[level-membership-for-neurology-category]
13 Autonomic Disorders and Syncope
Anatomy of the Autonomic System
The primary role of the autonomic system is the maintenance of homeostasis and this is done by two separate but complementary systems: the sympathetic and the parasympathetic systems. The central regulation of the autonomic system is mediated by neurons in the frontal lobe, limbic system and the hypothalamus. The preganglionic neurons of the sympathetic nervous system arise from the intermediolateral column of the thoracic spinal cord. These axons form the white communicating rami that synapse with the neurons of the sympathetic ganglia in the paravertebral chain; postganglionic fibers form the gray communicating rami that travel along with the spinal nerves to blood vessels and sweat glands (Fig. 13-1). Sympathetic innervation of the adrenal medulla is the exception as it receives preganglionic sympathetic fibers; the adrenal medulla is considered the equivalent of a sympathetic ganglion, and it secretes epinephrine and norepinephrine directly into the blood stream (Fig. 13-1).
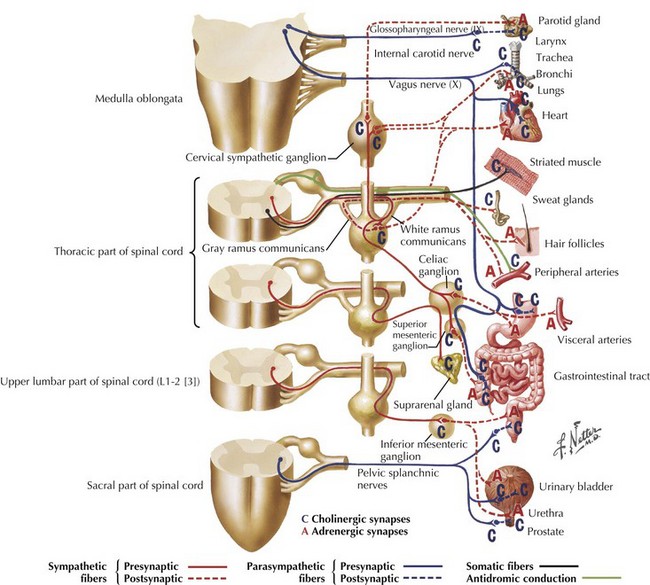
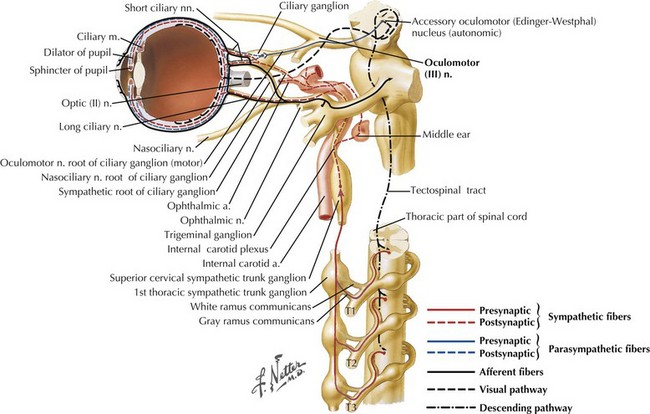
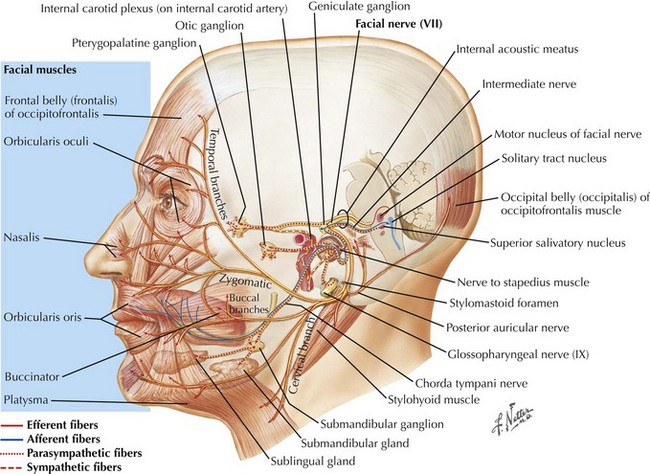
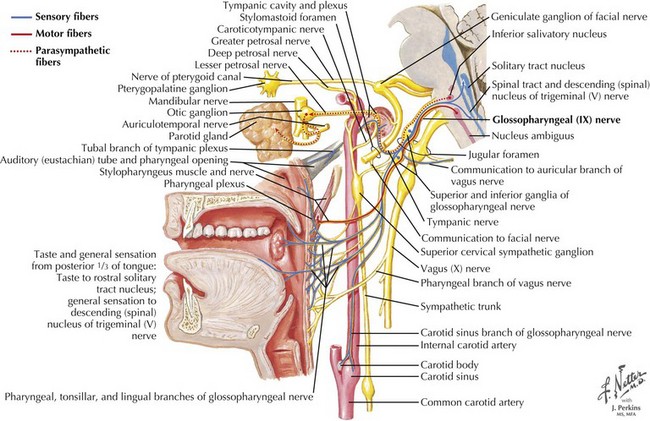
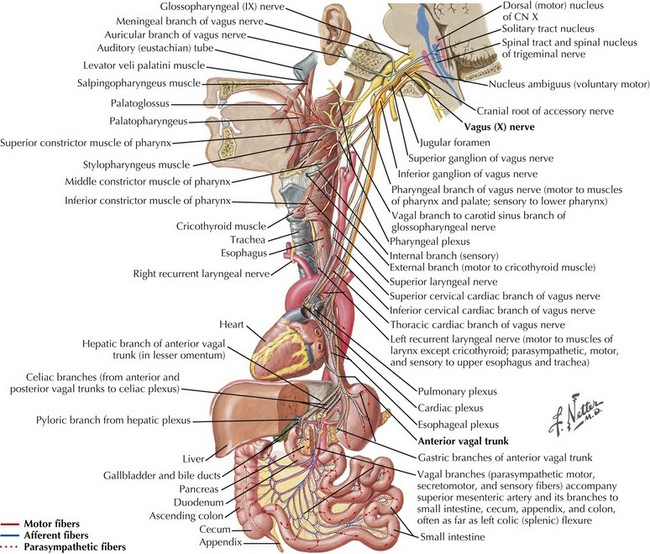
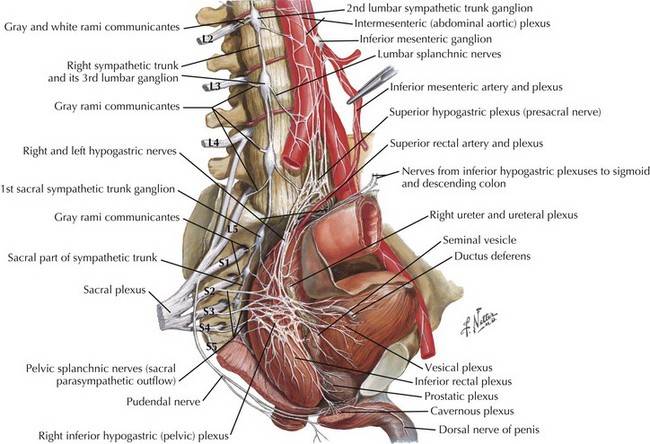
The parasympathetic system consists of the cranial and sacral output; the cranial output arises in the visceral nuclei of the cranial nerves III, VII, IX, and X, and the axons travel along with the respective cranial nerves to innervate target organs. The preganglionic fibers from the Edinger–Westphal nucleus travel in the oculomotor nerve (III) and synapse in the ciliary ganglion innervating the ciliary and pupillary muscles (Fig. 13-2). The preganglionic fibers from the superior salivatory nuclei travel along with the facial nerve (VII) as greater petrosal nerve and the chorda tympani, synapse in the sphenopalatine and submandibular ganglion, and innervate the lacrimal, submandibular, and sublingual glands (Fig. 13-3). The preganglionic fibers from the inferior salivatory nuclei travel along with the glossopharyngeal nerve (IX) and synapse in the otic ganglion and innervate the parotid gland (Fig. 13-4). The preganglionic fibers from the dorsal motor nucleus of the vagus travel along with the vagal nerve (X) and synapse in the ganglia in the walls of the viscera and innervate the visceral organs of the gastrointestinal, cardiac, and renal systems (Fig. 13-5). The sacral part of the parasympathetic system arises from the sacral spinal cord, synapses in the ganglia in the walls of the organs, and innervates the colon, bladder, and pelvic organs (Fig. 13-6).
Clinical Presentations
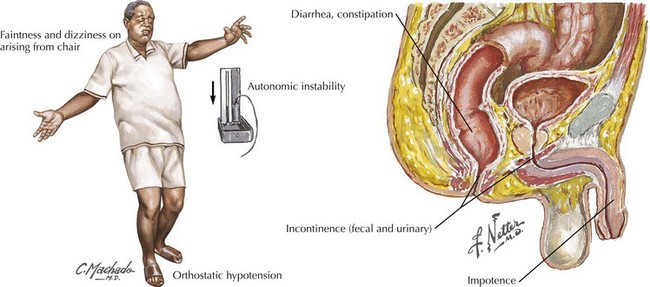
Typically, patients have combinations of both parasympathetic and sympathetic dysfunction (Fig. 13-7). The former is characterized by dry mucous membranes, particularly noticeable in the eyes and mouth, with varying gastrointestinal involvement manifested as early satiety, nausea, vomiting, constipation, diarrhea, urinary bladder dysmotility, and erectile dysfunction.
Syncope
Clinical Vignette
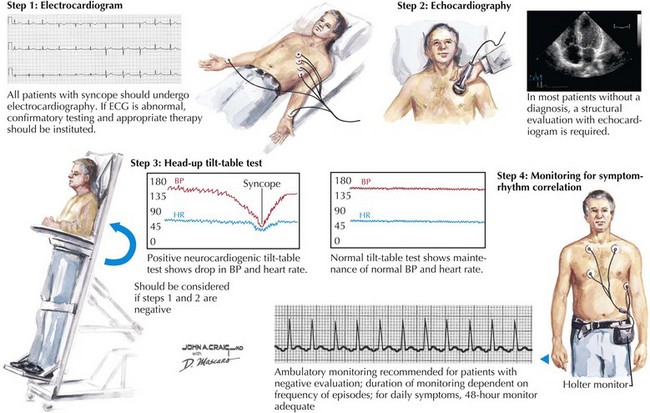
Syncope is defined as a brief and transient loss of consciousness from cerebral hypoperfusion. Lightheadedness, visual dimming, paleness, cold sweating, nausea, and a feeling of warmth are common premonitory symptoms (Fig. 13-8). These are followed by loss of consciousness and postural muscle tone, and, if the patient is standing, he or she will usually fall. Significant trauma and fractures occur in approximately 5% of these patients. In contrast to patients who lose consciousness from a convulsion, individuals who have syncope generally have no confusion after the episode. Typically, they have good recollection of premonitory symptoms. Loss of consciousness lasts just a few seconds. Occasionally, a few clonic twitches or a brief generalized seizure-like activity occurs at the end of the episode. EEG recorded during syncope demonstrates early depression of activity followed by slow wave activity in the theta and delta range. Transient EEG voltage depression may follow. Elderly patients may be amnestic for the event. Almost 20% of people have had a syncopal episode in their lifetime.
Benarroch EE, Smithson IL, Low PA, et al. Depletion of catecholaminergic neurons in the rostral ventrolateral medulla in multiple system atrophy with autonomic failure. Ann Neurol. 1998;43:56-163.
Cohen J, Low P, Fealey R, et al. Somatic and autonomic function in progressive autonomic failure and multiple system atrophy. Ann Neurol. 1987;22:692-699.
Lipp A, Sandroni P, Ahlskog JE, et al. Prospective differentiation of multiple system atrophy from Parkinson disease, with and without autonomic failure. Arch Neurol. 2009 Jun;66(6):742-750.
Low PA, Vernino S, Suarez G. Autonomic dysfunction in peripheral nerve disease. Muscle Nerve. 2003;27:646-661.
Vernino S, Low PA, Fealey RD, et al. Autoantibodies to ganglionic acetylcholine receptors in autoimmune autonomic neuropathies. N Engl J Med. 2000;343:847-855.
Winston N, Vernino S. Autoimmune autonomic ganglionopathy. Front Neurol Neurosci. 2009;26:85-93.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
13 Autonomic Disorders and Syncope
Anatomy of the Autonomic System
The primary role of the autonomic system is the maintenance of homeostasis and this is done by two separate but complementary systems: the sympathetic and the parasympathetic systems. The central regulation of the autonomic system is mediated by neurons in the frontal lobe, limbic system and the hypothalamus. The preganglionic neurons of the sympathetic nervous system arise from the intermediolateral column of the thoracic spinal cord. These axons form the white communicating rami that synapse with the neurons of the sympathetic ganglia in the paravertebral chain; postganglionic fibers form the gray communicating rami that travel along with the spinal nerves to blood vessels and sweat glands (Fig. 13-1). Sympathetic innervation of the adrenal medulla is the exception as it receives preganglionic sympathetic fibers; the adrenal medulla is considered the equivalent of a sympathetic ganglion, and it secretes epinephrine and norepinephrine directly into the blood stream (Fig. 13-1).
The parasympathetic system consists of the cranial and sacral output; the cranial output arises in the visceral nuclei of the cranial nerves III, VII, IX, and X, and the axons travel along with the respective cranial nerves to innervate target organs. The preganglionic fibers from the Edinger–Westphal nucleus travel in the oculomotor nerve (III) and synapse in the ciliary ganglion innervating the ciliary and pupillary muscles (Fig. 13-2). The preganglionic fibers from the superior salivatory nuclei travel along with the facial nerve (VII) as greater petrosal nerve and the chorda tympani, synapse in the sphenopalatine and submandibular ganglion, and innervate the lacrimal, submandibular, and sublingual glands (Fig. 13-3). The preganglionic fibers from the inferior salivatory nuclei travel along with the glossopharyngeal nerve (IX) and synapse in the otic ganglion and innervate the parotid gland (Fig. 13-4). The preganglionic fibers from the dorsal motor nucleus of the vagus travel along with the vagal nerve (X) and synapse in the ganglia in the walls of the viscera and innervate the visceral organs of the gastrointestinal, cardiac, and renal systems (Fig. 13-5). The sacral part of the parasympathetic system arises from the sacral spinal cord, synapses in the ganglia in the walls of the organs, and innervates the colon, bladder, and pelvic organs (Fig. 13-6).
Clinical Presentations
Typically, patients have combinations of both parasympathetic and sympathetic dysfunction (Fig. 13-7). The former is characterized by dry mucous membranes, particularly noticeable in the eyes and mouth, with varying gastrointestinal involvement manifested as early satiety, nausea, vomiting, constipation, diarrhea, urinary bladder dysmotility, and erectile dysfunction.
[/not-level-membership-for-neurology-category]
