Chapter 5 Autonomic Nervous System and Cardiac Arrhythmias
The autonomic nervous system (ANS) comprises the portion of the central nervous system that provides moment-to-moment regulation of the function of the cardiovascular system as well as that of all other organ systems. The ANS continuously monitors afferent neural signals from vascular beds and organ systems and coordinates efferent neural traffic to modify the responses of heart and blood vessels to ever-changing physiological and metabolic requirements. In this context, the sympathetic and parasympathetic components of the ANS are the dominant players (Figures 5-1 and 5-2).1 However, ANS cardiovascular control also incorporates actions of cardiac and extracardiac neurohumoral agents, intracardiac reflex arcs, and the contributions of certain less well-understood agents such as vasoactive intestinal peptide (VIP), neuropeptide Y, transmitters released by the so-called purinergic nerve endings, serotonin, inflammatory cytokines, vasopressin, and nitric oxide.2 Further, with respect to cardiovascular control, the ANS collaborates with the hypothalamic-pituitary-adrenal (HPA) axis. For its part, the HPA-axis, governed from the hypothalamus, participates by prompting the release of glucocorticoids, mainly cortisol and, to a lesser extent, mineralocorticoids. The HPA theater of operation therefore includes inflammatory, immune, metabolic, and pressor effects.3 Both systems (ANS and HPA) are involved in stress responses.2,3
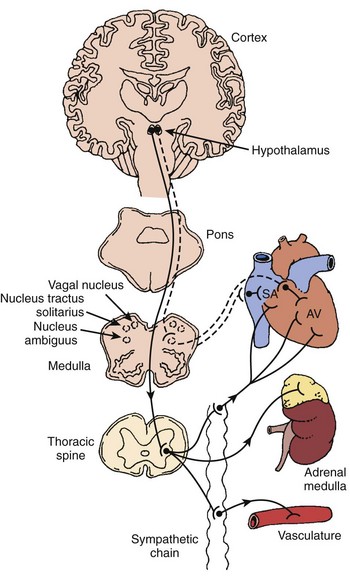
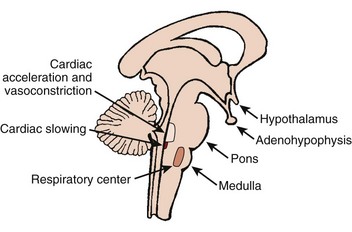
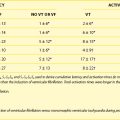
It is not unexpected that any disturbance of ANS function, given its wide-ranging impact, may lead to clinically important consequences. In terms of cardiac electrophysiology and arrhythmias, common clinical conditions in which ANS effects are evident include acute myocardial ischemia, heart failure, and neurally mediated reflex syncope (particularly the vasovagal faint). Furthermore, it is now widely acknowledged that the nervous system has the capacity to injure the heart acutely (e.g., stress-induced cardiomyopathy); serious acute cerebral disorders such as subarachnoid hemorrhage, intracerebral bleeds, infections, and seizures may induce electrocardiographic changes, myocardial damage, arrhythmias, and even sudden death.3–7 Perhaps the most publicized direct cardiac effects of presumed autonomic “storms” are the immediate, apparently stress-triggered, increases in the number of cardiovascular events; these include acute myocardial infarctions, sudden cardiac deaths, and presumed stress-induced cardiomyopathy (Table 5-1).3–5
Table 5-1 Epidemiologic Associations: Stress and Increased Cardiovascular Event Rates
MI, Myocardial infarction; SCD, sudden cardiac death; ICD, implantable cardioverter-defibrillator.
References
a Stalnikowicz R, Tsafrir A: Acute psychosocial stress and cardiovascular events, Am J Emerg Med 20:488–491, 2002.
b Brown DL: Disparate effects of the 1989 Loma Prieta and 1994 Northridge earthquakes on hospital admissions for acute myocardial infarction: Importance of superimposed triggers, Am Heart J 137:830–836, 1999.
c Watanabe H, Kodama M, Okura Y, et al: Impact of earthquakes on Takotsubo cardiomyopathy. JAMA 294:305–306, 2005.
d Zhang XQ, Chen M, Yang Q, et al: Effect of the Wenchuan earthquake in China on hemodynamically unstable ventricular tachyarrhythmia in hospitalized patients, Am J Cardiol 103(7):994–997, 2009.
e Wilbert-Lampen U, Leistner D, Greven S, et al: Cardiovascular events during World Cup soccer. N Engl J Med 358:475–483, 2008.
f Katz E, Metzker J-T, Marazzi A, Kappenberger L: Increased sudden cardiac deaths in Switzerland during the 2002 FIFA World Cup, Int J Cardiol 107:132–133, 2006.
g Brotman DJ, Golden SH, Wittstein IS: The cardiovascular toll of stress, Lancet 376: 1089–1100, 2007.
h Meisel SR, Kutz I, Dayan KI, et al: Effect of Iraqi missile war on incidence of acute myocardial infarction and sudden death in Israeli civilians, Lancet 338:660–661, 1991.
i Tofler GH, Muller JE: Triggering of acute cardiovascular disease and potential preventive strategies, Circulation 114:1863–1872, 2006.
Anatomic Nervous System and Cardiac Conduction System Physiology
Sinus Node, Atrioventricular Node, and His-Purkinje System
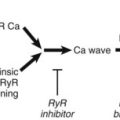
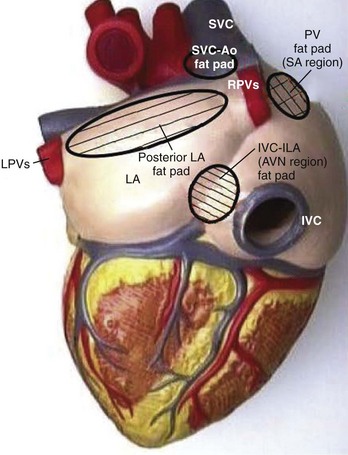
The sinus node (SN) and the atrioventricular (AV) node appear to be represented by separate cells within the nucleus ambiguus. However, it is uncertain whether the nodes are coordinated centrally; in fact, it seems increasingly likely that local circuits, often positioned within the epicardial fat pads of the heart, participate in the coordination of these structures (Figure 5-3).4,8
Atrioventricular Node and Cardiac Conduction System
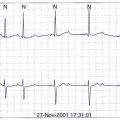
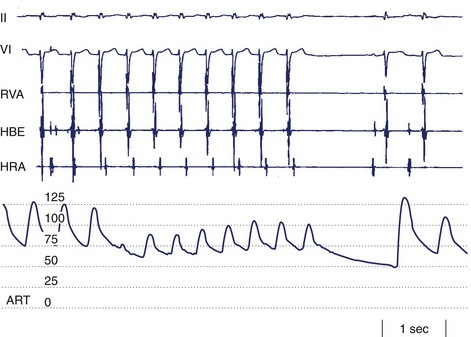
As a rule, AV nodal dromotropic responsiveness in the resting patient is under relatively balanced sympathetic and parasympathetic neural influence. However, this situation is readily altered by physiological events (e.g., exercise, sleep), the impact of disease states, drug effects, or during cardiac electrophysiology procedures when certain atrial regions are stimulated. Any tendency toward parasympathetic predominance markedly enhances the decremental properties of the AV node; in the extreme, this can be associated with transient complete AV nodal block (Figure 5-4). The latter is, in fact, a relatively common finding in sleeping patients and in very fit resting subjects. The relationship between ANS control of SN rate and AV conduction properties appears to foster both the maintenance of 1 : 1 AV conduction and a relatively optimal AV conduction interval.
Ventricular Myocardium
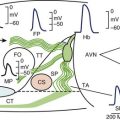
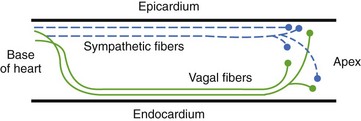
Ventricular sympathetics tend to lie within the subepicardial layer and follow the large coronary vessels as they spread out over the myocardium.9,10 The parasympathetics, in contrast, tend to penetrate the myocardium after crossing the AV groove and thereafter are subendocardial in location (Figure 5-5). The parasympathetic vagal efferents to the mycardium terminate not on the muscle cells themselves but on intracardiac ganglia. Evidence suggests that these ganglia not only form relay stations but also subserve certain local integrative functions, including the intracardiac reflex activity discussed earlier.
Autonomic Nervous System and Specific Bradyarrhythmias and Cardiac Conduction System Disturbances
Atrioventricular Conduction Disturbance
ANS-mediated higher degrees of AV block may also be observed (see Figure 5-4). These episodes of paroxysmal AV block are generally benign from a mortality perspective, although they may be associated with dizziness and syncope (e.g., vasovagal faint) and risk of physical injury. Sustained third-degree AV block is, however, not usually attributable to ANS effects. In adults, acquired complete heart block is almost always associated with structural heart disease.
Autonomic Nervous System and Specific Tachyarrhythmias
Atrial Fibrillation
Supraventricular Tachycardias (Other than Atrial Fibrillation)
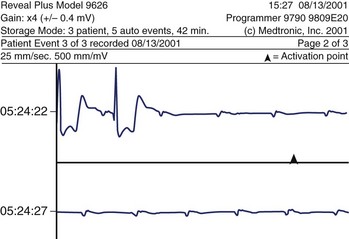
Supraventricular tachycardias are typically categorized into whether or not the tachycardia is dependent on AV node conduction. The first group is exemplified by AV nodal re-entry tachycardia (AVNRT) and AV re-entrant tachycardia (AVRT) using accessory AV connections. The AV nodal independent tachycardias include atrial re-entrant arrhythmias as well as those that are thought to be automatic or triggered in origin. In either case, the ventricular rate is determined by the ANS effect on the AV conduction system (particularly the AV node, see earlier). In certain cases, ANS effects may also play a role in terminating tachyarrhythmias, either spontaneously (Figure 5-6) or during medical interventions such as carotid sinus massage.
Ventricular Tachycardia
The importance of the ANS in determining susceptibility to ventricular tachyarrhythmias in certain disease states is well established. Acute ischemic heart disease is the best example.9,10 However, ANS influences may also be instrumental in triggering tachycardia events in patients with well-established long-standing substrates, such as those with pre-existing fibrotic areas as a consequence of prior myocardial infarction or remote cardiac surgery (e.g., childhood repairs). ANS participation in the triggering of arrhythmias is also almost certainly pertinent in other chronic states in which the arrhythmia substrate is present all the time and yet rhythm disturbances occur only sporadically. Among the best examples of the latter scenario are the abnormal ventricular repolarization syndromes (e.g., long QT syndrome [LQTS], Brugada syndrome).
Ischemic Heart Disease
The ANS contributes importantly to arrhythmogenesis in acute myocardial ischemia.11,12 In brief, the risk of potentially life-threatening arrhythmias and sudden death appears to increase in response to ischemia-associated increased sympathetic activity. Conversely, the risk of arrhythmia is diminished by sympathetic blockade, parasympathetic enhancement, and inhibition of the renin–angiotensin system.
Parasympathetic Neural Influences
Studies of baroreceptor sensitivity (BRS) (a measure of vagal influence on the heart) offer important insights into the potentially protective role played by the parasympathetic nervous system in patients with ischemic heart disease. In a prospective trial of relatively low-risk patients, at 2-year follow-up, BRS values were seen to be lower in those individuals who failed to survive, and the effect appeared to be independent of left ventricular function as assessed by ejection fraction measurement. In this regard, a multicenter trial, Autonomic Tone and Reflexes after Acute Myocardial Infarction (ATRAMI), provided convincing additional evidence.13 Patients were monitored for 21 ± 8 months. Cardiac mortality was higher (9% vs. 2%; 10% vs. 2%) among individuals with low BRS (<3 ms/mm Hg) or low standard deviation of normal NN intervals (SDNN) (<70 ms) than those with normal BRS or SDNN (>6.1 ms/mm Hg, >105 ms). Combining both indices resulted in recognition of even greater risk. Once again, the effect appeared to be independent of ejection fraction.
Long QT Syndrome, Brugada Syndrome, and Other Channelopathies
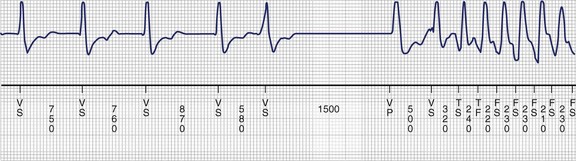
Disturbances of ventricular repolarization have been the subject of considerable interest in recent years, partly because of the recognition that they are a common cause of potentially life-threatening arrhythmias (Figure 5-7) but even more as a result of rapid progress in better understanding of why they occur.14 Currently, it is believed that underlying susceptibility to arrhythmia (primarily torsades de pointes ventricular tachycardia) is based on one or more genetically determined disturbances of the structure of cardiac membrane ionic channels, and/or disturbances of their function, or trafficking to the cell membrane.
Autonomic Responses and Treatment Considerations in Long QT Syndrome
According to the International Long-QT Syndrome Registry, patients with LQT3 have a higher incident of lethal cardiac events than those with LQT1 or LQT2.14 Patients with LQT3 are at a particularly high risk of a cardiac event during sleep when EADs may result from a slow heart rate rather than from sympathetic activation. In these patients, β-blockers may be relatively unfavorable, whereas pacing may be particularly desirable. Besides pacing, patients with LQT3 may benefit from LCSD, which will diminish norepinephrine release without slowing the heart rate.
Acquired Long QT Syndrome
Acquired LQTS is far more common than is congenital LQTS and is most frequently the result of QT interval–prolonging drugs. The impact of the ANS on initiating torsades in drug-induced LQTS is not well established, but circumstantial evidence clearly indicates an important link. Torsades in this setting is most often seen during periods of bradycardia (e.g., sleep) or following pauses in the cardiac rhythm (e.g., post–premature ventricular contraction long-short sequence) that accentuates the QT interval. Some of the best-known offending drugs are listed in Table 5-2. The majority of these drugs act by antagonizing outward (i.e., repolarizing) potassium (K+) currents (e.g., class 1A and class 3 antiarrhythmic drugs). Other agents in this list are reported to interfere with the metabolism of drugs that directly prolong the QT interval.
Table 5-2 Partial List of Drugs Known to Prolong the QT Interval
| ANTIARRHYTHMIC AGENTS |
| Class IA |
Autonomic Nervous System and Syncope
Syncope is best viewed as a syndrome characterized by transient loss of consciousness, usually associated with concomitant loss of postural tone and spontaneous recovery. (Table 5-3).15 Mechanistically, syncope is most often the result of transient disturbances of cerebral blood flow. In this regard, maintenance of cerebral blood flow is normally facilitated by several factors, all of which are, to some extent, significantly influenced by the ANS. Certain of these factors include (1) cardiac output, (2) baroreceptor-induced adjustments of heart rate and systemic vascular resistance, (3) cerebrovascular autoregulation (which is, in part, contributed to by the status of systemic arterial pressure as well as by local metabolic factors, particularly pCO2), and (3) regulation of vascular volume by the kidneys and by hormonal influences.
| NEURALLY MEDIATED REFLEX SYNCOPE |
Neurally Mediated Reflex Syncope
In the so-called vasovagal faint, and especially faints associated with stress or emotional upset, primary central nervous system stimuli are believed to be responsible for the trigger signals.4 However, receptors in any of the organ systems may contribute. For instance, mechanoreceptors and, to some extent, chemoreceptors located in the atrial myocardium and in the ventricular myocardium may participate in certain neurally mediated events by initiating afferent neural signals when subjected to increased wall tension or changes in the chemical environment (e.g., myocardial ischemia). Similarly, mechanoreceptors and chemoreceptors in the central great vessels and lungs may contribute, which accounts for the reported occurrence of vasovagal faints in heart transplant recipients. The basis for apparent variations in susceptibility to vasovagal syncope among seemingly otherwise well individuals and the factors causing a faint to occur at a certain point in time still remain unknown.
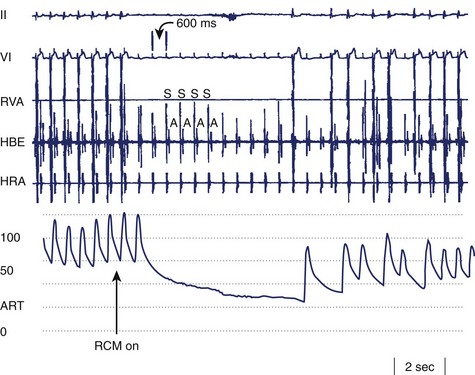
Bradycardia in neurally mediated reflex syncope is primarily the result of increased efferent parasympathetic tone mediated via the vagus nerve. It may manifest as asystole, sinus bradycardia, or even paroxysmal AV block. If the bradyarrhythmia is sufficiently severe, it may be the principal cause of the faint (i.e., cardioinhibitory syncope). However, most patients also exhibit a vasodepressor picture comprising inappropriate ANS-induced vasodilatation (Figure 5-8). The mechanism of the vasodilatation is believed to be mainly the result of abrupt peripheral sympathetic neural withdrawal, although potential contributions of excess β-adrenergic tone caused by frequently associated elevated circulating epinephrine levels or altered epinephrine-norepinephrine balance are certainly considerations.
Noncardiovascular Conditions
Noncardiovascular causes result in syncope mimics (Table 5-4) rather than true syncope. However, temporal lobe seizures may induce neurally mediated reflex bradycardia and hypotension (i.e., a vasovagal faint). Furthermore, certain central nervous system syncope mimics may cause worrisome ECG changes and even myocardial damage, as discussed earlier.
| METABOLIC/ENDOCRINE DISTURBANCES |
* Potential nervous system–mediated cardiac damage and sudden death.
Summary
Acknowledgment
1 Randall WC, Wurster RD. Peripheral innervation of the heart. In: Levy MN, Schwartz PJ, editors. Vagal control of the heart: Experimental basis and clinical implications. Armonk, NY: Futura Publishing, 1994.
2 Herring N, Paterson DJ. Neuromodulators of peripheral cardiac sympatho-vagal balance. Exp Physiol. 2009;94:46-53.
3 Brotman DJ, Goldman SH, Wittstein IS. The cardiovascular toll of stress. Lancet. 2007;370:1089-1100.
4 Samuels MA. The brain-heart connection. Circulation. 2007;116:77-84.
5 Shishehbor MH, Alves C, Rajajgopal V. Inflammation: Implications for understanding the heart-brain connection. Clev Clinic J Med. 2007;74(Supp 1):S37-S41.
6 Tomson T, Nashef L, Ryvlin P. Sudden unexpected death in epilepsy: Current knowledge and future directions. Lancet Neurol. 2008;7:1021-1031.
7 Oppenheimer S. Cortical control of the heart. Clev Clinic J Med. 2007;74(Supp 1):S27-S29.
8 Armour JA. Potential clinical relevance of the “little brain” on the mammalian heart. Exp Physiol. 2008;93:165-176.
9 Pauza DH, Skripka V, Pauziene N, Stropus R. Morphology, distribution, and variability of the epicardiac neural ganglionated subplexuses in the human heart. Anat Rec. 2000;259(4):353-382.
10 Zipes DP, Rubart M. Neural modulation of arrhythmias and sudden death. Heart Rhythm. 2006;3:108-113.
11 Vaseghi M, Shivkumar K. The role of the autonomic nervous system in sudden cardiac death. Prog Cardiovasc Dis. 2008;50(6):404-419.
12 Saffitz JE. Sympathetic neural activity and the pathogenesis of sudden cardiac death. Heart Rhythm. 2008;5:140-141.
13 La Rovere MT, Bigger JTJr, Marcus FI, Mortara A, Schwartz PJ. Baroreflex sensitivity and heart rate variability in prediction of total cardiac mortality after myocardial infarction for the ATRAMI Investigators. Lancet. 1998;351(9101):487-494.
14 Zareba W, Moss AJ, Schwartz PJ, et al. Influence of genotype on the clinical course of the long QT syndrome. International Long QT Registry Research Group. N Engl J Med. 1998;339:960-965.
15 Brignole M, Alboni P, Benditt D, et al. Task force on syncope, European Society of Cardiology: Guidelines on management (diagnosis and treatment) of syncope—update 2004. Europace. 2004;6:467-537.