[level-membership-for-allergy-and-immunology-category]
Autoimmunity
Learning Objectives
• Examine the relationship between thyroglobulin and iodine
• Identify the differences between T3 and T4
• Understand the hypothalamus–pituitary axis and its role in thyroid function
• Discuss the role of the thyrotropin-releasing hormone (TRH)
• Explain the function of the thyroid-stimulating hormone (TSH)
• Identify the target of the autoantibodies found in Graves’ disease
• Discuss the role(s) of cytokines and fibroblasts in Graves’ ophthalmopathy
• Define the term thyroid storm
• Design a therapy regimen to treat Graves’ disease
• Compare and contrast the symptoms of Graves’ disease and Hashimoto’s thyroiditis
• Discuss the immunologic mechanisms that contribute to Hashimoto’s thyroiditis
• Discuss the role of the intrinsic factor in vitamin B12 transport
• Identify the target of autoantibodies found in pernicious anemia
• Explain the role of vitamin B12 in red blood cell synthesis
• Design a therapy regimen to treat pernicious anemia
• Recognize the target of autoantibodies found in myasthenia gravis
• Identify the two mechanisms by which acetylcholine receptors are reduced in myasthenia gravis
• Compare and contrast ptosis and diplopia
• Design a therapy regimen to treat mild and severe myasthenia gravis
• Identify the clinical symptoms of systemic lupus erythematosus (SLE)
• List the viruses that may play a role in SLE
• Describe the two immunologic defects associated with SLE
• Design a therapy regimen to treat SLE
• Compare and contrast SLE and the “lupus-like” syndrome
• Recognize the autoantibody targets in Goodpasture syndrome
• Describe the genetic and immunologic factors that increase the risk of Goodpasture syndrome
• Design a therapy regimen to treat Goodpasture syndrome
• Differentiate between the function of pancreatic alpha, beta, and delta cells
• Relate the role(s) of insulin in the body
• List the targets of autoantibodies found in type 1a diabetes
• Identify the human leukocyte antigen (HLA) alleles that increase the risk of diabetes
• List the viruses that are implicated in the onset of diabetes
• Design a drug regimen to treat diabetes
• Define ankylosing spondylitis (AS)
• List the possible microbial triggers for AS
• Explain the role of HLA-B27 in the pathophysiology of AS
• Design a treatment regimen for AS
• Compare and contrast rheumatoid arthritis and reactive arthritis
• List the microbial agents that play a role in reactive arthritis
• Define rheumatoid arthritis (RA)
• Explain the relationship between HLA alleles and RA
• Identify the six viruses that may act as triggers for RA
• Compare and contrast the roles of macrophages and fibroblasts in the pathophysiology of RA
• Define rheumatoid factor (RF)
• Understand the relationship between RF and agalactosyl antibodies
• Design a treatment regimen to treat RA
• Discuss the role of the immune system in rheumatic heart disease
• Recognize the definition of autoimmune hemolytic anemia
• Contrast the mechanisms by which α-methyldopa, penicillin, and cephalosporin cause anemia
Key Terms
Acetylcholine
Acetylcholinesterase
Ankylosing spondylitis
Arthritis
Diabetes
Goiter
Goodpasture syndrome
Graves’ disease
Hashimoto’s thyroiditis
Intrinsic factor
Molecular mimicry
Myasthenia gravis
Pernicious anemia
RANKL (receptor activator of nuclear factor kappa-β ligand)
Reactive arthritis
Rheumatic fever
Rheumatoid factor
Systemic lupus erythematosus
Thyrotropin-releasing hormone
Thyroid-stimulating hormone
Introduction
Autoimmune diseases arise when an individual’s immune system attacks his or her own tissue and organs. The immune response may be mediated by antibodies or activated lymphocytes and macrophages. These diseases are found in 5% to 7% of the population, and the disease incidence is heavily skewed toward females. In women over 65 years of age, autoimmune diseases are one of the 10 leading causes of death.
The etiology of autoimmune disease in women is multifactorial. The factors include genetic, environmental, and hormonal triggers. Individuals expressing certain human leukocyte antigen (HLA) alleles are at high risk of developing autoimmune disease. For example, individuals expressing HLA B8 and DR3 have an increased risk of developing Graves’ disease. Because the onset of autoimmune diseases occurs shortly after puberty and just before the onset of menopause, estrogen particularly is thought to be one of the triggers for autoimmune disease. A wide range of different autoimmune diseases may affect a single organ (e.g., thyroid) or multiple organs (e.g., in systemic lupus erythematosus).
Autoimmune Diseases of the Thyroid
The function of the thyroid is to produce the hormones T3 and T4, which control physiologic functions in the cardiac, pulmonary, hematopoietic, gastrointestinal, skeletal, and endocrine systems. To produce the hormones, thyroglobulin (Tg) is synthesized by thyroid cells (thyrocytes) and stored in the follicular lumen. Iodine covalently binds to thyroglobulin tyrosine residues by using thyroperoxidase. Intracellular proteases digest thyroglobulin to produce functional triiodothyronine (T3) and thyroxin (T4), which differ only in the number of bound iodine molecules.
Synthesis of T3 and T4 is controlled by the hypothalamus–pituitary–thyroid axis. When the T3 and T4 levels fall below acceptable levels, the hypothalamus produces thyrotropin-releasing hormone (TRH) which travels through blood to the anterior pituitary gland. The anterior pituitary gland releases another hormone called thyroid-stimulating hormone (TSH). This hormone interacts with thyrotropin receptors and stimulates iodine uptake and the production of T3 and T4 (Figure 16-1).
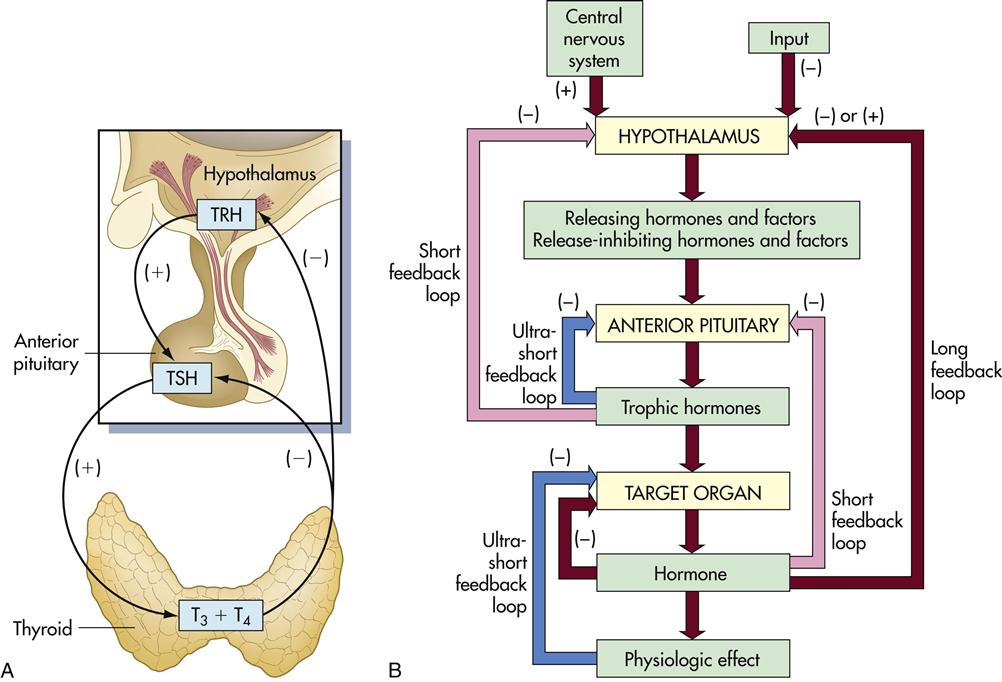
Thymic hormone levels are controlled by negative feedback loops. High levels of TSH act on the hypothalamus to downregulate the production of TRH. T3 and T4 also act on both the pituitary and the hypothalamus to downregulate hormone production. Autoantibodies directed at the thyroid results in either hypothyroidism or hyperthyroidism.
Graves’ Disease
Graves’ disease was first reported by Dr. Robert Graves in the 1830s and is the most common cause of hyperthyroidism in children and young women. The disease is caused by IgG1 antibodies reacting with thyrotropin or TSH receptors. What causes the immune system to recognize thyroid antigens is unclear. It is conceivable that a viral infection may expose a hidden epitope in thyroid receptors.
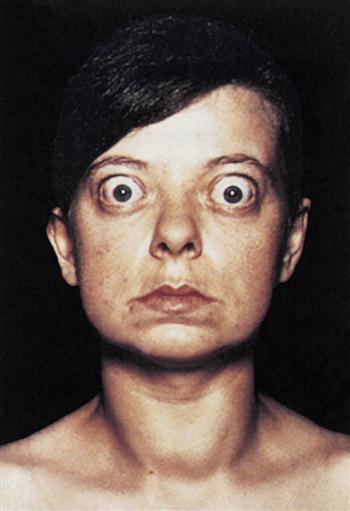
Constant stimulation of TSH receptors causes glandular hyperplasia (goiter) and accelerated synthesis of T3 and T4. Patients often exhibit a rapid heart rate, exophthalmos, tremors, sweating, and attention deficit disorders. TSH receptors are also found in orbital cells and muscles. Reaction between the autoantibody and the TSH orbital receptor causes an inflammatory reaction called Graves’ ophthalmopathy (Figure 16-2).
Graves’ Ophthalmopathy
In the pathogenesis of Graves’ ophthalmopathy, lymphocytes infiltrate the orbit of the eye and release cytokines such as tumor necrosis factor (TNF) and interleukin 1 (IL-1). These cytokines stimulate fibroblasts to secrete a glycosaminoglycan (GAG) mucopolysaccharide. GAG increases osmotic pressure in the eye, causing edema in the extraocular and retro-orbital muscles and in adipose tissue. These changes force the eyeball forward, creating bulging of the eye, or exophthalmos.
Thyroid Storm
In patients with undiagnosed Graves’ disease, thyroid hormones often increase dramatically, which precipitates a life-threatening clinical crisis. A thyroid storm is precipitated by excessive stress, surgery, or sepsis. High levels of thymic hormones cause extreme tachycardia and atrial fibrillation. The mortality rate is 20%, and death occurs within 48 hours.
Treatment for Graves’ Disease
Thiourea was the drug initially used to treat Graves’ disease; however, it had considerable toxicity. Second-generation drugs and derivatives such as propylthiouracil and methylthiouracil, methimazole, and carbimazole have less toxicity. With the exception of methimazole, which prevents the storage of thyroid hormones, these drugs inhibit thyroperoxidase, thereby reducing the synthesis of T4 and T3.
Oral administration of radiolabeled iodine-131 is now the most common therapy for Graves’ disease. The high-intensity radiation destroys thyroid cells and brings the levels of thyroid hormones into the normal range. Subtotal thyroidectomy was the treatment of choice before the advent of radioactive iodine. Methimazole therapy is indicated for those patients in whom radioactivity is contraindicated.
Hashimoto’s Thyroiditis
Hashimoto’s thyroiditis is the most common form of hypothyroidism, with a prevalence rate of 2% of the population of the United States. Individuals mount both an antibody response and a cellular response to thyroid tissue. The disease is caused by a break in peripheral tolerance to thyroid tissue, and the failure of regulatory cells to control auto-reactive T and B cells. About 85% to 90% of patients with Hashimoto’s thyroiditis develop anti-thyroid peroxidase and anti-thyroglobulin antibodies. The inactivation of thyroperoxidase prevents the incorporation of free iodine into thyroglobulin. Elevated intracellular iodine also stimulates the maturation of dendritic cells and macrophages, which present antigens to auto-reactive CD4 and CD8 cells that have infiltrated the thyroid (Figure 16-3).
CD4Th1 cells produce IL-12, tumor necrosis factor alpha (TNF-α) and interferon gamma (INF-γ), which induce apoptosis of thyrocytes. Individuals with Hashimoto’s thyroiditis also express a molecule called Fas on thyroid cells. The expression of the Fas molecules marks the cell for destruction by T lymphocytes.
Patients with Hashimoto’s thyroiditis present with nonspecific symptoms such as fatigue, dry skin, and weight gain. Other symptoms include decreased sweating, deafness, peripheral neuropathy, depression, and memory loss.
Treatment for Hashimoto’s Thyroiditis
Oral sodium levothyroxine, which is a synthetic thyroid hormone (T4), usually has to be taken for life by patients with Hashimoto’s thyroiditis. However, the dose must be carefully titrated on the basis of the patient’s needs. Some patients cannot tolerate the synthetic hormone. Natural porcine gland preparations of T3 and T4 can be used in these patients.
Pernicious Anemia
In pernicious anemia (PA), antibodies are directed at a glycoprotein known as intrinsic factor. These autoantibodies also neutralize any soluble intrinsic factor in the intestinal lumen.
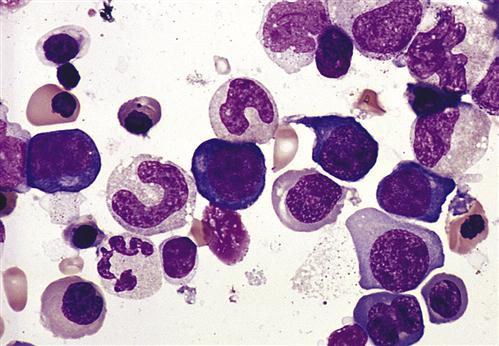
Neutralization of the soluble intrinsic factor prevents the transport of vitamin B12 across the intestinal mucosa to portal blood (Figure 16-4). Without this essential vitamin, red blood cell precursors cannot divide. This results in a megaloblastic anemia characterized by the presence of immature, dysfunctional red blood cells (megaloblasts) in the blood. If untreated, PA is usually fatal after 1 to 3 years (Figure 16-5).

Treatment for Pernicious Anemia
Vitamin replacement therapy combined with folate and iron supplements is used to treat PA. Cobalamin and hydroxocobalamin are natural forms of vitamin B12 produced by bacterial fermentation. Both can be administered parenterally. However, hydroxocobalamin is the drug of choice because it is bioavailable for longer periods. Folate and iron supplements are included in the treatment because restoration of normal B12 levels triggers a rapid increase in the synthesis of red blood cells, which depletes stores of folate and iron.
Myasthenia Gravis
Myasthenia gravis (MG) is a rare disorder in which the patient exhibits progressive weakness of skeletal muscles. Individuals with the disease produce antibodies directed at postsynaptic acetylcholine receptors at the neuromuscular junctions of skeletal muscle.
Surface receptors are destroyed as a result of complement activation or are internalized and destroyed by proteolytic enzymes (Figure 16-6).
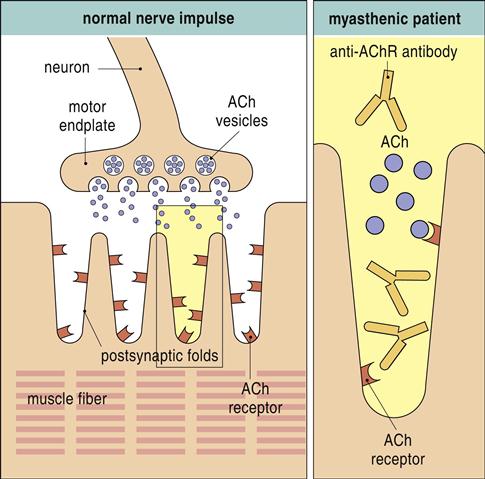
Both mechanisms decrease the number of receptors available for acetylcholine binding and the propagation of nerve impulses. Failure to propagate the nerve impulses causes fatigue and muscle weakness. Initially, muscle weakness is observed in the extraocular muscle group. Ptosis (abnormal drooping of one or both eyelids), diplopia (blurred or double vision), or both are characteristic signs of the disease (Figure 16-7).
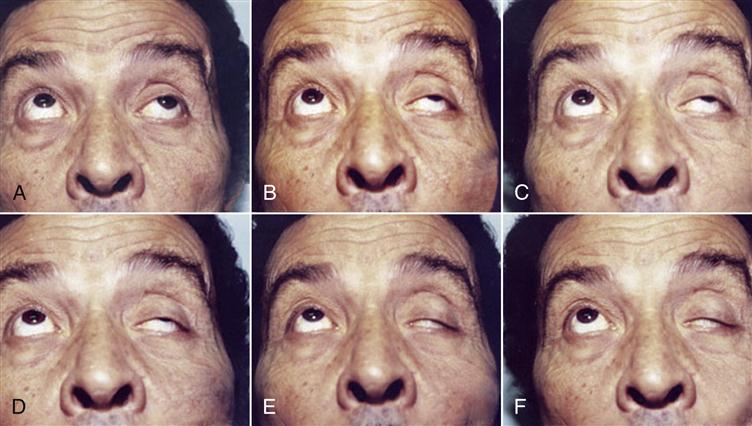
Over a period of months, the weakness progresses from the ocular muscles to facial, jaw, tongue, and throat muscles. Without treatment, the weakness may progress to the trunk and limb muscles. MG is fatal when impairment of respiratory muscles occurs.
Treatment for Myasthenia Gravis
Depending on the severity of the disease, several different treatment strategies are used to treat MG. In mild cases, corticosteroids and acetylcholinesterase inhibitors are used. Corticosteroids downregulate the immune response, and acetylcholine inhibitors slow cholinesterase inactivation of acetylcholine fixed to the functional motor plate receptors. In the treatment of more severe or difficult cases, immunosuppressive agents such as cyclosporine are used (see Chapter 7 for a discussion on the mechanism of action and adverse health effects of immunosuppressive agents).
Systemic Lupus Erythematosus
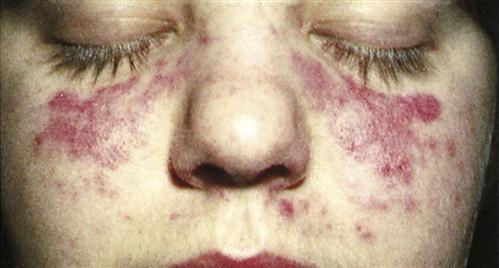
SLE is a multiple-organ disease occurring primarily in women of child-bearing age. It is characterized by multiple-organ inflammation. Almost any organ system can be involved, but the central nervous system (CNS), the renal system, and the pulmonary organs are common targets. Clinical manifestations include arthritis, glomerulonephritis, hemolytic anemia, neurologic disorders, and rashes. The classic SLE rash is a macular butterfly-shaped rash on the bridge of the nose and the cheeks or a discoid rash with red, raised patches on the face, scalp, and body (Figure 16-8).
SLE is considered an autoimmune disease because of the presence of autoantibodies directed at double-stranded deoxyribonucleic acid (anti-dsDNA) and core proteins of small nuclear ribonucleoproteins (anti-Sm). The presence of these antibodies is associated exclusively with SLE and is therefore included in the diagnostic criteria for the disease. Circulating immune complexes often settle out in the small vessels in the joints and kidneys causing arthritis, glomerulonephritis, and inflammatory vasculitis (Figure 16-9).
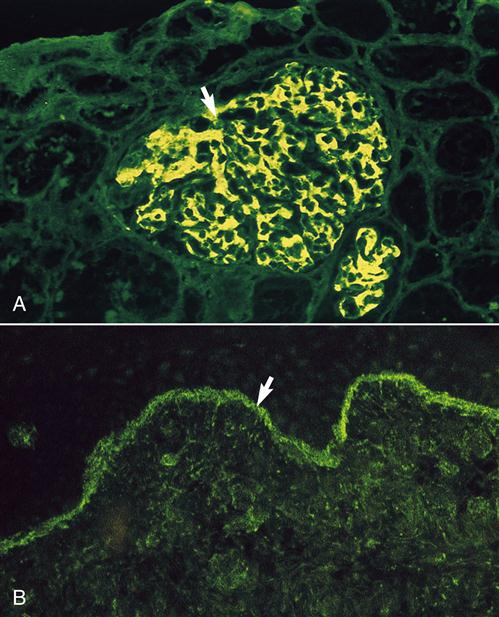
The pathophysiology of the disease is complex and may involve two immunologic defects. Evidence indicates that individuals with SLE have a defect in the innate killing Fas mechanism that normally destroys auto-reactive T and B cells. Hence, auto-reactive lymphocytes persist in the circulation. Individuals with SLE also have reduced synthesis of C1q, C2, and C4a and defective complement receptors on macrophages. Reduction in complement synthesis reduces the release of chemotactic factors and the influx of phagocytic cells, which normally remove immune complexes. Since immune complexes are not removed, they are deposited in the capillary bed and the glomeruli of the kidneys.
Treatment for Systemic Lupus Erythematosus
Several different drugs are indicated for the treatment for SLE. Nonacetylated salicylates and glucocorticoids are used to control the inflammatory response. Nonsteroidal anti-inflammatory drugs (NSAIDs) are indicated for the management of mild to moderate pain.
Drug-Induced “Lupus-Like” Disease
Drugs often cause a transient “lupus-like” syndrome. This syndrome differs from classic SLE in several ways. In the lupus-like syndrome, anti-DNA antibody production is transient, and memory cells are not generated. When the drug is discontinued, symptoms abate, and the patient returns to a normal state. Pharmaceuticals commonly associated with drug induced lupus-like syndrome are listed in Box 16-1.
The mechanisms involved in drug-induced lupus-like syndrome are unclear. However, agents inhibiting the methylation of T cell DNA (e.g., procainamide and hydralazine) cause the overexpression of eukocyte function–associated antigen 1 (LFA-1) or CD11a/CD18. T cells overexpressing LFA-1 adhere to the vascular cells and become auto-reactive. Clinical symptoms are dependent on cumulative drug dosage and liver CYA P-450 genotypes. For example, 30% to 60% of procainamide is excreted as N-acetylprocainamide, and the rate of metabolism is genetically determined. Slow metabolism of the drug is associated with the autoimmune response.
Goodpasture Syndrome
Goodpasture syndrome is characterized by pulmonary hemorrhage and a rapidly progressing glomerulonephritis. Patients present to the physicians with hemoptysis, dyspnea, and generalized weakness. The syndrome is characterized by antibodies directed at noncollagenous domains (NC1) of the alpha 3 chains of type IV collagen in the basement membranes of the lungs and kidneys (Figure 16-10).
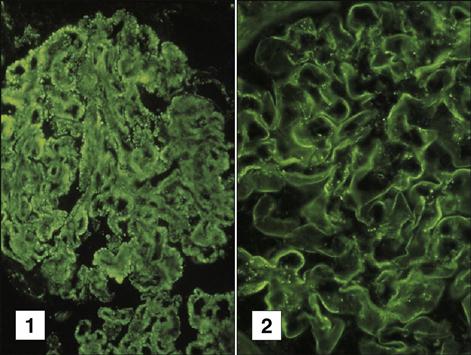
These epitopes are normally sequestered from immune recognition. Injury to the basement membrane from smoking, inhaled hydrocarbons, or viral infections may expose these antigens. Additional genetic and immunologic factors may be necessary for the autoimmune response. Goodpasture syndrome has a strong genetic component, and a positive association with HLA DRB1∗1501 is necessary for the presentation of collagen epitopes to auto-reactive T cells. In addition, individuals lack a critical B cell Fc receptor (FcγRIIB), which normally inhibits B cell activation, proliferation, and autoantibody production.
Treatment for Goodpasture Syndrome
Treatment for Goodpasture syndrome is focused on removal of circulating autoantibodies and downregulating the immune response. Autoantibodies are removed by plasmapheresis. To diminish the immune response, pulse-dosed glucocorticoids and cytotoxic alkylating agents are used. Auto-reactive B cells can also be reduced by treatment with anti-CD20 (rituximab). CD20 is present on immature B cells, but not antibody-producing plasma cells. Anti-CD20 selectively depletes the immature B cells and prevents the repopulation of antibody-producing plasma cells.
Molecular Mimicry
Microbial infections have been postulated as triggers for many autoimmune diseases. In theory, a genetically susceptible host is infected with a microbe that expresses antigens that are similar, but not identical, to the host’s self-markers (Figure 16-11).
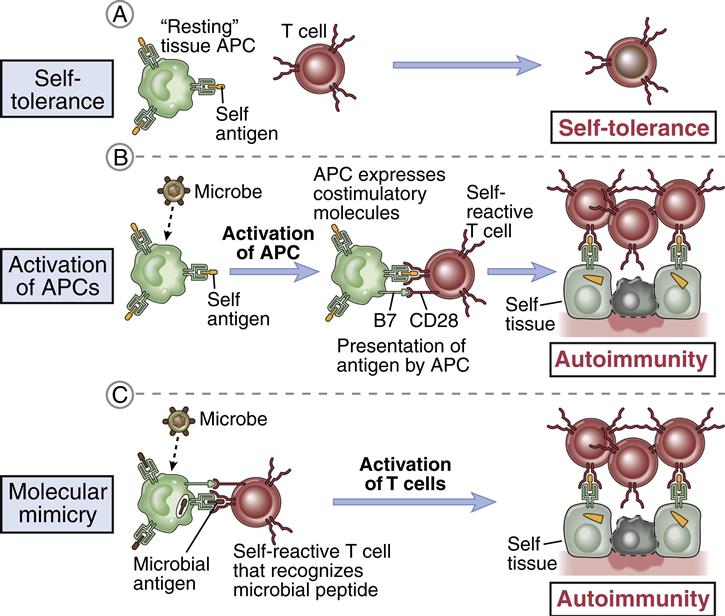
The immune response to the microbe generates antibodies that react with both the pathogen and the host tissue. Molecular mimicry may play a role in type 1a diabetes, ankylosing spondylitis, rheumatoid arthritis, reactive arthritis, and acute rheumatic fever.
Diabetes (Type 1a)
The pancreas functions both as an endocrine organ and an exocrine organ. As an exocrine organ, it produces digestive enzymes that are secreted into the small intestine. These enzymes include trypsin, chymotrypsin, lipases, and amylases. Pancreatic endocrine function is facilitated by three different pancreatic cell types found in the islets of Langerhans. These cells produce glucagon (α cells), insulin (β cells), and somatostatin and gastrin (δ cells).
Type 1a diabetes is a disease characterized by the destruction of the β cells and low or absent insulin in blood. Insulin is an anabolic hormone that reduces the levels of circulating glucose by allowing its entrance into muscle cells and conversion to a storage molecule called glycogen. When insulin is unavailable, glucose cannot be stored as glycogen and begins to accumulate in blood. Osmotic diuresis occurs when glucose enters the kidneys and changes the osmotic pressure in the renal tubule. The increase in osmotic pressure also causes water accumulation in the lumen and prevents water re-adsorption. This results in increased thirst, increased urinary output, and dehydration. In addition, individuals with type 1a diabetes have excessive weight loss despite an adequate caloric intake. Weight loss occurs because patients with diabetes cannot process many of the calories in their food.
The type 1a autoimmune form of diabetes has several synonyms, including insulin-dependent diabetes mellitus (IDDM), juvenile diabetes, and insulin-dependent diabetes. Autoimmune diabetes affects 0.3% of the population by puberty, making it the third most prevalent disease among children. After puberty, the incidence declines rapidly.
The onset of clinical disease is characterized by an infiltration of both CD4 and CD8 cells in the pancreas and by β cell destruction. Autoantibodies are also produced and directed at small forms of glutamate decarboxylase (GAD65), protein tyrosine phosphatase (IA-2), and insulin.
The pathophysiology of the disease may require a genetically susceptible population and a trigger for the initiation of the autoimmune response. A strong association exists between HLA-DR3, DR-4, and DQ1 and diabetes. DR3 or DR4 is present in 95% of patients with type 1a diabetes, and 30% have inherited both DR3 and DR4. In Caucasian populations, less than 50% have either DR3 or DR4 and less than 3% express both class II markers. Moreover, individuals who are heterozygous for DR3/DR4 have a 20- to 40-fold risk of developing diabetes compared with the general population.
Individuals with DR3 or DR4 also inherit closely linked DQ1 β alleles, which are critical to disease development. In Caucasians, DRB1∗0301/DQB1∗0201 or DRB1∗040101/DRB1∗0302 haplotypes have a high risk of developing type 1a diabetes. Resistance or susceptibility to diabetes has been mapped to residues at position 57 in the DR and DQ β-chains. In individuals with a low risk of diabetes, aspartate is found in position 57. Individuals with high-risk DR or DQ alleles have uncharged valine, serine, or alanine at the same position. It is speculated that pancreatic autoantigens are presented to T cells in the context of these DQ and DR molecules.
The trigger for fulminating diabetes may be a viral infection. In humans, these viruses include cytomegalovirus (CMV), EBV, Coxsackie virus, rubella virus, mumps virus, varicella zoster virus, and rotavirus. CMV may lyse β cells, releasing glutamate decarboxylase 65 (GAD65) from the cell surface. GAD65 binds to DQβ1 susceptibility alleles and is presented to the CD4Th2 cell, which stimulates B cells to produce autoantibodies.
Diabetes may also be triggered by molecular mimicry between viruses and β cell autoantigens. For example, EBV expresses a five-amino-acid sequence that is shared with high-risk DR and DQ alleles suggesting that the HLA alleles alone can act as antigens. Coxsackie virus capsid proteins and GAD65 share sequence homologies and cross-reactive antigens. Rotavirus, which is the most common cause of childhood diarrhea, also shares homologous amino acid sequences with GAD65 and IA-2.
Treatment for Diabetes
Insulin is the mainstay of treatment to control diabetes. The treatment depends on the medical status of the patient with regard to hypoglycemia, diabetic ketoacidosis, and complications such as macrovascular disease leading to myocardial ischemia.
Ankylosing Spondylitis
Ankylosing spondylitis (AS) is a chronic inflammatory disease of the spinal sacroiliac region, which connects spine to the pelvis. The disease appears to be mediated by CD8 cells. Constant inflammation causes fusion of the vertebrae, which results in chronic back pain and limited mobility. Occasionally, inflammation of the shoulders, hips, and knees occurs. In rare instances, the disease is associated with inflammation of the heart, lungs, and kidneys.
Like diabetes, the pathophysiology of AS has a strong genetic component and a possible microbial trigger. Over 90% of individuals with AS express the HLA B∗2704 and HLA B∗2705 markers. The nature of the microbial trigger is unclear. Early studies demonstrated an association between Klebsiella infections and the clinical onset of AS. Two Klebsiella proteins (nitrogenase enzyme and pulD secretion protein) can bind to the B27 and initiate T cell proliferation. However, the association between AS and Klebsiella is strictly circumstantial and has not yet been proven.
Treatment for Ankylosing Spondylitis
To control pain and stiffness, NSAIDs and analgesics are administered under the close supervision of a physician. If the disease is present in the peripheral joints, sulfasalazine is an effective therapy to reduce inflammation. TNF plays a key role in the pathophysiology of AS. Monoclonal antibodies directed at TNF (infliximab and golimumab) decrease inflammation by neutralizing TNF before it can reach its target. Methotrexate is often used for the treatment of severe disease.
Reactive Arthritis
Reactive arthritis is a unique arthritic condition characterized by skin plaques, conjunctivitis, urethritis, arthritis, and spondylitis. It usually occurs 1 month after infection with sexually transmitted microbes (Chlamydia or Neisseria gonorrhoeae) or select enteric organisms (Yersinia, Salmonella, Shigella, Campylobacter, and Clostridium). It is a classic example of a disease caused by molecular mimicry with the generation of auto antigen-specific CD8 cells. In the case of Chlamydia, evidence suggests that the microbe shares epitopes with HLA B∗2704 and HLA B∗2705. These B27 alleles bind a specific arthritogenic peptide that is recognized by CD8 cells. The evidence for molecular mimicry is much clearer with regard to the enterics. Extensive cross-reactivity occurs between B27 alleles and the molecules expressed by Yersinia, Salmonella, and Shigella. Cross-reactive antigens are usually outer membrane proteins (e.g., YadD), which are virulence factors or cationic membrane pore proteins (e.g., OmpH).
Acute Rheumatic Fever
Infection with group A Streptococcus species can progress from pharyngitis to acute rheumatic fever and permanent heart damage. Antibodies produced in response to the streptococcal infection cross-react with the myocardial heart valves. Acute interstitial valvulitis may cause edema. If untreated, destruction of the valves may cause valvular insufficiency.
Treatment for Rheumatic Fever
Therapy is directed at elimination of the streptococcal pharyngitis before it progresses to rheumatic fever and at downregulating the autoimmune inflammatory response. Penicillin or other cell wall inhibitors are used to eliminate the organism. NSAIDs or prednisone rapidly reduce the inflammatory response.
Rheumatoid Arthritis
Rheumatoid arthritis (RA) is a chronic disease characterized by inflammatory responses involving the synovial membranes and articular joint structures. However, often remote inflammatory responses occur in the heart, lung, liver, skin, and vasculature. While the disease onset can occur in the second decade of life, normal onset is between the fourth and fifth decades of life.
Genetics influences both the incidence and severity of the disease. A strong association exists between HLA-DR4 subsets and RA. Over 70% of patients with RA express the DR4 marker compared with 28% of normal controls. The risk of developing the disease increases in persons expressing DRB∗0401 and DRB∗0404. Disease severity is increased in individuals expressing DRB∗0402 or BRB∗0403. A skewed HLA pattern on lymphocytes in the synovium strongly suggests a genetic susceptibility related to the expression of class II DR alleles. A feature of these alleles is that they share a common five-amino-acid motif (QKRAA) in the β-chain of the DR molecule. These HLA alleles may bind auto-reactive antigens or act as cross-reactive antigens in the response to viral infections.
A viral etiology for RA has been suspected for many years. Parvovirus 19, human T cell leukemia virus (HTLV), herpes types 6 and 8, human endogenous virus 5, and EBV have all been implicated as causative agents of RA. EBV has attracted much attention because EBV-encoded protein gp-110 has a sequence homology with (QKRAA) epitopes.
Although the trigger is unknown, activated macrophages play a role in the initiation of RA lesions by secreting TNF and IL-2, which stimulate fibroblasts. In turn, fibroblasts produce a number of factors that act directly or indirectly to cause tissue damage. One of the major factors produced by fibroblasts is receptor-activated NF-κB ligand (RANKL), which stimulates osteoclasts to destroy bone tissue. Other fibroblast-produced factors are prostaglandin E2 (PGE2) and the cytokines IL-6 and IL-8. These cytokines stimulate polymorphonuclear leukocytes to produce additional proteases and oxygen-derived free radicals (ODFRs). ODFRs degrade hyaluronic acid, which acts as a lubricant in joints. In the absence of hyaluronic acid, inflammation causes articular nerve hyperexcitablity and pain.
Immune complexes, known as rheumatoid factors (RFs), are found in both the serum and synovial fluids of patients with RA. RF is an autoantibody against the Fc portion of another antibody. The production of RF by B cells is accelerated by macrophage-produced IL-6. Immunoglobulin M (IgM) antibodies are the predominant RFs, but IgG and IgA RFs are also found in the synovium. RFs are present in 70% to 80% of adults with RA but occur at a much lower frequency in juvenile rheumatoid arthritis.
The mechanism by which the Fc portion of antibodies becomes antigenic has recently been defined. Under normal conditions, antibodies are heavily glycosylated to sequester potential auto-reactive antigens in the Fc region. However, proteases in the synovial fluid of patients with RA remove sugars from the Fc portion of the IgG molecule, exposing auto-reactive antigens. RFs bind to the agalactosyl Fc domains, creating immune complexes. Deposition of these immune complexes in small blood vessels activates complement, which leads to joint destruction and pannus formation. Pannus is a layer of granulation tissue consisting of proliferating fibroblasts and inflammatory cells (Figure 16-12).
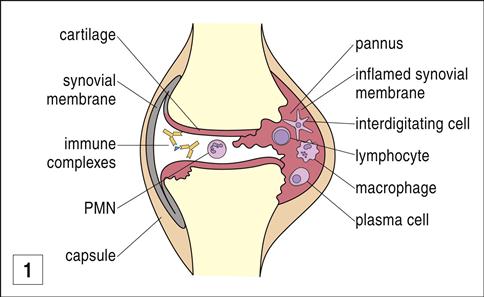

In the pannus, inflammatory cells secrete IL-1, PGA2 and PGE2, platelet-derived growth factor, and plasminogen activators, all of which accelerate the destruction of cartilage and synovial cells.
Treatment for Rheumatoid Arthritis
Multiple therapies are used for the treatment of RA. Early in the disease process, disease-modifying antirheumatic drugs (DMARDs) are used to prevent irreversible damage to joints. DMARDs include methotrexate, azathioprine, hydroxychloroquine sulfate, sulfasalazine, cyclosporine, and monoclonal antibodies. The therapy is designed to reduce the proliferation of auto-reactive cells and decrease inflammation. Methotrexate, azathioprine, and hydroxychloroquine sulfate interfere with DNA synthesis or the transcription of DNA to ribonucleic acid (RNA). Sulfasalazine, a folic acid pathway inhibitor, also inhibits DNA synthesis. Other agents such as cyclosporine inhibit T cell proliferation.
A new class of biologic response modifiers (BRMs) is also used in the treatment of RA. These products of recombinant technology are directed at the immunocompetent cells or soluble factors involved in RA. Rituximab is also used to decrease the number of plasma cells that produce autoantibodies. Other monoclonal antibodies such as infliximab or etanercept are used to neutralize soluble TNF, which indirectly plays a role in bone destruction. Anakinera, a nonglycosylated form of the human IL-1 receptor antagonist (IL-1Ra) is effective in reducing inflammation. In more advanced cases, NSAIDs are indicated for reduction of pain and inflammation. NSAIDs may be used in concert with COX-2 inhibitors which are also anti-inflammatory, analgesic, and anti-pyretic agents.
Autoimmune Hemolytic Anemia
Drugs can induce anemias by direct and indirect means. Some drugs such as α–methyldopa interact with Rh molecules on red cells creating a “neoantigen.” Penicillin and cephalosporin cause autoimmune hemolytic anemia by a different mechanism. At high doses, haptenic penicillin and cephalosporin molecules coat red blood cells, creating an antigen that elicits the production of IgG. Complement activation lyses red blood cells.
Treatment for Autoimmune Hemolytic Anemia
Mild autoimmune hemolytic anemia is treated with corticosteroids to slow red blood cell lysis. More severe forms are treated with immunosuppressive drugs (e.g., cyclophosphamide, azathioprine) and plasmapheresis.
Summary
[/level-membership-for-allergy-and-immunology-category][not-level-membership-for-allergy-and-immunology-category]
Autoimmunity
Learning Objectives
• Examine the relationship between thyroglobulin and iodine
• Identify the differences between T3 and T4
• Understand the hypothalamus–pituitary axis and its role in thyroid function
• Discuss the role of the thyrotropin-releasing hormone (TRH)
• Explain the function of the thyroid-stimulating hormone (TSH)
• Identify the target of the autoantibodies found in Graves’ disease
• Discuss the role(s) of cytokines and fibroblasts in Graves’ ophthalmopathy
• Define the term thyroid storm
• Design a therapy regimen to treat Graves’ disease
• Compare and contrast the symptoms of Graves’ disease and Hashimoto’s thyroiditis
• Discuss the immunologic mechanisms that contribute to Hashimoto’s thyroiditis
• Discuss the role of the intrinsic factor in vitamin B12 transport
• Identify the target of autoantibodies found in pernicious anemia
• Explain the role of vitamin B12 in red blood cell synthesis
• Design a therapy regimen to treat pernicious anemia
• Recognize the target of autoantibodies found in myasthenia gravis
• Identify the two mechanisms by which acetylcholine receptors are reduced in myasthenia gravis
• Compare and contrast ptosis and diplopia
• Design a therapy regimen to treat mild and severe myasthenia gravis
• Identify the clinical symptoms of systemic lupus erythematosus (SLE)
• List the viruses that may play a role in SLE
• Describe the two immunologic defects associated with SLE
• Design a therapy regimen to treat SLE
• Compare and contrast SLE and the “lupus-like” syndrome
• Recognize the autoantibody targets in Goodpasture syndrome
• Describe the genetic and immunologic factors that increase the risk of Goodpasture syndrome
• Design a therapy regimen to treat Goodpasture syndrome
• Differentiate between the function of pancreatic alpha, beta, and delta cells
• Relate the role(s) of insulin in the body
• List the targets of autoantibodies found in type 1a diabetes
• Identify the human leukocyte antigen (HLA) alleles that increase the risk of diabetes
• List the viruses that are implicated in the onset of diabetes
• Design a drug regimen to treat diabetes
• Define ankylosing spondylitis (AS)
• List the possible microbial triggers for AS
• Explain the role of HLA-B27 in the pathophysiology of AS
• Design a treatment regimen for AS
• Compare and contrast rheumatoid arthritis and reactive arthritis
• List the microbial agents that play a role in reactive arthritis
• Define rheumatoid arthritis (RA)
• Explain the relationship between HLA alleles and RA
• Identify the six viruses that may act as triggers for RA
• Compare and contrast the roles of macrophages and fibroblasts in the pathophysiology of RA
• Define rheumatoid factor (RF)
• Understand the relationship between RF and agalactosyl antibodies
• Design a treatment regimen to treat RA
• Discuss the role of the immune system in rheumatic heart disease
• Recognize the definition of autoimmune hemolytic anemia
• Contrast the mechanisms by which α-methyldopa, penicillin, and cephalosporin cause anemia
Key Terms
Acetylcholine
Acetylcholinesterase
Ankylosing spondylitis
Arthritis
Diabetes
Goiter
Goodpasture syndrome
Graves’ disease
Hashimoto’s thyroiditis
Intrinsic factor
Molecular mimicry
Myasthenia gravis
Pernicious anemia
RANKL (receptor activator of nuclear factor kappa-β ligand)
Reactive arthritis
Rheumatic fever
Rheumatoid factor
Systemic lupus erythematosus
Thyrotropin-releasing hormone
Thyroid-stimulating hormone
Introduction
Autoimmune diseases arise when an individual’s immune system attacks his or her own tissue and organs. The immune response may be mediated by antibodies or activated lymphocytes and macrophages. These diseases are found in 5% to 7% of the population, and the disease incidence is heavily skewed toward females. In women over 65 years of age, autoimmune diseases are one of the 10 leading causes of death.
The etiology of autoimmune disease in women is multifactorial. The factors include genetic, environmental, and hormonal triggers. Individuals expressing certain human leukocyte antigen (HLA) alleles are at high risk of developing autoimmune disease. For example, individuals expressing HLA B8 and DR3 have an increased risk of developing Graves’ disease. Because the onset of autoimmune diseases occurs shortly after puberty and just before the onset of menopause, estrogen particularly is thought to be one of the triggers for autoimmune disease. A wide range of different autoimmune diseases may affect a single organ (e.g., thyroid) or multiple organs (e.g., in systemic lupus erythematosus).
Autoimmune Diseases of the Thyroid
The function of the thyroid is to produce the hormones T3 and T4, which control physiologic functions in the cardiac, pulmonary, hematopoietic, gastrointestinal, skeletal, and endocrine systems. To produce the hormones, thyroglobulin (Tg) is synthesized by thyroid cells (thyrocytes) and stored in the follicular lumen. Iodine covalently binds to thyroglobulin tyrosine residues by using thyroperoxidase. Intracellular proteases digest thyroglobulin to produce functional triiodothyronine (T3) and thyroxin (T4), which differ only in the number of bound iodine molecules.
Synthesis of T3 and T4 is controlled by the hypothalamus–pituitary–thyroid axis. When the T3 and T4 levels fall below acceptable levels, the hypothalamus produces thyrotropin-releasing hormone (TRH) which travels through blood to the anterior pituitary gland. The anterior pituitary gland releases another hormone called thyroid-stimulating hormone (TSH). This hormone interacts with thyrotropin receptors and stimulates iodine uptake and the production of T3 and T4 (Figure 16-1).
Thymic hormone levels are controlled by negative feedback loops. High levels of TSH act on the hypothalamus to downregulate the production of TRH. T3 and T4 also act on both the pituitary and the hypothalamus to downregulate hormone production. Autoantibodies directed at the thyroid results in either hypothyroidism or hyperthyroidism.
Graves’ Disease
Graves’ disease was first reported by Dr. Robert Graves in the 1830s and is the most common cause of hyperthyroidism in children and young women. The disease is caused by IgG1 antibodies reacting with thyrotropin or TSH receptors. What causes the immune system to recognize thyroid antigens is unclear. It is conceivable that a viral infection may expose a hidden epitope in thyroid receptors.
Constant stimulation of TSH receptors causes glandular hyperplasia (goiter) and accelerated synthesis of T3 and T4. Patients often exhibit a rapid heart rate, exophthalmos, tremors, sweating, and attention deficit disorders. TSH receptors are also found in orbital cells and muscles. Reaction between the autoantibody and the TSH orbital receptor causes an inflammatory reaction called Graves’ ophthalmopathy (Figure 16-2).
Graves’ Ophthalmopathy
In the pathogenesis of Graves’ ophthalmopathy, lymphocytes infiltrate the orbit of the eye and release cytokines such as tumor necrosis factor (TNF) and interleukin 1 (IL-1). These cytokines stimulate fibroblasts to secrete a glycosaminoglycan (GAG) mucopolysaccharide. GAG increases osmotic pressure in the eye, causing edema in the extraocular and retro-orbital muscles and in adipose tissue. These changes force the eyeball forward, creating bulging of the eye, or exophthalmos.
Thyroid Storm
In patients with undiagnosed Graves’ disease, thyroid hormones often increase dramatically, which precipitates a life-threatening clinical crisis. A thyroid storm is precipitated by excessive stress, surgery, or sepsis. High levels of thymic hormones cause extreme tachycardia and atrial fibrillation. The mortality rate is 20%, and death occurs within 48 hours.
Treatment for Graves’ Disease
Thiourea was the drug initially used to treat Graves’ disease; however, it had considerable toxicity. Second-generation drugs and derivatives such as propylthiouracil and methylthiouracil, methimazole, and carbimazole have less toxicity. With the exception of methimazole, which prevents the storage of thyroid hormones, these drugs inhibit thyroperoxidase, thereby reducing the synthesis of T4 and T3.
Oral administration of radiolabeled iodine-131 is now the most common therapy for Graves’ disease. The high-intensity radiation destroys thyroid cells and brings the levels of thyroid hormones into the normal range. Subtotal thyroidectomy was the treatment of choice before the advent of radioactive iodine. Methimazole therapy is indicated for those patients in whom radioactivity is contraindicated.
Hashimoto’s Thyroiditis
Hashimoto’s thyroiditis is the most common form of hypothyroidism, with a prevalence rate of 2% of the population of the United States. Individuals mount both an antibody response and a cellular response to thyroid tissue. The disease is caused by a break in peripheral tolerance to thyroid tissue, and the failure of regulatory cells to control auto-reactive T and B cells. About 85% to 90% of patients with Hashimoto’s thyroiditis develop anti-thyroid peroxidase and anti-thyroglobulin antibodies. The inactivation of thyroperoxidase prevents the incorporation of free iodine into thyroglobulin. Elevated intracellular iodine also stimulates the maturation of dendritic cells and macrophages, which present antigens to auto-reactive CD4 and CD8 cells that have infiltrated the thyroid (Figure 16-3).
CD4Th1 cells produce IL-12, tumor necrosis factor alpha (TNF-α) and interferon gamma (INF-γ), which induce apoptosis of thyrocytes. Individuals with Hashimoto’s thyroiditis also express a molecule called Fas on thyroid cells. The expression of the Fas molecules marks the cell for destruction by T lymphocytes.
Patients with Hashimoto’s thyroiditis present with nonspecific symptoms such as fatigue, dry skin, and weight gain. Other symptoms include decreased sweating, deafness, peripheral neuropathy, depression, and memory loss.
Treatment for Hashimoto’s Thyroiditis
Oral sodium levothyroxine, which is a synthetic thyroid hormone (T4), usually has to be taken for life by patients with Hashimoto’s thyroiditis. However, the dose must be carefully titrated on the basis of the patient’s needs. Some patients cannot tolerate the synthetic hormone. Natural porcine gland preparations of T3 and T4 can be used in these patients.
Pernicious Anemia
In pernicious anemia (PA), antibodies are directed at a glycoprotein known as intrinsic factor. These autoantibodies also neutralize any soluble intrinsic factor in the intestinal lumen.
Neutralization of the soluble intrinsic factor prevents the transport of vitamin B12 across the intestinal mucosa to portal blood (Figure 16-4). Without this essential vitamin, red blood cell precursors cannot divide. This results in a megaloblastic anemia characterized by the presence of immature, dysfunctional red blood cells (megaloblasts) in the blood. If untreated, PA is usually fatal after 1 to 3 years (Figure 16-5).
[/not-level-membership-for-allergy-and-immunology-category]
