryoprecipitate in the absence of bleeding.
The use of low-dose heparin in patients with DIC has been suggested in an effort to reduce thrombin generation. This practice is controversial, however, because convincing data supporting its use are lacking.54,55
Summary of Agents Used to Treat Bleeding Disorders
Table 206.1 shows many of the agents used to correct various coagulopathies. FFP contains most coagulation factors in varying concentrations; some factor shave mildly to moderately decreased concentrations following thawing. Blood type (ABO) compatibility is required, but Rh factor is not. Each unit of plasma is approximately 200 to 250 mL of fluid. In contrast to FFP, which contains all clotting factors, cryoprecipitate contains fibrinogen, fibronectin, VWF, and factors VIII and XIII. It is the only widely form of concentrated fibrinogen currently available. Six bags of cryoprecipitate will raise the fibrinogen approximately 45 mg/dL. Finally, prothrombin complex concentrate (PCC) is a generic term for several different plasma-derived coagulation products that were designed as concentrates of factor IX and that contain varying amounts of factors II, VII, IX, X, and proteins C and S. Different formulations are available in different countries.
1 Dahlback B. Blood coagulation. Lancet. 2000;355:1627–1632.
2 Acharya SS, Coughlin A, Dimichele DM, et al. Rare bleeding disorder registry: deficiencies of factors II, V, VII, X, XIII, fibrinogen and dysfibrinogenemias. J Thromb Haemost. 2004;2:248–256.
3 Kaufman DW, Kelly JP, Rosenberg L, et al. Recent patterns of medication use in the ambulatory adult population of the United States. JAMA. 2002;287:337–344.
4 Singleton T, Kruse-Jarres R, Leissinger C. Emergency department care for patients with hemophilia and von Willebrand disease. J Emerg Med. 2010;39:158–165.
5 Shehab N, Sperling LS, Kegler SR, et al. National estimates of emergency department visits for hemorrhage-related adverse events from clopidogrel plus aspirin and from warfarin. Arch Intern Med. 2010;170:1926–1933.
6 Sharieff GQ, Trocinski DR, Thompson K. Pediatric patients with bleeding dyscrasias: what is the cause of delays in initiating replacement therapy? J Pediatr Health Care. 2001;15:10–13.
7 Wheeler AP, Rice TW. Coagulopathy in critically ill patients. Part 2: soluble clotting factors and hemostatic testing. Chest. 2010;137:185–194.
8 Mackman N. The role of tissue factor and factor VIIa in hemostasis. Anesth Analg. 2009;108:1147–1152.
9 Mackman N, Tilley RE, Key NS. Role of the extrinsic pathway and blood coagulation in hemostasis and thrombosis. Arterioscler Thromb Vasc Biol. 2007;27:1687–1693.
10 Vine AK. Recent advances in haemostasis and thrombosis. Retina. 2009;29:1–7.
11 Davi G, Patrono C. Platelet activation and thrombosis. N Engl J Med. 2007;357:2482–2494.
12 Ruggeri Z, Mendolicchio GL. Adhesion mechanisms in platelet function. Circ Res. 2007;100:1673–1685.
13 Ballas M, Kraut EH. Bleeding and bruising: a diagnostic work-up. Am Fam Physician. 2008;77:1117–1124.
14 Mannucci PM, Tuddenham EGD. The hemophilias: from royal genes to gene therapy. N Engl J Med. 2001;344:1773–1779.
15 Van den Berg HM. Issues surrounding therapeutic choices for hemophilia patients. Haematologica. 2004;89:645–650.
16 Josephson CD, Abshire TC. Clinical uses of plasma and plasma fractions: plasma-derived products for hemophiliac A and B, and for von Willebrand disease. Best Pract Res Clin Haematol. 2006;19:35–49.
17 Mannucci PM. Treatment of von Willebrand’s disease. N Engl J Med. 2004;351:683–694.
18 Lee JW. von Willebrand disease, hemophilia A and B, and other factor deficiencies. Int Anesthesiol Clin. 2004;42:59–76.
19 Duga S, Asselta R, Tenchini ML. Coagulation factor V. Int J Biochem Cell Biol. 2004;36:1393–1399.
20 Huang JM, Koerper MA. Factor V deficiency: a concise review. Haemophilia. 2008;14:1464–1469.
21 Uprichard J, Perry DJ. Factor X deficiency. Blood Rev. 2002;16:97–110.
22 Brown DL, Kouides PA. Diagnosis and treatment of inherited factor X deficiency. Haemophilia. 2008;14:1176–1182.
23 Gallais V, Bredoux H, le Roux G, et al. Acquired and transient factor X deficiency associated with sodium valproate treatment. Eur J Haematol. 1996;57:330.
24 Schmaier AH. The elusive physiologic role of factor XII. J Clin Invest. 2008;118:3006–3009.
25 Stavrou E, Schmaier AH. Factor XII: what does it contribute to our understanding of the physiology and pathophysiology of hemostasis and thrombosis. Thromb Res. 2010;125:210–215.
26 Emsley J, McEwan PA, Gailani D. Structure and function of factor XI. Blood. 2010;115:2569–2577.
27 Asselta R, Spena S, Duga S, et al. Molecular genetics of quantitative fibrinogen disorders. Cardiovasc Hematol Agents Med Chem. 2007;5:163–173.
28 Hirsh J, Dalen JE, Anderson DR, et al. Oral anticoagulants: mechanism of action, clinical effectiveness, and optimal therapeutic range. Chest. 2001;119(suppl):8S–21S.
29 Ansell J, Hirsh J, Hylek E, et al. Pharmacology and management of the vitamin K antagonists: American College of Chest Physicians evidence-based clinical practice guidelines. Chest. 2008;133:160S–198S.
30 Baker RL, Coughlin PB, Gallus AS, et al. Warfarin-reversal: consensus guidelines, on behalf of the Australasian Society of Thrombosis and Haemostasis. Med J Aust. 2004;181:492–497.
31 De Zee KJ, Shimeall WT, Douglas KM. Treatment of excessive anticoagulation with phytonadione (vitamin K): a meta-analysis. Arch Intern Med. 2006;166:391–397.
32 Gresham C, Levine M, Ruha AM. Case files of the medical toxicology fellowship at Banner Good Samaritan Medical Center in Phoenix, AZ: a non-warfarin anticoagulant overdose. J Med Toxicol. 2009;5:242–249.
33 Krishnaswamy A, Lincoff AM, Cannon CP. The use and limitations of unfractionated heparin. Crit Pathw Cardiol. 2010;9:35–40.
34 Hirsh J, Warkentin TE, Shaughnessy SG, et al. Heparin and low-molecular-weight heparin: mechanisms of action, pharmacokinetics, dosing, monitoring, efficacy, and safety. Chest. 2001;119(suppl):64S–94S.
35 Laposta M, Green D, Van Cott EM, et al. College of American Pathologists Conference XXXI on laboratory monitoring of anticoagulant therapy: the clinical use and laboratory monitoring of low-molecular-weight heparin, danaparoid, hirudin and related compounds, and argatroban. Arch Pathol Lab Med. 1998;122:799–807.
36 Levi MM, Eerenberg E, Lowenberg E, et al. Bleeding in patients using new anticoagulants or antiplatelet agents: risk factors and management. Neth J Med. 2010;68:68–72.
37 Bijsterveld NR, Moons AH, Boekholdt SM, et al. Ability of recombinant factor VIIa to reverse the anticoagulant effect of the pentasaccharide fondaparinux in healthy volunteers. Circulation. 2002;106:2550–2554.
38 Brohi K, Singh J, Heron M, et al. Acute traumatic coagulopathy. J Trauma. 2003;54:1127–1130.
39 Maani CV, DeSocio PA, Holcomb JB. Coagulopathy in trauma patients: what are the main influence factors? Curr Opin Anaesthesiol. 2009;22:255–260.
40 Hess JR, Brohi K, Dutton RP, et al. The coagulopathy of trauma: a review of mechanisms. J Trauma. 2008;65:748–754.
41 Lier H, Bottiger BW, Hinkelbein J, et al. Coagulation management in multiple trauma: a systematic review. Intensive Care Med. 2011;37:572–582.
42 Spahn DR, Cerny V, Coats TJ, et al. Management of bleeding following major trauma: a European guideline. Crit Care. 2007;11:R17.
43 Johansson PI, Osterowski SR, Scher NM. Management of major blood loss: an update. Acta Anaesthesiol Scand. 2010;54:1039–1049.
44 Alam HB, Rhee P. New developments in fluid resuscitation. Surg Clin North Am. 2007;87:55–72.
45 Hardy JF, deMoerloose P, Samama CM. The coagulopathy of massive transfusion. Vox Sang. 2005;89:123–127.
46 Sihler KC, Napolitano LM. Complications of massive transfusion. Chest. 2010;137:209–220.
47 Griffee MJ, DeLoughery TG, Thorborg PA. Coagulation management in massive bleeding. Curr Opin Anaesthesiol. 2010;23:263–268.
48 Greer SE, Rhynhart KK, Gupta R, et al. New developments in massive transfusion in trauma. Curr Opin Anaesthesiol. 2010;23:246–250.
49 Kitchens CS. Hemostatic aspects of envenomation by North American snakes. Hematol Oncol Clin North Am. 1992;6:1189–1195.
50 Budzynski AZ, Pandya BB, Rubin RN, et al. Fibrinogenolytic afibrinogenemia after envenomation by western diamondback rattlesnake (Crotalus atrox). Blood. 1984;63:1–14.
51 Lavonas EJ, Ruha AM, Banner W, et al. Unified treatment algorithm for the management of crotaline snakebite in the United States: results of an evidence-informed consensus workshop. BMC Emerg Med. 2011;11:2.
52 Levine M, Ruha AM, Graeme K, et al. Toxicology in the ICU. Part 3: natural toxins. Chest. 2011;140:1357–1370.
53 Ruha AM, Curry SC, Albrecht C, et al. Late hematologic toxicity following treatment of rattlesnake envenomation with Crotalidae polyvalent immune Fab antivenom. Toxicon. 2011;57:53–59.
54 Labelle CA, Kitchens GS. Disseminated intravascular coagulation: treat the cause, not the lab values. Cleve Clin J Med. 2005;72:377–378. 383–5, 390
55 Levi M. Disseminated intravascular coagulation. Crit Care Med. 2007;35:2191–2195.
56 Toh CH, Downey C. Back to the future: testing in disseminated intravascular coagulation. Blood Coagul Fibrinolysis. 2005;16:535–542.