Biotechnology
After reading this chapter, the student will be able to:
• Define biotechnology, recombinant DNA technology, and some goals of recombinant technology
• Describe the common steps and tools used in genetic engineering
• Illustrate the structure and function of restriction enzymes, and explain their source and naming; include the description of “blunt” and “sticky” ends, as well as the function of DNA ligase
• Describe gene libraries including the creation and screening of gene libraries, as well as cDNA libraries and synthetic DNA
• Explain the nature of plasmids and their usefulness in recombinant DNA technology
• Discuss the polymerase chain reaction and describe the main three steps of the procedure
• Describe the various types of vectors employed in biotechnology, their usefulness, and the criteria they all have in common
• Discuss human protein replacement, including recombinant insulin, human growth hormone, and factor VIII
• Discuss human disease therapies and vaccine development that have been developed through genetic engineering
• Describe transgenic plants and animals and discuss their usefulness as well as possible problems
• Removing certain traits in humans, animals, plants, and microorganisms that are undesirable for the specific organism
• Transferring genes from one organism to another in order to improve the quality or survival of the receiving organism
• Creating organisms that are capable of synthesizing products for human use
Tools of Genetic Engineering
• Isolation of the genes of interest
• Insertion of the genes into a vector
Because the genetic code is nearly universal, bacteria can express foreign DNA if it is attached to the bacterial DNA. This can be achieved through genetic engineering. Bacteria can also acquire fragments of DNA from the local environment and then express the proteins encoded by the genes in those fragments. This genetic recombination process is called transformation. Transformation reportedly takes place in less than 1% of the bacterial population, but it can bring about profound genetic changes. In this process several donor bacteria lyse, and their DNA bursts into fragments. If a recipient bacterium is present a segment of double-stranded DNA can pass through the wall and plasma membrane. An enzyme then dissolves one strand of the DNA, and the remaining strand incorporates itself into the recipient’s DNA (Figure 25.1). The transformation is complete when the foreign genes are expressed during protein synthesis. This process is one way by which bacteria can obtain drug resistance.
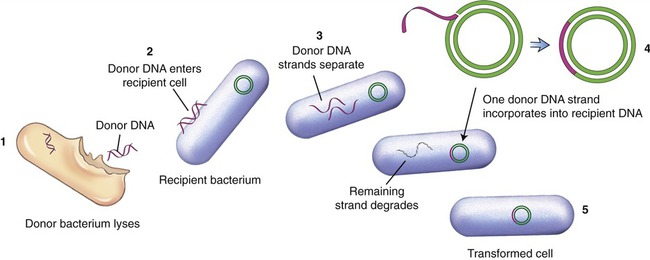
(1) The donor bacterium lyses and the DNA is released; (2) donor DNA enters the recipient bacterium; (3) the donor DNA strands separate; (4) one donor DNA strand incorporates into the recipient DNA, and the other strand degrades; (5) the result is a transformed cell containing recipient DNA and donor DNA.
Another process used by bacteria to obtain DNA fragments from other bacteria is conjugation (Figure 25.2). This genetic recombination process occurs between two living bacteria: a donor and a recipient. The donor and recipient form a cytoplasmic bridge by which single-stranded DNA of the donor crosses to the recipient. On rare occasions the DNA may be integrated into the recipient’s chromosome, but most often the DNA remains in the cytoplasm as a free-floating loop, called a plasmid (see Chapter 6, Bacteria and Archaea).
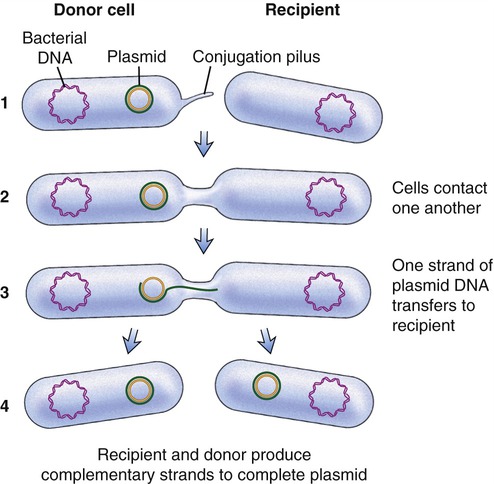
(1) A cytoplasmic bridge forms between the donor cell and the recipient; (2) the cells contact each other; (3) one strand of donor plasmid DA transfers to the recipient; and (4) a strand complementary to the plasmid is formed in both the recipient and donor cells.
A third form of recombination is transduction, which involves bacterial viruses called bacteriophages (see Chapter 7, Viruses). The virus enters a bacterial cell and integrates its DNA into the bacterial chromosome.
Viruses other than bacteriophages are also useful in genetic engineering experiments. Viruses (see Chapter 7) consist of nucleic acid fragments, either DNA or RNA, surrounded by a protein coat. They can replicate only in living cells, taking over the host cell machinery. Some viruses integrate their own DNA into the host cell’s DNA and become part of the cell’s genome. An example is herpesvirus, which integrates into a nerve cell genome and remains within that cell for years, causing recurrent herpes infections. DNA technologists realized that this was a way to bring genes into a cell of choice. The tools of genetic engineering include the following:
Restriction Enzymes
In the 1950s Salvador Luria and colleagues discovered that E. coli could resist destruction by bacteriophages; the organism could “restrict” the replication of the virus. Later, in 1962 Werner Arber and his research group identified the mechanism by which E. coli could achieve this restriction. They showed that an enzyme system was able to cleave the phage DNA before it could get to the bacterial cytoplasm. Subsequently they isolated the DNA-cleaving enzyme, which they called endonuclease, now known as a restriction enzyme. The enzyme is capable of cutting viral DNA but does not cut the host DNA because the organism modifies the bacterial DNA by adding a methyl group to it. Since the work of these two research groups other restriction enzymes have been discovered. More than 800 restriction enzymes have been identified so far, recognizing more than 100 different nucleotide sequences. Restriction enzymes are named after the bacterium from which they have been isolated. The name includes three letters from the bacterium, one from the strain, and roman numerals indicating the order in which enzymes from the same type of bacterium were identified (Box 25.1).
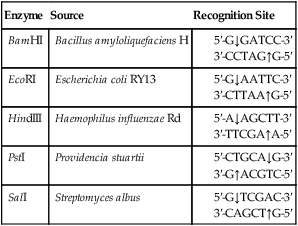
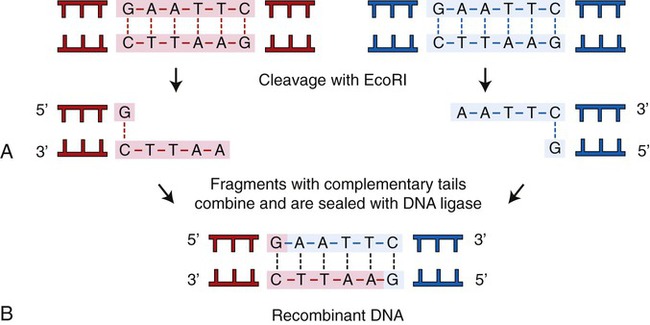
A restriction enzyme scans a DNA molecule, recognizing and cutting DNA only at a particular sequence of nucleotides. Some enzymes cut the DNA straight across both of the strands of the double helix and therefore produce “blunt” ends (Table 25.1). On the other hand, many restriction enzymes cut in an offset fashion and the ends of the cut have an overhanging piece of single-stranded DNA. These ends are called “sticky” ends (Table 25.2) because they are able to form base pairs with any DNA molecule that contains the complementary sticky end. Every cleavage performed with the same enzyme will occur at the same restriction site, regardless of the source of the DNA. When such molecules from different DNA sources are mixed together they will join with each other by base pairing between their sticky ends (Figure 25.3, A). This combination can be made permanent by DNA ligase, an enzyme that forms covalent bonds along the backbone of each strand. The result of this reaction is a molecule of recombinant DNA (rDNA) (Figure 25.3, B). The ability to produce rDNA molecules has revolutionized the study of genetics and formed the foundation for the biotechnology industry.
TABLE 25.1
Examples of Restriction Enzymes That Produce “blunt” Ends
| Enzyme | Source | Recognition Site |
| AluI | Arthrobacter luteus |
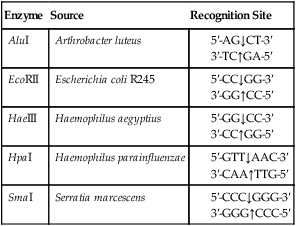
TABLE 25.2
Examples of Restriction Enzymes That Produce “Sticky” Ends
| Enzyme | Source | Recognition Site |
| BamHI | Bacillus amyloliquefaciens H | |
| EcoRI | Escherichia coli RY13 | |
| HindIII | Haemophilus influenzae Rd | |
| PstI | Providencia stuartii | |
| SalI | Streptomyces albus |
Gene Libraries
Creating a Gene Library
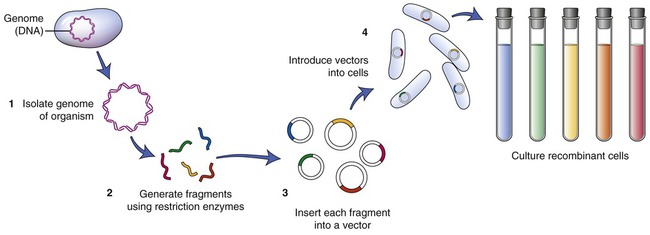
To establish a cell line containing the gene of interest, the particular DNA containing the gene must be isolated. A restriction enzyme is used to cut the cell’s DNA into fragments. After this process a host organism for the fragments needs to be chosen. An example of such an organism is Escherichia coli. The plasmids of E. coli are then isolated and the DNA fragments that were previously obtained are inserted into the plasmids (Figure 25.4). The recombined plasmids contain all the DNA fragments and are then inserted into E. coli cells, and together these E. coli cells now are a warehouse of all the DNA of the original cell of interest. The cell collection is the library, the individual cells represent the books of the library, and the individual DNA fragments contain the information in each of the books. This type of library is referred to as a genomic library. The next step is to find the particular gene of interest and therefore the library must be screened.
Screening the Gene Library
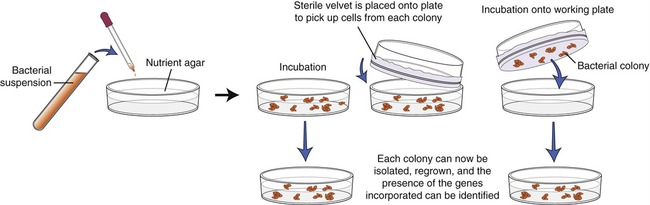
The screening process begins with growing the transformed E. coli cells on nutrient agar. Colonies derived from each individual cell will be visible within 24 to 48 hours, and each colony contains cells with the DNA insert. Now each colony needs to be examined for the presence of the insert that contains the gene of interest. To identify the specific colony a copy of the plate must be made for further work. This is done by a process called replica plating (Figure 25.5). This technique is performed by pressing a piece of sterile velvet onto the gel containing the colonies, followed by pressing the velvet to a new petri dish of nutrient agar to establish another plate, which then can be used for analysis of the target gene.
cDNA Libraries
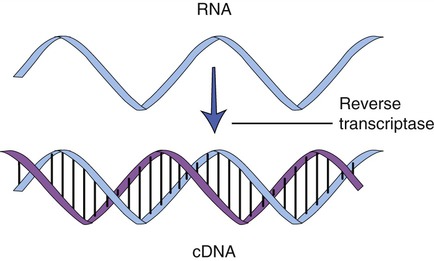
Cloning genes from a eukaryotic cell is more problematic than cloning genes from prokaryotes. As described in Chapter 3 (Cell Structure and Function), eukaryotic genes contain exons, the stretches of DNA that encode a protein, and introns, portions of DNA that do not encode a protein and are removed from the mRNA before leaving the nucleus. To clone eukaryotic genes it is best to use a version of the gene that lacks the introns, because once a gene is placed into a bacterial cell, the bacterium will be unable to remove these introns, and therefore will be unable to produce the intended protein. With the advances of genetic engineering it is now possible to create artificial genes that contain only exons. This is achieved with the aid of an enzyme called reverse transcriptase to synthesize complementary DNA (cDNA) from an mRNA template (Figure 25.6). This process is the reverse of the normal interaction between DNA and RNA, in that the isolated mRNA is used to produce a complementary DNA molecule. After the synthesis of the first strand of DNA, the mRNA is digested by reverse transcriptase, and the addition of DNA polymerase will produce the second, complementary strand of DNA, resulting in a double-stranded DNA molecule. These cDNA molecules can then be cloned to form a cDNA library.
Synthetic DNA
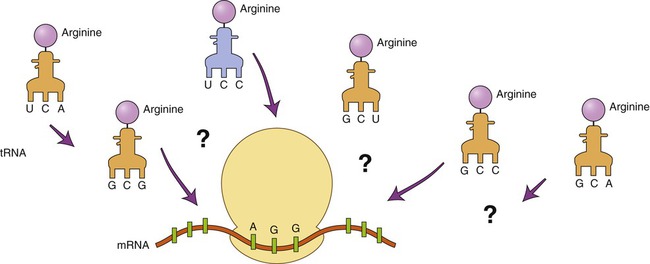
DNA technology has advanced to the point that DNA can be produced in the laboratory with instruments called DNA synthesis machines. The nucleotide sequence of the desired DNA is entered via keyboard, and a microprocessor controls the synthesis of the DNA from stored nucleotides and other vital reagents. Because only a relatively small number of nucleotides can be created in this way, several chains may have to be produced and then linked together to form a desired gene. To use this technique the nucleotide sequence of the gene must be known. If the gene has not been isolated previously the technique can still be used by predicting the DNA sequence if the amino acid sequence of the protein product of the gene is known. However, because the degeneracy of the genetic code prevents a definite outcome, the makeup of the gene cannot be 100% predicted. For example, how can it be determined which of the six codons for arginine is part of the gene (Figure 25.7)?
Plasmids
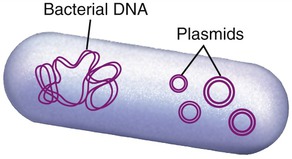
In the early 1970s, Stanley Cohen and his team at Stanford University studied small loops of DNA found in the cytoplasm of bacteria, which are now know as plasmids (Figure 25.8) (see Chapter 6, Bacteria and Archaea). At about the same time Herbert Boyer at the University of California San Francisco studied restriction enzymes found in E. coli. In 1973, Herbert Boyer and Stanley Cohen teamed up to produce the world’s first recombinant DNA organism, using gene splicing. They removed plasmids from E. coli and, using restriction enzymes, they cut the plasmid at precise positions. The segments could then be inserted into a different spliced plasmid and recombined using DNA ligase. The altered DNA (recombinant) could then be inserted into E. coli. With their studies with restriction enzymes and plasmids Boyer and Stanley set the foundation of modern biotechnology/genetic engineering (see Why You Need to Know at the beginning of this chapter).
Polymerase Chain Reaction
The polymerase chain reaction (PCR) is a technique, developed in 1986, that can produce DNA copies without a host cell. It is a technique by which small DNA fragments can quickly be amplified to quantities large enough to allow analysis. PCR is used for many applications such as in molecular biology, human genetics, evolution, development, conservation, and forensics. Before starting the PCR process with just one piece of DNA, generally the size of a gene, information concerning the nucleotide sequence of the target DNA is required. This information is necessary to be able to produce two nucleotide primers: one complementary to the 5′ end and one complementary to the 3′ end. PCR can be used to make billions of DNA copies in only a few hours and the amount of amplified DNA produced is limited only to the number of times the steps in the PCR process are repeated. These steps are as follows (Figure 25.9):
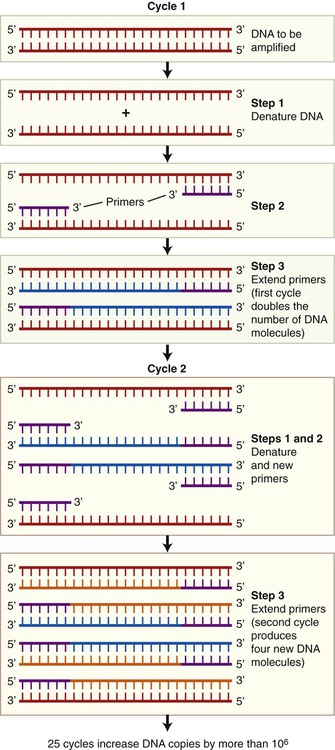
• Denaturation: The double-stranded target DNA to be cloned is denatured into two single strands. This process takes place when heating the double-stranded DNA from 90° C to 95° C for about 1 minute.
• Priming: Subsequently the temperature of the reactions is lowered to about 50° C to 70° C, which causes the primers to bind to the single-stranded DNA molecules. These primers, synthetic oligonucleotides, are complementary to the sequences flanking the DNA. They serve as the starting points for the synthesis of new DNA strands, complementary to the original strands.
• Extension: A heat-stable DNA polymerase is added and DNA synthesis takes place at temperatures between 70° C and 75° C.
Vectors in Biotechnology
• Vectors need to be small so that they can be manipulated in the laboratory. A large DNA molecule, such as an entire chromosome, is usually too frail to serve as a vector.
• Vectors need to survive in the target cell.
• Vectors need to contain a recognizable genetic marker so that the biotechnologist can identify the cells that have received the vector and the associated gene of interest.
• Vectors need to provide the required promoters to ensure genetic expression of the target gene in the cell.
Plasmid Vectors
The first vectors developed and still being used were genetically modified plasmids, derived from naturally occurring plasmids. Many plasmids have been genetically modified to serve as vectors and are now commercially available. As previously stated, once a plasmid enters a bacterial cell it will multiply and therefore enhance the number of DNA clones within that cell. In addition, these vectors have been genetically engineered and contain a number of different restriction enzyme recognition sequences, as well as marker genes that can express their presence in the host cell. In general, plasmids that are used in DNA technology should be as small as possible because they are less easily damaged than large plasmids. Furthermore, small plasmids are more easily taken up by the host cell. The ease of uptake of a plasmid into the host cell also requires that the inserted cDNA cannot be unreasonably large. A simplified version of the insertion of DNA into a plasmid and subsequent cloning is illustrated in Figure 25.10.
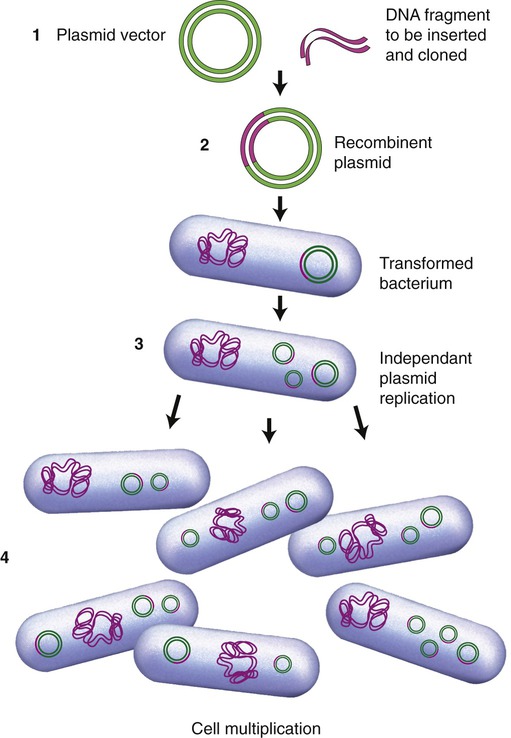
(1) A plasmid vector is cut by a specific restriction enzyme and the DNA fragment of interest is inserted to form a recombinant plasmid; (2) the recombinant plasmid is inserted into a bacterial cell (e.g., E. coli) and the bacterium is transformed; (3) within the bacteria the recombinant plasmids multiply independent of cell division; and (4) as the bacterial cells divide the plasmids continue to multiply and produce the desired gene product.
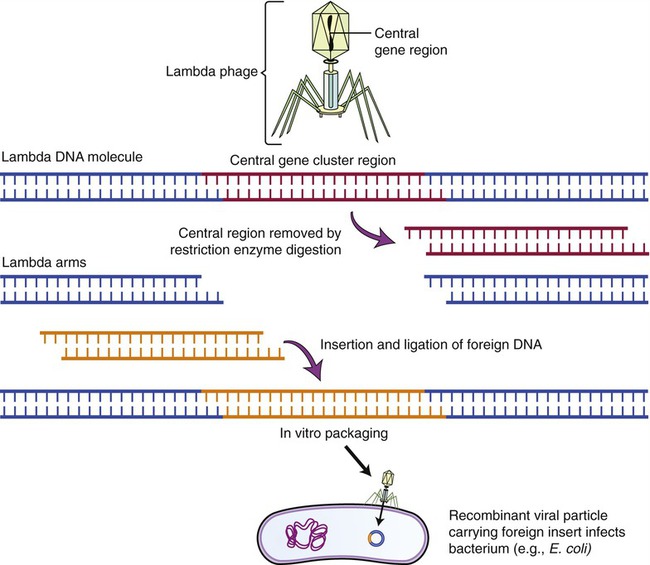
Lambda (λ) Phage Vectors
The lambda phage is a bacteriophage (see Chapter 7, Viruses) that infects E. coli. The genome of the phage has been completely mapped and sequenced, and experiments with this phage have shown that the central part of its chromosome can be replaced with foreign DNA without affecting the phage and its ability to infect E. coli. This makes this phage an ideal vector for larger DNA segments. To clone the phage and use it as a vector, the phage DNA needs to be isolated and cut with a restriction enzyme, to produce three chromosomal fragments, with the middle one being replaced by the DNA of another source. Once the foreign DNA has been inserted, DNA ligase is used to produce the recombinant λ vector that is then enclosed into the phage protein head (Figure 25.11). The recombinant phage will then be introduced to a culture of E. coli, and once in the bacteria the vectors replicate, forming many phages with the inserted DNA. As the phages reproduce, they lyse their bacterial host cells, and the cloned DNA can be recovered in multiple copies.
Biotechnology in Human Medicine
Today’s biotechnology allows the production of a variety of pharmaceutical and therapeutic substances to assist in a variety of medical tasks. These include protein replacement such as for insulin, aiding in therapy such as with interferons, vaccines such as the hepatitis B vaccine, screening for genetic diseases, and gene therapy, which for the most part is still in the developmental stage. Traditional biotechnology is now supplemented by recombinant DNA technology to produce a variety of pharmaceutical and therapeutic products, performing many medically important tasks. A summary of products produced by recombinant DNA technology and used in human medicine is shown in Table 25.3.
TABLE 25.3
Products From Recombinant DNA Technology Used in Human Medicine
| Product | Use |
| Insulin | Treatment for some type 1 and type 2 diabetes mellitus |
| Human growth hormone (hGH) | Dwarfism, growth hormone deficiency, adult growth hormone deficiency |
| Factor VIII | Replacement for blood-clotting factor in type A hemophilia |
| Tissue plasminogen activator (tPA) | Can dissolve blood clots in heart attack and stroke |
| Interferon | Treatment of viral infections such as hepatitis C and genital warts, some types of cancer, and multiple sclerosis |
| Interleukins | Cancer treatment |
| Orthoclone | Immune suppressant in transplant patients |
| Macrophage colony-stimulating factor | Stimulation of red bone marrow activity after bone marrow grafting |
| Tumor necrosis factor (TNF) | Treatment of cancer |
| Granulocyte colony-stimulating factor | Treatment of cancer patients experiencing low neutrophil counts |
| Antisense molecules | Possible way to treat genetic disorders |
| Erythropoietin | Treatment of anemia; hormone stimulates red blood cell production |
| rhDNase | Treatment that can break down thick lung secretions in cystic fibrosis |
| Antitrypsin | Replacement therapy to benefit emphysema patients |
| Vaccines | Hepatitis B, Haemophilus influenzae type b meningitis, experimental HPV and AIDS vaccine |
Human Protein Replacements
Insulin
• Isolation of the human insulin gene
• Preparation of the target DNA, the plasmid
• Insertion of the human gene into the plasmid
• Incorporation of the recombinant plasmid back into the bacteria
• Multiplication of the plasmid
Millions of diabetics now use the human insulin produced by bacteria.
Human Growth Hormone
In the past, generally only dwarfism was treated with growth hormone, which had to be obtained from cadavers. Approximately 80 cadavers had to be used to obtain enough HGH for a year’s therapy. This method of treatment unfortunately also brought the risk of the development of Creutzfeldt-Jakob syndrome, a central nervous system disease caused by a prion (see Chapter 7, Viruses). Because of the possibility of transmission of this disease, the use of cadaver tissue in the United States and Britain was restricted in 1985. HGH can now be produced by genetic engineering, using cDNA encoding growth hormone. The cDNA is then expressed in a bacterial expression vector. A problem with producing a short polypeptide hormone such as HGH is its susceptibility to protease digestion. This problem is being resolved by using bacterial host strains that lack several proteases.
Human Disease Therapies
Interferon
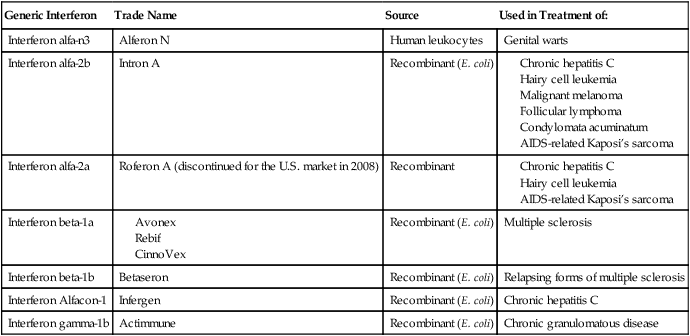
Interferon is a small protein produced naturally by certain white blood cells and by cells that have been infected with a virus (see Chapter 20, The Immune System). Originally, the interferon system was believed to be a defense system against viruses only, but it is now known to be involved in the defense against other microbes and also in immune regulation. The interferon system consists of three main groups: interferon-α, interferon-β, and interferon-γ. Interferon-α is primarily a product of lymphocytes and macrophages, interferon-β is produced by fibroblasts and epithelial cells, and interferon-γ is produced by T cells, B-cells, and NK cells. Interferon was not readily available until 1980, when the interferon gene was inserted into bacteria by recombinant DNA technology; this allowed mass cultivation and purification from bacterial cultures or from yeast. Synthetic interferons are now used in the treatment of viral infections, multiple sclerosis, and cancer (see Health Care Application: Examples of Interferon Therapy in Humans).
HEALTHCARE APPLICATION
Examples of Interferon Therapy in Humans
| Generic Interferon | Trade Name | Source | Used in Treatment of: |
| Interferon alfa-n3 | Alferon N | Human leukocytes | Genital warts |
| Interferon alfa-2b | Intron A | Recombinant (E. coli) |
Vaccines
Vaccines are preparations that provide/improve immunity against certain infectious diseases (see Chapter 20, The Immune System). Before the development of genetically engineered vaccines, vaccines included attenuated microbes, killed or fragmented microbes, and toxoids. In addition to genetically engineered vaccines, traditional vaccines are still available and in use to this date.
Biotechnology in Agriculture
Transgenic Plants
Fungus Tolerance/Resistance
• Introducing genes from other plants or bacteria that encode enzymes that break down essential components of fungal cell walls
• Introducing plant genes that enhance innate plant defense mechanisms
• Invoking a plant hypersensitivity reaction, resulting in the death of the infected cell, and thus stopping the fungal infection from spreading to other cells
Transgenic Animals
With the dramatic increase in the world’s population from 1.6 billion in 1900, to 6 billion at present, to an estimated 10 billion by 2030, the demand for agricultural animals will also increase. The use of genetic engineering has already impacted domesticated livestock and companion animals. More recently, steps have been taken to create transgenic animals. These animals received genes from other organisms to provide solutions for disease treatment, organ transplant shortage, and food production. Some mammals have been modified to produce therapeutic proteins in their milk for human use. These include agents for the treatment of cancer, heart attacks, hemophilia, rheumatoid arthritis, and many other diseases/conditions. Scientists are also attempting to create transgenic pigs for the purpose of transplanting their organs into humans (xenotransplants) (see Chapter 20, The Immune System).
Summary
• Modern biotechnology, a subdiscipline of biology, involves recombinant DNA technology, also referred to as genetic engineering, for industrial, medical, and agricultural purposes.
• The tools of genetic engineering include the use of restriction enzymes, plasmids, the polymerase chain reaction (PCR), and the creation of gene libraries.
• Restriction enzymes are molecular scissors, capable of cutting DNA at specific sites. Plasmids are capable of inserting these pieces of foreign DNA into their own, and PCR can produce multiple copies of DNA without a host cell.
• There are two different types of gene library: cDNA libraries and synthetic DNA. Both of these libraries must be created and then screened before they are usable in the laboratory.
• Vectors in biotechnology serve to insert foreign genes into a host cell to produce a new phenotype. Such vectors include plasmid vectors, lambda phage vectors, and cosmid vectors.
• With the help of recombinant DNA technology, human protein deficits such as insulin deficiency in diabetes mellitus, growth hormone deficiencies, and factor VIII disorder, can be treated.
• The use of recombinant DNA technology has allowed biotechnology companies to produce new drugs for human disease therapies. These include tissue plasminogen activator (tPA), various interferons, antisense molecules, and erythropoietin.
• Vaccines produced by traditional methods can now also be produced by recombinant DNA technology, typically genetically modified subunit vaccines and DNA vaccines consisting of genetically modified plasmids.
• Genetic engineering also has had a great impact in agriculture in the production of genetically modified plants and animals.
• The most widespread application of biotechnology in agriculture involves crops genetically modified to make them tolerant or resistant to herbicides, insects, viruses, and fungi. Moreover, certain mammals have been genetically modified to produce therapeutic proteins in their milk.
Review Questions
1. Which of the following is a true statement about recombinant DNA technology?
a. It is a single technique for genetic manipulation.
b. It involves the modification of an organism’s genome.
2. The genetic recombination process that occurs when bacteria acquire fragments of DNA from the local environment and then express the encoded proteins is called:
3. The term endonuclease refers to:
4. Small loops of DNA found in the cytoplasm of bacteria are:
5. All of the following are steps in the polymerase chain reaction except:
6. Which of the following is a vector created by the combination viral DNA fragments and fragments from a plasmid?
7. A portion of DNA, such as a gene, that is synthesized from an RNA template is:
8. Which of the following tools of genetic engineering is used to make multiple copies of DNA?
9. The recombinant protein that is used to help dissolve blood clots such as occur in heart attacks and stroke is
10. The synthetic interferon that is used in the treatment of multiple sclerosis is:
11. Recombinant DNA technology is also referred to as __________.
12. Enzymes that can cleave DNA at a particular site are __________.
13. Enzymes that cut DNA straight across both of the double helix strands produce __________ ends.
14. When a single bacterium can produce millions of identical cells, the population is called a(n) __________.
15. Synthetic proteins that can be used in the treatment of hepatitis C are __________.
16. Describe the steps and tools used in genetic engineering.
17. Describe a gene library and its usefulness.
18. In correct sequence, name and describe the steps used in PCR.
19. Name and describe the various vectors used in biotechnology.
20. Discuss transgenic plants used in the agricultural environment.






