AML, Acute myeloid leukemia.
∗ Determined among 1311 patients with de novo AML enrolled onto Study 8461 of the Cancer and Leukemia Group B.
† Partial tandem duplication of the MLL1.
‡ Three or more chromosomal aberrations in the absence of t(8;21), inv(16)/t(16;16), t(15;17), or t(9;11).
Source: Adapted from Grimwade D, Mrozek K. Hematology/Oncology. Clinics of North America. 2011, 25: 1135-1161.
Molecular Pathogenesis of AML
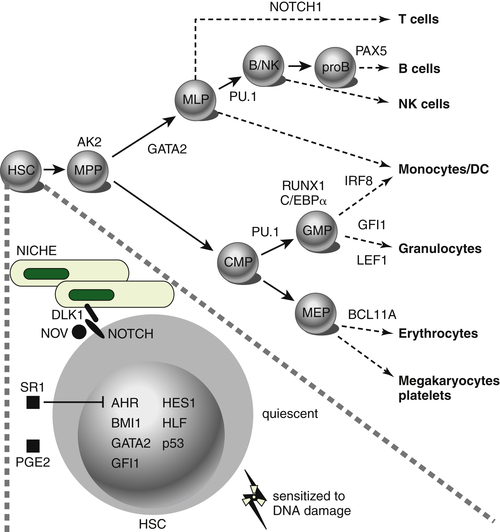
The most frequently mutated genes in AML encode transcription factors, which are typically implicated in chromosomal translocations that result in their inappropriate activation in the hematopoietic compartment, resulting in impairment of maturation and differentiation of myeloid cells (e.g., CBF translocations and
RARα and
MLL gene rearrangements) (
Figure 28-1 ). Frequent types of mutations are those that confer a proliferative and survival advantage to cells (i.e., activating mutations). An example of the latter include mutations in
KIT or
FLT3 (
Table 28-2 ). Mutations in AML are a concept in flux, both during the natural course of the disease and during the course of therapy (i.e., clonal evolution), as shown by whole-genome sequencing analyses performed in paired samples obtained at diagnosis and at relapse. Of note, in some cases, new mutations were found on relapse in the dominant clone in the primary leukemia sample, whereas in others, these newly acquired mutations occurred in small subclones of the founding clone, which resulted in expansion and clonal dominance in the relapse sample. Importantly, varying proportions of clonal cells from the founding clone persisted after chemotherapy in all cases.
Mutations Disrupting the Function of Transcription Factors in AML
Two different types of transcription factors play a major role in hematopoiesis and therefore in the pathogenesis of AML. The first group consists of master regulatory transcription factors, which, like AML1, are implicated in the development of all hematopoietic lineages. Impairment of the signaling stemming from these types of transcription factors results in complete hematopoietic failure. A second category of transcription factors is involved in the development of specific hematopoietic lineages. For instance, GATA-1 skews the development of hematopoietic progenitors toward the erythroid lineage, whereas C/EBPα promotes granulocytic differentiation.
PML-RARα Rearrangements
Translocations involving the
retinoic acid receptor (RAR) locus on chromosome 17, such as t(15;17)(q22;q11), drive the pathogenesis of acute promyelocytic leukemia (APL), which comprises 10%-15% of all cases of adult AML. Such translocations produce the
PML-RARα transcript that encodes a fusion protein containing most of the functional domains of RARα (including the
RAR binding domain and the DNA binding domain) and the majority of the
PML gene. The breakpoints within the
PML gene cluster locate to three different regions referred to as
breakpoint cluster region (bcr) 1, 2, and 3. In addition to t(15;17)(q22;q11), there are at least two other variant translocations involving
RARα associated with the APL phenotype. These include t(11;17)(q23;q21) and t(5;17)(q35;q21), which lead to the fusion of the
RAR? gene to the promyelocytic leukemia zinc finger
(PLZF) and
NPM1 genes, respectively. Transgenic mice expressing
PML-RARα, NPM/RARα, or
PLZF/RARα under the control of a human
Cathepsin G exhibit an APL phenotype after a variably long latency. Comparison of gene expression profiles set by
PML-RARα and
PLZF-RARα have demonstrated the inhibition of genes involved in DNA repair, repression of myeloid transcriptional regulators, and activation of the WNT/Catenin and Jagged/NOTCH pathways, which promote self-renewal of leukemic cells.
Patients with APL harboring
PLZF-RARα fail to respond to
all-trans retinoic acid (ATRA), although, paradoxically, both
PML-RARα and
PLZF-RARα contain identical
RAR sequences and inhibit ATRA-induced gene transcription as well as cell differentiation.
Figure 28-1 Transcription factors implicated in lineage specification of hematopoietic stem and progenitor cells. From Doulatov S et al. Hematopoiesis: a human perspective. Cell Stem Cell. 2012;10:120-136.
Table 28-2
Association of Karyotypic Aberrations with Molecular Findings in AML
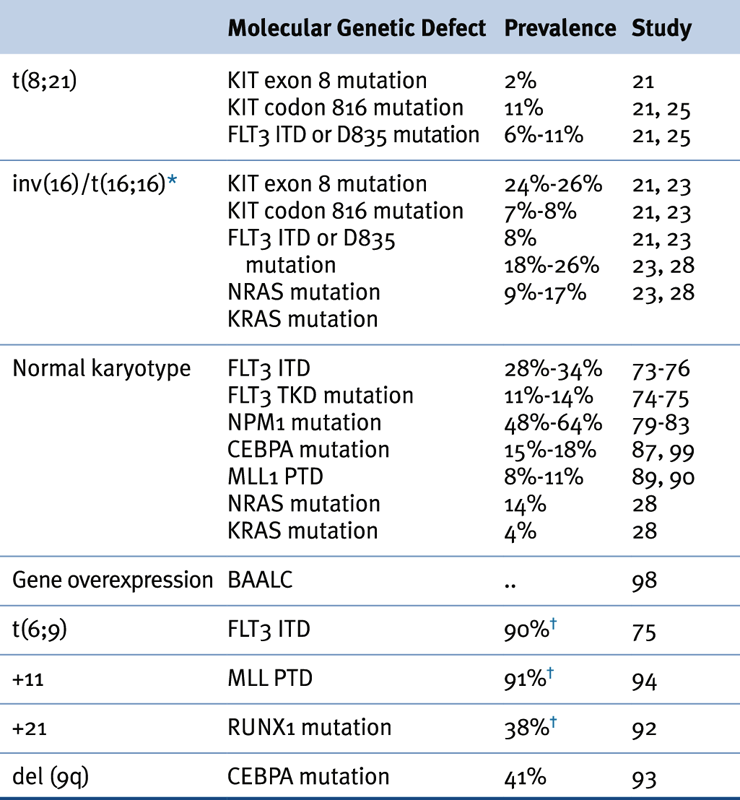
AML, Acute myeloid leukemia.
∗ ≤70% of inv(16) leukemias have mutations in receptor tyrosine kinase or RAS genes.
† Prevalence based on a limited number of cases.
Source: Adapted from Estey et al. Acute Myeloid Leukaemia. Lancet 2006;368:1894-1907, with permission.
The PML-RARα oncoprotein disrupts the interaction of retinoic acid and
RARα, which converts the latter into a transcription activator, resulting in maturation arrest of hematopoietic progenitors at the promyelocyte stage.
PML-RARα expression also disrupts PML localization, causing it to relocalize from discrete nuclear structures, the PML nuclear bodies, into microspeckled aberrant structures. PML-RARα acts as a dominant negative inhibitor of the PML protein, as well as the major heterodimeric partner of RARα, RXRα (retinoid X receptor). PML-RARα recruits several co-repressors, including the nuclear co-repressor (N-CoR). N-CoR inhibits transactivation from RARα target genes through the recruitment of the molecules sin3 and histone deacetylases, which in turn inhibit the binding of transcription factors and the binding of the transcriptional machinery to promoters, resulting in inhibition of gene expression for hematopoietic differentiation. Similarly, the PML moiety of the PML-RARα protein interacts with the DAXX co-repressor. Mutations preventing DAXX recruitment, although allowing PML-RARα dimerization,
abrogated the ability of PML-RARα to block hematopoietic differentiation and immortalize cells. ATRA is the mainstay of therapy in APL, inducing leukemic cell differentiation and remission in patients with t(15;17)/
PML-RARα or t(5;17)/
NPM-RARα. Similarly, arsenic trioxide (As
2O
3) has been demonstrated to be effective in the treatment of de novo as well as of ATRA-resistant t(15;17)/
PML-RARα APL. Current evidence suggests that the combination of ATRA and As
2O
3 represents a valid alternative to chemotherapy-containing regimens for the treatment of APL.
Core Binding Factor AMLs
CBF is a heterodimeric transcription factor that consists of a DNA binding α-subunit, encoded by one of three members of the RUNX family (RUNX1 or AML1, RUNX2, and RUNX3), and a β-subunit encoded by the CBFβ gene that increases DNA-binding affinity to the complex. Mutations involving CBF rearrangements occur in 15% of cases of AML and are associated with a favorable prognosis. CBF AML includes those carrying inv(16)/t(16;16), which gives rise to the fusion of CBFβ with the smooth muscle myosin heavy-chain gene (MYH11 or SMMHC), and t(8;21), which is associated with the fusion transcript composed of the AML1 and the eight-twenty-one (ETO) genes. Of note, in AML expressing the CBF translocation AML1-ETO, this fusion oncogene acts as a dominant negative inhibitor of native AML1. Similarly, the CBFβ-MYH11 oncoprotein is a dominant negative inhibitor of CBF both in transactivation assays and during development. In knockin CBFβ-MYH11 chimeric mice, CBFβ-MYH11 expression alters adult multilineage hematopoietic differentiation. CBFβ also modulates the effect of CBFβ-SMMHC in adult hematopoiesis and leukemogenesis.
Rearrangements of the MLL and HOX Genes
Approximately 4% of patients with de novo AML have balanced translocations or insertions involving the
mixed-lineage leukemia (
MLL) gene.
MLL-gene fusions are highly associated with previous therapy that includes topoisomerase-II inhibitors. The estimated incidence of this type of rearrangement in secondary treatment-related AML is 2% to 12%.
MLL is the human homologue of
Drosophila TRX and constitutes a maintenance factor for HOX proteins, which are central during development and hematopoiesis. The N terminus of
MLL, which contains the AT-hook DNA-binding motif and a region homologous to DNA methyltransferase, is always retained in the fusion protein arising from chromosomal translocations, whereas the C terminus, which contains the activation and SET domains, is always replaced by the fusion partner. The
MLL AT hooks bind to the minor groove of DNA, which facilitates the binding and recruitment of transcription factors to promoter elements.
1 MLL fusion genes can initiate both myeloid and lymphoid leukemogenic programs depending on the fusion partner, of which more than 65 have been thus far identified in AML. Differential activation of the Wnt/β-Catenin pathway is required for the maintenance of MLL leukemia stem cells, and MLL-AF9, one of the most frequent fusion transcripts, requires interacting with the Polycomb Group protein CBX8 to induce a leukemogenic transcriptional program. Approximately 5% to 10% of AML cases present with rearrangements of
MLL consisting of an in-frame partial tandem duplication (
MLL-PTD) of exons 11-5 or 12-5.
MLL-PTD promotes increased histone H3/H4 acetylation and methylation of H3 Lys4 at
cis-regulatory
HOXA sequences. Mislocalized activity of the H3K79 histone methyltransferase DOT1L has been proposed as a driver of leukemogenesis in AML carrying MLL rearrangements. Pharmacological inhibition of DOT1L with the selective DOT1L inhibitor EPZ004777 selectively inhibits H3K79 methylation and blocks expression of leukemogenic genes with little effect on non–MLL-translocated cells, suggesting that DOT1L inhibition represents a potential therapeutic option for patients with MLL rearrangements.
HOX genes are frequently overexpressed in leukemia. Constitutive
HOX gene activation is required for
MLL fusion protein-mediated AML. Gene expression profiling analysis showed that the
HOXA4, HOXA9,
HOXA10,
PBX3, and
MEIS1 homeobox genes are coexpressed across diverse cytogenetic groups but are undetectable in terminally differentiated hematopoietic cells. In AML,
HOX genes are mainly disrupted via chromosomal translocation, such as the fusion of
NUP98, NUP214 (also known as
CAN), or
MLL to
HOXA9,
HOXD13,
DEK, and
DDX10. Overexpression of
HOXA6,
HOXA7,
HOXA9, and the HOX cofactor
myeloid ecotropic viral integration site 1 (MEIS1) has also been correlated with chromosome 11q23 abnormalities involving the MLL protein, which directly regulates the expression of
HOX genes. The
caudal-type homeobox transcription factor 2 (CDX2) is overexpressed in 90% of patients with AML in spite of its lack of expression in hematopoietic progenitors.
CDX2 overexpression in primary murine hematopoietic progenitors resulted in transplantable AML in vivo, which was associated with upregulation of
HOXB6 expression, a protein that is overexpressed in 40% of cases of CN-AML.
2 This suggests the possibility that
CDX2-mediated deregulation of
HOX genes is a major pathway to leukemogenesis.
Mutations in the C/EBPα and PU.1 Genes
The
C/EBPα gene, which encodes the CCAAT/enhancer-binding-protein-alpha, is a member of the family of leucine-zipper (bZIP) transcription factors that couples lineage commitment to terminal differentiation and cell cycle arrest in the process of myeloid differentiation.
C/EBPα initiates growth arrest through induction of
p21 and by disrupting
the E2F transcriptional complexes during the G1 phase of the cell cycle. Mutations in the
C/EBPα gene occur in 15% to 19% of patients with AML and normal cytogenetics.
C/EBPα mutations increase the capacity of bone marrow myeloid progenitors to proliferate and predispose mice to a granulocytic myeloproliferative disorder. In the absence of specific
C/EBPα mutations, decreased expression may serve as an alternative mechanism that disrupts
C/EBPα gene function. For example,
AML1-ETO appears to indirectly suppress
C/EBPα expression by inhibiting positive autoregulation of the
C/EBPα promoter. Notably,
C/EBPα mutations, when biallelic, are associated with a favorable prognosis in patients with cytogenetically normal (CN)-AML.
The transcription factor PU.1 is indispensable for myelomonocytic differentiation during normal hematopoiesis and for regulating the commitment of multipotent hematopoietic progenitors. The course of AML in mice after knockdown of PU.1 includes a preleukemic stage during which immature myelomonocytic precursors accumulate in the bone marrow, followed by a leukemic phase with elevated leukemic blasts in peripheral blood. Also, PU.1-induced upregulation of CSF1R is crucial for leukemia stem cell potential induced by MOZ-TIF2. Despite its leukemogenic potential, PU.1 mutations have been rarely found in human AML.
Mutations Altering Signal Transduction
Mutations at several oncogenes promoting cell growth have been shown to participate in the pathogenesis of AML.
FLT3 expression is restricted to CD34
+ cells and a subset of dendritic precursors, where it regulates proliferation, differentiation, and apoptosis.
3 Internal tandem duplication (ITD) within the
FLT3 juxtamembrane domain (exons 14 and 15) is among the most prevalent mutations in patients with CN-AML, being detected in about 30% of cases. Moreover, 7% of patients with AML harbor missense point mutations affecting the activation loop of the tyrosine kinase domain of FLT3 coded by exon 20, typically involving residue 835. There is evidence suggesting that
FLT3 mutations occur in leukemic stem cells. In fact, 84% of patients with AML carrying
FLT3-ITD mutations exhibit the same mutation at relapse.
FLT3-ITD mutations activate aberrant signaling, including STAT5, PI3K/AKT, and MAPK pathways, and repress
PU.1 and
C/EBPα.
FLT3 gene mutations are associated with high relapse rates and poor prognosis. Small-molecule tyrosine kinase inhibitors directed against the constitutively activated FLT3 protein such as midostaurin, sorafenib, or quizartinib have shown encouraging results in clinical trials. Further development of these agents is being pursued in the context of chemotherapy or hypomethylation-based combinatorial approaches.
Activation of the KIT tyrosine kinase by somatic mutation has been documented in a variety of human malignancies, including core binding factor AML, clustering within exon 17 and exon 8. KIT mutations confer a higher risk of relapse in patients with CBF AML. Gain-of-function KIT mutations may serve as a target for tyrosine kinase inhibitors (e.g., dasatinib), and their activity warrants further investigation in CBF AML harboring such mutations.
RAS oncogenes encode a family of membrane-associated proteins that regulate proliferation, differentiation, and apoptosis. The RAS proteins oscillate between a guanosine triphosphate (GTP)- and a guanosine diphosphate (GDP)-bound state. GDP-bound RAS is incapable of activating signal transduction pathways. NRAS mutations can be detected in 10% of patients with AML. Mutated RAS proteins are constitutively activated, which is held in their GTP-bound status, and efficiently induce an AML-like disorder in a mouse bone marrow transplantation model. The prognostic impact of RAS mutations in AML remains controversial. MEK inhibitors are currently being tested in patients with AML carrying RAS mutations.
Other Genetic Events Implicated in the Pathogenesis of AML
Mutations of the Nucleophosmin Gene
The
NPM1 gene encodes a nucleus-cytoplasm shuttling protein implicated in preventing nucleolar protein aggregation, regulation of ribosomal protein assembly, initiation of centrosome duplication, and regulation of
p53, p19ARF, and HDM2.
NPM1 is involved in the control of primitive hematopoiesis, and mice lacking
NPM1 alleles develop a syndrome reminiscent of human myelodysplasia. Translocations involving the
NPM gene cause cytoplasmic dislocation of the NPM protein.
NPM1 mutations are found in approximately 35% to 40% of adult patients with primary AML and are associated with high sensitivity to cytarabine-based therapy and to higher complete remission rates. Most of the cases (60% to 80%) carrying mutant
NPM1 alleles correspond to patients with CN-AML.
NPM1 mutations are typically heterozygous and almost exclusively map to exon 12.
4–6 A duplication of a TCTG tetranucleotide at position 956 to 959 accounts for 75% to 80% of cases.
6,7 Notably, NPM1 mutations are associated with a very favorable prognosis in the presence of wild-type FLT3 alleles, particularly when associated with IDH1 mutations. The recent development of experimental mouse models lacking NPM1 alleles or carrying NMP1 mutations will aid in understanding the role of
NPM1 in
the pathogenesis of AML and in developing novel targeted agents.
Mutations in Genes Involved in Epigenetic Patterning and Chromatin Conformation
DNA methylation is a common mode of epigenetic regulation, typically involving cytosine residues that reside within GC-rich promoter regions called
CpG islands. Another means of epigenetic regulation is via histone modifications. Histones, like DNA, can be methylated, but they are also acetylated, phosphorylated, sumoylated, and ubiquitinated. Histone modifications can result in either gene activation or repression. Mutations in a series of genes encoding proteins involved in epigenetic pathways are frequently found in patients with CN-AML, including
ASXL1 and
MLL, which encode histone modifiers, and
DNMT3A,
TET2, and
IDH1/2, which encode proteins that regulate cytosine modifications (
Figure 28-2 ).
DNMT3A is an enzyme that catalyzes cytosine methylation, which is critical in DNA imprinting and modulation of gene expression. An analysis of 281 AML samples reported that 22% of cases carried mutations in DNMT3A, a frequency that increased to 34% among those with CN-AML. DNMT3a mutations were associated with a markedly shorter overall survival compared to that of patients carrying wild-type DNMT3A alleles (12.3 vs. 41.1 months).
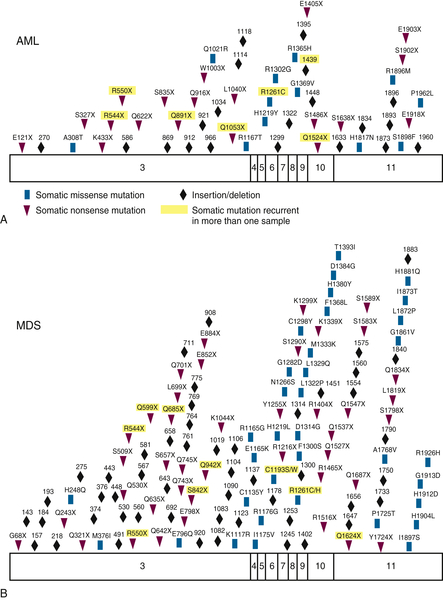
Mutations in the
TET2 gene are found in 8% to 23% of patients with AML and have been associated with a poor prognosis (
Figure 28-3 ). The role of TET2 mutations in the pathogenesis of AML is not well understood. TET proteins catalyze the conversion of 5-methylcytosine, which acts as a transcriptional repressor, to 5-hydroxymethylcytosine, thus potentially promoting transcription. The enzymatic activity of TET2 is inhibited by 2-hydroxyglutarate (2-HG), which is found at high levels in cells carrying mutant
IDH1/
2 alleles. Mutations at the
IDH alleles have been described at frequencies ranging from 15% to 20% overall and from 25% to 30% among those with CN-AML. The most frequent mutations in AML take place at the arginine 132 residue (R132) in
IDH1, and the corresponding arginine 172 (R172) residue in
IDH2, as well as at the arginine 140 (R140) residue in
IDH2. Available data suggest that
IDH2 R140Q is associated with a favorable prognosis, whereas
IDH1 R132 confers a worse prognosis.
IDH2 R172 does not appear to affect the prognosis of patients with AML. Of note,
TET2 and
IDH mutations are mutually exclusive.
IDH proteins convert isocitrate to α-KG, and
IDH mutations result in neomorphic alleles that encode proteins that produce 2-HG in excess, which in turn inhibit the activity of
TET2. The end result of both
TET2 and
IDH mutations is the inability of cells to metabolize 5-methylcytosine to 5-hydroxymethylcytosine, thus inducing a hypermethylation phenotype. This suggests that possibility of using hypomethylating agents for the treatment of
TET2- or
IDH-mutated AML, although so far small trials have not shown any significant benefit. In addition, small molecules are being developed to inhibit the activity of mutated
IDH1/2.
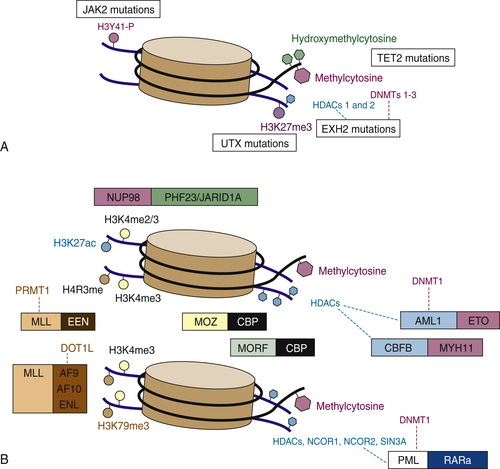
Figure 28-2 Simple mutations and chromosomal translocations in the epigenetic machinery in AML and MDS (A) Somatic mutations that affect the epigenetic machinery include gain-of-function mutations in JAK2 (which can phosphorylate histone 3 tyrosine 41) and loss-of-function mutations in UTX, EZH2, and TET2. (B) Epigenetic modifying genes can also be altered via chromosomal translocations such as those involving MLL fusions that lose H3K4 methyltransferase and gain H3K79 methyltransferase activity, JARID1A/PHF23 fusions that regulate H3K4 di/tri-methylation, and fusions such as PML-RARA or those involving core binding factors, which interact with histone deacetylases and modulate chromatin state. From Abdel-Wahab O, Levine RL. EZH2 mutations: mutating the epigenetic machinery in myeloid malignancies. Cancer Cell. 2010;18:105-107.
Overexpression of Specific Genes in AML
Overexpression of
BAALC, the brain and acute leukemia gene, mRNA in the cytoplasm of peripheral blood blasts portends an adverse clinical outcome in terms of both failure to achieve complete response and shorter overall survival. The
MN1 gene is occasionally overexpressed in AML and is associated with worse overall survival. A similar phenomenon
has been demonstrated among patients whose AML cells overexpress the v-ets erythroblastosis virus E26 oncogene homologue
(ERG) gene, particularly among patients with low
BAALC expression, as well as among those overexpressing the ecotropic viral integration site 1
(EVI1) gene. Elevated FOXO expression is present in 40% of cases of AML and is necessary to maintain leukemia-initiating cells. Resistance to FOXO depletion is mediated by JNK/c-JUN signaling. Over the past few years, a number of studies have correlated certain gene mutations with different patterns of microRNA (miR) expression. For instance,
NPM1 mutations associate with upregulation of miR-10a and miR-196a, whereas
FLT3 mutations associate with upregulation of miR-155 and
C/EBPα with upregulation of miR-181 in CN-AML. Also, miR-10a, miR10b, and miR-196a-1 correlate with expression of
HOX genes in CN-AML.
Figure 28-3 Somatic acquired TET2 mutations Patients with (A) AML or (B) MDS acquire somatic missense, nonsense, and frameshift mutations in TET2. From Cimmino L, Abdel-Wahab O, Levine RL, et al. TET family proteins and their role in stem cell differentiation and transformation. Cell Stem Cell. 2011;9:193-204.
Myelodysplastic Syndromes
MDS is a heterogeneous group of clonal disorders of the hematopoietic stem cell, characterized by excessive apoptosis, maturation abnormalities of hematopoietic precursors manifested as dysplastic changes, and ineffective hematopoiesis. Unlike other malignant hematologic disorders, the biologic hallmark of the stem cell in MDS is a limited ability for self-renewal and differentiation. Furthermore, MDS has a tendency to transform into AML. Indeed, approximately 30% to 40% of cases evolve to AML. The survival of patients with MDS is quite heterogeneous, ranging from weeks to years. In an attempt to incorporate clinical features associated with prognosis in MDS, the International Prognostic Scoring System (IPSS) was developed. This system identified the presence of specific cytogenetic aberrancies, the percentage of blasts in the bone marrow, and the number of cytopenias as the most important variables in disease outcome.
8 Patients with therapy-related MDS are usually refractory to standard chemotherapy-based therapies, and their prognosis is very poor.
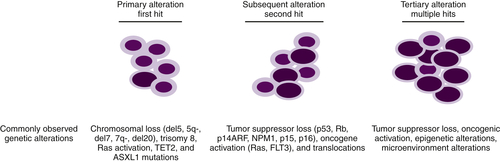
Although it is established that MDs arises from primitive hematopoietic progenitors, our understanding of the pathobiology that drives these diseases is incomplete. Some of the difficulties in determining the critical defects responsible for MDS can be explained by the fact that several programs such as cellular differentiation, apoptosis, and/or proliferation contribute to the etiology of this disease. MDS, like other cancers, results from multiple genetic alterations, likely acquired in a stepwise fashion, which frequently result in transformation to AML (
Figure 28-4 ). This is in consonance with other models of molecular progression described in solid tumors such as colon or pancreatic cancer.
Cytogenetic Abnormalities in MDS
The main prognostic factors in patients with MDS are chromosomal abnormalities. An abnormal karyotype is present in approximately 50% of patients at diagnosis. Although multiple chromosomal lesions have been associated with MDS, losses of chromosomes 5 and/or 7 are associated with a worse prognosis among patients with de novo MDS. The precise genomic regions and genes responsible for the phenotypes observed in patients with specific chromosomal abnormalities are being revealed. Two commonly deleted regions (CDRs) have been defined: 5q33.1, which is associated with the 5q
− syndrome, and 5q31, which is associated with therapy-related MDS and progression to AML.
RPS14 has been shown to be critical for the erythroid phenotype observed in the 5q
− syndrome; miR-145 and miR-146 have been associated with an elevated platelet count. Approximately 10% of patients will present
with abnormalities of chromosome 7, where three CDRs have been identified. Importantly, EZH2, which acts as the catalytic component of the histone H3 lysine 27 methyltransferase polycomb repressive complex 2 (PRC2), maps to 7q36, which may explain why chromosome 7 losses are so frequent in MDS. Trisomy 8 occurs in 8% of patients and is also associated with poor prognosis in MDS. Other chromosomal abnormalities have been associated with a more favorable prognosis such as 20q
− or
−Y.
Figure 28-4 Progression of MDS to AML Primary somatic mutations in hematopoietic stem cells or myeloid progenitors result in dysplastic phenotypes. Subsequent genetic lesions that confer proliferation or anti-apoptotic advantages aid in the clonal evolution of the disease. Following numerous genetic anomalies, the dysplastic clone is rapidly amplified and becomes dominant.
Loss of miRNAs 145/146
The miRNAs miR-143, miR-145, and miR-146a mapping at 5q33 are significantly reduced in bone marrow cells isolated from 5q− syndrome patients. miR-145 and miR-146a regulate the toll-interleukin-1 receptor domain-containing adaptor protein (TIRAP) and tumor necrosis factor receptor–associated factor 6 (TRAF6). TIRAP is a regulator of TRAF6, an E3 ubiquitin ligase, required for nuclear factor κB (NFκB) activation. Collectively, these proteins regulate innate immunity. Haploinsufficiency of miR-145 and miR-146a resulted in increased TIRAP and TRAF6 expression and activity in hematopoietic stem/progenitor cells. Transplantation of haploinsufficient miRNA145/146a cells or TRAF6-overexpressing cells into recipient mice resulted in several hallmark MDS/5q− syndrome phenotypes, such as thrombocytosis, neutropenia, dysplastic megakaryopoiesis, and propensity to transform to AML. These findings link innate immunity to MDS pathogenesis.
NUP98-HOX13 Translocation
NUP98 maps to 11p15.5 and can partner with multiple genes by chromosomal translocation in MDS. In t(2;11)(q31;p15), NUP98 fuses with HOXD11 or HOXD13.
9 Transgenic mice expressing NUP98-HOXD13 exhibit anemia, decreased neutrophil and lymphocyte counts, dysplastic changes, and increased bone marrow cellularity, thus recapitulating an MDS phenotype. Furthermore, a subset of
NUP98-HOX13 transgenic mice also transforms to AML after a latency of approximately 1 year, suggesting the need for secondary hits for transformation, which have been shown to consist of activating RAS mutations.
Gene Mutations in MDS
Mutations in the Spliceosome Machinery
Spliceosomes are complexes composed of small nuclear RNA (snRNA) that remove introns in protein-encoding genes. Several groups, by means of next-generation sequencing platforms, have unveiled the presence of somatic mutations in genes encoding proteins involved in the spliceosome machinery in 45% to 85% of patients with MDS or chronic myelomonocytic leukemia (CMML). Collectively, eight different splicing genes—SF3B1, SRSF2, U2AF35 (also known as U2AF1), ZRSR2, SF3A1, PRP40B, SF1, and U2AF65—can be found mutated in MDS, constituting, as a group, the most frequently detected mutations in MDS (
Figure 28-5 ). These splicing factors are highly expressed in hematopoietic lineages and are involved in recognition of the canonical 3′ DNA splice elements. Similar gene mutations can also be found at low frequencies in chronic lymphocytic leukemia (9% to 15%) or myeloproliferative neoplasms (3% to 9%), and in rare instances of solid cancers (e.g., breast or renal cancer). In MDS, these mutations are always present in the heterozygous state and are almost mutually exclusive. Across studies, mutations in SF3B1 have been described in 20% to 45% of patients and in 65% to 85% of those with MDS with ringed sideroblasts. It is hypothesized that altered splicing of key cancer genes might result in oncogenic gain or loss of function. In addition, genes such as SF3B1 might have oncogenic nonsplicing mechanisms because they have been shown to be implicated in cell cycle control and Hox gene regulation. The impact of these genes has not been clearly delineated, but SF3B1 mutations appear to confer a more favorable prognosis, whereas U2AF1 mutations are associated with poor prognosis among patients with MDS.
Gene Mutations Altering Epigenetic Regulation in MDS: TET2, IDH1/2, and ASXL1
There is now growing evidence that in addition to genetic changes, epigenetic modifications play a critical role in the pathogenesis of MDS.
10,11 Loss-of-function mutations in
TET2, a protein involved in global DNA demethylation, occurs in approximately 20% of patients with MDS.
12,13 However, TET2 mutations are not responsible for the dysplastic changes observed in the bone marrow of patients with MDS. The complete loss of
Tet2 resulted in myeloproliferation, increased repopulation of transplanted cells in recipient mice, and expansion of the hematopoietic progenitor pool.
Tet2 −et mice display phenotypes reminiscent of MDS with a short latency. The impact of
TET2 mutations on the outcome of patients with MDS remains controversial, with more recent reports indicating no impact on overall survival.
Much like in AML,
IDH1/2 mutations in MDS are heterozygous and appear to act as dominant negatives. However, IDH1/2 mutations are significantly less frequent in MDS than in AML, with frequencies ranging from 5% to 10%. A recent genetically engineered mouse model in which the
IDH1 R132H mutation was expressed in the hematopoietic system has been reported. Cells carrying the mutation exhibit high 2-HG levels, which are associated with an expansion of the stem cell pool and epigenetic changes frequently seen in MDS and AML (i.e., DNA and histone
hypermethylation). Recent studies suggest that
IDH1 mutations may portend an adverse prognosis in MDS, whereas
IDH2 mutations had no impact.
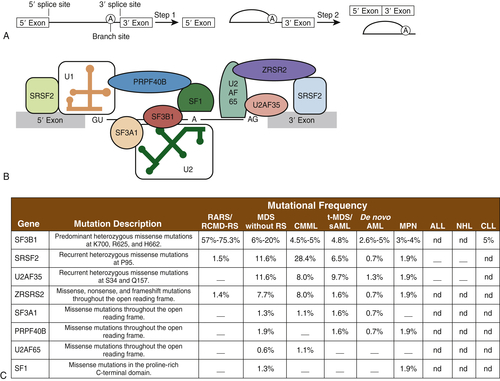
Figure 28-5 Mutations in genes encoding spliceosomal proteins Five small ribonuclear proteins (snRNPs) and more than 50 accessory proteins complex to form the spliceosome at the exon/intron junction of pre-mRNA molecules. Following U1 snRNP and U2AF assembly, the U2 snRNP, the U4-6 tri-snRNP, and other splicing factors are assembled sequentially to form the spliceosome. SF3B1 and SF3A1 are components of U2 snRNP. SR proteins bind to an exonic-splicing enhancer region to directly recruit splicing machinery by interacting with U2AF35 and ZRSR2. This interaction is critical in defining exon/intron boundaries. (A) Spliceosomal proteins found to be mutated in myeloid malignancies. (B) Frequency of mutations in spliceosomal proteins in different hematological malignancies including AML and MDS. ALL, Acute lymphoblastic leukemia; CLL, chronic lymphocytic leukemia; CMML, chronic myelomonocytic leukemia; MPN, myeloproliferative neoplasms; NHL, non-Hodgkin lymphoma; RARS, refractory anemia with ring sideroblasts; RCMD-RS, refractory cytopenia with multilineage dysplasia and ring sideroblasts; sAML, secondary AML; t-MDS, therapy-related MDS. A dash indicates that mutations have not been detected; nd indicates that sequencing was not done. A and B from Padgett RA. New connections between splicing and human disease. Trends Genet. 2012;28:147-154. C from Abdel-Wahab O, Levine RL. The spliceosome as an indicted conspirator in myeloid malignancies. Cancer Cell. 2011;20:420-422.
ASXL1 (additional sex-comb like-1) encodes a member of the polycomb family of chromatin-binding proteins that, depending on the cellular context, either activates or silences genes.
ASXL1 alleles are mutated at the C terminus of the protein, and mutations can be detected in approximately 10% to 20% of patients with MDS and in 17% of those with AML. Loss of ASXL1 function results in aberrant repression or activation of numerous genes. Deletion of
Asxl1 in the germline of mice resulted in an embryonic lethal phenotype in some animals. Those surviving exhibited disrupted expression of the Homeobox family of
Hox genes and defects in differentiation of lymphoid and myeloid progenitor cells but not in multipotent progenitors. The lack of an informative phenotype in the
Asxl1 − / − mice could be due in part to compensatory activities of the
ASXL1 homologues,
ASXL2 and
ASXL3. However, Asxl1 functions are most frequently disrupted by DNA mutations that result in frameshift mutations. These types of mutations often confer dominant negative activities, which result in phenotypic changes quite different from those resulting from allelic loss. Knockin mice harboring
ASXL1 mutations are warranted
to fully examine the role of these mutations in the pathogenesis of MDS.
ASXL1 frameshift mutations have been associated with a reduced time to progression to AML and shorter survival.
RUNX1 Mutations
RUNX1 (also known as AML1) is a transcription core binding factor and represents the third most frequently mutated gene in MDS, being detected in 7% to 15% of cases. Runx1 knockout mice die at the embryonic stage with no evidence of definitive hematopoiesis. When Runx1 is deleted in adult hematopoietic tissues, mice expand their myeloid compartment and exhibit inefficient platelet production. Mutations in the Runt domain of Runx1 in mice are associated with a dominant negative/gain-of-function phenotype characterized by increased blast burden and AML-like disease.
NPM1 Mutations
NPM1 maps to chromosome 5 and is mutated in 5% of cases of MDS. NPM1 regulates the p53 pathway via p14ARF. Genetic deletion of both NPM1 alleles in mice results in embryonic lethality due to severe anemia. However, Npm1 +/- mice are viable and present with several hematopoietic anomalies consistent with MDS, including abnormal platelet counts and erythroid or megakaryocytic dysplasia. In addition, aged Npm1 +/- mice had an increased rate of incidence of AML. This model clearly establishes NPM1 as a true tumor suppressor in MDS. Npm1 mutant mice harboring a humanized Npm1 mutation driven by a hematopoietic promoter have also been developed. These mice also exhibit myeloid-specific phenotypes, but they are more reminiscent of a myeloproliferative neoplasm.
TP53 Mutations
TP53 maps to 17p and is found mutated in 5% to 15% of patients with de novo MDS. TP53 mutations portend a very poor prognosis regardless of IPSS score, and they are associated with advanced disease and complex karyotypes. TP53 mutations to chromothripsis and complex DNA rearrangements in myeloid malignancies. TP53 activation has been demonstrated to be critical for the erythroid phenotype induced by RPS14 haploinsufficiency in 5q- syndrome patients.
Alterations in the Bone Marrow Microenvironment
Although genetic and epigenetic factors clearly play a role in the pathogenesis of MDS and its progression to AML, little is known about the impact of alterations in the bone marrow niche. The importance of such signals has been recently demonstrated in one study that used Osterix-mediated Cre expression to delete the
Dicer gene in osteoprogenitor cells.
14 Mice harboring
Dicer deletions in osteoprogenitor cells exhibited MDS features such as cytopenias and morphologic changes in hematopoietic cells, demonstrating that bone marrow alterations may play an important role in MDS pathogenesis
.
Concluding Remarks
The increasing sophistication of genome-wide sequencing techniques as well as of genetically engineered murine models has provided important insights into the pathogenesis of myeloid malignancies. These advances have also opened new avenues for the development of targeted therapies directed against mutant proteins present in AML or MDS cells, such as FLT3, KIT, IDH1/2, or RAS. However, a better understanding of the various cellular functions and signaling pathways subverted by these mutant proteins is warranted to extend the concept of targeted therapy in AML beyond the isolated paradigm that constitutes the success of ATRA and arsenic trioxide therapy in PML expressing PML-RARα. The alterations in transcription factor expression observed in AML or MDS are truly “tumor specific” and, as such, provide novel targets for therapy. However, designing therapeutic modalities aimed at modulating transcription factors remains challenging.
References
1. Dash A. , Gilliland D.G. Molecular genetics of acute myeloid leukaemia . Best Pract Res Clin Haematol . 2001 ; 14 : 49 – 64 .
2. Giampaolo A. , Felli N. , Diverio D. et al. Expression pattern of HOXB6 homeobox gene in myelomonocytic differentiation and acute myeloid leukemia . Leukemia . 2002 ; 16 : 1293 – 1301 .
3. Stirewalt D.L. , Radich J.P. The role of FLT3 in haematopoietic malignancies . Nat Rev Cancer . 2003 ; 3 : 650 – 665 .
4. Falini B. , Mecucci C. , Tiacci E. et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype . N Engl J Med . 2005 ; 352 : 254 – 266 .
5. Thiede C. , Koch S. , Creutzig E. et al. Prevalence and prognostic impact of NPM1 mutations in 1485 adult patients with acute myeloid leukemia (AML) . Blood . 2006 ; 107 : 4011 – 4020 .
6. Boissel N. , Renneville A. , Biggio V. et al. Prevalence, clinical profile, and prognosis of NPM mutations in AML with normal karyotype . Blood . 2005 ; 106 : 3618 – 3620 .
7. Falini B. , Nicoletti I. , Martelli M.F. , Mecucci C. Acute myeloid leukemia carrying cytoplasmic/mutated nucleophosmin (NPMc+ AML): biologic and clinical features . Blood . 2007 ; 109 : 874 – 885 .
8. Greenberg P. , Cox C. , LeBeau M.M. et al. International scoring system for evaluating prognosis in myelodysplastic syndromes . Blood . 1997 ; 89 : 2079 – 2088 .
9. Abramovich C. , Pineault N. , Ohta H. , Humphries R.K. Hox Genes: From leukemia to hematopoietic stem cell expansion . Ann N Y Acad Sci . 2005 ; 1044 : 109 – 116 .
10. Fathi A.T. , Abdel-Wahab O. Mutations in epigenetic modifiers in myeloid malignancies and the prospect of novel epigenetic-targeted therapy . Adv Hematol . 2012 ; 2012 : 469592 .
11. Wei Y. , Gañán-Gómez I. , Salazar-Dimicoli S. , McCay S.L. , Garcia-Manero G. Histone methylation in myelodysplastic syndromes . Epigenomics . 2011 ; 3 : 193 – 205 .
12. Bacher U. , Haferlach C. , Schnittger S. , Kohlmann A. , Kern W. , Haferlach T. Mutations of the TET2 and CBL genes: novel molecular markers in myeloid malignancies . Ann Hematol . 2010 ; 89 : 643 – 652 .
13. Bejar R. , Levine R. , Ebert B.L. Unraveling the molecular pathophysiology of myelodysplastic syndromes . J Clin Oncol . 2011 ; 29 : 504 – 515 .
14. Raaijmakers M.H.G.P. , Mukherjee S. , Guo S. et al. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia . Nature . 2010 ; 464 : 852 – 857 .