[level-membership-for-basic-science-category]
Bioavailability – physicochemical and dosage form factors
Marianne Ashford
Chapter contents
Physicochemical factors influencing bioavailability
Dosage form factors influencing bioavailability
Key points
• Lipid solubility and the drug dissociation affect drug absorption.
Introduction
As discussed in Chapter 19, the rate and extent of drug absorption are influenced by the physiological factors associated with the structure and function of the gastrointestinal tract. This chapter discusses the physicochemical properties of the drug and dosage form factors that influence bioavailability. For a drug to be absorbed, it needs to be in solution and to be able to pass across the membrane. In the case of orally administered drugs, this is the gastrointestinal epithelium. The physicochemical properties of the drug that will influence its passage into solution and transfer across membranes include its dissolution rate, pKa, lipid solubility, chemical stability and complexation potential.
Physicochemical factors influencing bioavailability
Dissolution and solubility
Solid drugs need to dissolve before they can be absorbed. The dissolution of drugs can be described by the Noyes–Whitney equation (Eqn 20.1). This equation, first proposed in 1897, describes the rate of diffusion of solute through boundary layers surrounding a dissolving spherical particle. When the dissolution process is diffusion controlled and involves no chemical reaction then this also equates to the rate of dissolution:
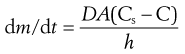 (20.1)
(20.1)
where dm/dt is the rate of dissolution of the drug particles, D is the diffusion coefficient of the drug in solution in the gastrointestinal fluids, A is the effective surface area of the drug particles in contact with the gastrointestinal fluids, h is the thickness of the diffusion layer around each drug particle, Cs is the saturation solubility of the drug in solution in the diffusion layer and C is the concentration of the drug in the gastrointestinal fluids.
More detail regarding Noyes–Whitney equation and its limitations in describing the dissolution of drug particles are outlined in Chapter 2. Despite these limitations, the equation serves to illustrate and explain how various physicochemical and physiological factors can influence the rate of dissolution in the gastrointestinal tract. These are summarized in Table 20.1 and are discussed in more detail in the next section.
Table 20.1
| Factor | Physicochemical parameter | Physiological parameter |
| Effective surface area of drug | Particle size, wettability | Surfactants in gastric juice and bile. pH, buffer capacity, bile, food components |
| Solubility in diffusion layer | Hydrophilicity, crystal structure | |
| Amount of drug already dissolved | Solubilization | Permeability, transit |
| Diffusivity of drug | Molecular size | Viscosity of luminal contents |
| Boundary layer thickness | Motility patterns and flow rate | |
| Volume of solvent available | Gastrointestinal secretions, co-administered fluids |
Figure 20.1 illustrates the dissolution of a spherical drug particle in the gastrointestinal fluids.
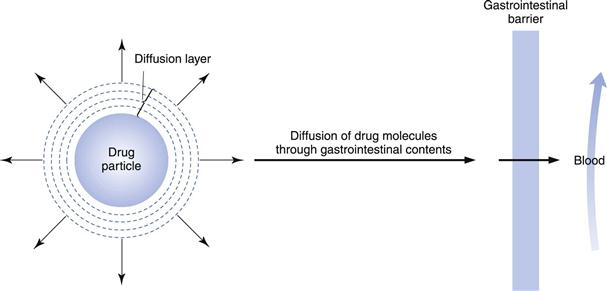
Fig. 20.1 Schematic representation of the dissolution of a drug particle in the gastrointestinal fluids.
Physiological factors affecting the dissolution rate of drugs
The environment of the gastrointestinal tract can affect the parameters of the Noyes–Whitney equation (Eqn 20.1) and hence the dissolution rate of a drug. For instance, the diffusion coefficient, D, of the drug in the gastrointestinal fluids may be decreased by the presence of substances that increase the viscosity of the fluids. Hence the presence of food in the gastrointestinal tract may cause a decrease in drug dissolution rate by reducing the rate of diffusion of the drug molecules away from the diffusion layer surrounding each undissolved drug particle. Surfactants in gastric juice and bile salts will affect both the wettability of the drug, and hence its effective surface area, A, exposed to gastrointestinal fluids, and the solubility of the drug in the gastrointestinal fluids via micellization. The thickness of the diffusion layer, h, will be influenced by the degree of agitation experienced by each drug particle in the gastrointestinal tract. Hence, an increase in gastric and/or intestinal motility may increase the dissolution rate of a sparingly soluble drug by decreasing the thickness of the diffusion layer around each drug particle.
The concentration of drug in solution in the bulk of the gastrointestinal fluids, C, will be influenced by such factors as the rate of removal of dissolved drug by absorption through the gastrointestinal-blood barrier and by the volume of fluid available for dissolution, which in turn will be dependent on the location of the drug in the gastrointestinal tract and the timing with respect to meal intake. In the stomach, the volume of fluid will be influenced by the intake of fluid in the diet. According to the Noyes–Whitney equation, a low value of C will favour more rapid dissolution of the drug by virtue of increasing the value of the term (Cs − C). In the case of drugs whose absorption is dissolution-rate limited, the value of C is normally kept very low by absorption of the drug. Hence dissolution occurs under sink conditions; that is, under conditions such that the value of (Cs − C) approximates to Cs. Thus for the dissolution of a drug in the gastrointestinal tract under sink conditions, the Noyes–Whitney equation can be expressed as:
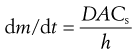 (20.2)
(20.2)
Drug factors affecting dissolution rate
Drug factors that can influence the dissolution rate are the particle size, the wettability, the solubility and the form of the drug (whether a salt or a free form).
Surface area and particle size.
According to Equation 20.1, an increase in the total surface area of drug in contact with the gastrointestinal fluids will cause an increase in dissolution rate. Provided that each particle of drug is intimately wetted by the gastrointestinal fluids, the effective surface area exhibited by the drug will be inversely related to the particle size of the drug. Hence the smaller the particle size, the greater the effective surface area exhibited by a given mass of drug and the higher the dissolution rate. Particle size reduction is thus likely to result in increased bioavailability, provided that the absorption of the drug is dissolution rate limited.
One of the classic examples of particle size effects on the bioavailability of poorly soluble compounds is that of griseofulvin, where a reduction of particle size from about 10 µm (specific surface area = 0.4 m2 g−1) to 2.7 µm (specific surface area = 1.5 m2 g−1) was shown to produce approximately double the amount of drug absorbed in humans. Many poorly soluble, slowly dissolving drugs are routinely presented in micronized form to increase their surface area.
Examples of drugs where a reduction in particle size has been shown to improve the rate and extent of oral absorption and hence bioavailability are shown in Table 20.2. Such improvements in bioavailability can result in an increased incidence of side-effects; thus for certain drugs it is important that the particle size is well controlled, and many pharmacopoeias state a requirement for particle size.
Table 20.2
Examples of drugs where a reduction in particle size has led to improvements in bioavailability
| Drug | Therapeutic class |
| Digoxin | Cardiac glycoside |
| Nitrofurantoin | Antifungal |
| Medroxyprogesterone | Hormone acetate |
| Danazol | Steroid |
| Tolbutamide | Antidiabetic |
| Aspirin | Analgesic |
| Sulfadiazine | Antibacterial |
| Naproxen | Non-steroidal anti-inflammatory |
| Ibuprofen | Non-steroidal anti-inflammatory |
| Phenacetin | Analgesic |
| Griseofulvin | Antifungal |
| Fenofibrate | Lipid regulating agent |
| Megestrol acetate | Apetite loss |
| Aprepitant | Anti-emetic |
| Rapamycin | Immunosuppressant |
| Lapinovir/ritonavir | HIV protease inhibitors |
For some drugs, particularly those that are hydrophobic in nature, micronization and other dry particle size reduction techniques can result in aggregation of the material. This will cause a consequent reduction in the effective surface area of the drug exposed to the gastrointestinal fluids and hence a reduction in its dissolution rate and bioavailability. Aspirin, phenacetin and phenobarbital are all prone to aggregation during particle size reduction. One approach that may overcome this problem is to micronize or mill the drug with a wetting agent or hydrophilic carrier. To overcome aggregation and to achieve particle sizes in the nano-size region, wet milling in the presence of stabilizers has been used. The relative bioavailability of danazol has been increased 400% by administering particles in the nanometre rather than the micrometre size range.
There are now several specialized drug delivery companies who can produce solid dosage forms with the drug stabilized in the nano-size range to afford greater bioavailaility. Examples of commercialized products are the immunosuppressant Rapamune®, the anti-emetic Emend® and the lipid regulating agent TriCor® containing fenofibrate. Megace® ES is an oral nano-suspension of megestrol acetate for the treatment of appetite loss, severe malnutrition, or unexplained significant weight loss in AIDS patients. It is a reformulation of the oral suspension using Nanocrystal® technology to improve the dissolution rate, absorption rate and bioavailability of the original formulation. The formulation is less viscous and allows a quarter of the volume to be dosed, thus aiding patient swallowing and compliance.
As well as milling with wetting agents, the effective surface area of hydrophobic drugs can be increased by the addition of a wetting agent to the formulation. The presence of polysorbate 80 in a fine suspension of phenacetin (particle size less than 75 µm) greatly improved the rate and extent of absorption of the phenacetin in human volunteers compared to the same-size suspension without a wetting agent. Polysorbate 80 helps by increasing the wetting and solvent penetration of the particles and by minimizing aggregation of suspended particles, thereby maintaining a large effective surface area. Wettability effects are highly drug specific; however wetting agents are routinely added to many formulations.
If an increase in the effective surface area of a drug does not increase its absorption rate, it is likely that the dissolution process is not rate limiting. For drugs such as penicillin G and erythromycin, which are unstable in gastric fluids, their chemical degradation will be minimized if they remain in the solid state. Thus, particle size reduction would not only serve to increase their dissolution rate but would simultaneously increase chemical degradation and therefore reduce the amount of intact drug available for absorption.
Solubility in the diffusion layer, Cs.
The dissolution rate of a drug under sink conditions, according to the Noyes–Whitney equation (Eqn 20.2), is directly proportional to its intrinsic solubility in the diffusion layer surrounding each dissolving drug particle, Cs. The aqueous solubility of a drug is dependent on the interactions between molecules within the crystal lattice, intermolecular interactions with the solution in which it is dissolving, and the entropy changes associated with fusion and dissolution. In the case of drugs that are weak electrolytes, their aqueous solubility is dependent on pH (as discussed in Chapter 2). Hence in the case of an orally administered solid dosage form containing a weak electrolyte drug, the dissolution rate of the drug will be influenced by its solubility and the pH in the diffusion layer surrounding each dissolving drug particle. The pH in the diffusion layer – the microclimate pH – for a weak electrolyte will be affected by the pKa and solubility of the dissolving drug and the pKa and solubility of the buffers in the bulk gastrointestinal fluids. Thus differences in dissolution rate will be expected in different regions of the gastrointestinal tract.
The solubility of weakly acidic drugs increases with pH and so as a drug moves down the gastrointestinal tract from the stomach to the intestine, its solubility will increase. Conversely, the solubility of weak bases decreases with increasing pH, i.e. as the drug moves down the gastrointestinal tract. It is important therefore for poorly soluble weak bases to dissolve rapidly in the stomach, as the rate of dissolution in the small intestine will be much slower. The antifungal drug ketoconazole, a weak base, is particularly sensitive to gastric pH. Dosing ketoconazole 2 hours after the administration of the H2 blocker cimetidine, which reduces gastric acid secretion, results in a significantly reduced rate and extent of absorption Similarly, in the case of the antiplatelet drug dipyrimidole, pretreatment with the H2 blocker famotidine reduces the peak plasma concentration by a factor of up to 10.
Salts.
The solubility of a weakly acidic drug in gastric fluid (pH 1–3.5) will be relatively low however it will be much greater in the higher pH of the intestine. The sodium salt of a weak acid will dissociate as shown:
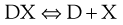 (20.3)
(20.3)
where D is the drug and X is the counter ion. The concentrations of the drug multiplied by the counter ion concentration at any pH will give the solubility product Ksp, i.e.:
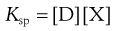 (20.4)
(20.4)
The pH solubility profile of a weak acid in the presence of counterions depends on the solubility product of the ionized drug and its counter-ions and is depicted for both a weak acid and a weak base.
Many examples can be found of the effects of salts improving the rate and extent of absorption. The dissolution rate of the oral hypoglycaemic drug, tolbutamide sodium in 0.1 M HC1 is 5000 times faster than that of the free acid. Oral administration of a non-disintegrating disc of the more rapidly dissolving sodium salt of tolbutamide produces a very rapid decrease in blood sugar level (a consequence of the rapid rate of drug absorption), followed by a rapid recovery. In contrast, a non-disintegrating disc of the tolbutamide free acid produces a much slower rate of decrease of blood sugar (a consequence of the slower rate of drug absorption) that is maintained over a longer period of time. The barbiturates are often administered in the form of sodium salts to achieve a rapid onset of sedation and provide more predictable effects.
The non-steroidal anti-inflammatory drug naproxen was originally marketed as the free acid for the treatment of rheumatoid and osteoarthritis. However, the sodium salt (naproxen sodium) is absorbed faster, due to faster dissolution of the dosage from, and hence is more effective, and thus has now largely replaced the free form. Conversely, strongly acidic salt forms of weakly basic drugs, for example chlorpromazine hydrochloride, dissolve more rapidly in gastric and intestinal fluids than do the free bases (e.g. chlorpromazine). The presence of strongly acidic anions (e.g. Cl− ions) in the diffusion layer around each drug particle ensures that the pH in that layer is lower than the bulk pH in either the gastric or the intestinal fluid. This lower pH will increase the solubility of the drug in the diffusion layer.
The oral administration of a salt form of a weakly basic drug in a solid oral dosage form generally ensures that dissolution occurs in the gastric fluid before the drug passes into the small intestine where pH conditions are unfavourable. Thus the drug should be delivered to the major absorption site, the small intestine, in solution. If absorption is fast enough, precipitation of the dissolved drug is unlikely to significantly affect bioavailability. It is important to be aware that hydrochloride salts may experience a common ion effect owing to the presence of chloride ions in the stomach (also discussed in Chapter 2). The in vitro dissolution of a sulfate salt of an HIV protease inhibitor analogue is significantly greater in hydrochloric acid than that of the hydrochloride salt. The bioavailability of the sulfate salt is more than three times greater than that of the hydrochloride salt. These observations are attributed to the common ion effect of the hydrochloride.
The sodium salts of acidic drugs and the hydrochloride salts of basic drugs are by far the most common. However, many other salt forms are increasingly being employed (there is further information in Chapter 23). Some salts have a lower solubility and dissolution rate than the free form, for example aluminium salts of weak acids and palmoate salts of weak bases. In these cases, insoluble films of either aluminium hydroxide or palmoic acid are found to coat the dissolving solids when the salts are exposed to a basic or an acidic environment, respectively. In general, poorly soluble salts delay absorption and may therefore be used to sustain the release of the drug. A poorly soluble salt form is generally employed for suspension dosage forms.
Although salt forms are often selected to improve bioavailability, other factors such as chemical stability, hygroscopicity, manufacturability and crystallinity will all be considered during salt selection and may preclude the choice of a particular salt. The sodium salt of aspirin, sodium acetylsalicylate, is much more prone to hydrolysis than is aspirin, acetylsalicylic acid, itself. One way to overcome chemical instabilities or other undesirable features of salts is to form the salt in situ or to add basic/acidic excipients to the formulation of a weakly acidic or weakly basic drug. The presence of the basic excipients in the formulation of acidic drugs ensures that a relatively basic diffusion layer is formed around each dissolving particle. The inclusion of the basic ingredients aluminium dihydroxyaminoacetate and magnesium carbonate in aspirin tablets was found to increase their dissolution rate and bioavailability.
Crystal form
Polymorphism
Many drugs can exist in more than one crystalline form. This property is referred to as polymorphism and each crystalline form is known as a polymorph (discussed further in Chapter 8). As discussed in Chapters 2 and 8, a metastable polymorph usually exhibits a greater dissolution rate than the corresponding stable polymorph. Consequently, the metastable polymorphic form of a poorly soluble drug may exhibit an increased bioavailability compared to the stable polymorphic form.
A classic example of the influence of polymorphism on drug bioavailability is provided by chloramphenicol palmitate. This drug exists in three crystalline forms designated A, B and C. At normal temperature and pressure, A is the stable polymorph, B is the metastable polymorph and C is the unstable polymorph. Polymorph C is too unstable to be included in a dosage form but polymorph B, the metastable form, is sufficiently stable. The plasma profiles of chloramphenicol from orally administered suspensions containing varying proportions of the polymorphic forms A and B were investigated. The extent of absorption of chloramphenicol increases as the proportion of the polymorphic form B of chloramphenicol palmitate is increased in each suspension. This was attributed to the more rapid in vivo rate of dissolution of the metastable polymorphic form, B, of chloramphenicol palmitate. Following dissolution, chloramphenicol palmitate is hydrolysed to give free chloramphenicol in solution, which is then absorbed. The stable polymorphic form A of chloramphenicol palmitate dissolves so slowly and consequently is hydrolysed so slowly to chloramphenicol in vivo that this polymorph is virtually ineffective. The importance of polymorphism to the gastrointestinal bioavailability of chloramphenicol palmitate is reflected by a limit being placed on the content of the inactive polymorphic form A, in chloramphenicol palmitate mixture.
Amorphous solids
In addition to different polymorphic crystalline forms, a drug may exist in an amorphous form (see Chapter 8). Because the amorphous form usually dissolves more rapidly than the corresponding crystalline form(s), the possibility exists that there will be significant differences in the bioavailabilities exhibited by the amorphous and crystalline forms of drugs that show dissolution rate-limited bioavailability.
A classic example of the influence of amorphous versus crystalline forms of a drug on its gastrointestinal bioavailability is provided by the antibiotic novobiocin. The more soluble and rapidly dissolving amorphous form of novobiocin was readily absorbed following oral administration of an aqueous suspension. However, the less soluble and slower dissolving crystalline form was not absorbed to any significant extent. The crystalline form was thus therapeutically ineffective. A further important observation was made in the case of aqueous suspensions of novobiocin. The amorphous form slowly converts to the more thermodynamically stable crystalline form, with an accompanying loss of therapeutic effectiveness. Thus, unless adequate precautions are taken to ensure the stability of the less stable, more therapeutically effective amorphous form of a drug in a dosage form, then unacceptable variations in therapeutic effectiveness may occur.
Several delivery technologies for poorly soluble drugs rely on stabilizing the drug in its amorphous form to increase its dissolution and bioavailability. An example of this is Kaletra® which is a combination tablet of the protease inhibitors lopinavir and ritinovir used for HIV infection, in combination with other antiretroviral drugs. These drugs are stabilized in their amorphous form by a polymer, copovidone, following melt extrusion of the drug with the polymer. The tablets provide a significant improvement in bioavailability and variability such that two medium sized tablets are equivalent to three large capsules of the old formulation.
Solvates
Another variation in the crystalline form of a drug can occur if the drug is able to associate with solvent molecules to produce crystalline forms known as solvates (discussed further in Chapter 8). When water is the solvent, the solvate formed is called a hydrate. Generally, the greater the solvation of the crystal, the lower is the solubility and dissolution rate in a solvent identical to the solvation molecules. As the solvated and non-solvated forms usually exhibit differences in dissolution rates, they may also exhibit differences in bioavailability, particularly in the case of poorly soluble drugs that exhibit dissolution rate-limited bioavailability.
An example is that of the antibiotic ampicillin. The faster dissolving anhydrous form of ampicillin is absorbed to a greater extent from both hard gelatin capsules and an aqueous suspension than is the slower dissolving trihydrate form. The anhydrous form of the hydrochloride salt of a protease inhibitor, an analogue of indinavir, has a much faster dissolution rate than the hydrated form in water. This is reflected in a significantly greater rate and extent of absorption and a more than doubling of the bioavailability of the anhydrous form.
Factors affecting the concentration of drug in solution in the gastrointestinal fluids
The rate and extent of absorption of a drug depend on the effective concentration of that drug, i.e. the concentration of drug in solution in the gastrointestinal fluids, which is in an absorbable form. Complexation, micellar solubilization, adsorption and chemical stability are the principal physicochemical properties that can influence the effective drug concentration in the gastrointestinal fluids.
Complexation.
Complexation of a drug may occur within the dosage form and/or in the gastrointestinal fluids, and can be beneficial or detrimental to absorption.
Mucin which is present in gastrointestinal fluids forms complexes with some drugs. The antibiotic streptomycin binds to mucin, thereby reducing the available concentration of the drug for absorption. It is thought that this may contribute to its poor bioavailability. Another example of complexation is that between drugs and dietary components, as in the case of the tetracyclines, which is discussed in Chapter 19.
The bioavailability of some drugs can be reduced by the presence of some excipients within its dosage form. The presence of calcium (e.g. from the diluent dicalcium phosphate) in the dosage form of tetracycline reduces its bioavailability via the formation of a poorly soluble complex. Other examples of complexes that reduce drug bioavailability are those between amphetamine and sodium carboxymethylcellulose, and between phenobarbital and polyethylene glycol 4000. Complexation between drugs and excipients probably occurs quite often in liquid dosage forms and may be beneficial to the physical stability of the dosage form.
Complexation is sometimes used to increase drug solubility, particularly of poorly water-soluble drugs. One class of complexing agents that is increasingly being employed is the cyclodextrin family (see Chapter 24). Cyclodextrins are enzymatically modified starches composed of glucopyranose units which form a ring of either six (α-cyclodextrin), seven (β-cyclodextrin) or eight (γ-cyclodextrin) units. The outer surface of the ring is hydrophilic and the inner cavity is hydrophobic. Lipophilic molecules can fit into the ring to form soluble inclusion complexes. The ring of β-cyclodextrin is the correct size for the majority of drug molecules, and normally one drug molecule will associate with one cyclodextrin molecule to form reversible complexes, although other stoichiometries are possible. For example, the antifungal miconazole shows poor oral bioavailability owing to its poor solubility, but in the presence of cyclodextrin, the solubility and dissolution rate of miconazole are significantly enhanced (by up to 55– and 255–fold, respectively). This enhancement of dissolution rate resulted in a more than doubling of the oral bioavailability in a study in rats. There are numerous examples in the literature of drugs whose solubility, and hence bioavailability, have been increased by the use of cyclodextrins: they include piroxicam, itraconazole, indometacin, pilocarpine, naproxen, hydrocortisone, diazepam and digitoxin. The first product on the UK market containing a cyclodextrin includes the poorly soluble antifungal itraconazole, which has been formulated as a liquid dosage form with the more soluble derivative of β-cyclodextrin, hydroxypropyl-β-cyclodextrin.
Micellar solubilization.
Micellar solubilization can also increase the solubility of drugs in the gastrointestinal tract. The ability of bile salts to solubilize drugs depends mainly on the lipophilicity of the drug. Further information on solubilization and complex formation can be found in Chapter 5 and in Florence & Attwood (2011).
Adsorption.
The concurrent administration of drugs and medicines containing solid adsorbents (e.g. antidiarrhoeal mixtures) may result in the adsorbents interfering with the absorption of drugs from the gastrointestinal tract. The adsorption of a drug onto solid adsorbents such as kaolin or charcoal may reduce its rate and/or extent of absorption owing to a decrease in the effective concentration of the drug in solution available for absorption. A consequence of the reduced concentration of free drug in solution at the site of absorption will be a reduction in the rate of drug absorption. Whether there is also a reduction in extent of absorption will depend on whether the drug-adsorbent interaction is readily reversible. If the absorbed drug is not readily released from the solid adsorbent in order to replace the free drug that has been absorbed from the gastrointestinal tract, there will also be a reduction in the extent of absorption from the gastrointestinal tract.
An example of a drug-adsorbent interaction that gives reduced extent of absorption is promazine-charcoal. The adsorbent properties of charcoal have been exploited as an antidote to orally administered drug overdoses.
Care also needs to be taken when insoluble excipients are included in dosage forms to ensure that the drug will not adsorb to them. Talc, which can be included in tablets as a glidant, is claimed to interfere with the absorption of cyanocobalamin by virtue of its ability to adsorb this vitamin.
Chemical stability of the drug in the gastrointestinal fluids.
If the drug is unstable in the gastrointestinal fluids, the amount of drug that is available for absorption will be reduced as will its bioavailability. Instability in gastrointestinal fluids is usually caused by acidic or enzymatic hydrolysis. When a drug is unstable in gastric fluid, its extent of degradation will be minimized (and hence its bioavailability improved) if it remains in the solid state in gastric fluid and dissolves only in intestinal fluid.
The concept of delaying the dissolution of a drug until it reaches the small intestine has been employed to improve the bioavailability of erythromycin in the gastrointestinal tract. Gastro-resistant coating of tablets containing the free base erythromycin has been used to protect the drug from gastric fluid. The gastro-resistant coating resists gastric fluid but disrupts or dissolves at the less acid pH range of the small intestine (discussed later in this chapter and in Chapters 31 and 32). An alternative method of protecting a susceptible drug from gastric fluid, which has been employed for erythromycin, is the administration of chemical derivatives of the parent drug. These derivatives, or prodrugs, exhibit limited solubility (and hence minimal dissolution) in gastric fluid but once in the small intestine, liberate the parent drug to be absorbed. For instance, erythromycin stearate, after passing through the stomach undissolved, dissolves and dissociates in the intestinal fluid, yielding the free base erythromycin that is absorbed.
The proton pump inhibitors omeprazole and esomeprazole are acid labile and are therefore formulated in a multi-unit gastro-resistant coated pelleted system. Instability in gastrointestinal fluids is one of the reasons why many peptide-like drugs are poorly absorbed when delivered via the oral route.
Poorly soluble drugs
Poorly water-soluble drugs present a problem in terms of obtaining the satisfactory dissolution within the gastrointestinal tract that is necessary for good bioavailability. It is not only existing drugs that cause problems but it is the challenge of medicinal chemists to ensure that new drugs are not only active pharmacologically but also have sufficient solubility to ensure fast enough dissolution at the site of administration, often the gastrointestinal tract. This is a particular problem for certain classes of drugs, such as the HIV protease inhibitors, many antiinfective drugs and anticancer drugs where the targets are very lipophilic and thus designing potency and water solubility are challenging. Medicinal chemists are using approaches such as introducing ionizable groups, reducing melting points, changing polymorphs or introducing prodrugs to improve solubility.
Pharmaceutical scientists, as alluded to in earlier parts of this chapter, are also applying a wide range of formulation approaches to improve the dissolution rate of poorly soluble drugs. These include formulating in the nano-size range, formulating in a solid solution or dispersion or self-emulsifying drug delivery system, stabilizing the drug in the amorphous form or formulating with cyclodextrins. Many drug delivery companies thrive on technologies designed to improve the delivery of poorly soluble drugs.
Drug absorption
Once the drug has successfully passed into solution, it is available for absorption. In Chapter 19, many physiological factors were described that influence drug absorption. Absorption, and hence the bioavailability of a drug once in solution, is also influenced by many drug factors, in particular the pKa and hence charge, lipid solubility, molecular weight, the number of hydrogen bonds in the molecule and its chemical stability.
Drug dissociation and lipid solubility
The dissociation constant and lipid solubility of a drug and the pH at the absorption site often influence the absorption characteristics of a drug throughout the gastrointestinal tract. The interrelationship between the degree of ionization of a weak electrolyte drug (which is determined by its dissociation constant and the pH at the absorption site) and the extent of absorption is embodied in the pH-partition hypothesis of drug absorption, first proposed by Overton in 1899. Although it is an oversimplification of the complex process of absorption, the pH-partition hypothesis still provides a useful framework for understanding the transcellular passive route of absorption, which is that favoured by the majority of drugs.
pH-partition hypothesis of drug absorption.
According to the pH-partition hypothesis, the gastrointestinal epithelium acts as a lipid barrier to drugs which are absorbed by passive diffusion, and those that are lipid soluble will pass across the barrier. As most drugs are weak electrolytes, the unionized form of weakly acidic or basic drugs (i.e. the lipid-soluble form) will pass across the gastrointestinal epithelium, whereas it is impermeable to the ionized (i.e. poorly lipid-soluble) form of such drugs. Consequently, according to the pH-partition hypothesis, the absorption of a weak electrolyte will be determined chiefly by the extent to which the drug exists in its unionized form at the site of absorption.
The extent to which a weakly acidic or basic drug ionizes in solution in the gastrointestinal fluid may be calculated using the appropriate form of a Henderson-Hasselbalch equation (discussed further in Chapter 3). For a weakly acidic drug having a single ionizable group (e.g. aspirin, phenobarbital, ascorbic acid (vitamin C) ), the equation takes the form of:
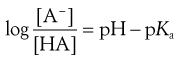 (20.5)
(20.5)
This is a slightly rearranged form of Equation 3.16 where pKa is the negative logarithm of the acid dissociation constant of the drug, and [HA] and [A−] are the respective concentrations of the unionized and ionized forms of the weakly acidic drug, which are in equilibrium and in solution in the gastrointestinal fluid. pH refers to the pH of the environment of the ionized and unionized species, i.e. the gastrointestinal fluids.
For a weakly basic drug possessing a single ionizable group (e.g. chlorpromazine, erythromycin, morphine), the analogous equation is:
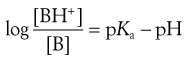 (20.6)
(20.6)
This is a slightly rearranged form of Equation 3.19 where [BH+] and [B] are the respective concentrations of the ionized and unionized forms of the weak basic drug, which are in equilibrium and in solution in the gastrointestinal fluids.
Therefore, according to these equations, a weakly acidic drug, pKa 3.0, will be predominantly (98.4%) unionized in gastric fluid at pH 1.2 and almost totally (99.98%) ionized in intestinal fluid at pH 6.8, whereas a weakly basic drug, pKa 5, will be almost entirely (99.98%) ionized at gastric pH of 1.2 and predominantly (98.4%) unionized at intestinal pH of 6.8. This means that, according to the pH-partition hypothesis, a weakly acidic drug is more likely to be absorbed from the stomach where it is unionized and a weakly basic drug from the intestine where it is predominantly unionized. However, in practice, very little absorption occurs in the stomach and many other factors need to be taken into consideration.
Limitations of the pH-partition hypothesis.
The extent to which a drug exists in its unionized form is not the only factor determining the rate and extent of absorption of a drug molecule from the gastrointestinal tract. Despite their high degree of ionization, weak acids are still quite well absorbed from the small intestine. In fact, the rate of intestinal absorption of a weak acid is often higher than its rate of absorption in the stomach, even though the drug is unionized in the stomach. The significantly larger surface area that is available for absorption in the small intestine more than compensates for the high degree of ionization of weakly acidic drugs at intestinal pH values. In addition, a longer small intestinal residence time and a microclimate pH (that exists at the surface of the intestinal mucosa and is lower than that of the luminal pH of the small intestine) are thought to aid the absorption of weak acids from the small intestine.
The mucosal unstirred layer is another recognized component of the gastrointestinal barrier to drug absorption that is not accounted for in the pH-partition hypothesis. During absorption, drug molecules must diffuse across this layer and then on through the lipid layer. Diffusion across this layer is liable to be a significant component of the total absorption process for those drugs that cross the lipid layer very quickly. Diffusion across this layer will also depend on the molecular weight of the drug.
A physiological factor that causes deviations from the pH-partition hypothesis is convective flow or solvent drag. The movement of water molecules into and out of the gastrointestinal tract will affect the rate of passage of small water-soluble molecules across the gastrointestinal barrier. Water movement occurs because of differences in osmotic pressure between blood and the luminal contents and because of differences in hydrostatic pressure between the lumen and the perivascular tissue. The absorption of water-soluble drugs will be increased if water flows from the lumen to the blood, provided that the drug and water are using the same route of absorption. This will have greatest effect in the jejunum, where water movement is at its greatest. Water flow also affects the absorption of lipid-soluble drugs. It is thought that this is because the drug becomes more concentrated as water flows out of the intestine, thereby favouring a greater drug concentration gradient and increased absorption.
Lipid solubility.
A number of drugs are poorly absorbed from the gastrointestinal tract despite the fact that their unionized forms predominate. For example, the barbiturates: barbitone and thiopentone have similar dissociation constants – pKa of 7.8 and 7.6 respectively – and therefore similar degrees of ionization at intestinal pH. However, thiopentone is absorbed much better than barbitone. The reason for this difference is that the absorption of drugs is also affected by the lipid solubility of the drug. Thiopentone, being more lipid soluble than barbitone, exhibits a greater affinity for the gastrointestinal membrane and is thus far better absorbed.
An indication of the lipid solubility of a drug, and therefore whether that drug is liable to be transported across membranes, is given by its ability to partition between a lipid-like solvent and water or an aqueous buffer. This is known as the drug’s partition coefficient and is a measure of its lipophilicity. The value of the partition coefficient P is determined by measuring the drug partitioning between water and a suitable non-water miscible solvent at constant temperature. As this ratio normally spans several orders of magnitude it is usually expressed as the logarithm, log P. The solvent that is usually selected to mimic the biological membrane, because of its many similar properties, is octanol.
 (20.7)
(20.7)
The effective partition coefficient, taking into account the degree of ionization of the drug, is known as the distribution coefficient and again is normally expressed as the logarithm (log D); it is given by the following equations for acids and bases:
For acids:
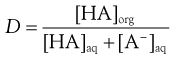 (20.8)
(20.8)
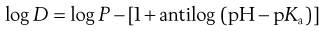 (20.9)
(20.9)
For bases:
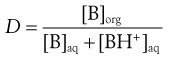 (20.10)
(20.10)
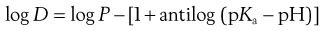 (20.11)
(20.11)
The lipophilicity of a drug is critical in the drug discovery process. Polar molecules, i.e. those that are poorly lipid soluble (log P < 0) and relatively large, such as gentamicin, ceftriaxone, heparin and streptokinase, are poorly absorbed after oral administration and therefore have to be given by injection. Smaller molecules that are poorly lipid soluble and hydrophilic in nature, such as the β-blocker atenolol, can be absorbed via the paracellular route. Lipid-soluble drugs with favourable partition coefficients (i.e. log P > 0) are usually absorbed after oral administration. Drugs which are very lipid soluble (log P > 3) tend to be well absorbed but are also more likely to be susceptible to metabolism and biliary clearance. Although there is no general rule that can be applied to all drug molecules, within a homologous series, such as the barbiturates or β-blockers, drug absorption usually increases as the lipophilicity rises.
Sometimes, if the structure of a compound cannot be modified to yield lipid solubility while maintaining pharmacological activity, medicinal chemists may investigate the possibility of making lipid soluble prodrugs to improve absorption. A prodrug is a chemical modification, frequently an ester of an existing drug, which converts back to the parent compound as a result of metabolism by the body. A prodrug itself has no pharmacological activity. Examples of prodrugs which have been successfully used to improve the lipid solubility and hence absorption of their parent drugs are shown in Table 20.3.
Table 20.3
| Prodrug | Active drug | Ester |
| Pivampicillin | Ampicillin | Pivaloyloxymethyl |
| Bacampicillin | Ampicillin | Carbonate |
| Indanylcarbenicillin | Carbenicillin | Indanyl |
| Cefuroxime axetil | Cefuroxime | Acetylethyl |
| Enalapril | Enalaprilat | Ester of 1-carboxylic acid |
| Ibuterol | Terbutaline | Dibutyl |
| Valaciclovir | Aciclovir | L-valyl (amino acid) |
| Fosamprenavir | Amprenavir | Phosphate |
Molecular size and hydrogen bonding.
Two other drug properties that are important in permeability are the number of hydrogen bonds within the molecule and the molecular size.
For paracellular absorption, the molecular weight should ideally be less than 200 Da; however, there are examples where larger molecules (up to molecular weights of 400 Da) have been absorbed via this route. Shape is also an important factor for paracellular absorption.
In general, for transcellular passive diffusion, a molecular weight of less than 500 Da is preferable. Drugs with molecular weights above this are absorbed less efficiently. There are few examples of drugs with molecular weights above 700 Da being well absorbed.
Too many hydrogen bonds within a molecule are detrimental to its absorption. In general, no more than five hydrogen bond donors and no more than 10 hydrogen bond acceptors (the sum of nitrogen and oxygen atoms in the molecule is often taken as a rough measure of hydrogen bond acceptors) should be present if the molecule is to be well absorbed. The large number of hydrogen bonds within peptides is one of the reasons why peptide drugs are poorly absorbed.
Summary
There are many properties of the drug itself that will influence its passage into solution in the gastrointestinal tract and across the gastrointestinal membrane, and hence its overall rate and extent of absorption.
Dosage form factors influencing bioavailability
Introduction
The rate and/or extent of absorption of a drug from the gastrointestinal tract have been shown to be influenced by many physiological factors and by many physicochemical properties associated with the drug itself. The bioavailability of a drug can also be influenced by factors associated with the formulation and production of the dosage form. Increasingly, many dosage forms are being designed to affect the release and absorption of drugs, for example controlled-release systems (see Chapter 31) and delivery systems for poorly soluble drugs. This section summarizes how the type of dosage form and the excipients used in conventional oral dosage forms can affect the rate and extent of drug absorption.
Influence of the type of dosage form
The type of dosage form and its method of preparation or manufacture can influence bioavailability; that is whether a particular drug administered in the form of a solution, a suspension or solid dosage form can influence its rate and/or extent of absorption from the gastrointestinal tract. The type of oral dosage form will influence the number of possible intervening steps between administration and the appearance of dissolved drug in the gastrointestinal fluids, i.e. it will influence the release of drug into solution in the gastrointestinal fluids (Fig. 20.2).
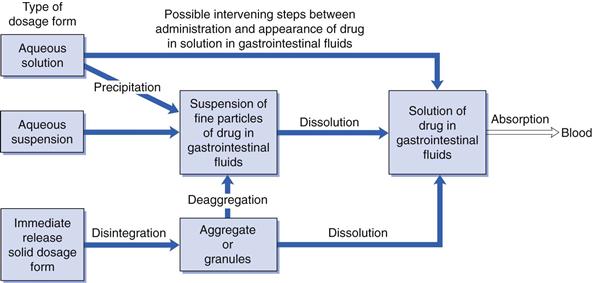
In general, drugs must be in solution in the gastrointestinal fluids before absorption can occur. Thus the greater the number of intervening steps, the greater will be the number of potential obstacles to absorption and the greater will be the likelihood of that type of dosage form reducing the bioavailability exhibited by the drug. Hence the bioavailability of a given drug tends to decrease in the following order of types of dosage form: aqueous solutions > aqueous suspensions > solid dosage forms (e.g. hard gelatin capsules or tablets). Although this ranking is not universal, it does provide a useful guideline. In general, solutions and suspensions are the most suitable for administering drugs intended to be rapidly absorbed. However, it should be noted that other factors (e.g. stability, patient acceptability, etc.) can also influence the type of dosage form in which a drug is administered via the gastrointestinal route.
Aqueous solutions
For drugs that are water soluble and chemically stable in aqueous solution, formulation as a solution normally eliminates the in vivo dissolution step and presents the drug in the most readily available form for absorption. However, dilution of an aqueous solution of a poorly water-soluble drug whose aqueous solubility had been increased by formulation techniques such as cosolvency, complex formation or solubilization can result in precipitation of the drug in the gastric fluids. Similarly, exposure of an aqueous solution of a salt of a weak acidic compound to gastric pH can also result in precipitation of the free acid form of the drug. In most cases the extremely fine nature of the resulting precipitate permits a more rapid rate of dissolution than if the drug had been administered in other types of oral dosage forms, such as an aqueous suspension, hard gelatin capsule or tablet. However, for some drugs this precipitation can have a major effect on bioavailability. For example, the same dose of an experimental drug was given to dogs in three different solution formulations: a polyethylene glycol solution and two different concentrations of hydroxypropyl-β-cyclodextrin. Bioavailabilities of 19%, 57% and 89% were obtained for polyethylene glycol, the lower concentration and the higher concentration of hydroxypropyl-β-cyclodextrin, respectively. The difference in bioavailability of the three solutions was attributed to the difference in precipitation rates of the candidate drug from the three solutions on dilution. The experimental drug was observed to precipitate most quickly from the polyethylene glycol solution, and slowest from the most concentrated hydroxypropyl-β-cyclodextrin solution.
Factors associated with the formulation of aqueous solutions that can influence drug bioavailability include:
Information concerning the potential influence of each of the above factors was given earlier in this chapter. Further details concerning the formulation and uses of oral solution dosage forms are given in Chapter 24.
Aqueous suspensions
An aqueous suspension is a useful dosage form for administering an insoluble or poorly water-soluble drug. Usually the absorption of a drug from this type of dosage form is dissolution rate limited. The oral administration of an aqueous suspension results in a large total surface area of dispersed drug being immediately presented to the gastrointestinal fluids. This facilitates dissolution and hence absorption of the drug. In contrast to powder-filled hard gelatin capsule and tablet dosage forms, dissolution of all drug particles commences immediately on dilution of the suspension in the gastrointestinal fluids. A drug contained in a tablet or hard gelatin capsule may ultimately achieve the same state of dispersion in the gastrointestinal fluids but only after a delay. Thus a well-formulated, finely subdivided aqueous suspension is regarded as being an efficient oral drug delivery system, second only to a non-precipitating solution-type dosage form.
Factors associated with the formulation of aqueous suspension dosage forms that can influence the bioavailabilities of drugs from the gastrointestinal tract include:
• the particle size and effective surface area of the dispersed drug
• the crystal form of the drug
• the inclusion of a surfactant as a wetting, flocculating or deflocculating agent
Information concerning the potential influence of the above factors on drug bioavailability is given in earlier sections of this chapter. Further information concerning the formulation and uses of suspensions as dosage forms is given in Chapter 26.
Liquid-filled capsules
Liquids can be filled into capsules made from soft or hard gelatin or hydroxypropylmethylcellulose (HPMC). Both types combine the convenience of a unit dosage form with the potentially rapid drug absorption associated with aqueous solutions and suspensions. Drugs encapsulated in liquid-filled capsules for peroral administration are dissolved or dispersed in non-toxic, non-aqueous vehicles. Sometimes the vehicles have thermal properties such that they can be filled into capsules while hot but are solids at room temperature.
The release of the contents of capsules is affected by dissolution and breaking of the shell. Following release, a water-miscible vehicle disperses and/or dissolves readily in the gastrointestinal fluids, liberating the drug (depending on its aqueous solubility) as either a solution or a fine suspension, which is conducive to rapid absorption. In the case of capsules containing drugs in solution or suspension in water-immiscible vehicles, release of the contents will almost certainly be followed by dispersion in the gastrointestinal fluids. Dispersion is facilitated by emulsifiers included in the vehicle, and also by bile. Once dispersed, the drug may end up as an emulsion, a solution, a fine suspension or a nano/microemulsion.
Well-formulated liquid-filled capsules are designed to improve the absorption of poorly soluble drugs and will ensure that no precipitation of drug occurs from the nano-or microemulsion formed in the gastrointestinal fluids. If the lipophilic vehicle is a digestible oil and the drug is highly soluble in the oil, it is possible that the drug will remain in solution in the dispersed oil phase and be absorbed (along with the oil) by fat absorption processes. For a drug that is less lipophilic or is dissolved in a non-digestible oil, absorption probably occurs following partitioning of the drug from the oily vehicle into the aqueous gastrointestinal fluids. In this case the rate of drug absorption appears to depend on the rate at which drug partitions from the dispersed oil phase. The increase in interfacial area of contact resulting from dispersion of the oily vehicle in the gastrointestinal fluids will facilitate partitioning of the drug across the oil/aqueous interface. For drugs suspended in an oily vehicle, release may involve dissolution in the vehicle, diffusion to the oil/aqueous interface and partition across the interface.
Many poorly water-soluble drugs have been found to exhibit greater bioavailabilities from liquid-filled capsule formulations. The cardiac glycoside digoxin, when formulated as a solution in a mixture of polyethylene glycol, ethanol and propylene glycol in a soft gelatin capsule, has been shown to be absorbed faster than from the standard commercial tablets.
More recently, far more complex capsule formulations have been investigated to improve the absorption of poorly soluble drugs. Ciclosporin is a large hydrophobic drug with poor permeability and solubility in gastrointestinal fluids. It showed low and variable oral bioavailability from its original liquid-filled soft gelatin capsule formulation (Sandiinmun) and was particularly sensitive to the presence of fat in diet and bile acids. In its newer formulation (Sandiinmun Neoral), which is a complex mixture of hydrophilic and lipophilic phases, surfactants, cosurfactants and a cosolvent, it forms a non-precipitating microemulsion on dilution with gastrointestinal fluids. It has a significantly improved bioavailability with reduced variability that is independent of the presence of food.
Many protease inhibitors (antiviral drugs) are peptidomimetic in nature. They have high molecular weights and low aqueous solubility, are susceptible to degradation in the lumen and extensive hepatic metabolism, and consequently have poor bioavailability. Saquinavir has been reformulated from a powder-filled hard gelatin capsule (Invirase) to a complex soft gelatin capsule formulation (Fortovase). The latter shows a significant improvement in bioavailability (3–4 times greater) over the standard hard gelatin capsule formulation and, as a consequence, a significantly greater viral load reduction
Factors associated with the formulation of liquid-filled capsules that can influence the bioavailabilities of drugs from this type of dosage form include:
• the solubility of the drug in the vehicle (and gastrointestinal fluids)
• the particle size of the drug (if suspended in the vehicle)
• the inclusion of a suspending agent (viscosity-enhancing agent) in the vehicle
• the complexation, i.e. formation, of a non-absorbable complex between the drug and any excipient.
More information on liquid-filled hard capsules and soft capsules can be found in Chapters 33 and 34, respectively.
Powder-filled capsules
Generally the bioavailability of a drug from a well-formulated powder-filled hard gelatin or HPMC capsule dosage form will be similar to that from the same drug in a well formulated compacted tablet. Provided the capsule shell dissolves rapidly in the gastrointestinal fluids and the encapsulated mass disperses rapidly and efficiently, a relatively large effective surface area of drug will be exposed to the gastrointestinal fluids, thereby facilitating dissolution. However, it is incorrect to assume that a drug formulated as a hard gelatin capsule is in a finely divided form surrounded by a water-soluble shell and that no bioavailability problems can occur. The overall rate of dissolution of drugs from capsules appears to be a complex function of the rates of different processes – such as the dissolution rate of the capsule shell, the rate of penetration of the gastrointestinal fluids into the encapsulated mass, the rate at which the mass deaggregates (i.e. disperses) in the gastrointestinal fluids and the rate of dissolution of the dispersed drug particles.
The inclusion of excipients (e.g. diluents, lubricants and surfactants) in a capsule formulation can have a significant effect on the rate of dissolution of drugs, particularly those that are poorly soluble and hydrophobic. Figure 20.3 shows that a hydrophilic diluent (e.g. sorbitol, lactose) often serves to increase the rate of penetration of the aqueous gastrointestinal fluids into the contents of the capsule and to aid the dispersion and subsequent dissolution of the drug in these fluids. However, the diluent should exhibit no tendency to adsorb or complex with the drug as either can impair absorption from the gastrointestinal tract.
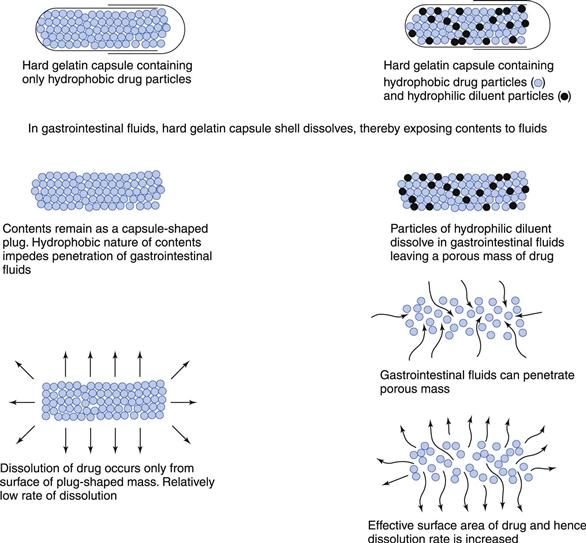
Both the formulation and the type and process conditions of the capsule-filling process can affect the packing density and liquid permeability into the capsule contents. In general, an increase in packing density (i.e. a decrease in porosity) of the encapsulated mass will result in a decrease in liquid permeability into the capsule mass and dissolution rate, particularly if the drug is hydrophobic or if a hydrophilic drug is mixed with a hydrophobic lubricant such as magnesium stearate. If the encapsulated mass is tightly packed and the drug is hydrophobic in nature, then a decrease in dissolution rate would be expected unless a surfactant had been included to facilitate liquid penetration into the mass.
In summary, formulation factors that can influence the bioavailabilities of drugs from capsules include:
• the use of the salt form of a drug in preference to the parent weak acid or base
• the crystal form of the drug
• the chemical stability of the drug (in the dosage form and in gastrointestinal fluids)
• the nature and quantity of the diluent, lubricant and wetting agent
• drug-excipient interactions (e.g. adsorption, complexation)
• the type and conditions of the filling process
• the packing density of the capsule contents
• the composition and properties of the capsule shell (including gastro-resistant capsules)
More information on powder-filled hard capsules can be found in Chapter 33.
Tablets
Uncoated tablets.
Tablets are the most widely used dosage form. When a drug is formulated as a compacted tablet there is an enormous reduction in the effective surface area of the drug, owing to the compaction processes involved in tablet making. These processes necessitate the addition of excipients, which serve to return the surface area of the drug back to its original precompacted state. Bioavailability problems can arise if a fine, well-dispersed suspension of drug particles in the gastrointestinal fluids is not generated following the administration of a tablet. Because the effective surface area of a poorly soluble drug is an important factor influencing its dissolution rate, it is especially important that tablets containing such drugs should disintegrate rapidly and completely in the gastrointestinal fluids if rapid release, dissolution and absorption are required. The overall rate of tablet disintegration is influenced by several interdependent factors, which include the concentration and type of drug, diluent, binder, disintegrant, lubricant and wetting agent, as well as the compaction pressure (discussed in Chapter 30).
The dissolution of a poorly soluble drug from an intact tablet is usually extremely limited because of the relatively small effective surface area of drug exposed to the gastrointestinal fluids. Disintegration of the tablet into granules causes a relatively large increase in effective surface area of drug and the dissolution rate may be likened to that of a coarse, aggregated suspension. Further disintegration into small, primary drug particles produces a further large increase in effective surface area and dissolution rate. The dissolution rate is probably comparable to that of a fine, well-dispersed suspension. Disintegration of a tablet into primary particles is thus important, as it ensures that a large effective surface area of a drug is generated in order to facilitate dissolution and subsequent absorption.
However, simply because a tablet disintegrates rapidly, does not necessarily guarantee that the liberated primary drug particles will dissolve in the gastrointestinal fluids and that the rate and extent of absorption are adequate. In the case of poorly water-soluble drugs, the rate-controlling step is usually the overall rate of dissolution of the liberated drug particles in the gastrointestinal fluids. The overall dissolution rate and bioavailability of a poorly soluble drug from an uncoated conventional tablet is influenced by many factors associated with the formulation and manufacture of this type of dosage form. These include:
• the nature and quantity of the diluent, binder, disintegrant, lubricant and any wetting agent
• drug-excipient interactions (e.g. complexation)
• the size of the granules and their method of manufacture
• the compaction pressure and speed of compaction used in tableting
Because drug absorption and hence bioavailability are dependent upon the drug being in the dissolved state, suitable dissolution characteristics can be an important property of a satisfactory tablet, particularly if it contains a poorly soluble drug. On this basis, specific in vitro dissolution test conditions and dissolution limits are included in many pharmacopoeias for tablets (and capsules) for certain drugs. That a particular drug product meets the requirements of a compendial dissolution standard provides a greater assurance that the drug will be released satisfactorily from the formulated dosage form in vivo and be absorbed adequately (also discussed in Chapters 21 and 35).
More information on drug release from tablets can be found in Chapter 30.
Coated tablets.
Tablet coatings may be used simply for aesthetic reasons, to improve the appearance of a tablet or to add a company identity, to mask an unpleasant taste or odour, to protect an ingredient from decomposition during storage or to protect health workers from the drug. Currently, the most common type of tablet coat is that created with a polymer film. However, several older preparations, such as tablets containing vitamins, ibuprofen and conjugated oestrogens, still have sugar coats.
The presence of a coating presents a physical barrier between the tablet core and the gastrointestinal fluids. Coated tablets therefore not only possess all the potential bioavailability problems associated with uncoated conventional tablets but are subject to the additional potential problem of being surrounded by a physical barrier. In the case of a coated tablet which is intended to disintegrate/dissolve and release drug rapidly into solution in the gastrointestinal fluids, the coating must dissolve or disrupt before these processes can begin. The physicochemical nature and thickness of the coating can thus influence how quickly a drug is released from a tablet.
In the process of sugar coating, the tablet core is usually sealed with a thin continuous film of a poorly water-soluble polymer such as shellac or cellulose acetate phthalate. This sealing coat serves to protect the tablet core and its contents from the aqueous fluids used in the subsequent steps of the sugar-coating process. The presence of this water-impermeable sealing coat can potentially retard drug release from sugar-coated tablets. In view of this potential problem, annealing agents such as polyethylene glycols or calcium carbonate, which do not substantially reduce the water impermeability of the sealing coat during sugar coating but which dissolve readily in gastric fluid, may be added to the sealer coat in order to reduce the barrier effect and to aid rapid drug release.
The film coating of a tablet core by a thin film of a water-soluble polymer, such as hydroxypropyl methycellulose, should have no significant effect on the rate of disintegration of the tablet core and subsequent drug dissolution, provided that the film coat dissolves rapidly and independently of the pH of the gastrointestinal fluids. However, if hydrophobic water-insoluble film-coating materials, such as ethylcellulose or certain acrylic resins, are used (Chapter 32), the resulting film coat acts as a barrier which delays and/or reduces the rate of drug release. Thus, these types of film-coating materials form barriers which can have a significant influence on drug absorption. Although the formation of such barriers would be disadvantageous in the case of film-coated tablets intended to provide rapid rates of drug absorption, the concept of barrier coating has been used (along with other techniques) to obtain more precise control over drug release than is possible with conventional uncoated tablets (see Chapters 31 and 32).
Gastro-resistant tablets.
The use of barrier coating to control the site of release of an orally administered drug is well illustrated by gastro-resistant tablets (formerly known as enteric-coated tablets). A gastro-resistant coat is designed to resist the low pH of gastric fluids but to disrupt or dissolve when the tablet enters the higher pH of the duodenum. Polymers such as cellulose acetate phthalate, hydroxypropyl methylcellulose phthalate, some copolymers of methacrylic acid and their esters and polyvinyl acetate phthalate can be used as gastro-resistant coatings. These materials do not dissolve over the gastric pH range but dissolve rapidly at the less acidic pH (about 5) associated with the small intestine. Gastro-resistant coatings should preferably begin to dissolve at pH 5 in order to ensure the availability of drugs which are absorbed primarily in the proximal region of the small intestine. Gastro-resistant coating thus provides a means of delaying the release of a drug until the dosage form reaches the small intestine. Such delayed release provides a means of protecting drugs which would otherwise be destroyed if released into gastric fluid. Hence, gastro-resistant coating serves to improve the oral bioavailability exhibited by such drugs from uncoated conventional tablets. Gastro-resistant coating also protects the stomach against drugs which can produce nausea or mucosal irritation (e.g. aspirin, ibuprofen) if released at this site.
In addition to the protection offered by gastro-resistant coating, the delayed release of drug also results in a significant delay in the onset of the therapeutic response of a drug. The onset of the therapeutic response is largely dependent on the residence time of the gastro-resistant tablet in the stomach. Gastric emptying of such tablets is an all-or-nothing process, i.e. the tablet is either in the stomach or in the duodenum. Consequently, drug is either not being released or is being released. The residence time of an intact gastro-resistant tablet in the stomach can vary from about 5 minutes to several hours (discussed further in Chapter 19). Hence there is considerable intra- and intersubject variation in the onset of therapeutic action exhibited by drugs administered as gastro-resistant tablets.
The formulation of a gastro-resistant product in the form of small individually coated granules or pellets (multiparticulates) contained in a rapidly dissolving capsule or a rapidly disintegrating tablet largely eliminates the dependency of this type of dosage form on the all-or-nothing gastric emptying process associated with intact (monolith) gastro-resistant tablets. Provided the coated granules or pellets are sufficiently small (around 1 mm diameter), they will be able to empty from the stomach with liquids. Hence gastro-resistant granules and pellets exhibit a gradual but continual release from the stomach into the duodenum. This type of release also avoids the complete dose of drug being released into the duodenum, as occurs with a gastro-resistant tablet. The intestinal mucosa is thus not exposed locally to a potentially toxic concentration of drug.
Further information on coated tablets and multiparticulates is given in Chapter 32.
Influence of excipients for conventional dosage forms
Drugs are almost never administered alone but rather in dosage forms that generally consist of a drug (or drugs) together with a varying number of other substances (excipients). Excipients are added to the formulation in order to facilitate the preparation, patient acceptability and functioning of the dosage form as a drug delivery system. Excipients include disintegrating agents, diluents, lubricants, suspending agents, emulsifying agents, flavouring agents, colouring agents, chemical stabilizers, etc. Although historically excipients were considered to be inert in that they themselves should exert no therapeutic or biological action or modify the biological action of the drug present in the dosage form, they are now regarded as having the ability to influence the rate and/or extent of absorption of the drug. For instance, the potential influence of excipients on drug bioavailability has already been illustrated by the formation of poorly soluble, non-absorbable drug-excipient complexes between tetracyclines and dicalcium phosphate, amphetamine and sodium carboxymethylcellulose, and phenobarbital and polyethylene glycol 4000.
Diluents.
An important example of the influence that excipients employed as diluents can have on drug bioavailability is provided by the observed increase in the incidence of phenytoin intoxication which occurred in epileptic patients in Australia as a consequence of the diluent in sodium phenytoin capsules being changed. Many epileptic patients who had been previously stabilized with sodium phenytoin capsules containing calcium sulfate dihydrate as the diluent developed clinical features of phenytoin overdosage when given sodium phenytoin capsules containing lactose as the diluent, even though the quantity of drug in each capsule formulation was identical. The experimental data from this study are shown in Figure 33.6. It was later shown that the excipient calcium sulfate dihydrate had been responsible for decreasing the gastrointestinal absorption of phenytoin, possibly because part of the administered dose of drug formed a poorly absorbable calcium phenytoin complex. Hence, although the size of dose and frequency of administration of the sodium phenytoin capsules containing calcium sulfate dihydrate gave therapeutic blood levels of phenytoin in epileptic patients, the efficiency of absorption of phenytoin had been lowered by the incorporation of this excipient in the hard gelatin capsules. Hence, when the calcium sulfate dihydrate was replaced by lactose, without any alteration in the quantity of drug in each capsule, or in the frequency of administration, an increased bioavailability of phenytoin was achieved. In many patients, the higher plasma levels exceeded the maximum safe concentration for phenytoin and produced toxic side-effects.
Surfactants.
Surfactants are often used in dosage forms as emulsifying agents, solubilizing agents, suspension stabilizers or wetting agents. However, surfactants in general cannot be assumed to be ‘inert’ excipients as they have been shown to be capable of increasing, decreasing or exerting no effect on the transfer of drugs across biological membranes.
Surfactant monomers can potentially disrupt the integrity and function of a biological membrane. Such an effect would tend to enhance drug penetration and hence absorption across the gastrointestinal barrier, but may also result in toxic side-effects. Inhibition of absorption may occur as a consequence of a drug being incorporated into surfactant micelles. If such surfactant micelles are not absorbed, which appears usually to be the case, then solubilization of a drug may result in a reduction of the concentration of ‘free’ drug in solution in the gastrointestinal fluids that is available for absorption. Inhibition of drug absorption in the presence of micellar concentrations of surfactant would be expected to occur in the case of drugs that are normally soluble in the gastrointestinal fluids, i.e. in the absence of surfactant. Conversely, in the case of poorly soluble drugs whose absorption is dissolution rate limited, the increase in saturation solubility of the drug by solubilization in surfactant micelles could result in more rapid rates of dissolution and hence absorption.
The release of poorly soluble drugs from tablets and capsules may be increased by the inclusion of surfactants in their formulations. The ability of a surfactant to reduce the solid/liquid interfacial tension will permit the gastrointestinal fluids to wet the solid more effectively and thus enable it to come into more intimate contact with the solid dosage forms. This wetting effect may thus aid the penetration of gastrointestinal fluids into the mass of capsule contents that often remains when the hard gelatin shell has dissolved, and/or reduce the tendency of poorly soluble drug particles to aggregate in the gastrointestinal fluids. In each case the resulting increase in the total effective surface area of drug in contact with the gastrointestinal fluids would tend to increase the dissolution and absorption rates of the drug. It is interesting to note that the enhanced gastrointestinal absorption of phenacetin in humans resulting from the addition of polysorbate 80 to an aqueous suspension of this drug was attributed to the surfactant preventing aggregation and thus increasing the effective surface area and dissolution rate of the drug particles in the gastrointestinal fluids.
The possible mechanisms by which surfactants can influence drug absorption are varied and it is likely that only rarely will a single mechanism operate in isolation. In most cases, the overall effect on drug absorption will probably involve a number of different actions of the surfactant (some of which will produce opposing effects on drug absorption) and the observed effect on drug absorption will depend on which of the different actions is the overriding one. The ability of a surfactant to influence drug absorption will also depend on the physicochemical characteristics and concentration of the surfactant, the nature of the drug and the type of biological membrane involved.
Lubricants.
Both tablets and capsules require lubricants in their formulation to reduce friction between the powder and metal surfaces during their manufacture. Lubricants are often hydrophobic in nature. Magnesium stearate is commonly included as a lubricant during tablet compaction and capsule-filling operations. Its hydrophobic nature often retards liquid penetration into capsule ingredients so that after the shell has dissolved in the gastrointestinal fluids, a capsule-shaped plug often remains, especially when the contents have been machine-filled as a consolidated plug (see Chapter 33). Similar reductions in dissolution rate are observed when magnesium stearate is included in tablets. Alternatively, they can be overcome by the simultaneous addition of a wetting agent (i.e. a water-soluble surfactant) and the use of a hydrophilic diluent (e.g. stearic acid) or minimized by decreasing the magnesium stearate content of the formulation.
Disintegrants.
Disintegrants are required to break up capsules, tablets and granules into primary powder particles in order to increase the surface area of the drug exposed to the gastrointestinal fluids. A tablet that fails to disintegrate or disintegrates slowly may result in incomplete absorption or a delay in the onset of action of the drug. The compaction force used in tablet manufacture can affect disintegration. In general, the higher the force, the slower the disintegration time. Even small changes in formulation may result in significant effects on dissolution and bioavailability. A classic example is that of tolbutamide where two formulations, the commercial product and the same formulation but containing half the amount of disintegrant, were administered to healthy volunteers. Both tablets disintegrated in vitro within 10 minutes, meeting pharmacopoeial specifications, but the commercial tablet had a significantly greater bioavailability and hypoglycaemic response.
Viscosity-enhancing agents.
Viscosity-enhancing agents are often employed in the formulation of liquid dosage forms for oral use in order to control such properties as palatability, ease of pouring and, in the case of suspensions, the rate of sedimentation of the dispersed particles. Viscosity-enhancing agents are often hydrophilic polymers.
There are a number of mechanisms by which a viscosity-enhancing agent may produce a change in the gastrointestinal absorption of a drug. Complex formation between a drug and a hydrophilic polymer could reduce the concentration of drug in solution that is available for absorption. The administration of viscous solutions or suspensions may produce an increase in viscosity of the gastrointestinal contents. In turn, this could lead to a decrease in dissolution rate and/or a decrease in the rate of movement of drug molecules to the absorbing membrane.
Normally, a decrease in the rate of dissolution would not be applicable to solution dosage forms unless dilution of the administered solution in the gastrointestinal fluids caused precipitation of the drug.
In the case of suspensions containing drugs with bioavailabilities that are dissolution rate dependent, an increase in viscosity could also lead to a decrease in the rate of dissolution of the drug in the gastrointestinal tract.
Summary
As well as physiological and drug factors, the dosage form can play a major role in influencing the rate and extent of absorption. Often this is by design. However, even with conventional dosage forms, it is important to consider whether changing the dosage form or excipients will affect the bioavailability of the drug. Some drugs will be more susceptible to changes in rate and extent of absorption through dosage form changes than others; this will depend on the biopharmaceutical properties of the drug, which form the basis of the next chapter (Chapter 21).
Bibliography
1. Avdeeff A, Voloboy D, Foreman A. Dissolution and solubility. Taylor JB, Triggle DJ, eds. Comprehensive Medicinal Chemistry II. 2007;5:399–423.
2. Buggins T, Dickinson P, Taylor G. The effect of pharmaceutical excipients on drug disposition. Advanced Drug Delivery Reviews. 2007;59:1482–1503.
3. Florence AT, Attwood D. Physicochemical Principles of Pharmacy. 5th edn London: Pharmaceutical Press; 2011.
4. Stahl H, Wermuth CG. Pharmaceutical Salts: Properties, Selection and Use. Germany: International Union of Pure and Applied Chemistry, Wiley-VCH; 2002.
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
Bioavailability – physicochemical and dosage form factors
Marianne Ashford
Chapter contents
Physicochemical factors influencing bioavailability
Dosage form factors influencing bioavailability
Key points
• Lipid solubility and the drug dissociation affect drug absorption.
Introduction
As discussed in Chapter 19, the rate and extent of drug absorption are influenced by the physiological factors associated with the structure and function of the gastrointestinal tract. This chapter discusses the physicochemical properties of the drug and dosage form factors that influence bioavailability. For a drug to be absorbed, it needs to be in solution and to be able to pass across the membrane. In the case of orally administered drugs, this is the gastrointestinal epithelium. The physicochemical properties of the drug that will influence its passage into solution and transfer across membranes include its dissolution rate, pKa, lipid solubility, chemical stability and complexation potential.
Physicochemical factors influencing bioavailability
Dissolution and solubility
Solid drugs need to dissolve before they can be absorbed. The dissolution of drugs can be described by the Noyes–Whitney equation (Eqn 20.1). This equation, first proposed in 1897, describes the rate of diffusion of solute through boundary layers surrounding a dissolving spherical particle. When the dissolution process is diffusion controlled and involves no chemical reaction then this also equates to the rate of dissolution:
(20.1)
where dm/dt is the rate of dissolution of the drug particles, D is the diffusion coefficient of the drug in solution in the gastrointestinal fluids, A is the effective surface area of the drug particles in contact with the gastrointestinal fluids, h is the thickness of the diffusion layer around each drug particle, Cs is the saturation solubility of the drug in solution in the diffusion layer and C is the concentration of the drug in the gastrointestinal fluids.
More detail regarding Noyes–Whitney equation and its limitations in describing the dissolution of drug particles are outlined in Chapter 2. Despite these limitations, the equation serves to illustrate and explain how various physicochemical and physiological factors can influence the rate of dissolution in the gastrointestinal tract. These are summarized in Table 20.1 and are discussed in more detail in the next section.
Table 20.1
| Factor | Physicochemical parameter | Physiological parameter |
| Effective surface area of drug | Particle size, wettability | Surfactants in gastric juice and bile. pH, buffer capacity, bile, food components |
| Solubility in diffusion layer | Hydrophilicity, crystal structure | |
| Amount of drug already dissolved | Solubilization | Permeability, transit |
| Diffusivity of drug | Molecular size | Viscosity of luminal contents |
| Boundary layer thickness | Motility patterns and flow rate | |
| Volume of solvent available | Gastrointestinal secretions, co-administered fluids |
Figure 20.1 illustrates the dissolution of a spherical drug particle in the gastrointestinal fluids.
Fig. 20.1 Schematic representation of the dissolution of a drug particle in the gastrointestinal fluids.
Physiological factors affecting the dissolution rate of drugs
The environment of the gastrointestinal tract can affect the parameters of the Noyes–Whitney equation (Eqn 20.1) and hence the dissolution rate of a drug. For instance, the diffusion coefficient, D, of the drug in the gastrointestinal fluids may be decreased by the presence of substances that increase the viscosity of the fluids. Hence the presence of food in the gastrointestinal tract may cause a decrease in drug dissolution rate by reducing the rate of diffusion of the drug molecules away from the diffusion layer surrounding each undissolved drug particle. Surfactants in gastric juice and bile salts will affect both the wettability of the drug, and hence its effective surface area, A, exposed to gastrointestinal fluids, and the solubility of the drug in the gastrointestinal fluids via micellization. The thickness of the diffusion layer, h, will be influenced by the degree of agitation experienced by each drug particle in the gastrointestinal tract. Hence, an increase in gastric and/or intestinal motility may increase the dissolution rate of a sparingly soluble drug by decreasing the thickness of the diffusion layer around each drug particle.
The concentration of drug in solution in the bulk of the gastrointestinal fluids, C, will be influenced by such factors as the rate of removal of dissolved drug by absorption through the gastrointestinal-blood barrier and by the volume of fluid available for dissolution, which in turn will be dependent on the location of the drug in the gastrointestinal tract and the timing with respect to meal intake. In the stomach, the volume of fluid will be influenced by the intake of fluid in the diet. According to the Noyes–Whitney equation, a low value of C will favour more rapid dissolution of the drug by virtue of increasing the value of the term (Cs − C). In the case of drugs whose absorption is dissolution-rate limited, the value of C is normally kept very low by absorption of the drug. Hence dissolution occurs under sink conditions; that is, under conditions such that the value of (Cs − C) approximates to Cs. Thus for the dissolution of a drug in the gastrointestinal tract under sink conditions, the Noyes–Whitney equation can be expressed as:
(20.2)
Drug factors affecting dissolution rate
Drug factors that can influence the dissolution rate are the particle size, the wettability, the solubility and the form of the drug (whether a salt or a free form).
Surface area and particle size.
According to Equation 20.1, an increase in the total surface area of drug in contact with the gastrointestinal fluids will cause an increase in dissolution rate. Provided that each particle of drug is intimately wetted by the gastrointestinal fluids, the effective surface area exhibited by the drug will be inversely related to the particle size of the drug. Hence the smaller the particle size, the greater the effective surface area exhibited by a given mass of drug and the higher the dissolution rate. Particle size reduction is thus likely to result in increased bioavailability, provided that the absorption of the drug is dissolution rate limited.
One of the classic examples of particle size effects on the bioavailability of poorly soluble compounds is that of griseofulvin, where a reduction of particle size from about 10 µm (specific surface area = 0.4 m2 g−1) to 2.7 µm (specific surface area = 1.5 m2 g−1) was shown to produce approximately double the amount of drug absorbed in humans. Many poorly soluble, slowly dissolving drugs are routinely presented in micronized form to increase their surface area.
Examples of drugs where a reduction in particle size has been shown to improve the rate and extent of oral absorption and hence bioavailability are shown in Table 20.2. Such improvements in bioavailability can result in an increased incidence of side-effects; thus for certain drugs it is important that the particle size is well controlled, and many pharmacopoeias state a requirement for particle size.
Table 20.2
Examples of drugs where a reduction in particle size has led to improvements in bioavailability
| Drug | Therapeutic class |
| Digoxin | Cardiac glycoside |
| Nitrofurantoin | Antifungal |
| Medroxyprogesterone | Hormone acetate |
| Danazol | Steroid |
| Tolbutamide | Antidiabetic |
| Aspirin | Analgesic |
| Sulfadiazine | Antibacterial |
| Naproxen | Non-steroidal anti-inflammatory |
| Ibuprofen | Non-steroidal anti-inflammatory |
| Phenacetin | Analgesic |
| Griseofulvin | Antifungal |
| Fenofibrate | Lipid regulating agent |
| Megestrol acetate | Apetite loss |
| Aprepitant | Anti-emetic |
| Rapamycin | Immunosuppressant |
| Lapinovir/ritonavir | HIV protease inhibitors |
For some drugs, particularly those that are hydrophobic in nature, micronization and other dry particle size reduction techniques can result in aggregation of the material. This will cause a consequent reduction in the effective surface area of the drug exposed to the gastrointestinal fluids and hence a reduction in its dissolution rate and bioavailability. Aspirin, phenacetin and phenobarbital are all prone to aggregation during particle size reduction. One approach that may overcome this problem is to micronize or mill the drug with a wetting agent or hydrophilic carrier. To overcome aggregation and to achieve particle sizes in the nano-size region, wet milling in the presence of stabilizers has been used. The relative bioavailability of danazol has been increased 400% by administering particles in the nanometre rather than the micrometre size range.
There are now several specialized drug delivery companies who can produce solid dosage forms with the drug stabilized in the nano-size range to afford greater bioavailaility. Examples of commercialized products are the immunosuppressant Rapamune®, the anti-emetic Emend® and the lipid regulating agent TriCor® containing fenofibrate. Megace® ES is an oral nano-suspension of megestrol acetate for the treatment of appetite loss, severe malnutrition, or unexplained significant weight loss in AIDS patients. It is a reformulation of the oral suspension using Nanocrystal® technology to improve the dissolution rate, absorption rate and bioavailability of the original formulation. The formulation is less viscous and allows a quarter of the volume to be dosed, thus aiding patient swallowing and compliance.
As well as milling with wetting agents, the effective surface area of hydrophobic drugs can be increased by the addition of a wetting agent to the formulation. The presence of polysorbate 80 in a fine suspension of phenacetin (particle size less than 75 µm) greatly improved the rate and extent of absorption of the phenacetin in human volunteers compared to the same-size suspension without a wetting agent. Polysorbate 80 helps by increasing the wetting and solvent penetration of the particles and by minimizing aggregation of suspended particles, thereby maintaining a large effective surface area. Wettability effects are highly drug specific; however wetting agents are routinely added to many formulations.
If an increase in the effective surface area of a drug does not increase its absorption rate, it is likely that the dissolution process is not rate limiting. For drugs such as penicillin G and erythromycin, which are unstable in gastric fluids, their chemical degradation will be minimized if they remain in the solid state. Thus, particle size reduction would not only serve to increase their dissolution rate but would simultaneously increase chemical degradation and therefore reduce the amount of intact drug available for absorption.
Solubility in the diffusion layer, Cs.
The dissolution rate of a drug under sink conditions, according to the Noyes–Whitney equation (Eqn 20.2), is directly proportional to its intrinsic solubility in the diffusion layer surrounding each dissolving drug particle, Cs. The aqueous solubility of a drug is dependent on the interactions between molecules within the crystal lattice, intermolecular interactions with the solution in which it is dissolving, and the entropy changes associated with fusion and dissolution. In the case of drugs that are weak electrolytes, their aqueous solubility is dependent on pH (as discussed in Chapter 2). Hence in the case of an orally administered solid dosage form containing a weak electrolyte drug, the dissolution rate of the drug will be influenced by its solubility and the pH in the diffusion layer surrounding each dissolving drug particle. The pH in the diffusion layer – the microclimate pH – for a weak electrolyte will be affected by the pKa and solubility of the dissolving drug and the pKa and solubility of the buffers in the bulk gastrointestinal fluids. Thus differences in dissolution rate will be expected in different regions of the gastrointestinal tract.
The solubility of weakly acidic drugs increases with pH and so as a drug moves down the gastrointestinal tract from the stomach to the intestine, its solubility will increase. Conversely, the solubility of weak bases decreases with increasing pH, i.e. as the drug moves down the gastrointestinal tract. It is important therefore for poorly soluble weak bases to dissolve rapidly in the stomach, as the rate of dissolution in the small intestine will be much slower. The antifungal drug ketoconazole, a weak base, is particularly sensitive to gastric pH. Dosing ketoconazole 2 hours after the administration of the H2 blocker cimetidine, which reduces gastric acid secretion, results in a significantly reduced rate and extent of absorption Similarly, in the case of the antiplatelet drug dipyrimidole, pretreatment with the H2 blocker famotidine reduces the peak plasma concentration by a factor of up to 10.
Salts.
The solubility of a weakly acidic drug in gastric fluid (pH 1–3.5) will be relatively low however it will be much greater in the higher pH of the intestine. The sodium salt of a weak acid will dissociate as shown:
(20.3)
where D is the drug and X is the counter ion. The concentrations of the drug multiplied by the counter ion concentration at any pH will give the solubility product Ksp, i.e.:
(20.4)
The pH solubility profile of a weak acid in the presence of counterions depends on the solubility product of the ionized drug and its counter-ions and is depicted for both a weak acid and a weak base.
Many examples can be found of the effects of salts improving the rate and extent of absorption. The dissolution rate of the oral hypoglycaemic drug, tolbutamide sodium in 0.1 M HC1 is 5000 times faster than that of the free acid. Oral administration of a non-disintegrating disc of the more rapidly dissolving sodium salt of tolbutamide produces a very rapid decrease in blood sugar level (a consequence of the rapid rate of drug absorption), followed by a rapid recovery. In contrast, a non-disintegrating disc of the tolbutamide free acid produces a much slower rate of decrease of blood sugar (a consequence of the slower rate of drug absorption) that is maintained over a longer period of time. The barbiturates are often administered in the form of sodium salts to achieve a rapid onset of sedation and provide more predictable effects.
The non-steroidal anti-inflammatory drug naproxen was originally marketed as the free acid for the treatment of rheumatoid and osteoarthritis. However, the sodium salt (naproxen sodium) is absorbed faster, due to faster dissolution of the dosage from, and hence is more effective, and thus has now largely replaced the free form. Conversely, strongly acidic salt forms of weakly basic drugs, for example chlorpromazine hydrochloride, dissolve more rapidly in gastric and intestinal fluids than do the free bases (e.g. chlorpromazine). The presence of strongly acidic anions (e.g. Cl− ions) in the diffusion layer around each drug particle ensures that the pH in that layer is lower than the bulk pH in either the gastric or the intestinal fluid. This lower pH will increase the solubility of the drug in the diffusion layer.
The oral administration of a salt form of a weakly basic drug in a solid oral dosage form generally ensures that dissolution occurs in the gastric fluid before the drug passes into the small intestine where pH conditions are unfavourable. Thus the drug should be delivered to the major absorption site, the small intestine, in solution. If absorption is fast enough, precipitation of the dissolved drug is unlikely to significantly affect bioavailability. It is important to be aware that hydrochloride salts may experience a common ion effect owing to the presence of chloride ions in the stomach (also discussed in Chapter 2). The in vitro dissolution of a sulfate salt of an HIV protease inhibitor analogue is significantly greater in hydrochloric acid than that of the hydrochloride salt. The bioavailability of the sulfate salt is more than three times greater than that of the hydrochloride salt. These observations are attributed to the common ion effect of the hydrochloride.
The sodium salts of acidic drugs and the hydrochloride salts of basic drugs are by far the most common. However, many other salt forms are increasingly being employed (there is further information in Chapter 23). Some salts have a lower solubility and dissolution rate than the free form, for example aluminium salts of weak acids and palmoate salts of weak bases. In these cases, insoluble films of either aluminium hydroxide or palmoic acid are found to coat the dissolving solids when the salts are exposed to a basic or an acidic environment, respectively. In general, poorly soluble salts delay absorption and may therefore be used to sustain the release of the drug. A poorly soluble salt form is generally employed for suspension dosage forms.
Although salt forms are often selected to improve bioavailability, other factors such as chemical stability, hygroscopicity, manufacturability and crystallinity will all be considered during salt selection and may preclude the choice of a particular salt. The sodium salt of aspirin, sodium acetylsalicylate, is much more prone to hydrolysis than is aspirin, acetylsalicylic acid, itself. One way to overcome chemical instabilities or other undesirable features of salts is to form the salt in situ or to add basic/acidic excipients to the formulation of a weakly acidic or weakly basic drug. The presence of the basic excipients in the formulation of acidic drugs ensures that a relatively basic diffusion layer is formed around each dissolving particle. The inclusion of the basic ingredients aluminium dihydroxyaminoacetate and magnesium carbonate in aspirin tablets was found to increase their dissolution rate and bioavailability.
Crystal form
Polymorphism
Many drugs can exist in more than one crystalline form. This property is referred to as polymorphism and each crystalline form is known as a polymorph (discussed further in Chapter 8). As discussed in Chapters 2 and 8, a metastable polymorph usually exhibits a greater dissolution rate than the corresponding stable polymorph. Consequently, the metastable polymorphic form of a poorly soluble drug may exhibit an increased bioavailability compared to the stable polymorphic form.
A classic example of the influence of polymorphism on drug bioavailability is provided by chloramphenicol palmitate. This drug exists in three crystalline forms designated A, B and C. At normal temperature and pressure, A is the stable polymorph, B is the metastable polymorph and C is the unstable polymorph. Polymorph C is too unstable to be included in a dosage form but polymorph B, the metastable form, is sufficiently stable. The plasma profiles of chloramphenicol from orally administered suspensions containing varying proportions of the polymorphic forms A and B were investigated. The extent of absorption of chloramphenicol increases as the proportion of the polymorphic form B of chloramphenicol palmitate is increased in each suspension. This was attributed to the more rapid in vivo rate of dissolution of the metastable polymorphic form, B, of chloramphenicol palmitate. Following dissolution, chloramphenicol palmitate is hydrolysed to give free chloramphenicol in solution, which is then absorbed. The stable polymorphic form A of chloramphenicol palmitate dissolves so slowly and consequently is hydrolysed so slowly to chloramphenicol in vivo that this polymorph is virtually ineffective. The importance of polymorphism to the gastrointestinal bioavailability of chloramphenicol palmitate is reflected by a limit being placed on the content of the inactive polymorphic form A, in chloramphenicol palmitate mixture.
Amorphous solids
In addition to different polymorphic crystalline forms, a drug may exist in an amorphous form (see Chapter 8). Because the amorphous form usually dissolves more rapidly than the corresponding crystalline form(s), the possibility exists that there will be significant differences in the bioavailabilities exhibited by the amorphous and crystalline forms of drugs that show dissolution rate-limited bioavailability.
A classic example of the influence of amorphous versus crystalline forms of a drug on its gastrointestinal bioavailability is provided by the antibiotic novobiocin. The more soluble and rapidly dissolving amorphous form of novobiocin was readily absorbed following oral administration of an aqueous suspension. However, the less soluble and slower dissolving crystalline form was not absorbed to any significant extent. The crystalline form was thus therapeutically ineffective. A further important observation was made in the case of aqueous suspensions of novobiocin. The amorphous form slowly converts to the more thermodynamically stable crystalline form, with an accompanying loss of therapeutic effectiveness. Thus, unless adequate precautions are taken to ensure the stability of the less stable, more therapeutically effective amorphous form of a drug in a dosage form, then unacceptable variations in therapeutic effectiveness may occur.
Several delivery technologies for poorly soluble drugs rely on stabilizing the drug in its amorphous form to increase its dissolution and bioavailability. An example of this is Kaletra® which is a combination tablet of the protease inhibitors lopinavir and ritinovir used for HIV infection, in combination with other antiretroviral drugs. These drugs are stabilized in their amorphous form by a polymer, copovidone, following melt extrusion of the drug with the polymer. The tablets provide a significant improvement in bioavailability and variability such that two medium sized tablets are equivalent to three large capsules of the old formulation.
Solvates
Another variation in the crystalline form of a drug can occur if the drug is able to associate with solvent molecules to produce crystalline forms known as solvates (discussed further in Chapter 8). When water is the solvent, the solvate formed is called a hydrate. Generally, the greater the solvation of the crystal, the lower is the solubility and dissolution rate in a solvent identical to the solvation molecules. As the solvated and non-solvated forms usually exhibit differences in dissolution rates, they may also exhibit differences in bioavailability, particularly in the case of poorly soluble drugs that exhibit dissolution rate-limited bioavailability.
An example is that of the antibiotic ampicillin. The faster dissolving anhydrous form of ampicillin is absorbed to a greater extent from both hard gelatin capsules and an aqueous suspension than is the slower dissolving trihydrate form. The anhydrous form of the hydrochloride salt of a protease inhibitor, an analogue of indinavir, has a much faster dissolution rate than the hydrated form in water. This is reflected in a significantly greater rate and extent of absorption and a more than doubling of the bioavailability of the anhydrous form.
Factors affecting the concentration of drug in solution in the gastrointestinal fluids
The rate and extent of absorption of a drug depend on the effective concentration of that drug, i.e. the concentration of drug in solution in the gastrointestinal fluids, which is in an absorbable form. Complexation, micellar solubilization, adsorption and chemical stability are the principal physicochemical properties that can influence the effective drug concentration in the gastrointestinal fluids.
Complexation.
Complexation of a drug may occur within the dosage form and/or in the gastrointestinal fluids, and can be beneficial or detrimental to absorption.
Mucin which is present in gastrointestinal fluids forms complexes with some drugs. The antibiotic streptomycin binds to mucin, thereby reducing the available concentration of the drug for absorption. It is thought that this may contribute to its poor bioavailability. Another example of complexation is that between drugs and dietary components, as in the case of the tetracyclines, which is discussed in Chapter 19.
[/not-level-membership-for-basic-science-category]
