Chapter 26 Basic Mechanisms of Pathological Retinal and Choroidal Angiogenesis
Introduction
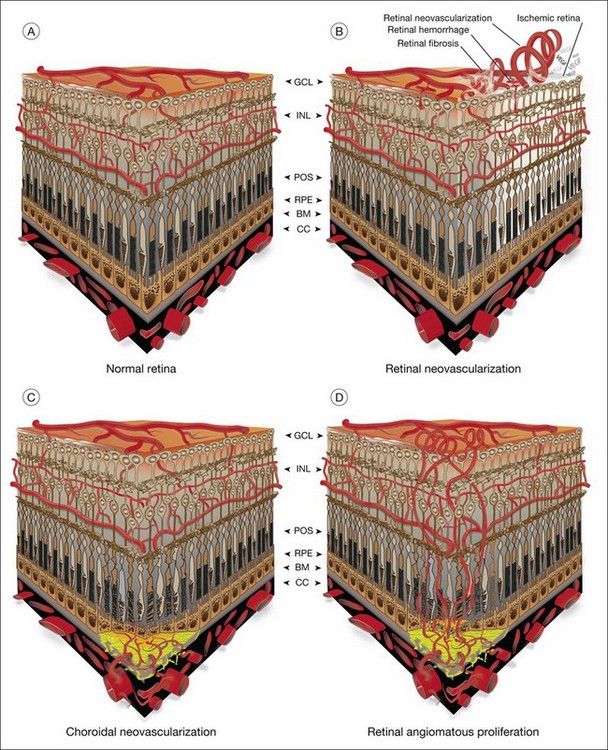
The vasculature of the eye is completely formed shortly after birth and is normally quiescent and nonproliferating in adults.1 An active balance of pro- and antiangiogenic influences is required to maintain this state of homeostasis. Neovascularization, or angiogenesis, occurs when environmental changes result in tissue expression of molecules that favor the growth of abnormal vessels prone to leak, bleed, and cause fibrous proliferation. In the retina, neovascularization causes vitreous hemorrhage and traction retinal detachment in diseases such as diabetic retinopathy (DR) and retinopathy of prematurity (ROP). Choroidal neovascularization (CNV) denotes the pathologic growth of new blood vessels from pre-existing choroidal vessels into the subretinal space. The newly formed vessels lie between the choroid and the retinal pigment epithelium (RPE) or between the native RPE and the neurosensory retina; thus, CNV is also referred to as subretinal neovascularization. In some patients neovascularization forms in the neurosensory retina, and at later stages may extend to the choroidal space. This is also known as retinal angiomatous proliferation or retinochoroidal anastomosis. Neovascularization leads to destruction of the retina and formation of a disciform scar in diseases such as neovascular age-related macular degeneration (AMD) (Fig. 26.1).
Pathogenesis
In angiogenesis, capillary endothelial cells proliferate from pre-existing blood vessels. Activated endothelial cells secrete proteolytic enzymes that dissolve basement membrane around the parent vessel and align themselves to form a new capillary sprout. By curving and elongating, the sprouts form tubes with lumens which anastomose to form loops. Mesenchymal cells are recruited to form smooth-muscle cells, or pericytes, and new basement membrane is deposited.2 More recently it has been demonstrated that hematopoietic stem cells may potentially differentiate into various cellular lineages, including endothelial progenitor cells, which migrate and contribute to sites of choroidal and retinal neovascularization.3–6
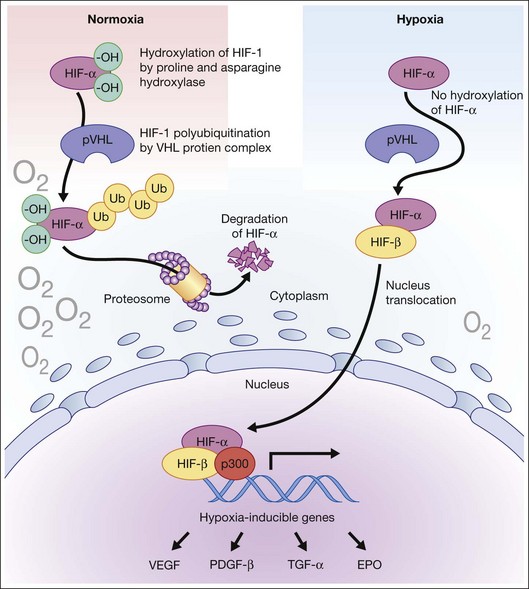
Hypoxia is a primary stimulus for angiogenesis. In early studies of retinal development, growing vessels were noted to invade the developing retina, giving rise to the hypothesis that a diffusible hypoxia-induced growth factor initiated this blood vessel growth.7,8 In DR and other adult vasculopathies, neovascularization was associated with retinal capillary nonperfusion.9–11 Experimental laser vein occlusion in the miniature pig and monkey models have demonstrated this.12,13 Hypoxia prevents the ubiquitin proteosome system degradation of the transcription factor hypoxia-inducible factor 1-alpha (HIF-1α). Stable HIF-1α binds to the promoter region of its major target gene, vascular endothelial growth factor (VEGF), resulting in transcriptional upregulation of VEGF and causing angiogenesis (Fig. 26.2).14–16
Response to tissue injury also plays a key role in angiogenesis. CNV occurs both experimentally and clinically after injury to the outer retina, iatrogenic laser injury, traumatic choroidal ruptures, and outer retinal choroidopathies.17,18 Sublethal cellular injury may release growth factors such as basic fibroblast growth factor (bFGF or FGF-2).19 Growth factor-secreting monocytes may be recruited to the site of tissue injury, which may increase the expression of growth factors causing angiogenesis.20
Free radicals in the form of reactive oxygen species (ROS) or intermediates have been implicated in the pathogenesis of numerous vascular diseases. ROS initiate the repair process by stimulating an angiogenic response. There is evidence that, over time, continual ROS stress depletes tissue of its antioxidant capabilities. Oxidative stress and inflammation may play a role in the pathogenesis of AMD.21 Increase in free radicals is believed to result in an overexpression of growth factors by the RPE, causing a proinflammatory state, damage to Bruch’s membrane, and ultimately CNV.22 The Age-Related Eye Disease Study (AREDS), a multicentered randomized clinical trial, showed a clinical benefit of high-dose antioxidants in reducing visual loss associated with macular degeneration.23
CNV associated with AMD (Fig. 26.3)
Aging and senescence of the RPE
The incidence and progression of nearly every feature of AMD, including CNV, relate to age.24 Lipofuscin, a byproduct of photoreceptor outer-segment digestion by lysosomes, increases with age in RPE as lysosomal activity decreases in RPE with aging. Progressive accumulation of lipofuscin is thought to result in disturbance of RPE function. Aged RPE cells may decrease production of antiangiogenic molecules such as pigment epithelium-derived growth factor (PEDF) as successive passage of cultured RPE cells results in diminished production of PEDF.25–27
Drusen, basal laminar/linear deposit formation
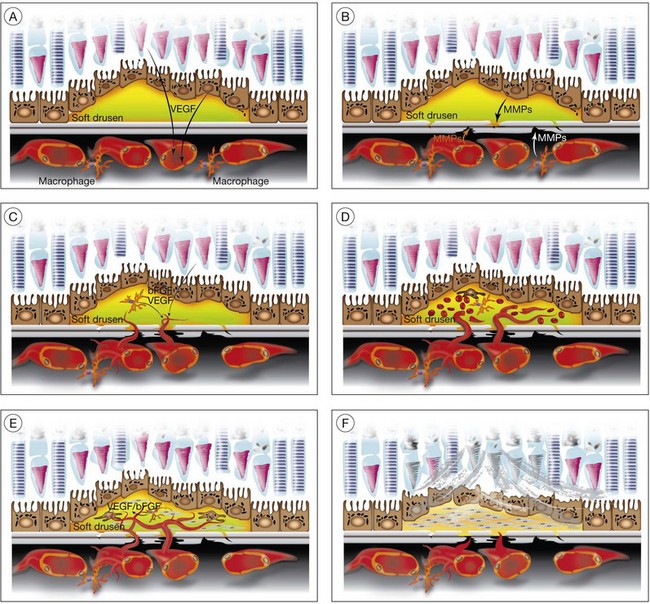
Soft drusen, unlike hard drusen, appear to be an important associated and predisposing feature of CNV. It is thought that membranous accumulation of debris as part of a diffuse disturbance of the RPE, softening of hard drusen, and cleavage in basal laminar/linear deposits may aid in the formation of soft drusen.28–32
Histopathologic studies also reveal that basal laminar deposits (accumulating between the plasma and basement membrane of the RPE) and basal linear deposits (with a thickening of the inner collagenous zone of the Bruch’s membrane) also have important associations with CNV.20 Therefore, abnormal deposits that occur with diffuse distribution pattern between the RPE layer and Bruch’s membrane are predisposing features of CNV. It is hypothesized that deposits between the RPE layer and Bruch’s membrane may block the diffusion of oxygen and nutrients from choriocapillaris to the RPE monolayer and photoreceptors.33 This speculative localized cellular hypoxia could result in overexpression of angiogenic growth factors such as VEGF, which, in turn, induce neovascularization from the choroidal vasculature. Additionally, the deposits are known to contain components of the immune response, and thus may act as initiators of inflammation.34–37 The deposits may also serve as a reservoir for sequestration of factors such as advanced glycation end products (AGE)38,39 that may affect the function of adjacent RPE and choroidal endothelial cells.
Enzymatic and mechanical disruption of Bruch’s membrane
In CNV, activated endothelial cells migrate through Bruch’s membrane; this process occurs by degradation of an intact Bruch’s membrane, or growth through an existing Bruch’s membrane break. Clinicopathologic studies suggest that classic CNVs are predominantly subretinal in location, whereas occult CNVs are predominantly sub-RPE.40 Bruch’s membrane may be disrupted when the balance between proteolytic enzymes such as matrix metalloproteinases (MMPs) and their inhibitors, the tissue inhibitors of metalloproteinases (TIMPs), favors a proteolytic environment. RPE cells express MMP-1 (interstitial collagenase),41 MMP-2 (72-kDa gelatinase),41,42 MMP-3 (stromolysin), and MMP-9 (92-kDa gelatinase),41 as well as TIMP-1,42,43 TIMP-2, and TIMP-3.43 Thus, proteolysis of Bruch’s membrane may potentially result from reduced expression of TIMPs or increased expression of MMPs. MMPs also play an important role in degrading the extracellular matrix at the leading edge of neovascular fronds. It has been shown that decreases in thickness and integrity of the elastic layer of Bruch’s membrane are seen in the macula of eyes with AMD but not in controls.44 Lysyl oxidase-like (LOXL) protein 1 has been shown to guide the spatially defined deposition of elastin and is essential for the maintenance of elastic fibers. LOXL1-deficient mice have been shown to develop increased neovascularization after laser injury.45 Impairment in other components of the Bruch’s membrane such as collagen XVIII has been shown to affect CNV formation in animals. Collagen XVIII knockout mice develop normal choroidal vasculature; however, they demonstrate increased size and leakage of laser-induced CNV.46
Transgenic mice with an intact Bruch’s membrane that overexpress VEGF in photoreceptors develop subretinal neovascularization; however, the subretinal vessels extend from retinal vessels rather than the choroidal vasculature.47 In contrast, transgenic mice that overexpress VEGF in RPE cells show intrachoroidal CNV.48 These findings support the notion that CNV requires both the expression of an angiogenic factor and a break in Bruch’s membrane (by proteolysis, physical disruption, or pre-existing break).
Macrophages are an alternative source of enzymes (such as MMPs) that could cause focal disruption of Bruch’s membrane.49 Histopathologic studies reveal that macrophages accumulate near thinned segments of Bruch’s membrane.50 In AMD, the RPE shows increased expression of monocyte chemoattractant protein-1 (MCP-1), a factor critical for macrophage recruitment.51 Macrophages in the choroid may subsequently degrade Bruch’s membrane, thus forming a passage that can be used by activated choroidal endothelial cells to gain entrance to the sub-RPE space.
Complement, AMD, and CNV
Recent studies suggest that complement dysregulation may mediate AMD pathogenesis. Complement components have been detected in drusen29,30,34–37 and an animal model of CD59 knockout leads to increased CNV.52 Most recently, a Y402H polymorphism in complement factor H (CFH) has been associated with increased risk of AMD and likely wet AMD.53–56 This particular allele of CFH leads to decrease in its activity and thus an increased complement-mediated inflammation since CFH is a negative regulator of the alternative pathway. Other human studies have identified protective haplotypes in two other genes encoding complement proteins, factor B (BF) and complement component 2 (C2).57
Studies of human donor eyes have shown increased levels of C-reactive protein and decreased CFH in the RPE/Bruch’s membrane/choriocapillaris region of eyes with AMD compared to controls, suggesting a role of complement and inflammation in the progression of the disease.58
Inflammation, bone marrow-derived cells, and CNV
Macrophages are an additional source of angiogenic growth factors that may promote the development of CNV. Activated macrophages show increased expression of inflammatory cytokines such as tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β), which may promote angiogenesis by stimulating VEGF expression in RPE.59 Depletion of macrophages diminishes the lesion size and severity in experimental laser-induced CNV.60,61 Ccr-2 knockout mice, characterized by hampered macrophage recruitment, also show inhibition of laser-induced CNV.62
Microglial cells (MC) are involved in phagocytosis of injured and dead cells. Dysfunction of phagocytosis of accumulating debris may lead to further inflammation.63 CX3CR1 is a cytokine produced by MC, and an association between AMD and CX3CR1 has been reported.64–66 A double knockout of MCP1 and CX3CR1 in an animal model leads to an AMD phenotype with spontaneous CNV formation.67,68
The contribution of bone marrow-derived endothelial cells to CNV has been evaluated by inducing CNV in irradiated mice that have received bone marrow transplants from green fluorescent protein (GFP)-expressing mice. These studies show that GFP+ endothelial cells are incorporated into the laser-induced CNV, suggesting that bone marrow-derived progenitor cells provide an additional source of endothelial cells in CNV.6,69,70
CNV membrane formation
In a series of 760 eyes with AMD from 450 patients; 310 eyes (~40%) demonstrated a disciform membrane.20 The mean diameter of these membranes was 3.73 mm, and the mean thickness was 0.27 mm. Preservation of photoreceptor cells was seen when the thickness of the disciform scars was 0.2 mm or less. Histologic studies of highly vascular CNV show that they contain endothelial cell-lined channels, RPE cells, and macrophages within the extracellular matrix-rich stroma. The stromal RPE are immunoreactive for smooth-muscle actin, indicating a transdifferentiated phenotype. The transdifferentiated RPE often expresses VEGF, suggesting that these RPE have a proangiogenic role, and that VEGF is a mediator of AMD-related CNV.71
CNV likely occurs when there is an imbalance between proangiogenic and antiangiogenic growth factors. Angiogenic growth factors, such as VEGF, angiopoietins (Ang 1, Ang 2), and bFGF, released from RPE cells and/or other retinal cells, promote CNV. Choroidal angiogenesis is inhibited by antiangiogenic growth factors such as thrombospondin-1 and PEDF72 and by Fas-mediated killing of new vessels by Fas-ligand-positive RPE or leukocytes.73
Cicatricial membrane formation
Cellular and highly vascularized membranes gradually evolve into paucicellular cicatricial membranes. The loss of cellularity is most likely due to cell death of stromal cells.74 Surgically excised AMD-related CNV contains apoptotic stromal RPE, endothelial cells, and macrophages. Cell death may be associated with a local decrease in the expression of angiogenic growth factors that promote survival of activated cells. Fas and Fas ligand expression may also be involved in the induction of apoptosis in these cells.75 Little is known about the mediators of collagenous scar formation in CNV. Studies have shown that connective tissue growth factor (CTGF), a proangiogenic and profibrotic growth factor, is expressed in stromal RPE cells in surgically excised CNV.76 The development of a cicatricial disciform lesion promotes overlying photoreceptor cell loss.77
Neovascularization associated with diabetic retinopathy
Neovascularization in DR can be thought of as a two-step process. An initial vascular dropout/occlusion phase is followed by abnormal vascular growth. Chronic hyperglycemia leads to vascular injury with basement membrane thickening, pericyte loss, microaneurysms, and vascular leakage.78,79 Many biochemical pathways (protein kinase C, nuclear factor (NF)-κB, mitogen activated protein kinase) have been involved in the pathogenesis of early DR but the exact mechanism remains ill defined. Oxidative injury, microthrombi formation, inflammatory mediator upregulation, and leukostasis have been observed. Proinflammatory cytokines IL-1β and TNF-α are elevated in animal models of DR, recruiting leukocytes and accelerating vascular dropout by promoting further inflammation as well as activating apoptosis directly or indirectly.80–82 This leads to vascular leakage as well as microvascular occlusion. As a consequence of microvascular occlusion there is inadequate supply of oxygen and nutrients for proper cellular metabolism. This leads neurons and supporting astrocytes to secrete proangiogenic factors such as VEGF and insulin-like growth factor (IGF)-1. Clinical studies have substantiated the role of VEGF in DR.83,84 VEGF levels are elevated in patients with proliferative DR (PDR), and although recent reports have not been able to show decreases of VEGF levels in the short term after panretinal photocoagulation,85,86 successful laser retinal ablation decreases them over the long term.84 In addition, several studies have shown the beneficial effects of anti-VEGF intervention in DR.87–90 Before the discovery of VEGF and its important role in DR, hypophysectomy studies had revealed a role of growth hormone (GH) and its associated factors in DR. IGF-1 levels have been shown to be elevated in the vitreous of patients with PDR relative to control eyes.91–93 Although there may be a role for IGF-1 in retinal neovascularization, our understanding of IGF-1 in DR remains unclear.94 IGF-1 may act indirectly via VEGF. Studies of cultured RPE cells demonstrated that adding IGF-1 in vitro increased VEGF mRNA and secreted protein.95 The accumulation of AGE after long-term exposure to hyperglycemia caused an increase in IGF-1 synthesis in human monocytes, suggesting a role for inflammatory pathways.96
Angiopoietins are molecules that bind endothelial Tie receptors and are involved in angiogenesis during development. Vitreous level of Ang 2 and VEGF were significantly higher in patients with PDR than in controls or those with inactive PDR and the levels of Ang 2 correlated significantly with that of VEGF, suggesting an association of Ang 2 and VEGF with angiogenic activity in PDR.97 A nuclease-resistant RNA aptamer that binds and inhibits Ang 2 but not the related Tie2 agonist, Ang 1, inhibited bFGF-mediated neovascularization in the rat corneal micropocket angiogenesis assay, demonstrating that a specific inhibitor of Ang 2 can act as an antiangiogenic agent.98
More recently, a role of erythropoietin (Epo) in DR has been discovered.99 Epo is involved in the generation of red blood cells but has been shown to be synthesized in astrocytes and its receptors have been detected in photoreceptors. Epo has been shown to have neuroprotective effects in various models.100,101 It has also been shown to be proangiogenic. Epo levels are higher in the vitreous of patients with PDR, and inhibition of both Epo and VEGF leads to greater inhibition of neovascularization.99 However, since Epo has neuroprotective effects, its inhibition for neovascularization may come with significant collateral damage.
In addition to elevation of proangiogenic factors, there is also an imbalance of antiangiogenic factors. PEDF, now known as serpin peptidase inhibitor clade F member 1(SERPINF1), is considered to be the most potent inhibitor of neovascularization, inhibiting endothelial cell migration with a median effective dose of 0.4 nM.102 It specifically interferes with neovasculature and not with established vessels due to its mechanism of action through Fas/FasL and its cooperation with angiogenic factors. SERPINF1 upregulates FasL on endothelial cells, whereas angiogenic factors induce its essential partner Fas receptor on neovessels but not on established vessels.103 In PDR, it has been found that PEDF levels are downregulated compared to controls.104 PEDF also has neuroprotective effects, and thus seems to be an ideal candidate for therapeutic intervention.
Inflammation is another important player in the pathogenesis of proliferative DR. Inflammatory cytokines such as TNF-α, IL-1β, intercellular adhesion molecule (ICAM), inducible nitric oxide synthase (iNOS), and IL-6 are elevated in patients with PDR.105–108 Localized inflammation is thought to lead to leukostasis through the interaction of ICAM and CD18.81,109–111 This leukostasis leads to local vaso-occlusion, nonperfusion, and ischemia, which leads to upregulation of angiogenic factors. IL-1β can promote endothelial cell proliferation, propagate inflammation, and upregulate HIF-1α.112,113 TNF-α can promote angiogenesis, and macrophage-induced angiogenesis is mediated through TNF-α.114,115 TNF-receptor p55-deficient animals are protected from retinal neovascularization in animal models.116 Inhibitors of inflammation such as cyclo-oxygenase 2 inhibitors and aspirin have been shown to curtail vascular pathology and neovascularization.81,117,118
The proinflammatory environment is characterized by elevated MMPs, which degrade the extracellular matrix, a step necessary for angiogenesis. Additionally, MMP-9 not only is induced by IL-8119 but also activates IL-8,120 which recruits more inflammatory cells, feeding a destructive positive-feedback loop. Insulin121 and PEDF122 are degraded by MMP-9, leading to a state of more insulin resistance and neuronal peril. Beránek et al. in 2008 showed MMP2 and MMP9 to be elevated in patients with PDR.123
Neovascularization associated with retinopathy of prematurity
Retinal vasculature development begins at about the fourth month of gestation and is complete at about 40 weeks. When an infant is born prematurely, it moves from a hypoxic (Pao2 ~30 mmHg) to relative hyperoxic (Pao2 ~ 60–100 mmHg) environment.124,125 The increase in oxygen tension, coupled with weak antioxidant defenses, leads to production of toxic ROS and decreased HIF. The combination of these effects leads to damage of the vascular endothelial cells and decreased production of VEGF and Epo, with resultant cessation of normal vascular development. As the peripheral avascular retina continues to develop in the absence of a developing vascular bed, it becomes relatively hypoxic and secretes VEGF and Epo at levels above normal physiologic values. Accordingly, the new vascular growth becomes exaggerated, misguided, and abnormal, leading to physical traction and potential retinal detachment. It is important to know that there is no direct evidence of the above-described processes from eyes of premature babies, but it is rather an extrapolation from animal data and other vascular eye diseases. However, the importance of VEGF upregulation in the proliferative phase of this disease has been shown in humans, since blockade of VEGF with anti-VEGF antibodies (bevacizumab) showed significant efficacy in infants with stage 3+ zone I disease.126
Oxygen level changes cannot fully account for disease progression and prematurity is the strongest risk factor for ROP development. Growth factors such as IGF-1 have been implicated by epidemiological and animal studies. Babies with decreased levels of IGF-1 have slower vascular development in the first phase of the disease and animals that lack IGF-1 have substantial reduction in neovascularization in the proliferative phase of the disease.127
Neovascularization in vascular occlusions
Ischemia and subsequent elevation of angiogenic factors are thought to be major and important contributors in neovascularization after central retinal vein occlusion (CRVO). The role of VEGF in particular has been well established. Aiello et al. in 1994 showed increased VEGF levels in the vitreous of patients with active neovascular CRVO;84 in the same year, Miller et al. showed evidence that VEGF is temporally and spatially correlated with ocular angiogenesis in a primate model of CRVO and that blockade of VEGF could inhibit neovascularization.128 Obviously, VEGF is not the only cytokine elevated in CRVO, and elevated levels of VEGF can be seen without neovascularization. Several interleukins have been shown to be elevated in CRVO as well as MCP-1 and ICAM-1.129–131 Additionally, antiangiogenic factors such as PEDF have been shown to be downregulated in CRVO.132 The alterations of cytokines seen in CRVO not only lead to potential neovascularization but are major contributors to vascular leakage and macular edema. Thus far, only anti-VEGF blockade has been studied as a targeted therapy in human patients with CRVO, and several studies have shown significant (but not absolute) success of such a strategy, suggesting that combination therapies may be needed for improved outcomes.133
Neovascularization in uveitis
Retinal neovascularization and CNV in uveitis are well recognized, although uncommon. Ischemia from nonperfusion and inflammation may contribute to the formation of new vessels. Peripheral ischemia and the extent of neovascularization seem to correlate in predominantly ischemic vasculitides such as Eales disease.134 However, neovascularization of the disc has been seen in other uveitic cases without apparent ischemia.135 Given the fact that neovascularization can regress with corticosteroid and other immune-suppressive treatments, it seems that inflammation alone may be sufficient for retinal neovascularization and CNV in this heterogeneous group of diseases.136–138 Nevertheless, VEGF is still a key molecule in uveitis-associated neovascularization, and several studies have shown benefit (albeit partial) of anti-VEGF therapy.139,140
Genetic aspects of neovascularization
Age-related macular degeneration
AMD has a significant genetic component, as there is higher concordance among monozygotic twins compared to dizygotic twins.141 Family members of individuals with AMD are more likely to develop the disease.
The protein ApoE is involved in the regulation of blood lipids, and has been found to be involved in AMD. The ε2 allele of the APOE gene may increase the risk, whereas the ε4 allele may be protective.142 However, other studies find opposite or no association between ApoE and AMD.143–145
The chemokine receptor CX3CR1 and the Toll-like receptor 4 (TLR4) have been implicated in several, but not all, studies as risk factors for progression of the disease.146–149 Conflicting data exist as well on TLR3 as a risk factor for AMD progression.150–153
Convincing genetic association data exist for a locus of chromosome 10q26, which harbors the pleckstrin homology domain-containing protein 1 (PLEKHA1), the ARMS2 gene product of unknown function, and the trypsin-like protease HTRA1. This locus is associated with a seven- to tenfold increased risk of the disease.154–156 In addition, very strong evidence exists for the CFH gene, whereas the single nucleotide polymorphism Y402H can increase the risk of the disease three- to sevenfold.157–159 However, the variation in these genomic regions alone is unable to predict disease development with high accuracy.
Several other studies have implicated various other components of the complement pathway in disease progression. Variations in complement 2, complement factor B, and CFH-related protein 1 and 3 are thought to have protective odds ratios whereas complement 3 may be associated with increased susceptibility.57,160–163
Mutations in other genes have been associated with macular degenerations other than AMD, such as the ABCR gene, a rod-specific ATP-binding cassette (ABC) transporter seen in Stargardt disease.164,165 Mutations in TIMP-3, an inhibitor of proteolysis found within Bruch’s membrane, are seen in Sorsby fundus dystrophy, an autosomal dominant disorder with histologic changes similar to those of neovascular AMD.166–169
Mutation in the vitelliform macular dystrophy (VMD)2 gene that encodes the protein bestrophin has been found in Best macular dystrophy, and mutation in the epidermal growth factor-containing, fibrillin-like extracellular matrix protein (EFEMP1) gene has been seen in patients with malattia levantinese and Doyne honeycomb retinal dystrophy, which are disorders associated with drusen formation.170,171
Diabetic retinopathy
Although the Diabetes Control and Complications Trial and the UK Prospective Diabetes Study demonstrate the beneficial effects of tight glucose and blood pressure control, patients with good glucose control still develop DR. The recent ADVANCE study reported that intensive glucose control to reduce A1c to 6.5% or less had no effect on retinopathy rates.172,173 In addition, it is apparent that some patients with poor control of their disease may not develop DR even over long periods of time, while others develop DR in a short time despite good disease control. This is exemplified in the Joslin Medalist study, which found that a significant number of elderly patients with type 1 diabetes had no evidence of retinopathy despite surviving over 50 years with diabetes.174,175 These results suggest that genetic factors may play a role in the progression of this disease.
Candidate gene approaches have shown fairly consistent associations with genes encoding aldose reductase (ALR2), VEGF, and receptor for AGE (RAGE) in the progression of DR (reviewed by Liew et al.176). Aldose reductase is involved in the metabolism of glucose into sorbitol inside the cells. Sorbitol accumulation leads to osmotic stress and cell injury. Unfortunately, clinical trials with aldose reductase inhibitors failed to show benefit. It is important to note that in these trials no stratification on ALR2 gene polymorphism status was undertaken.177,178 Polymorphism in the VEGF promoter region has been shown to be associated with DR progression and there may be ethnic variation in the “at-risk” haplotype (reviewed by Liew et al.176).
Retinopathy of prematurity
ROP is a multifactorial disease that has many phenotypic similarities with a genetic disease called familial exudative vitreoretinopathy (FEVR). At least four genes have been implicated in FEVR (norrin–FZD4–LRP5–TSPAN12 signaling pathway), and a candidate gene approach has shown that three of them (NDP, FZD4, and LRP5) are mutated in a small percentage of severe ROP patients.179 Association between VEGF, IGF-1 receptor, and angiotensin-converting enzyme gene polymorphisms have been implicated in some (but not all) studies.180–191 Other suggestive associations have been reported between ROP and several candidate genes such as angiotensin II type I receptor, Indian hedgehog, T-box 5, glycoprotein Ibα polypeptide, and cholesterol ester transfer protein, but confirmation is still needed.192
Environmental factors
Age-related macular degeneration
Several modifiable risk factors have been associated with progression of AMD. Amongst them, smoking has been the most consistently reported risk factor, elevating the risk of disease progression by about twofold.193–195 Additionally, a health-promoting diet (rich in seafood, vegetables, and fruits), physical activity, and a nonsmoker status are thought to lead to a 70% reduction in the risk of developing AMD.196 The AREDS study showed that supplementation with vitamins A and C, zinc, and copper in patients with moderate-severity AMD reduces the risk of progression by about 25%.197 Unfortunately, this was not one of the a priori questions set to be answered by the study, so the benefits of this supplementation are not universally accepted. More recently, the Women’s Antioxidant and Folic Acid Cardiovascular Study discovered that supplementation with folate, vitamins B6 and B12 reduced the risk of advanced AMD by 30–40% in women with high-risk cardiovascular factors.198
Diabetic retinopathy
Smoking, hypercholesterolemia, and hypertension are known factors that worsen disease progression. Smoking increases oxidative stress, and may increase TNF levels as well as alter high-density lipoprotein and low-density lipoprotein levels.199,200 Smoking has also been shown to elevate VEGF levels acutely in about one-quarter of diabetic patients.201 Several studies have shown the link between high blood pressure and cholesterol in the progression of DR. Recent studies show that there is greater benefit in reducing the progression of the disease by lowering cholesterol levels compared to lowering high blood pressure.202,203
Retinopathy of prematurity
Besides early birth and reduced birth weight, other factors have been shown to contribute to development of ROP. Excessively high levels of oxygen in incubators, used to save the lives of premature infants, led to an ROP epidemic and the realization that reducing the level of oxygen given to premature babies reduces the incidence of ROP.204,205 Although oxygen levels and ROP are linked, the exact mechanism is not clear. It is possible that the rate of change of oxygenation rather than the absolute level may be more important for disease progression, and there have been reports that return to high oxygen levels for prolonged time with gradual decline to normal oxygen levels may halt and reverse the disease.206–210
Light exposure has also been investigated, but the LIGHT-ROP study concluded that a reduction in ambient light exposure does not alter the incidence of ROP.211
The diet of premature babies is usually poorer in omega-3 fatty acids compared to mother’s milk. Omega-3 fatty acid supplementation in animal models of ROP has shown reduction in neovascularization.212 Human studies with docosahexaenoic acid supplementation of infant formula at 0.32% of total fatty acids showed improved visual acuity as measured by visual-evoked potential, but this study did not investigate neovascularization.213
Angiogenic and antiangiogenic factors in neovacularization
Vascular endothelial growth factor
VEGF was first identified in tumor models as a vasopermeability factor and initiator of angiogenesis upregulated by hypoxia. In addition to being the strongest endothelial cell mitogen, VEGF has been shown to induce the expression of plasminogen activators in microvascular endothelial cells, which is important in the extracellular proteolysis necessary for capillary formation.214 VEGF induces the expression of endothelial cell α1β1 and α2β1 integrins, which are important in migration.215 There is also in vitro evidence that VEGF upregulates endothelial cell fenestrations in kidney glomerulus, choroid plexus, and even the choriocapillaris.216 Leukocyte adhesion has been shown to be important in vascular leakage. VEGF increases the expression of ICAM-1 on endothelial cells, resulting in increased leukostasis, which mediates the breakdown of the blood–retinal barrier.217,218
VEGF, also known as VEGF-A or VEGF-1, is expressed as five mRNA splice variants in humans: isoforms 121, 145, 165, 189, and 206.219 VEGF is a heparin-binding dimeric glycoprotein with disulfide-linked subunits, which share significant sequence homology with the A and B chains of platelet-derived growth factor (PDGF).220 VEGF 121 is entirely soluble and unbound to extracellular matrix. VEGF 165 is intermediate and binds somewhat to extracellular matrix. VEGF 189 is almost entirely sequestered to extracellular matrix and cell surface sites.221,222 VEGF 165 is the predominantly expressed isoform when human cDNA libraries are screened, and is optimal for bioavailability and biologic potency. It is the critical isoform for both developmental and pathologic retinal angiogenesis.222–224 Two other related endothelial growth factors, VEGF-B and VEGF-C, have structural homology to VEGF and appear to play roles in tumor angiogenesis and in the development of the lymphatic system, as does VEGF-D.225 VEGF-C, also known as VEGF-2, has been shown to promote angiogenesis in the rabbit ischemic hind limb model.226 VEGF-E has similar angiogenic activity to VEGF-A, primarily through VEGF receptor 2.227
Both high- and low-affinity VEGF receptors have been identified on not only endothelial cells, but also on bone marrow-derived and retinal epithelial cells.228–230 They belong to the family of tyrosine kinases requiring phosphorylation to be activated upon ligand binding. VEGFR-1 (FMS-like tyrosine kinase or FLT-1 in human) and VEGFR-2 (fetal liver kinase 1 or Flk-1 in the mouse; TKR-C in the rat; kinase insert domain receptor or KDR in the human) are expressed on endothelial cells, whereas VEGFR-3 (FLT4) is primarily found on lymphatic endothelial cells.231 Although VEGF binds to both VEGFR-1 and VEGFR-2, the latter is primarily responsible for endothelial cell mitogenesis, survival, and permeability.232 VEGFR-1 may be important in development by sequestering VEGF, preventing it from interacting with VEGFR-2.233 VEGFR-1 has an established role in monocyte chemotaxis.234,235 VEGF-C has also been shown to be a ligand for VEGFR-2 and VEGFR-3.226 Synergism between FGF-2 and VEGF has been demonstrated by the finding that FGF-2 increases VEGFR-2 expression in microvascular endothelial and aortic endothelial cells.236 In addition to the receptor tyrosine kinases, VEGF also interacts with a family of coreceptors, the neuropilins, which may enhance its angiogenic function.237
VEGF is expressed by retinal cells in vitro and is upregulated by hypoxia.238,239 Other modulators of VEGF expression are hypoglycemia,234 beta-estradiol, and ROS.240–242 Several growth factors cause upregulation of VEGF gene expression, including epidermal growth factor, transforming growth factor (TGF)-α and β, IGF-1, FGF, and PDGF, implicating both paracrine and autocrine release of these factors in the hypoxic regulation of VEGF.228,243
In vivo work has demonstrated VEGF to be spatially and temporally correlated with iris neovascularization in a monkey model of ischemic retinopathy and iris neovascularization.128 VEGF protein levels in serial aqueous samples have been shown to correlate with the severity of induced retinal ischemia and iris neovascularization. In situ hybridization identified the inner retina as the source of VEGF.128,244 Work in models of ROP also implicates the importance of VEGF in the development of retinal neovascularization.245–247
In vitro, VEGF has been shown to be sufficient to produce neovascularization. VEGF results in neovascularization in the corneal micropocket and in chick chorioallantoic membrane (CAM) bioassay.248,249 A single intra-arterial bolus of VEGF was sufficient to stimulate angiogenesis and collateralization in a rabbit ischemic hind limb model, leading to studies of the therapeutic use of VEGF in peripheral vascular and coronary artery disease.250 Injections of recombinant human VEGF in normal monkey eyes led to iris neovascularization and neovascular glaucoma, as well as many of the changes of DR, including vessel dilation, tortuosity, microaneurysm formation, hemorrhage, edema, capillary dropout, and intraretinal neovascularization.251,252 However, VEGF alone may not be sufficient in vivo to induce retinal neovascularization.
To demonstrate the causal role of VEGF in neovascularization secondary to ischemia, VEGF activity was specifically blocked using anti-VEGF antibodies, soluble VEGF receptors, or antisense oligonucleotides to VEGF. Intravitreous injection of anti-VEGF antibodies completely prevented the development of iris neovascularization in the monkey model.253 In a mouse ROP model, dominant-negative VEGF receptors and VEGF antisense oligonucleotides substantially decreased retinal neovascularization.254,255 This work has confirmed the central role of VEGF in pathologic retinal neovascularization.
VEGF also plays an important role in the development of CNV. Immunostaining of surgical specimens of choroidal neovascular membranes showed increased VEGF expression.71,256 In situ hybridization studies have demonstrated a correlation between VEGF expression and the development of CNV in laser injury models in the rat and monkey.257 Oxidative stress may stimulate the overexpression of growth factors from the RPE, a possible inflammatory state, and subsequent damage and thickening of Bruch’s membrane from recruited macrophages.21,22 It has been suggested that the abnormally thickened Bruch’s membrane may interfere with polarized RPE secretion of VEGF necessary for maintenance of the choriocapillaris. Atrophy of the choriocapillaris, often seen clinically, may result in a state of outer retinal hypoxia, stimulating VEGF-induced angiogenesis.258 Studies have also shown that despite the absence of hypoxia in the laser injury models, specific compounds that bind VEGF and its receptors virtually eliminate CNV.259,260 These data have important clinical implications, as specific pharmacologic inhibitors of VEGF have been approved for the treatment of neovascular AMD and shown to be effective.261–264
Insulin-like growth factor-1
The role of GH and its associated factors in DR was first suggested by the clinical observation of regression of neovascularization in the retina following infarction of the pituitary during pregnancy.265 This was followed by experimental and clinical observations that hypophysectomy led to remission of DR.266,267 The complications of hypophysectomy were frequent, severe, and often lethal; with the advent of laser photocoagulation, the therapy was largely abandoned. Subsequently, it was demonstrated that GH mediated its effects through IGF-1 and IGF-2, and investigations were directed towards these factors. The role of IGF-1 in ROP is more developed. Studies of transgenic mice have shown that IGF-1 is important for normal retinal vessel development, and allows for VEGF to enhance endothelial cell survival and promote early vascular development.268 Transgenic mice expressing a GH antagonist demonstrated decreased retinal neovascularization when subjected to hyperoxia/normoxia in the murine model of ischemic retinopathy. Adding exogenous IGF-1 completely restored the amount of retinal neovascularization seen in controls. Inhibition of retinal neovascularization in these mice was inversely proportional to serum levels of GH and IGF-1.269 It has been shown that a low serum level of IGF-1 is a predictor for ROP in infants.268,270
Fibroblast growth factor-2
Acidic and basic FGF are prototype members of the FGF family. Basic FGF or FGF-2 is a fibroblast mitogen, and one of the first angiogenic factors identified and suspected in ocular neovascularization. FGF-2 has been called “the stored growth factor” because much of the cell-associated FGF-2 is found in the extracellular matrix.271
FGF-2 has been implicated in various aspects of angiogenesis. It stimulates endothelial cell proliferation and migration and induces the production of proteases.272 FGF-2 is angiogenic in vivo in the chick CAM and corneal micropocket bioassays at very low levels.273,274 FGF-2 has been isolated from many normal tissues and tumors of mesodermal and neuroectodermal origin, as well as in CNV membranes.275–278
One of the arguments against FGF-2’s role in pathologic angiogenesis is its lack of a definitive signal peptide for secretion.279 However, a number of alternative pathways for FGF-2 release have been postulated, including the ATP-binding cassette (ABC) transport proteins, selective exocytosis, and cell death or injury.19,280–282 During sublethal injury, cells may transiently release FGF-2. Using cultured aortic endothelial cells, McNeil and colleagues have demonstrated that mechanical wounding by scraping leads to efficient release of FGF-2 from injured cells19 (also reviewed by D’Amore281). In an experimental model of optic nerve crush in the mouse, FGF-2 immunostaining is dramatically increased in the retinal photoreceptor layer.283 Finally, injury to the corneal epithelium leads to release of FGF-2, which binds to basement membrane.284 These and other observations suggest that FGF-2 may act as a “wound hormone” both in maintenance of tissue integrity and repair after injury.
Integrins
The integrins are a family of cell adhesion proteins that are heterodimer combinations of 15 α and 8 β subunits, and are important regulators of angiogenesis. They mediate endothelial cell adhesion to the extracellular matrix, which facilitates proliferation, migration, and response to prosurvival or apoptotic signals in the formation of new vessels.285 Different integrins can bind to the same ligand, but initiate different intracellular signaling pathways. Also, one integrin can bind to multiple ligands; in addition to extracellular matrix components, ICAMs important for leukocyte adhesion can also be bound to integrins.286
αvβ3 has been demonstrated to inhibit endothelial cell apoptosis in newly sprouting blood vessels through the regulation of NF-κB.287 αvβ3 and αvβ5 are highly expressed on angiogenic endothelial cells and have been demonstrated in neovascular tissue of experimental models and in clinical specimens.288,289 Friedlander and colleagues found both αvβ3 and αvβ5 expressed in neovascular tissue from eyes with proliferative DR, whereas only αvβ3 was expressed in neovascular tissue from AMD and ocular histoplasmosis syndrome.289 Blocking the integrins halts angiogenesis in the chick CAM and corneal neovascularization models, and integrin binding also partially suppresses retinal neovascularization.290,291
Ang and Tie2
Ang-1 is a 70-kDa glycoprotein that is chemotactic for endothelial cells, and is postulated to play a role in the assembly of nonendothelial cell components and the formation of capillary sprouts.292,293 Ang-1 binds to Tie2, a tyrosine kinase receptor, which is expressed on endothelial cells and early hematopoietic cells.294,295 Knockouts of the gene encoding Ang-1 or its receptor Tie2 are embryologically lethal, with failure to recruit smooth-muscle and pericyte precursors. Tie2 has also been demonstrated in quiescent and angiogenic vasculature in adults, and may play a role in vascular maintenance.295,296 Ang-2 also binds strongly to Tie2 but does not result in phosphorylation of the receptor; instead, it acts as a competitive inhibitor preventing Ang-1 binding.297 It has been postulated that Ang-2 binding mediates endothelial cell survival signals by making them more responsive to VEGF, resulting in neovascularization. However, with VEGF inhibition, Ang-2 binding results in apoptosis.297 In the rabbit cornea micropocket model, neither Ang-1 or Ang-2 alone induced corneal neovascularization, but either Ang-1 or Ang-2 added to VEGF promoted neovascularization, with Ang-2 having the more potent effect.298 Transgenic mice overexpressing Ang-2 demonstrate disruption of blood vessel formation, and are similar in phenotype to Ang-1-deficient mice.297 Additional studies are needed to understand better the role of the Ang and Tie2 in ocular angiogenesis.
Pigment epithelium-derived factor
PEDF is a 50-kDa serpin protease that was first discovered as a secreted protein from human fetal RPE cells.299 PEDF is the most potent antiangiogenic growth factor identified to date.300 PEDF has been demonstrated to promote apoptosis in proliferating endothelial cells through increasing the interaction of Fas ligand with its receptor Fas. This seems to be the only cell type where PEDF promotes apoptosis.301 PEDF has also been shown to signal cellular differentiation of retinoblastoma cells in vitro.299 Furthermore, a neuroprotective role for PEDF has been identified in neural cerebellar granule cells where PEDF caused the expression of antiapoptotic genes through the activation of NF-κB.302 In retinal degeneration slow (RDS) mutant mice, PEDF was shown to protect photoreceptors from undergoing apoptosis.303 Thus, the signaling cascades that are activated by PEDF lead to cell-specific actions to promote both survival and cell death.
Immunohistochemical studies have shown PEDF to be expressed in the RPE cells, the interphotoreceptor matrix, ganglion cells, and in the ciliary neuroepithelium.304,305 PEDF is also bound to components of the extracellular matrix. In vitro, hypoxia causes a decrease in PEDF expression while VEGF is increased.300 In vivo, levels of PEDF were shown to be decreased in the vitreous of patients with proliferative DR and wet AMD, such that PEDF levels correlated inversely with neovascularization.306–308
PEDF may have therapeutic potential as both a neuroprotective and antiangiogenic agent. The fact that it is both differentiation-promoting and antiangiogenic has popularized its study in tumor research. Using gene therapy approaches to administer PEDF may also be important in treating ocular neovascularization. Both intravitreal and subretinal adenoviral-associated PEDF have been used to treat retinal ischemia and CNV in mouse models.309,310
Matrix metalloproteinases
The extracellular matrix of the microvasculature is a highly dynamic structure containing collagens, laminins, fibronectins, proteoglycans, and other proteins. Growth factors such as FGF-2 are localized there and integrins mediate interactions between cells and the extracellular matrix. The extracellular matrix is remodeled during development and angiogenesis, with degradation of existing matrix and synthesis of new matrix material permitting migration and proliferation of endothelial cells. Degradation is accomplished by 16 known MMPs and plasmin. Plasmin is synthesized as a latent proenzyme, plasminogen, which requires proteolytic activation by enzymes such as urokinase-type plasminogen activator. This activation can be inhibited by plasminogen activator inhibitors. The MMPs are likewise synthesized as latent proenzymes and require proteolytic activation. The active MMPs can be inhibited by four known specific tissue inhibitors (TIMPS) and α-macroglobulin. For a review of the MMPs and their inhibitors, see Hadler-Olsen et al.311
VEGF has been shown to induce tissue factor and MMP production in endothelial cells, and MMP production in smooth-muscle cells via Flt-1.312,313 AGE also caused increased MMP-2 mRNA in choroidal endothelial cells in vitro.314 Another interaction has been demonstrated between MMP-2 and the integrin αvβ3, which are functionally associated on the surface of angiogenic blood vessels; upon their binding, collagenolytic activity is increased. A naturally occurring fragment of MMP-2, termed PEX, has also been shown to prevent binding of MMP-2 to αvβ3, acting to decrease the proteolytic activity.315 Thus, endogenous MMP fragments and TIMPs serve to regulate the invasive new vessels. MMP-9 is one of the major inducible MMPs. Its levels in the basal state seem to be suppressed by the energy sensor AMP-activated protein kinase (AMPK).316 Human neutrophils have been shown uniquely to release TIMP-free MMP-9 to provide a potent catalytic stimulator of angiogenesis.317
Angiostatin and endostatin
Angiostatin is a 38-kDa peptide identified in a murine model of Lewis lung carcinoma, where it inhibited tumor metastasis at distant sites.318 It was found to be an internal fragment of plasminogen. This fragment contains kringle structures, which based on their disulfide architecture, have been shown to inhibit angiogenesis. Angiostatin contains four out of the five plasminogen kringle structures. Once angiostatin is cleaved from the parent plasminogen molecule, it has been shown specifically to inhibit endothelial cell proliferation.318 It inhibited neovascularization in the chick CAM assay and inhibited neovascularization and growth of metastases in a mouse tumor model.
Endostatin is a 20-kDa peptide isolated from murine hemangioendothelioma, and has been identified as the C-terminal fragment of collagen XVIII.319 Endostatin inhibits endothelial cell proliferation and angiogenesis in vivo, including corneal neovascularization. Mice that lack collagen XVIII form larger and leakier laser-induced CNV.46 Finally, circulating endostatin has been identified in human plasma.320 Both angiostatin and endostatin have been proposed as potential therapeutic agents to treat ocular neovascularization; however, up to now they have not found success in clinical use.
Advanced glycosolation end products
AGEs are formed by nonenzymatic protein glycation. They have been localized in basal laminar/linear deposits or soft drusen in AMD.39 In surgically excised CNV, AGE deposition is spatially associated with RPE that express VEGF.39 AGEs induce VEGF expression by RPE cells in vitro,321 and growth of RPE on AGE leads to the upregulation of genes associated with RPE aging322 and downregulation of cathepsin D expression.323 AGEs are also involved in lipofuscin formation in RPE cells.324 Reactive oxygen intermediates are generated in parallel with AGEs either directly or through AGE–RAGE interaction.325 Consistent with the ability of AGEs to induce VEGF expression in vitro,321 AGEs can also induce angiogenesis in vivo.326
Other growth factors
PDGF expression has been reported in the outer nuclear layer of the macula from patients with AMD.77 RPE cells in CNV are strongly immunoreactive for TGF-β, and may act by modulating the effects of other growth factors such as bFGF and VEGF.275 This hypothesis is supported by the fact that TGF-β and IL-1 induce VEGF expression in cultured choroidal fibroblasts.327 CTGF is a proangiogenic and profibrotic growth factor that is expressed in stromal cells in human CNV.76 It also plays a role in mediating and modulating the effects of other growth factors; CTGF is upregulated in vitro by VEGF in choroidal endothelial cells and by TGF-β in RPE cells. However, the relative importance of these growth factors, in comparison with VEGF, has not been determined.
1 Denekamp J. Vascular endothelium as the vulnerable element in tumours. Acta Radiol Oncol. 1984;23:217–225.
2 Ausprunk DH, Folkman J. Migration and proliferation of endothelial cells in preformed and newly formed blood vessels during tumor angiogenesis. Microvasc Res. 1977;14:53–65.
3 Grant MB, Caballero S, Brown GA, et al. The contribution of adult hematopoietic stem cells to retinal neovascularization. Adv Exp Med Biol. 2003;522:37–45.
4 Grant MB, May WS, Caballero S, et al. Adult hematopoietic stem cells provide functional hemangioblast activity during retinal neovascularization. Nat Med. 2002;8:607–612.
5 Tomita M, Adachi Y, Yamada H, et al. Bone marrow-derived stem cells can differentiate into retinal cells in injured rat retina. Stem Cells. 2002;20:279–283.
6 Tomita M, Yamada H, Adachi Y, et al. Choroidal neovascularization is provided by bone marrow cells. Stem Cells. 2004;22:21–26.
7 Ashton N. Retinal vascularization in health and disease. Am J Ophthalmol. 1957;44:7–24.
8 Michaelson I. The mode of development of the vascular system of the retina, with some observations on its significance for certain retinal disease. Trans Ophthalmol Soc UK. 1948;68:137–180.
9 Gartner S, Henkind N. Neovascularization of the iris (rubeosis iridis). Surv Ophthalmol. 1978;22:291–312.
10 Henkind P. Ocular neovascularization. The Krill memorial lecture. Am J Ophthalmol. 1978;85:287–301.
11 Wise G. Retinal neovascularization. Trans Ophthalmol Soc UK. 1956;54:729–826.
12 Pournaras CJ, Miller JW, Gragoudas ES, et al. Systemic hyperoxia decreases vascular endothelial growth factor gene expression in ischemic primate retina. Arch Ophthalmol. 1997;115:1553–1558.
13 Pournaras CJ, Tsacopoulos M, Strommer K, et al. Experimental retinal branch vein occlusion in miniature pigs induces local tissue hypoxia and vasoproliferative microangiopathy. Ophthalmology. 1990;97:1321–1328.
14 Liu Y, Cox SR, Morita T, et al. Hypoxia regulates vascular endothelial growth factor gene expression in endothelial cells. Identification of a 5’ enhancer. Circ Res. 1995;77:638–643.
15 Maxwell PH, Dachs GU, Gleadle JM, et al. Hypoxia-inducible factor-1 modulates gene expression in solid tumors and influences both angiogenesis and tumor growth. Proc Natl Acad Sci USA. 1997;94:8104–8109.
16 Tsuzuki Y, Fukumura D, Oosthuyse B, et al. Vascular endothelial growth factor (VEGF) modulation by targeting hypoxia-inducible factor-1alpha – hypoxia response element – VEGF cascade differentially regulates vascular response and growth rate in tumors. Cancer Res. 2000;60:6248–6252.
17 Ryan SJ. Subretinal neovascularization after argon laser photocoagulation. Albrecht Von Graefes Arch Klin Exp Ophthalmol. 1980;215:29–42.
18 Wallow I, Johns K, Chandra BS, et al. Chorioretinal and choriovitreal neovascularization after photocoagulation for proliferative diabetic retinopathy. A clinicopathologic correlation. Ophthalmology. 1985;92:523–532.
19 McNeil PL, Muthukrishnan L, Warder E, et al. Growth factors are released by mechanically wounded endothelial cells. J Cell Biol. 1989;109:811–822.
20 Green WR, Enger C. Age-related macular degeneration histopathologic studies. The 1992 Lorenz E. Zimmerman lecture. Ophthalmology. 1993;100:1519–1535.
21 Beatty S, Koh H, Phil M, et al. The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv Ophthalmol. 2000;45:115–134.
22 Penfold PL, Madigan MC, Gillies MC, et al. Immunological and aetiological aspects of macular degeneration. Prog Retin Eye Res. 2001;20:385–414.
23 AREDS. A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E, beta carotene, and zinc for age-related macular degeneration and vision loss: AREDS report no. 8. Arch Ophthalmol. 2001;119:1417–1436.
24 Klein R, Klein BE, Jensen SC, et al. The five-year incidence and progression of age-related maculopathy: the Beaver Dam Eye Study [see comments]. Ophthalmology. 1997;104:7–21.
25 Wilcox DK. Vectorial accumulation of cathepsin D in retinal pigmented epithelium: effects of age. Invest Ophthalmol Vis Sci. 1988;29:1205–1212.
26 Kennedy CJ, Rakoczy PE, Constable IJ. Lipofuscin of the retinal pigment epithelium: a review. Eye. 1995;9:763–771.
27 Tombran-Tink J, Shivaram SM, Chader GJ, et al. Expression, secretion, and age-related downregulation of pigment epithelium-derived factor, a serpin with neurotrophic activity. J Neurosci. 1995;15:4992–5003.
28 Rakoczy PE, Zhang D, Robertson T, et al. Progressive age-related changes similar to age-related macular degeneration in a transgenic mouse model. Am J Pathol. 2002;161:1515–1524.
29 Hageman GS, Mullins RF. Molecular composition of drusen as related to substructural phenotype. Mol Vis. 1999;5:28.
30 Mullins RF, Russell SR, Anderson DH, et al. Drusen associated with aging and age-related macular degeneration contain proteins common to extracellular deposits associated with atherosclerosis, elastosis, amyloidosis, and dense deposit disease. FASEB J. 2000;14:835–846.
31 Sarks JP, Sarks SH, Killingsworth MC. Evolution of soft drusen in age-related macular degeneration. Eye. 1994;8:269–283.
32 Abdelsalam A, Del Priore L, Zarbin MA. Drusen in age-related macular degeneration: pathogenesis, natural course, and laser photocoagulation-induced regression. Surv Ophthalmol. 1999;44:1–29.
33 Starita C, Hussain AA, Patmore A, et al. Localization of the site of major resistance to fluid transport in Bruch’s membrane. Invest Ophthalmol Vis Sci. 1997;38:762–767.
34 Russell SR, Mullins RF, Schneider BL, et al. Location, substructure, and composition of basal laminar drusen compared with drusen associated with aging and age-related macular degeneration. Am J Ophthalmol. 2000;129:205–214.
35 Mullins RF, Aptsiauri N, Hageman GS. Structure and composition of drusen associated with glomerulonephritis: implications for the role of complement activation in drusen biogenesis. Eye (Lond). 2001;15:390–395.
36 Hageman GS, Luthert PJ, Victor Chong NH, et al. An integrated hypothesis that considers drusen as biomarkers of immune-mediated processes at the RPE-Bruch’s membrane interface in aging and age-related macular degeneration. Prog Retin Eye Res. 2001;20:705–732.
37 Anderson DH, Mullins RF, Hageman GS, et al. A role for local inflammation in the formation of drusen in the aging eye. Am J Ophthalmol. 2002;134:411–431.
38 Lutty GA, McLeod DS, Merges C, et al. Localization of vascular endothelial growth factor in human retina and choroid. Arch Ophthalmol. 1996;114:971–977.
39 Ishibashi T, Murata T, Hangai M, et al. Advanced glycation end products in age-related macular degeneration. Arch Ophthalmol. 1998;116:1629–1632.
40 Lafaut BA, Bartz-Schmidt KU, Vanden Broecke C, et al. Clinicopathological correlation in exudative age related macular degeneration: histological differentiation between classic and occult choroidal neovascularisation. Br J Ophthalmol. 2000;84:239–243.
41 Alexander JP, Bradley JM, Gabourel JD, et al. Expression of matrix metalloproteinases and inhibitor by human retinal pigment epithelium. Invest Ophthalmol Vis Sci. 1990;31:2520–2528.
42 Padgett LC, Lui GM, Werb Z, et al. Matrix metalloproteinase-2 and tissue inhibitor of metalloproteinase-1 in the retinal pigment epithelium and interphotoreceptor matrix: vectorial secretion and regulation. Exp Eye Res. 1997;64:927–938.
43 Vranka JA, Johnson E, Zhu X, et al. Discrete expression and distribution pattern of TIMP-3 in the human retina and choroid. Curr Eye Res. 1997;16:102–110.
44 Chong NH, Keonin J, Luthert PJ, et al. Decreased thickness and integrity of the macular elastic layer of Bruch’s membrane correspond to the distribution of lesions associated with age-related macular degeneration. Am J Pathol. 2005;166:241–251.
45 Yu HG, Liu X, Kiss S, Connolly E, et al. Increased choroidal neovascularization following laser induction in mice lacking lysyl oxidase-like 1. Invest Ophthalmol Vis Sci. 2008;49:2599–2605.
46 Marneros AG, She H, Zambarakji H, et al. Endogenous endostatin inhibits choroidal neovascularization. FASEB J. 2007;21:3809–3818.
47 Okamoto N, Tobe T, Hackett SF, et al. Transgenic mice with increased expression of vascular endothelial growth factor in the retina: a new model of intraretinal and subretinal neovascularization. Am J Pathol. 1997;151:281–291.
48 Schwesinger C, Yee C, Rohan RM, et al. Intrachoroidal neovascularization in transgenic mice overexpressing vascular endothelial growth factor in the retinal pigment epithelium. Am J Pathol. 2001;158:1161–1172.
49 Goetzl EJ, Banda MJ, Leppert D. Matrix metalloproteinases in immunity. J Immunol. 1996;156:1–4.
50 Killingsworth MC, Sarks JP, Sarks SH. Macrophages related to Bruch’s membrane in age-related macular degeneration. Eye. 1990;4:613–621.
51 Grossniklaus HE, Ling JX, Wallace TM, et al. Macrophage and retinal pigment epithelium expression of angiogenic cytokines in choroidal neovascularization. Mol Vis. 2002;8:119–126.
52 Bora NS, Kaliappan S, Jha P, et al. CD59, a complement regulatory protein, controls choroidal neovascularization in a mouse model of wet-type age-related macular degeneration. J Immunol. 2007;178:1783–1790.
53 Klein RJ, Zeiss C, Chew EY, et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308:385–389.
54 Edwards AO, Ritter R, Abel KJ, et al. Complement factor H polymorphism and age-related macular degeneration. Science. 2005;308:421–424.
55 Hageman GS, Anderson DH, Johnson LV, et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci U S A. 2005;102:7227–7232.
56 Haines JL, Hauser MA, Schmidt S, et al. Complement factor H variant increases the risk of age-related macular degeneration. Science. 2005;308:419–421.
57 Gold B, Merriam JE, Zernant J, et al. Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat Genet. 2006;38:458–462.
58 Johnson PT, Betts KE, Radeke MJ, et al. Individuals homozygous for the age-related macular degeneration risk-conferring variant of complement factor H have elevated levels of CRP in the choroid. Proc Natl Acad Sci U S A. 2006;103:17456–17461.
59 Oh H, Takagi H, Takagi C, et al. The potential angiogenic role of macrophages in the formation of choroidal neovascular membranes. Invest Ophthalmol Vis Sci. 1999;40:1891–1898.
60 Espinosa-Heidmann DG, Suner IJ, Hernandez EP, et al. Macrophage depletion diminishes lesion size and severity in experimental choroidal neovascularization. Invest Ophthalmol Vis Sci. 2003;44:3586–3592.
61 Sakurai E, Anand A, Ambati BK, et al. Macrophage depletion inhibits experimental choroidal neovascularization. Invest Ophthalmol Vis Sci. 2003;44:3578–3585.
62 Tsutsumi C, Sonoda KH, Egashira K, et al. The critical role of ocular-infiltrating macrophages in the development of choroidal neovascularization. J Leukoc Biol. 2003;74:25–32.
63 Gupta N, Brown KE, Milam AH. Activated microglia in human retinitis pigmentosa, late-onset retinal degeneration, and age-related macular degeneration. Exp Eye Res. 2003;76:463–471.
64 Tuo J, Smith B, Bojanowski CM, et al. The involvement of sequence variation and expression of CX3CR1 in the pathogenesis of age-related macular degeneration. FASEB J. 2004;18:1297–1299.
65 Chan CC, Tuo J, Bojanowski CM, et al. Detection of CX3CR1 single nucleotide polymorphism and expression on archived eyes with age-related macular degeneration. Histol Histopathol. 2005;20:857–863.
66 Combadière C, Feumi C, Raoul W, et al. CX3CR1-dependent subretinal microglia cell accumulation is associated with cardinal features of age-related macular degeneration. J Clin Invest. 2007;117:2920–2928.
67 Tuo J, Bojanowski CM, Zhou M, et al. Murine ccl2/cx3cr1 deficiency results in retinal lesions mimicking human age-related macular degeneration. Invest Ophthalmol Vis Sci. 2007;48:3827–3836.
68 Chan C-C, Ross RJ, Shen D, et al. Ccl2/Cx3cr1-deficient mice: an animal model for age-related macular degeneration. Ophthalmic Res. 2008;40:124–128.
69 Espinosa-Heidmann DG, Caicedo A, Hernandez EP, et al. Bone marrow-derived progenitor cells contribute to experimental choroidal neovascularization. Invest Ophthalmol Vis Sci. 2003;44:4914–4919.
70 Sengupta N, Caballero S, Mames RN, et al. The role of adult bone marrow-derived stem cells in choroidal neovascularization. Invest Ophthalmol Vis Sci. 2003;44:4908–4913.
71 Lopez PF, Sippy BD, Lambert HM, et al. Transdifferentiated retinal pigment epithelial cells are immunoreactive for vascular endothelial growth factor in surgically excised age-related macular degeneration-related choroidal neovascular membranes. Invest Ophthalmol Vis Sci. 1996;37:855–868.
72 Miyajima-Uchida H, Hayashi H, Beppu R, et al. Production and accumulation of thrombospondin-1 in human retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. 2000;41:561–567.
73 Kaplan HJ, Leibole MA, Tezel T, et al. Fas ligand (CD95 ligand) controls angiogenesis beneath the retina. Nat Med. 1999;5:292–297.
74 Wyllie AH. Glucocorticoid-induced thymocyte apoptosis is associated with endogenous endonuclease activation. Nature. 1980;284:555–556.
75 Hinton DR, He S, Lopez PF. Apoptosis in surgically excised choroidal neovascular membranes in age-related macular degeneration. Arch Ophthalmol. 1998;116:203–209.
76 He S, Jin ML, Worpel V, et al. Connective tissue growth factor and its role in the pathogenesis of choroidal neovascularization. Arch Ophthalmol. 2003;121:1283–1288.
77 Kliffen M, Sharma HS, Mooy CM, et al. Increased expression of angiogenic growth factors in age-related maculopathy. Br J Ophthalmol. 1997;81:154–162.
78 Engerman RL. Pathogenesis of diabetic retinopathy. Diabetes. 1989;38:1203–1206.
79 Engerman RL, Kern TS. Retinopathy in animal models of diabetes. Diabetes Metab Rev. 1995;11:109–120.
80 Kowluru RA, Odenbach S. Role of interleukin-1beta in the pathogenesis of diabetic retinopathy. Br J Ophthalmol. 2004;88:1343–1347.
81 Joussen AM, Poulaki V, Mitsiades N, et al. Nonsteroidal anti-inflammatory drugs prevent early diabetic retinopathy via TNF-alpha suppression. FASEB J. 2002;16:438–440.
82 Adamis AP, Berman AJ. Immunological mechanisms in the pathogenesis of diabetic retinopathy. Semin Immunopathol. 2008;30:65–84.
83 Adamis AP, Miller JW, Bernal MT, et al. Increased vascular endothelial growth factor levels in the vitreous of eyes with proliferative diabetic retinopathy. Am J Ophthalmol. 1994;118:445–450.
84 Aiello LP, Avery RL, Arrigg PG, et al. Vascular endothelial growth factor in ocular fluid of patients with diabetic retinopathy and other retinal disorders. N Engl J Med. 1994;331:1480–1487.
85 Shimura M, Yasuda K, Nakazawa T, et al. Panretinal-photocoagulation before pars plana vitrectomy influences vitreous level of interleukin-6 but not of vascular endothelial growth factor in patients with diabetic retinopathy. Int J Biomed Sci. 2007;3:31–37.
86 Shimura M, Yasuda K, Nakazawa K, et al. Panretinal photocoagulation induces pro-inflammatory cytokines and macular thickening in high-risk proliferative diabetic retinopathy. Graefes Arch Clin Exp Ophthalmol. 2009;247:1617–1624.
87 Moradian S, Ahmadieh H, Malihi M, et al. Intravitreal bevacizumab in active progressive proliferative diabetic retinopathy. Graefes Arch Clin Exp Ophthalmol. 2008;246:1699–1705.
88 Cho WB, Oh SB, Moon JW, et al. Panretinal photocoagulation combined with intravitreal bevacizumab in high-risk proliferative diabetic retinopathy. Retina. 2009;29:516–522.
89 Tonello M, Costa RA, Almeida FP, et al. Panretinal photocoagulation versus PRP plus intravitreal bevacizumab for high-risk proliferative diabetic retinopathy (IBeHi study). Acta Ophthalmol. 2008;86:385–389.
90 Jorge R, Costa RA, Calucci D, et al. Intravitreal bevacizumab (Avastin) for persistent new vessels in diabetic retinopathy (IBEPE study). Retina. 2006;26:1006–1013.
91 Boulton M, Gregor Z, McLeod D, et al. Intravitreal growth factors in proliferative diabetic retinopathy: correlation with neovascular activity and glycaemic management. Br J Ophthalmol. 1997;81:228–233.
92 Dills DG, Moss SE, Klein R, et al. Association of elevated IGF-I levels with increased retinopathy in late-onset diabetes. Diabetes. 1991;40:1725–1730.
93 Grant M, Russell B, Fitzgerald C, et al. Insulin-like growth factors in vitreous. Studies in control and diabetic subjects with neovascularization. Diabetes. 1986;35:416–420.
94 Wang Q, Dills DG, Klein R, et al. Does insulin-like growth factor I predict incidence and progression of diabetic retinopathy? Diabetes. 1995;44:161–164.
95 Punglia RS, Lu M, Hsu J, et al. Regulation of vascular endothelial growth factor expression by insulin-like growth factor I. Diabetes. 1997;46:1619–1626.
96 Kirstein M, Aston C, Hintz R, et al. Receptor-specific induction of insulin-like growth factor I in human monocytes by advanced glycosylation end product-modified proteins. J Clin Invest. 1992;90:439–446.
97 Watanabe D, Suzuma K, Suzuma I, et al. Vitreous levels of angiopoietin 2 and vascular endothelial growth factor in patients with proliferative diabetic retinopathy. Am J Ophthalmol. 2005;139:476–481.
98 White RR, Shan S, Rusconi CP, et al. Inhibition of rat corneal angiogenesis by a nuclease-resistant RNA aptamer specific for angiopoietin-2. Proc Natl Acad Sci U S A. 2003;100:5028–5033.
99 Watanabe D, Suzuma K, Matsui S, et al. Erythropoietin as a retinal angiogenic factor in proliferative diabetic retinopathy. N Engl J Med. 2005;353:782–792.
100 Sakanaka M, Wen T-C, Matsuda S, et al. In vivo evidence that erythropoietin protects neurons from ischemic damage. Proc Nat Acad Sci U S A. 1998;95:4635–4640.
101 Becerra SP, Amaral J. Erythropoietin – an endogenous retinal survival factor. N Engl J Med. 2002;347:1968–1970.
102 Dawson DW, Volpert OV, Gillis P, et al. Pigment epithelium-derived factor: a potent inhibitor of angiogenesis. Science. 1999;285:245–248.
103 Volpert OV, Zaichuk T, Zhou W, et al. Inducer-stimulated Fas targets activated endothelium for destruction by anti-angiogenic thrombospondin-1 and pigment epithelium-derived factor. Nat Med. 2002;8:349–357.
104 Ogata N, Matsuoka M, Matsuyama K, et al. Plasma concentration of pigment epithelium-derived factor in patients with diabetic retinopathy. J Clin Endocrinol Metab. 2007;92:1176–1179.
105 Spranger J, Meyer-Schwickerath R, Klein M, et al. TNF-alpha level in the vitreous body. Increase in neovascular eye diseases and proliferative diabetic retinopathy. Med Klin (Munich). 1995;90:134–137.
106 Abu el Asrar AM, Maimone D, Morse PH, et al. Cytokines in the vitreous of patients with proliferative diabetic retinopathy. Am J Ophthalmol. 1992;114:731–736.
107 Funatsu H, Yamashita H, Shimizu E, et al. Relationship between vascular endothelial growth factor and interleukin-6 in diabetic retinopathy. Retina. 2001;21:469–477.
108 Meleth AD, Agron E, Chan CC, et al. Serum inflammatory markers in diabetic retinopathy. Invest Ophthalmol Vis Sci. 2005;46:4295–4301.
109 Miyamoto K, Khosrof S, Bursell SE, et al. Prevention of leukostasis and vascular leakage in streptozotocin-induced diabetic retinopathy via intercellular adhesion molecule-1 inhibition. Proc Natl Acad Sci U S A. 1999;96:10836–10841.
110 Kern TS. Contributions of inflammatory processes to the development of the early stages of diabetic retinopathy. Exp Diabetes Res. 2007;2007:95103.
111 Iliaki E, Poulaki V, Mitsiades N, et al. Role of α4 integrin (cd49d) in the pathogenesis of diabetic retinopathy. Invest Ophthalmol Vis Sci. 2009;50:4890–4904.
112 Dinarello CA. Biologic basis for interleukin-1 in disease. Blood. 1996;87:2095–2147.
113 Jung YJ, Isaacs JS, Lee S, et al. IL-1beta-mediated up-regulation of HIF-1alpha via an NFkappaB/COX-2 pathway identifies HIF-1 as a critical link between inflammation and oncogenesis. FASEB J. 2003;17:2115–2117.
114 Frater-Schroder M, Risau W, Hallmann R, et al. Tumor necrosis factor type, a potent inhibitor of endothelial cell growth in vitro, is angiogenic in vivo. Proc Natl Acad Sci U S A. 1987;84:5277–5281.
115 Leibovich SJ, Polverini PJ, Shepard HM, et al. Macrophage-induced angiogenesis is mediated by tumour necrosis factor-alpha. Nature. 1987;329:630–632.
116 Kociok N, Radetzky S, Krohne TU, et al. Pathological but not physiological retinal neovascularization is altered in TNF-Rp55-receptor-deficient mice. Invest Ophthalmol Vis Sci. 2006;47:5057–5065.
117 Joussen AM, Poulaki V, Qin W, et al. Retinal vascular endothelial growth factor induces intercellular adhesion molecule-1 and endothelial nitric oxide synthase expression and initiates early diabetic retinal leukocyte adhesion in vivo. Am J Pathol. 2002;160:501–509.
118 Joussen AM, Poulaki V, Le ML, et al. A central role for inflammation in the pathogenesis of diabetic retinopathy. FASEB J. 2004;18:1450–1452.
119 Pruijt JFM, Fibbe WE, Laterveer L, et al. Prevention of interleukin-8-induced mobilization of hematopoietic progenitor cells in rhesus monkeys by inhibitory antibodies against the metalloproteinase gelatinase B (MMP-9). Proc Natl Acad Sci U S A 1999;96:10863–8.
120 Van den Steen PE, Proost P, Wuyts A, et al. Neutrophil gelatinase B potentiates interleukin-8 tenfold by aminoterminal processing, whereas it degrades CTAP-III, PF-4, and GRO-alpha and leaves RANTES and MCP-2 intact. Blood. 2000;96:2673–2681.
121 Descamps FJ, Van den Steen PE, Martens E, et al. Gelatinase B is diabetogenic in acute and chronic pancreatitis by cleaving insulin. FASEB J. 2003;17:887–889.
122 Notari L, Miller A, Martínez A, et al. Pigment epithelium-derived factor is a substrate for matrix metalloproteinase type 2 and type 9: implications for downregulation in hypoxia. Invest Ophthalmol Vis Sci. 2005;46:2736–2747.
123 Beránek M, Kolar P, Tschoplova S, et al. Genetic variations and plasma levels of gelatinase A (matrix metalloproteinase-2) and atinase B (matrix metalloproteinase-9) in proliferative diabetic retinopathy. Mol Vis. 2008;14:1114–1121.
124 Bell EF, Klein JM. Comments on oxygen toxicity and retinopathy (ROP) in the premature infant. In: Bell EF, Segar JL. Iowa neonatology handbook: pulmonary. Iowa: University of Iowa, Children’s Hospital, Department of Pediatrics, 1994.
125 Ashton N. Oxygen and the growth and development of retinal vessels. In vivo and in vitro studies. The XX Francis I. Proctor Lecture. Am J Ophthalmol. 1966;62:412–435.
126 Mintz-Hittner HA, Kennedy KA, Chuang AZ, et al. Efficacy of intravitreal bevacizumab for stage 3+ retinopathy of prematurity. N Engl J Med. 2011;364:603–615.
127 Hellstrom A, Perruzzi C, Ju M, et al. Low IGF-I suppresses VEGF-survival signaling in retinal endothelial cells: direct correlation with clinical retinopathy of prematurity. Proc Natl Acad Sci U S A. 2001;98:5804–5808.
128 Miller JW, Adamis A, Shima DT, et al. Vascular endothelial growth factor/vascular permeability factor is temporally and spatially correlated with ocular angiogenesis in a primate model. Am J Pathol. 1994;145:574–584.
129 Noma H, Funatsu H, Mimura T, et al. Vitreous levels of interleukin-6 and vascular endothelial growth factor in macular edema with central retinal vein occlusion. Ophthalmology. 2009;116:87–93.
130 Suzuki Y, Nakazawa M, Suzuki K, et al. Expression profiles of cytokines and chemokines in vitreous fluid in diabetic retinopathy and central retinal vein occlusion. Jpn J Ophthalmol. 2011;55:256–263.
131 Noma H, Funatsu H, Harino S, et al. Vitreous inflammatory factors in macular edema with central retinal vein occlusion. Jpn J Ophthalmol. 2011;55:248–255.
132 Noma H, Funatsu H, Mimura T, et al. Vitreous levels of pigment epithelium-derived factor and vascular endothelial growth factor in macular edema with central retinal vein occlusion. Curr Eye Res. 2011;36:256–263.
133 Brown DM, Campochiaro PA, Singh RP, et al. Ranibizumab for macular edema following central retinal vein occlusion: six-month primary end point results of a phase III study. Ophthalmology. 2010;117:1124–1133.
134 Brockhurst R, Schepens C, Okamura I. Uveitis. II. Peripheral uveitis: clinical description, complications and differential diagnosis. Am J Ophthalmol. 1960;49:1257–1266.
135 Shorb S, Irvine A, Kimura S. Optic disc neovascularization associated with chronic uveitis. Am J Ophthalmol. 1976;82:175–178.
136 Dees C, Arnold JJ, Forrester JV, et al. Immunosuppressive treatment of choroidal neovascularization associated with endogenous posterior uveitis. Arch Ophthalmol. 1998;116:1456–1461.
137 Flaxel CJ, Owens SL, Mulholland B, et al. The use of corticosteroids for choroidal neovascularisation in young patients. Eye. 1998;12:266–272.
138 Martidis A, Miller DG, Ciulla TA, et al. Corticosteroids as an antiangiogenic agent for histoplasmosis-related subfoveal choroidal neovascularisation. J Ocul Pharmacol Ther. 1999;15:425–428.
139 Adan A, Mateo C, Navarro R, et al. Intravitreal bevacizumab (Avastin) injection as primary treatment of inflammatory choroidal neovascularisation. Retina. 2007;27:1180–1186.
140 Chan WM, Lai TM, Liu DT, et al. Intravitreal bevacizumab (Avastin) for choroidal neovascularisation secondary to central serous retinopathy, secondary to punctuate inner choroidopathy, or of idiopathic origin. Am J Ophthalmol. 2007;143:977–983.
141 Hammond CJ, Webster AR, Snieder H, et al. Genetic influence on early age-related maculopathy: a twin study. Ophthalmology. 2002;109:730–736.
142 Baird PN, Guida E, Chu DT, et al. The epsilon2 and epsilon4 alleles of the apolipoprotein gene are associated with age-related macular degeneration. Invest Ophthalmol Vis Sci. 2004;45:1311–1315.
143 Asensio-Sanchez VM, Rodriguez-Martin T, Gala-Molina I, et al. Age-related macular degeneration: its association with the epsilon4 allele of the apolipoprotein E gene. Arch Soc Esp Oftalmol. 2006;81:9–12.
144 Utheim OA, Ritland JS, Utheim TP, et al. Apolipoprotein E genotype and risk for development of cataract and age-related macular degeneration. Acta Ophthalmol. 2008;86:401–403.
145 Losonczy G, Fekete Á, Vokó Z, et al. Analysis of complement factor H Y402H, LOC387715, HTRA1 polymorphisms and ApoE alleles with susceptibility to age-related macular degeneration in Hungarian patients. Acta Ophthalmol. 2011;89:255–262.
146 Brión M, Sanchez-Salorio M, Cortón M, et al. Genetic association study of age-related macular degeneration in the Spanish population. Acta Ophthalmol. 2011;89:e12–e22.
147 Yang X, Hu J, Zhang J, et al. Polymorphisms in CFH, HTRA1 and CX3CR1 confer risk to exudative age-related macular degeneration in Han Chinese. Br J Ophthalmol. 2010;94:1211–1214.
148 Zerbib J, Puche N, Richard F, et al. No association between the T280M polymorphism of the CX3CR1 gene and exudative AMD. Exp Eye Res. 2011;93:382–386.
149 Liu MM, Agrón E, Chew E, et al. Copy number variations in candidate genes in neovascular age-related macular degeneration. Invest Ophthalmol Vis Sci. 2011;52:3129–3135.
150 Yang Z, Stratton C, Francis P, et al. Toll-like receptor 3 and geographic atrophy in age-related macular degeneration. N Engl J Med. 2008;359:1456–1463.
151 Sng CC, Cackett PD, Yeo IY, et al. Toll-like receptor 3 polymorphism rs3775291 is not associated with choroidal neovascularization or polypoidal choroidal vasculopathy in Chinese subjects. Ophthalmic Res. 2011;45:191–196.
152 Klein ML, Ferris FL, 3rd., Francis PJ, et al. Progression of geographic atrophy and genotype in age-related macular degeneration. Ophthalmology. 2010;117:1554–1559. 1559.e1
153 Edwards AO, Chen D, Fridley BL, et al. Toll-like receptor polymorphisms and age-related macular degeneration. Invest Ophthalmol Vis Sci. 2008;49:1652–1659.
154 Gotoh N, Yamashiro K, Nakanishi H, et al. Haplotype analysis of the ARMS2/HTRA1 region in Japanese patients with typical neovascular age-related macular degeneration or polypoidal choroidal vasculopathy. Jpn J Ophthalmol. 2010;54:609–614.
155 Friedrich U, Myers CA, Fritsche LG, et al. Risk- and non-risk-associated variants at the 10q26 AMD locus influence ARMS2 mRNA expression but exclude pathogenic effects due to protein deficiency. Hum Mol Genet. 2011;20:1387–1399.
156 Hadley D, Orlin A, Brown G, et al. Analysis of six genetic risk factors highly associated with AMD in the region surrounding ARMS2 and HTRA1 on chromosome 10, region q26. Invest Ophthalmol Vis Sci. 2010;51:2191–2196.
157 Haines JL, Hauser MA, Schmidt S, et al. Complement factor H variant increases the risk of age-related macular degeneration. Science. 2005;308:419–421.
158 Edwards AO, Ritter R, III., Abel KJ, et al. Complement factor H polymorphism and age-related macular degeneration. Science. 2005;308:421–424.
159 Klein RJ, Zeiss C, Chew EY, et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308:385–389.
160 Francis PJ, Hamon SC, Ott J, et al. Polymorphisms in C2, CFB and C3 are associated with progression to advanced age related macular degeneration associated with visual loss. J Med Genet. 2009;46:300–307.
161 Yates JR, Sepp T, Matharu BK, et al. Complement C3 variant and the risk of age-related macular degeneration. N Engl J Med. 2007;357:553–561.
162 Maller JB, Fagerness JA, Reynolds RC, et al. Variation in complement factor 3 is associated with risk of age-related macular degeneration. Nat Genet. 2007;39:1200–1201.
163 Spencer KL, Hauser MA, Olson LM, et al. Deletion of CFHR3 and CFHR1 genes in age-related macular degeneration. Hum Mol Genet. 2008 1;17:971–977.
164 Allikmets R, Singh N, Sun H, et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy [see comments]. Nat Genet. 1997;15:236–246.
165 Webster AR, Heon E, Lotery AJ, et al. An analysis of allelic variation in the ABCA4 gene. Invest Ophthalmol Vis Sci. 2001;42:1179–1189.
166 Weber BH, Vogt G, Pruett RC, et al. Mutations in the tissue inhibitor of metalloproteinases-3 (TIMP3) in patients with Sorsby’s fundus dystrophy. Nat Genet. 1994;8:352–356.
167 Fariss RN, Apte SS, Olsen BR, et al. Tissue inhibitor of metalloproteinases-3 is a component of Bruch’s membrane of the eye. Am J Pathol. 1997;150:323–328.
168 Felbor U, Doepner D, Schneider U, et al. Evaluation of the gene encoding the tissue inhibitor of metalloproteinases-3 in various maculopathies. Invest Ophthalmol Vis Sci. 1997;38:1054–1059.
169 De La Paz MA, Pericak-Vance MA, Lennon F, et al. Exclusion of TIMP3 as a candidate locus in age-related macular degeneration. Invest Ophthalmol Vis Sci. 1997;38:1060–1065.
170 Petrukhin K, Koisti MJ, Bakall B, et al. Identification of the gene responsible for Best macular dystrophy. Nat Genet. 1998;19:241–247.
171 Marmorstein LY, Munier FL, Arsenijevic Y, et al. Aberrant accumulation of EFEMP1 underlies drusen formation in malattia leventinese and age-related macular degeneration. Proc Natl Acad Sci USA. 2002;99:13067–13072.
172 Heller SR, ADVANCE Collaborative Group. A summary of the ADVANCE Trial. Diabetes Care. 2009;32(Suppl 2):S357–S361.
173 Beulens JWJ, Patel A, Vingerling JR, et al. Effects of blood pressure lowering and intensive glucose control on incidence and progression of retinopathy in patients with type 2 diabetes mellitus: a randomised controlled trial. Diabetologia. 2009;52:2027–2036.
174 Sun JK, Keenan HA, Cavallerano JD, et al. Protection from retinopathy and other complications in patients with type 1 diabetes of extreme duration: the Joslin 50-year Medalist study. Diabetes Care. 2011;34:968–974.
175 Keenan HA, Costacou T, Sun JK, et al. Clinical factors associated with resistance to microvascular complications in diabetic patients of extreme disease duration: the 50-year Medalist study. Diabetes Care. 2007;30:1995–1997.
176 Liew G, Klein R, Wong TY. The role of genetics in susceptibility to diabetic retinopathy. Int Ophthalmol Clin. 2009;49:35–52.
177 Sorbinil Retinopathy Trial Research Group. A randomized trial of sorbinil, an aldose reductase inhibitor, in diabetic retinopathy. Arch Ophthalmol. 1990;108:1234–1244.
178 Lorenzi M. The polyol pathway as a mechanism for diabetic retinopathy: attractive, elusive, and resilient. Exp diabetes res. 2007;2007:61038.
179 Ells A, Guemsey DL, Wallace K, et al. Severe retinopathy of prematurity associated with FZD4 mutations. Ophthalmic Genet. 2010;31:37–43.
180 Haider MZ, Devarajan LV, Al-Essa M, et al. Angiotensin-converting enzyme gene insertion/deletion polymorphism in Kuwaiti children with retinopathy of prematurity. Biol Neonate. 2002;82:84–88.
181 Cooke RW, Drury JA, Mountford R, et al. Genetic polymorphisms and retinopathy of prematurity. Invest Ophthalmol Vis Sci. 2004;45:1712–1715.
182 Vannay AV, Dunai G, Banyasz I, et al. Association of genetic polymorphisms of vascular endothelial growth factor and risk of proliferative retinopathy of prematurity. Pediatr Res. 2005;57:396–398.
183 Shastry BS, Qu X. Lack of association of the VEGF gene promoter (-634 G to C and -460 C to T) polymorphism and the risk of advanced retinopathy of prematurity. Graefes Arch Clin Exp Ophthalmol. 2007;245:741–743.
184 Shastry BS. Lack of association of VEGF (-2578 C to A) and ANG2 (-35 G to C) gene polymorphisms with the progression of retinopathy of prematurity. Graefes Arch Clin Exp Ophthalmol. 2009;247:859–860.
185 Kwinta P, Bik-Multanowski M, Mitkowska Z, et al. The clinical role of vascular endothelial growth factor (VEGF) system in the pathogenesis of retinopathy of prematurity. Graefes Arch Clin Exp Ophthalmol. 2008;246:1467–1475.
186 Banyasz I, Bokodl G, Vannay A, et al. Genetic polymorphisms of vascular endothelial growth factor and angiopoietin 2 in retinopathy of prematurity. Curr Eye Res. 2006;31:685–690.
187 Dunai G, Vasarhelyi B, Szabo M, et al. Published genetic variants in retinopathy of prematurity: random forest analysis suggests a negligible contribution to risk and severity. Curr Eye Res. 2008;33:501–505.
188 Shastry BS. Assessment of the contribution of insulin-like growth factor -I receptor 3174 G to A polymorphism to the progression of advanced retinopathy of prematurity. Eur J Ophthalmol. 2007;17:950–953.
189 Balogh A, Derzbach L, Vannay A, et al. Lack of association between insulin-like growth factor – I receptor G3174A polymorphism and retinopathy of prematurity. Graefes Arch Clin Exp Ophthalmol. 2006;244:1035–1038.
190 Spiegler J, Gilhaus A, Konig IR, et al. Polymorphism in the renin–angiotensin system and outcome of very low birth weight infants. Neonatology. 2009;97:10–14.
191 John BR, Loggins J, Yanamandra K. Angiotensin converting enzyme insertion/deletion polymorphism does not alter sepsis outcome in ventilated very low birth weight infants. J Perinatol. 2005;25:205–209.
192 Mohamed S, Schaa K, Cooper ME, et al. Genetic contributions to the development of retinopathy of prematurity. Pediatr Res. 2009;65:193–197.
193 Christen WG, Glynn RJ, Manson JE, et al. A prospective study of cigarette smoking and risk of age-related macular degeneration in men. JAMA. 1996;276:1147–1151.
194 Seddon JM, Willett WC, Speizer FE, et al. A prospective study of cigarette smoking and age-related macular degeneration in women. JAMA. 1996;276:1141–1146.
195 Klein R, Klein BE, Moss SE. Relation of smoking to the incidence of age-related maculopathy: the Beaver Dam Eye Study. Am J Epidemiol. 1998;147:103–110.
196 Mares JA, Voland RP, Sondel SA, et al. Healthy lifestyles related to subsequent prevalence of age-related macular degeneration. Arch Ophthalmol. 2011;129:470–480.
197 Age-Related Eye Disease Study Research Group. A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E, beta carotene, and zinc for age-related macular degeneration and vision loss: AREDS report No. 8. Arch Ophthalmol. 2001;119:1417–1436.
198 Christen WG, Glynn RJ, Chew EY, et al. Folic acid, pyridoxine, and cyanocobalamin combination treatment and age-related macular degeneration in women: the Women’s Antioxidant and Folic Acid Cardiovascular Study. Arch Intern Med. 2009;169:335–341.
199 Zoppini G, Faccini G, Muggeo M, et al. Elevated plasma levels of soluble receptors of TNF-alpha and their association with smoking and microvascular complications in young adults with type 1 diabetes. J Clin Endocrinol Metab. 2001;86:3805–3808.
200 Mol MJ, de Rijke YB, Demacker PN, et al. Plasma levels of lipid and cholesterol oxidation products and cytokines in diabetes mellitus and cigarette smoking: effects of vitamin E treatment. Atherosclerosis. 1997;129:169–176.
201 Wasada T, Kawahara R, Katsumori K, et al. Plasma concentration of immunoreactive vascular endothelial growth factor and its relation to smoking. Metabolism. 1998;47:27–30.
202 Keech AC, Mitchell P, Summanen PA, et al. Effect of fenofibrate on the need for laser treatment for diabetic retinopathy (FIELD study): a randomised controlled trial. Lancet. 2007;370:1687–1697.
203 ACCORD Study Group. Effects of medical therapies on retinopathy progression in type 2 diabetes. et al. N Engl J Med. 2010;363:233–244.
204 Forrester RM, Jefferson E, Naunton WJ. Oxygen and retrolental fibroplasia; a seven-year survey. Lancet. 1954;ii:258–260.
205 Oxygen restriction and retinopathy of prematurity. Editorial. Lancet. 1992;339:961–962.
206 Szewczyk TS. Retrolental fibroplasia: etiology and prophylaxis. Am J Ophthalmol. 1951;34:1609.
207 Bedrossian RH, Carmichael P, Ritter J. Retinopathy of prematurity (retrolental fibroplasia) and oxygen. Am J Ophthalmol. 1954;37:78.
208 Kinsey VE. Retrolental fibroplasia. Co-operative study of retrolental fibroplasia and the use of oxygen. Arch Ophthalmol. 1955;59:481–542.
209 Forrester RM. Oxygen cerebral palsy and retrolental fibroplasia. Dev Med Child Neurol. 1964;6:648–650.
210 Swart-Van Der Hoeven JT, Mak TMB. Effects of oxygen on retrolental fibroplasia in premature infants, with report of two cases. Maadschr Kindergeneesk. 1952;20:276.
211 Reynolds JD, Hardy RJ, Kennedy KA, et al. Lack of efficacy of light reduction in preventing retinopathy of prematurity. Light Reduction in Retinopathy of Prematurity (LIGHT-ROP) Cooperative Group. N Engl J Med. 1998;338:1572–1576.
212 Connor KM, SanGiovanni JP, Lofqvist C, et al. Increased dietary intake of ω3 polyun- saturated fatty acids reduces pathological retinal angiogenesis. Nat Med. 2007;7:868–873.
213 Birch EE, Carlson SE, Hoffman DR, et al. The DIAMOND (DHA Intake And Measurement Of Neural Development) study: a double-masked, randomized controlled clinical trial of the maturation of infant visual acuity as a function of the dietary level of docosahexaenoic acid. Am J Clin Nutr. 2010;91:848–859.
214 Pepper MS, Ferrara N, Orci L, et al. Vascular endothelial growth factor (VEGF) induces plasminogen activators and plasminogen activator inhibitor-1 in microvascular endothelial cells. Biochem Biophys Res Commun. 1991;181:902–906.
215 Senger DR, Claffey K, Benes JE, et al. Angiogenesis promoted by vascular endothelial growth factor: regulation through alpha1beta1 and alpha2beta1 integrins. Proc Natl Acad Sci U S A. 1997;94:13612–13617.
216 Esser S, Wolburg K, Wolburg H, et al. Vascular endothelial growth factor induces endothelial fenestrations in vitro. J Cell Biol. 1998;140:947–959.
217 Lu M, Perez VL, Ma N, et al. VEGF increases retinal vascular ICAM-1 expression in vivo. Invest Ophthalmol Vis Sci. 1999;40:1808–1812.
218 Miyamoto K, Khosrof S, Bursell SE, et al. Vascular endothelial growth factor (VEGF)-induced retinal vascular permeability is mediated by intercellular adhesion molecule-1 (ICAM-1). Am J Pathol. 2000;156:1733–1739.
219 Hutchings H, Ortega N, Plouet J. Extracellular matrix-bound vascular endothelial growth factor promotes endothelial cell adhesion, migration, and survival through integrin ligation. FASEB J. 2003;17:1520–1522.
220 Tischer E, Gospodarowicz D, Mitchell R, et al. Vascular endothelial growth factor: a new member of the platelet-derived growth factor gene family. Biochem Biophys Res Commun. 1989;165:1198–1206.
221 Ferrara N, Houck K, Jakeman L, et al. Molecular and biological properties of the vascular endothelial growth factor family of proteins. Endocr Rev. 1992;13:18–32.
222 Houck KA, Leung DW, Rowland AM, et al. Dual regulation of vascular endothelial growth factor bioavailability by genetic and proteolytic mechanisms. J Biol Chem. 1992;267:26031–26037.
223 Carmeliet P, Ng YS, Nuyens D, et al. Impaired myocardial angiogenesis and ischemic cardiomyopathy in mice lacking the vascular endothelial growth factor isoforms VEGF164 and VEGF188. Nat Med. 1999;5:495–502.
224 Stalmans I, Ng YS, Rohan R, et al. Arteriolar and venular patterning in retinas of mice selectively expressing VEGF isoforms. J Clin Invest. 2002;109:327–336.
225 Salven P, Lymboussaki A, Heikkila H, et al. Vascular endothelial growth factors VEGF-B and VEGF-C are expressed in human tumors. Am J Pathol. 1998;153:103–108.
226 Witzenbichler B, Asahara T, Murohara T, et al. Vascular endothelial growth factor-C (VEGF-C/VEGF-2) promotes angiogenesis in the setting of tissue ischemia. Am J Pathol. 1998;153:381–394.
227 Meyer M, Clauss M, Lepple-Wienhues A, et al. A novel vascular endothelial growth factor encoded by Orf virus, VEGF-E, mediates angiogenesis via signalling through VEGFR-2 (KDR) but not VEGFR-1 (Flt-1) receptor tyrosine kinases. EMBO J. 1999;18:363–374.
228 Ferrara N, Davis-Smyth T. The biology of vascular endothelial growth factor. Endocr Rev. 1997;18:4–25.
229 Jakeman LB, Winer J, Bennett GL, et al. Binding sites for vascular endothelial growth factor are localized on endothelial cells in adult rat tissues. J Clin Invest. 1992;89:244–253.
230 Kim I, Ryan AM, Rohan R, et al. Constitutive expression of VEGF, VEGFR-1, and VEGFR-2 in normal eyes. Invest Ophthalmol Vis Sci. 1999;40:2115–2121.
231 Ferrara N, Gerber H, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–676.
232 Matsumoto T, Claesson-Welsh L. VEGF receptor signal transduction. Sci STKE. 2001;2001:RE21.
233 Park JE, Chen HH, Winer J, et al. Placenta growth factor. Potentiation of vascular endothelial growth factor bioactivity, in vitro and in vivo, and high affinity binding to Flt-1 but not to Flk-1/KDR. J Biol Chem. 1994;269:25646–25654.
234 Barleon B, Sozzani S, Zhou D, et al. Migration of human monocytes in response to vascular endothelial growth factor (VEGF) is mediated via the VEGF receptor Flt-1. Blood. 1996;87:3336–3343.
235 Hiratsuka S, Minowa O, Kuno J, et al. Flt-1 lacking the tyrosine kinase domain is sufficient for normal development and angiogenesis in mice. Proc Natl Acad Sci U S A. 1998;95:9349–9354.
236 Pepper MS, Mandriota SJ. Regulation of vascular endothelial growth factor receptor-2 (Flk-1) expression in vascular endothelial cells. Exp Cell Res. 1998;241:414–425.
237 Lee P, Goishi K, Davidson AJ, et al. Neuropilin-1 is required for vascular development and is a mediator of VEGF-dependent angiogenesis in zebrafish. Proc Natl Acad Sci U S A. 2002;99:10470–10475.
238 Shima DT, Adamis A, Ferrara N, et al. Hypoxic induction of endothelial cell growth factors in retinal cells: identification and characterization of vascular endothelial growth factor (VEGF) as the mitogen. Mol Med. 1995;1:182–193.
239 Shweiki D, Itin A, Soffer D, et al. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature. 1992;359:843–845.
240 Huang JC, Liu DY, Dawood MY. The expression of vascular endothelial growth factor isoforms in cultured human endometrial stromal cells and its regulation by 17beta-oestradiol. Mol Hum Reprod. 1998;4:603–607.
241 Kuroki M, Voest EE, Amano S, et al. Reactive oxygen intermediates increase vascular endothelial growth factor expression in vitro and in vivo. J Clin Invest. 1996;98:1667–1675.
242 Satake S, Kuzuya M, Miura H, et al. Up-regulation of vascular endothelial growth factor in response to glucose deprivation. Biol Cell. 1998;90:161–168.
243 Neufeld G, Cohen T, Gengrinovitch S, et al. Vascular endothelial growth factor (VEGF) and its receptors. FASEB J. 1999;13:9–22.
244 Shima DT, Gougos A, Miller JW, et al. Cloning and mRNA expression of vascular endothelial growth factor in ischemic retinas of Macaca fascicularis. Invest Ophthalmol Vis Sci. 1996;37:1334–1340.
245 Dorey CK, Aouididi S, Reynaud X, et al. Correlation of vascular permeability factor/vascular endothelial growth factor with extraretinal neovascularization in the rat. Arch Ophthalmol. 1996;114:1210–1217.
246 Pierce EA, Avery RL, Foley ED, et al. Vascular endothelial growth factor/ vascular permeability factor expression in a mouse model of retinal neovascularization. Proc Natl Acad Sci U S A. 1995;92:905–909.
247 Stone J, Chan-Ling T, Pe’er J, et al. Roles of vascular endothelial growth factor and astrocyte degeneration in the genesis of retinopathy of prematurity. Invest Ophthalmol Vis Sci. 1996;37:290–299.
248 Connolly DT, Heuvelman DM, Nelson R, et al. Tumor vascular permeability factor stimulates endothelial cell growth and angiogenesis. J Clin Invest. 1989;84:1470–1478.
249 Plouet J, Schilling J, Gospodarowicz D. Isolation and characterization of a newly identified endothelial cell mitogen produced by AtT-20 cells. EMBO J. 1989;8:3801–3806.
250 Takeshita S, Zheng L, Brogi E, et al. Therapeutic angiogenesis. A single intraarterial bolus of vascular endothelial growth factor augments revascularization in a rabbit ischemic hind limb model. J Clin Invest. 1994;93:662–670.
251 Tolentino MJ, Miller JW, Gragoudas ES, et al. Vascular endothelial growth factor is sufficient to produce iris neovascularization and neovascular glaucoma in a nonhuman primate. Arch Ophthalmol. 1996;114:964–970.
252 Tolentino MJ, Miller JW, Gragoudas ES, et al. Intravitreous injections of vascular endothelial growth factor produce retinal ischemia and microangiopathy in an adult primate. Ophthalmology. 1996;103:1820–1828.
253 Adamis AP, Shima DT, Tolentino MJ, et al. Inhibition of vascular endothelial growth factor prevents retinal ischemia-associated iris neovascularization in a nonhuman primate. Arch Ophthalmol. 1996;114:66–71.
254 Aiello LP, Pierce EA, Foley ED, et al. Suppression of retinal neovascularization in vivo by inhibition of vascular endothelial growth factor (VEGF) using soluble VEGF-receptor chimeric proteins. Proc Natl Acad Sci U S A. 1995;92:10457–10461.
255 Robinson GS, Pierce EA, Rook SL, et al. Oligodeoxynucleotides inhibit retinal neovascularization in a murine model of proliferative retinopathy. Proc Natl Acad Sci U S A. 1996;93:4851–4856.
256 Kvanta A, Algvere PV, Berglin L, et al. Subfoveal fibrovascular membranes in age-related macular degeneration express vascular endothelial growth factor. Invest Ophthalmol Vis Sci. 1996;37:1929–1934.
257 Ishibashi T, Hata Y, Yoshikawa H, et al. Expression of vascular endothelial growth factor in experimental choroidal neovascularization. Graefes Arch Clin Exp Ophthalmol. 1997;235:159–167.
258 Schlingemann RO. Role of growth factors and the wound healing response in age-related macular degeneration. Graefes Arch Clin Exp Ophthalmol. 2004;242:92–101.
259 Kwak N, Okamoto N, Wood JM, et al. VEGF is major stimulator in model of choroidal neovascularization. Invest Ophthalmol Vis Sci. 2000;41:3158–3164.
260 Seo MS, Kwak N, Ozaki H, et al. Dramatic inhibition of retinal and choroidal neovascularization by oral administration of a kinase inhibitor. Am J Pathol. 1999;154:1743–1753.
261 Gragoudas ES, Adamis AP, Cunningham ET, Jr., et al. Pegaptanib for neovascular age-related macular degeneration. N Engl J Med. 2004;351:2805–2816.
262 CATT Research Group. Ranibizumab and bevacizumab for neovascular age-related macular degeneration. N Engl J Med. 2011;364:897–908.
263 Rosenfeld PJ, Brown DM, Heier JS, et al. Ranibizumab for neovascular age-related macular degeneration. N Engl J Med. 2006;355:1419–1431.
264 Brown DM, Kaiser PK, Michels M, et al. Ranibizumab versus verteporfin for neovascular age-related macular degeneration. N Engl J Med. 2006;355:1432–1444.
265 Poulsen J. The Houssay phenomenon in man: recovery from retinopathy in a case of diabetes with Simmonds’ disease. Diabetes. 1953;2:7–12.
266 Luft R, Olivecrona H, Ikkos D. Hypophysectomy in man: further experiences in severe diabetes mellitus. Br Med J. 1955;2:752–756.
267 Sharp PS, Fallon TJ, Brazier OJ, et al. Long-term follow-up of patients who underwent yttrium-90 pituitary implantation for treatment of proliferative diabetic retinopathy. Diabetologia. 1987;30:199–207.
268 Hellstrom A, Perruzzi C, Ju M, et al. Low IGF-I suppresses VEGF-survival signaling in retinal endothelial cells: direct correlation with clinical retinopathy of prematurity. Proc Natl Acad Sci U S A. 2001;98:5804–5808.
269 Smith LE, Kopchick JJ, Chen W, et al. Essential role of growth hormone in ischemia-induced retinal neovascularization. Science. 1997;276:1706–1709.
270 Hellstrom A, Engstrom E, Hard AL, et al. Postnatal serum insulin-like growth factor I deficiency is associated with retinopathy of prematurity and other complications of premature birth. Pediatrics. 2003;112:1016–1020.
271 Vlodavsky I, Folkman J, Sullivan R, et al. Endothelial cell-derived basic fibroblast growth factor: synthesis and deposition into subendothelial extracellular matrix. Proc Natl Acad Sci U S A. 1987;84:2292–2296.
272 Mignatti P, Tsuboi R, Robbins E, et al. In vitro angiogenesis on the human amniotic membrane: requirement for basic fibroblast growth factor-induced proteinases. J Cell Biol. 1989;108:671–682.
273 Lobb RR, Alderman EM, Fett JW. Induction of angiogenesis by bovine brain derived class 1 heparin-binding growth factor. Biochemistry. 1985;24:4969–4973.
274 Shing Y, Folkman J, Haudenschild C, et al. Angiogenesis is stimulated by a tumor-derived endothelial cell growth factor. J Cell Biochem. 1985;29:275–287.
275 Amin R, Puklin JE, Frank RN. Growth factor localization in choroidal neovascular membranes of age-related macular degeneration. Invest Ophthalmol Vis Sci. 1994;35:3178–3188.
276 Baird A. Fibroblast growth factors. In: Roberts A, ed. Handbook of experimental pharmacology: peptide growth factors and their receptors. Berlin: Springer-Verlag, 1990.
277 Gospodarowicz D. Fibroblast growth factors. In: Pimental E, Perucho M. Critical reviews in oncogenesis. Boca Raton, FL: CRC Press, 1989.
278 Kitaoka T, Morse LS, Schneeberger S, et al. Expression of FGF5 in choroidal neovascular membranes associated with ARMD. Curr Eye Res. 1997;16:396–399.
279 Abraham JA, Mergia A, Whang JL, et al. Nucleotide sequence of a bovine clone encoding the angiogenic protein, basic fibroblast growth factor. Science. 1986;233:545–548.
280 Cooper DN, Barondes SH. Evidence for export of a muscle lectin from cytosol to extracellular matrix and for a novel secretory mechanism. J Cell Biol. 1990;110:1681–1691.
281 D’Amore PA. Modes of FGF release in vivo and in vitro. Cancer Metastasis Rev. 1990;9:227–238.
282 Kuchler K, Dohlman HG, Thorner J. The a-factor transporter (STE6 gene product) and cell polarity in the yeast Saccharomyces cerevisiae. J Cell Biol. 1993;120:1203–1215.
283 Kostyk SK, D’Amore PA, Herman IM, et al. Optic nerve injury alters basic fibroblast growth factor localization in the retina and optic tract. J Neurosci. 1994;14:1441–1449.
284 Adamis AP, Meklir B, Joyce NC. In situ injury-induced release of basic-fibroblast growth factor from corneal epithelial cells. Am J Pathol. 1991;139:961–967.
285 Joseph-Silverstein J, Silverstein RL. Cell adhesion molecules: an overview. Cancer Invest. 1998;16:176–182.
286 McEver RP. Adhesive interactions of leukocytes, platelets, and the vessel wall during hemostasis and inflammation. Thromb Haemost. 2001;86:746–756.
287 Scatena M, Almeida M, Chaisson ML, et al. NF-kappaB mediates alphavbeta3 integrin-induced endothelial cell survival. J Cell Biol. 1998;141:1083–1093.
288 Brooks PC, Clark RA, Cheresh DA. Requirement of vascular integrin alpha v beta 3 for angiogenesis. Science. 1994;264:569–571.
289 Friedlander M, Theesfeld CL, Sugita M, et al. Involvement of integrins alpha v beta 3 and alpha v beta 5 in ocular neovascular diseases. Proc Natl Acad Sci U S A. 1996;93:9764–9769.
290 Friedlander M, Brooks PC, Shaffer RW, et al. Definition of two angiogenic pathways by distinct alpha v integrins. Science. 1995;270:1500–1502.
291 Hammes HP, Brownlee M, Jonczyk A, et al. Subcutaneous injection of a cyclic peptide antagonist of vitronectin receptor-type integrins inhibits retinal neovascularization. Nat Med. 1996;2:529–533.
292 Folkman J, D’Amore PA. Blood vessel formation: what is its molecular basis? Cell. 1996;87:1153–1155.
293 Koblizek TI, Weiss C, Yancopoulos GD, et al. Angiopoietin-1 induces sprouting angiogenesis in vitro. Curr Biol. 1998;8:529–532.
294 Davis S, Aldrich TH, Jones PF, et al. Isolation of angiopoietin-1, a ligand for the TIE2 receptor, by secretion-trap expression cloning. Cell. 1996;87:1161–1169.
295 Witzenbichler B, Maisonpierre PC, Jones GD, et al. Chemotactic properties of angiopoietin-1 and -2, ligands for the endothelial-specific receptor tyrosine kinase Tie2. J Biol Chem. 1998;273:18514–18521.
296 Wong AL, Haroon ZA, Werner S, et al. Tie2 expression and phosphorylation in angiogenic and quiescent adult tissues. Circ Res. 1997;81:567–574.
297 Maisonpierre PC, Suri C, Jones PF, et al. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science. 1997;277:55–60.
298 Asahara T, Chen D, Takahashi T, et al. Tie2 receptor ligands, angiopoietin-1 and angiopoietin-2, modulate VEGF-induced postnatal neovascularization. Circ Res. 1998;83:233–240.
299 Tombran-Tink J, Johnson LV. Neuronal differentiation of retinoblastoma cells induced by medium conditioned by human RPE cells. Invest Ophthalmol Vis Sci. 1989;30:1700–1707.
300 Dawson DW, Volpert OV, Gillis SE, et al. Pigment epithelium-derived factor: a potent inhibitor of angiogenesis. Science. 1999;285:245–248.
301 Volpert OV, Zaichuk T, Zhou W, et al. Inducer-stimulated Fas targets activated endothelium for destruction by anti-angiogenic thrombospondin-1 and pigment epithelium-derived factor. Nat Med. 2002;8:349–357.
302 Yabe T, Wilson D, Schwartz JN, Fkappa B. Activation is required for the neuroprotective effects of pigment epithelium-derived factor (PEDF) on cerebellar granule neurons. J Biol Chem. 2001;276:43313–43319.
303 Cayouette M, Smith SB, Becerra S, et al. Pigment epithelium-derived factor delays the death of photoreceptors in mouse models of inherited retinal degenerations. Neurobiol Dis. 1999;6:523–532.
304 Karakousis PC, John SK, Behling KC, et al. Localization of pigment epithelium derived factor (PEDF) in developing and adult human ocular tissues. Mol Vis. 2001;7:154–163.
305 Ogata N, Wada M, Otsuji T, et al. Expression of pigment epithelium-derived factor in normal adult rat eye and experimental choroidal neovascularization. Invest Ophthalmol Vis Sci. 2002;43:1168–1175.
306 Holekamp NM, Bouck N, Volpert O. Pigment epithelium-derived factor is deficient in the vitreous of patients with choroidal neovascularization due to age-related macular degeneration. Am J Ophthalmol. 2002;134:220–227.
307 Ogata N, Nishikawa M, Nishimura T, et al. Inverse levels of pigment epithelium-derived factor and vascular endothelial growth factor in the vitreous of eyes with rhegmatogenous retinal detachment and proliferative vitreoretinopathy. Am J Ophthalmol. 2002;133:851–852.
308 Ogata N, Tombran-Tink J, Nishikawa M, et al. Pigment epithelium-derived factor in the vitreous is low in diabetic retinopathy and high in rhegmatogenous retinal detachment. Am J Ophthalmol. 2001;132:378–382.
309 Mori K, Gehlbach S, Yamamoto E, et al. AAV-mediated gene transfer of pigment epithelium-derived factor inhibits choroidal neovascularization. Invest Ophthalmol Vis Sci. 2002;43:1994–2000.
310 Raisler BJ, Berns KI, Grant MB, et al. Adeno-associated virus type-2 expression of pigmented epithelium-derived factor or Kringles 1–3 of angiostatin reduce retinal neovascularization. Proc Natl Acad Sci U S A. 2002;99:8909–8914.
311 Hadler-Olsen E, Fadnes B, Sylte I, et al. Regulation of matrix metalloproteinase activity in health and disease. FEBS J. 2011;278:28–45.
312 Wang H, Keiser JA. Vascular endothelial growth factor upregulates the expression of matrix metalloproteinases in vascular smooth muscle cells: role of flt-1. Circ Res. 1998;83:832–840.
313 Zucker S, Mirza H, Conner CE, et al. Vascular endothelial growth factor induces tissue factor and matrix metalloproteinase production in endothelial cells: conversion of prothrombin to thrombin results in progelatinase A activation and cell proliferation. Int J Cancer. 1998;75:780–786.
314 Hoffmann S, Friedrichs U, Eichler W, et al. Advanced glycation end products induce choroidal endothelial cell proliferation, matrix metalloproteinase-2 and VEGF upregulation in vitro. Graefes Arch Clin Exp Ophthalmol. 2002;240:996–1002.
315 Brooks PC, Silletti S, von Schalscha TL, et al. Disruption of angiogenesis by PEX, a noncatalytic metalloproteinase fragment with integrin binding activity. Cell. 1998;92:391–400.
316 Morizane Y, Thanos A, Takeuchi K, et al. AMP-activated protein kinase suppresses matrix metalloproteinase-9 expression in mouse embryonic fibroblasts. J Biol Chem. 2011;286:16030–16038.
317 Ardi VC, Kupriyanova TA, Deryugina EI, et al. Human neutrophils uniquely release TIMP-free MMP-9 to provide a potent catalytic stimulator of angiogenesis. Proc Natl Acad Sci U S A. 2007;104:20262–20267.
318 O’Reilly MS, Holmgren L, Shing Y, et al. Angiostatin novel angiogenesis inhibitor that mediates the suppression of metastases by a Lewis lung carcinoma. Cell. 1994;79:315–328.
319 O’Reilly MS, Boehm T, Shing Y, et al. Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell. 1997;88:277–285.
320 Standker L, Schrader M, Kanse SM, et al. Isolation and characterization of the circulating form of human endostatin. FEBS Lett. 1997;420:129–133.
321 Lu M, Kuroki M, Amano S, et al. Advanced glycation end products increase retinal vascular endothelial growth factor expression. J Clin Invest. 1998;101:1219–1224.
322 Honda S, Farboud B, Hjelmeland LM, et al. Induction of an aging mRNA retinal pigment epithelial cell phenotype by matrix-containing advanced glycation end products in vitro. Invest Ophthalmol Vis Sci. 2001;42:2419–2425.
323 McFarlane S, McMullen CB, Henry DN. Advanced glycation end products (AGEs) modulate cathepsin D expression in retinal pigment epithelium (RPE): implications for age-related macular dysfunction. Invest Ophthalmol Vis Sci. 2001;42:S414.
324 Schutt F, Bergmann M, Holz FG, et al. Proteins modified by malondialdehyde, 4-hydroxynonenal, or advanced glycation end products in lipofuscin of human retinal pigment epithelium. Invest Ophthalmol Vis Sci. 2003;44:3663–3668.
325 Yan SD, Schmidt AM, Anderson GM, et al. Enhanced cellular oxidant stress by the interaction of advanced glycation end products with their receptors/binding proteins. J Biol Chem. 1994;269:9889–9897.
326 Okamoto T, Tanaka S, Stan AC, et al. Advanced glycation end products induce angiogenesis in vivo. Microvasc Res. 2002;63:186–195.
327 Kvanta A. Expression and regulation of vascular endothelial growth factor in choroidal fibroblasts. Curr Eye Res. 1995;14:1015–1020.