[level-membership-for-surgery-category]
Chapter 39 Assisted Reproductive Technology: Laboratory Aspects
HUMAN SEMEN: PREPARATION AND EVALUATION
Semen Collection
Many clinicians limit male infertility evaluation to a single semen analysis if the initial sample is found to be normal. However, a thorough infertility evaluation requires analysis of two ejaculates collected as least 1 month apart because semen quality varies over time.1 If either one of the two ejaculates are abnormal, additional samples should be evaluated. Some clinicians recommend that all fertility patients undergo semen analysis once or twice yearly during the duration of their treatment. More frequent analysis may be indicated in postsurgical patients as a part of routine follow-up.
It is preferable for samples to be collected near the andrology assessment area. Rooms designated for production of semen samples should be located in a quiet part of the facility and doors should be labeled with DO NOT DISTURB signs. Reading materials and/or video aids should be available for those men requiring them.
Semen Parameters
Semen Volume
The suffix -spermia refers to the volume of seminal fluid. A man is aspermic if he produces no semen after orgasm, hypospermic if he produces less than 0.55 mL of ejaculate, and hyperspermic if ejaculate volume is above 6.0 mL. The most common cause of low ejaculate volume is incomplete collection. Any abnormalities or interruptions during collection should be noted. Sample loss often compounds interpretation because distinct sperm-rich and sperm-poor fractions can be emitted. Short duration of abstinence from ejaculation may also lead to low semen volume. Hyperspermia is a rare clinical entity with unknown significance. Although spermatozoa density may be decreased, spermatozoa motility is unchanged.2
Semen pH
The semen pH should be recorded because it may be a useful parameter for diagnosis of some conditions affecting semen quality. Normal semen is alkaline, with a pH between 7.2 and 8.0.3 Values up to 8.5 have been noted in otherwise normal samples. Measurement is most easily performed with pH paper.
Semen Liquefaction
Fresh semen quickly coagulates into a gelatinous mass after ejaculation before liquefying over 5 to 25 minutes at room temperature. Coagulation is due to coagulating proteins contributed by the seminal vesicles in the final portion of the ejaculate; secretions from the bulbourethral glands of Cowper and prostate present in the initial fraction of the ejaculate are responsible for liquefaction.4
An absence of coagulation may indicate absent or hypoplastic seminal vesicles, or ejaculatory duct obstruction. Conversely, prolonged liquefaction time may be due to a lack of prostate-supplied, liquefying proteases such as prostate-specific antigen, amylase, and plasminogen activators.5 Absence of liquefaction may cause infertility, although impaired liquefaction is frequently associated with normal fertility.
Spermatozoa Concentration
The Makler counting chamber (Sefi Medical Instruments, Haifa, Israel) is a specialized instrument designed for rapid semen analysis. Since its inception in 1980, the Makler counting chamber has gained wide acceptance and is used in many laboratories. The main advantage of the Makler counting chamber over the Neubauer hemocytometer is that except in cases of extremely high spermatozoa concentration, no dilution of the semen specimen is needed. Additionally, motility and forward progression may be assessed at the time of spermatozoa concentration determination. Although the Makler Chamber simplifies determination of spermatozoa concentration in semen analysis, the method lacks the accuracy of traditional hemocytometry and may be prone to errors.6
Spermatozoa Motility and Forward Progression
The World Health Organization (WHO) 1999 progression system classifies spermatozoa into one of four categories: A represents spermatozoa with rapid progressive motility; B, slow or sluggish progressive motility; C, nonprogressive motility; and D, no motility.7 The percentage of spermatozoa in each category is reported. A normal semen specimen contains at least 50% motile spermatozoa with the majority exhibiting good (modal progression class >2, WHO class A and B) forward progression. Athenozoospermia is defined as less than 50% of spermatozoa with good motility.
Spermatozoa Morphology
In 1986, Kruger and associates proposed a strict criteria for classification of normal and abnormal spermatozoa morphology.8 Objective measurements of individual spermatozoa components are included in this system, also known as the Tygerberg strict criteria classification system. According to this system, a spermatozoon is considered normal only if: (1) the head measures 5 to 6 microns in length and 2.5 to 3.5 microns in width; (2) acrosomal staining covers 40% to 70% of the anterior aspect of the sperm head; (3) the midpiece is 1.5 times as long as the head length and less than 1 micron in width; (4) the tail is approximately 45 microns long, uniform, uncoiled, and free from kinks; and (5) cytoplasmic droplets are no more than half the size of the spermatozoa head and occupy the midpiece only. Borderline forms are considered abnormal in this classification scheme.
In men with spermatozoa counts greater than 20 million/mL and motility greater than 30%, fertilization rates during IVF cycles were 91% for men with greater than 14% normal spermatozoa by Tygerberg strict criteria and 37% for men with less than 14% normal forms.8 This study established the reference value for normal morphology as 15% or greater by strict criteria.
In 1992, the WHO adopted a grading system based on the Tygerberg strict characterization of distinct spermatozoon components.3 Under these guidelines, a spermatozoon is considered normal if: (1) the head measures 4 to 5.5 microns in length and 2.5 to 3.5 microns in width; (2) acrosomal staining covers 40% to 70% of the anterior aspect of the sperm head; (3) there are no neck, midpiece, or tail defects; and (4) cytoplasmic droplets are less than one third the size of the head. Although these parameters appear quite similar to Tygerberg criteria, slightly looser standards increase the reference value for normal morphology to 30%. Use of both grading schemes varies from laboratory to laboratory.
Spermatozoa Viability
Another method for assessing spermatozoa viability is the hypo-osmotic swelling test. The test relies on the ability of live spermatozoa to tolerate moderate hypo-osmotic stress as evidenced by controlled swelling. Dead spermatozoa do not show discernible swelling when exposed to hypo-osmotic conditions, whereas viable spermatozoa exhibit a characteristic coiling pattern in the tail.9
Functional Spermatozoa Tests
Zona Pellucida Binding Assay
The zona pellucida binding assay is another adjunctive test of spermatozoa function. This test assesses the capacity of spermatozoa to bind to the outer surface of the zona pellucida, a prerequisite for binding and penetrating the egg. Microscopically, the zona pellucida is divided and each half is mixed with a sample of the patient’s spermatozoa or a sample of a fertile donor. Spermatozoa bound to the zona pellucida outer surface are counted. A hemizona index is calculated by dividing the total number of patient spermatozoa bound by the total number of donor spermatozoa bound. Successful IVF is associated with an index greater than 60%.10
Hamster Egg Penetration Assay
The zona pellucida, the acellular glycoprotein layer surrounding the ovum, regulates species-specific fertilization. Removal of this layer allows evaluation of interspecies, spermatozoa–oocyte interaction using the sperm penetration assay (SPA). This procedure assesses the ability of human spermatozoa to penetrate a hamster oocyte as a test of the fertilizing capability of the human spermatozoa.11 Human spermatozoa penetration of zona-free hamster oocytes has been shown to correlate with human spermatozoa penetration of human ova.12
Sperm Chromatin Structure Assay/Single-cell Gel Electrophoresis
Assays utilize direct staining and evaluation of spermatozoa chromatin to assess the integrity of the genetic material. The tests provide useful information on the presence of genetic damage to spermatozoa DNA. Briefly, both procedures evaluate shifts from intact, double-stranded DNA to denatured, single-stranded DNA.13 These assays can be used to assess male spermatozoa DNA integrity as related to fertility potential and embryo development as well as effects of reproductive toxins.14
Collection-Specific Spermatozoa Evaluation and Processing
Processing of Semen Collected by Masturbation
Spermatozoa Migration Techniques (Swim-up Method)
Spermatozoa washing of a sample before enrichment for motile spermatozoa (swim-up from pellet) has been criticized because dead and abnormal spermatozoa present in the original sample remain in the pellet. Aitken and Clarkson showed that the functional fertilizing capacity of motile spermatozoa isolated from such a pellet can be impaired when compared to that of motile spermatozoa isolated before spermatozoa washing and pelleting.15 The authors propose that the production of oxygen free radicals by abnormal spermatozoa in the pellet may result in cellular damage in motile spermatozoa, leading to abnormal spermatozoa function. Additionally, the centrifuged pellet has a relatively small interface with the overlying layer, making it difficult for motile spermatozoa in the bottom of the pellet to migrate into the media.
Processing of Spermatozoa Collected Following Retrograde Ejaculation
Viable spermatozoa may be retrieved from the urine of patients failing medical therapy.16 Because urine’s acidity makes it naturally toxic to spermatozoa, it is first suggested to alter the chemical milieu of the bladder to minimize its deleterious effects on spermatozoa quality. Alkalinization of the urine above a pH of 7.5 can be achieved by instructing the patient to ingest sodium bicarbonate for several days prior to collection. The patient empties the bladder before masturbation. Immediately after orgasm, the patient urinates into a sterile container, which is taken to the laboratory for spermatozoa extraction. Alternatively, for men unable to volitionally void, the bladder may be catheterized and washed with fresh, pH-adjusted, spermatozoa processing medium. After irrigation, a small volume of medium is left in the bladder. The patient masturbates and ejaculates, and the bladder is catheterized again to obtain fluid containing retrograde ejaculate, which is collected and transported to the laboratory for processing. Laboratory procedures begin with centrifugation and removal of urine from the spermatozoa. The resuspended pellet can then be processed as previously described for samples produced from masturbation.
Processing of Spermatozoa Collected Following Vibratory Stimulation/Electroejaculation
Failure of emission is termed anejaculation. Causes of anejaculation are similar to those of retrograde ejaculation, although spinal cord injury is a much more common cause. Because most of these patients do not respond to sympathomimetic therapy, they may require the use of external stimulatory devices to achieve emission. The most common methods of stimulation are penile vibratory stimulation and rectal electroejaculation. Penile vibratory stimulation entails the application of a penile vibrator to the glans penis, leading to a reflexive ejaculation. This technique induces ejaculation in approximately 70% of anejaculatory men with spinal cord injuries.17
Men with lesions above the level of T10 are more likely to respond than those with lesions below that level. Electroejaculation may be used in men who do not respond to vibratory stimulation. This procedure involves carefully inserting a probe into the rectum, followed by the intermittent application of electrical current to the periprostatic nerve plexus. The current stimulates the muscular tissue of the prostate, seminal vesicle, vas deferens, and epididymis to contract and propel semen into the urethra. It is successful in approximately 75% of patients.18,19 If antegrade ejaculation ensues, it may be collected from the urethra. If retrograde ejaculation occurs, then the urine is alkalinized beforehand and the bladder irrigated with medium and catheterized to remove any spermatozoa-bearing fluid.
Risks of electroejaculation include rectal thermal injury or perforation. The rectum should be examined with proctoscopy before and after the procedure. Life-threatening autonomic dysreflexia, or massive, uncoordinated sympathetic release, may occur in patients with spinal cord injuries above the level of T6. Blood pressure monitoring is imperative in this patient population. Semen obtained by vibratory stimulation and electroejaculation frequently has low volume, high spermatozoa concentration, and poor motility, likely owing to stasis within the genital tract.20 Again, processing of samples produced with vibratory stimulation or electroejaculation continues as in samples produced with masturbation.
Processing of Spermatozoa Collected Following Surgical Spermatozoa Collection
The management of severe male factor infertility has been dramatically changed with the introduction of intracytoplasmic sperm injection (ICSI). Because only one spermatozoa is needed to inject into each oocyte, few spermatozoa are needed to ensure fertilization of retrieved oocytes with ICSI. Advances in surgical techniques of spermatozoa retrieval combined with ICSI have allowed men with certain male infertility factors previously considered to be incompatible with fertility to father biologic progeny. Spermatozoa may be retrieved in men with both nonobstructive and obstructive azoospermia. Chapter 53 discusses the technical details of the specific surgical procedures.
Microsurgical Epididymal Sperm Aspiration
Microsurgical epididymal sperm aspiration (MESA) is indicated for men with reconstructible or nonreconstructible obstructive azoospermia. It is a relatively simple procedure requiring regional or general anesthesia. MESA involves direct exposure of the epididymis with microscopic retrieval of spermatozoa from that structure. An incision is made through the scrotal skin, dartos, and tunica vaginalis to expose the testis and epididymis. A small incision is made in the wall of an epididymal tubule, and a needle or hollow glass rod is used to aspirate fluid from the lumen.
Although pregnancy rates are similar, MESA is preferred to the other, less-invasive method of epididymal spermatozoa extraction, percutaneous epididymal sperm aspiration (PESA), because it provides the setting to correct reconstructible etiologies of azoospermia identified at the time of exploration. Additionally more spermatozoa are recovered with MESA than PESA.21
Testicular Sperm Extraction/Testicular Sperm Aspiration
Testicular sperm extraction (TESE) is an effective procedure for the retrieval of spermatozoa for ICSI. It is indicated in men with nonobstructive azoospermia as well as men with obstructive azoospermia in whom spermatozoa could not be retrieved from the epididymis.22 TESE entails testicular biopsy with retrieval of spermatozoa. An incision is made through the scrotal skin, dartos, and tunica vaginalis to expose the testis. The tunica albuginea is then opened. The testicle is gently squeezed to deliver seminiferous tubules through the incision. Several tubules are cut and immediately placed in medium for processing.
Microscopic Testicular Sperm Extraction
Microscopic testicular sperm extraction (microTESE) is a relatively new procedure designed to maximize the yield of testicular spermatozoa from men with nonobstructive azoospermia while minimizing the amount of tissue excised.23 Testicular microdissection has several advantages over the standard, open procedure. Use of the operating microscope allows for visualization of small blood vessels and minimizes the risk of injury to blood supply. Additionally, spermatozoa production in patients with nonobstructive azoospermia is not constant throughout the testis, with isolated areas showing spermatogenesis even while the majority of the tissue is quiescent.
Schlegel’s discovery that tubules with active spermatogenesis appear larger and fuller than nonactive tubules with microscopy make microTESE a much more sensitive procedure than TESE for identifying active areas. Much smaller volumes of tissue are needed for spermatozoa retrieval, avoiding the impairment in testosterone production that may occur after removal of larger amounts of tissue with TESE.24
Spermatozoa found in the epididymis and testis are not fully mature. Those isolated from these structures may exhibit some movement. However, specimens retrieved from the testis are frequently immotile. Motility of epididymal- and testicular-derived spermatozoa, reflecting viability, is a critical factor in predicting the success of ICSI. Additionally, testicular spermatozoa are enclosed within the surgical specimen and must first be released before subsequent processing. Methods for recovery and maturation of surgically retrieved spermatozoa have been described.25 In MESA, the aspirate is flushed in a small volume of medium. If spermatozoa are present, an aliquot should be used for concentration and motility determination. If present, fresh epididymal spermatozoa may be used for injection or cryopreservation.
Spermatozoa Preservation
Cryopreservation of human spermatozoa became a feasible method of storage in 1949, when Polge discovered that fowl spermatozoa could be protected from the effects of freezing by the addition of glycerol. Soon after, Sherman described successful cryopreservation and thawing of human spermatozoa with subsequent pregnancy.26 Since that time, a large body of research has focused on understanding the physical and chemical changes that occur during freezing and in evaluating methods and agents to optimize freezing. Although many advanced aspects of cryobiology are beyond the scope of this text, several basic principles are important to discuss.
Cryopreservation has become an important component of ART. Patients with ejaculatory dysfunction requiring vibratory stimulation or electroejaculation may freeze spermatozoa and use aliquots to attempt fertilization over several cycles without the need for repeat procedures. It has been recommended that surgically retrieved spermatozoa be cryopreserved for future cycles of IVF with ICSI to obviate the need for repeat procedures.27 Donor insemination relies on the reliable, prolonged storage of large numbers of samples. Patients facing potentially fertility-damaging medical therapies may choose to store spermatozoa for future use.
OOCYTES: PREPARATION AND EVALUATION
Advances in culture medium composition have significantly influenced embryo quality and pregnancy rates over the years. All media for ART can now be purchased commercially. Before use they must be tested for toxins and their ability to support the growth and development of embryos. Media should only be opened in a laminar flow hood, with attention to maintaining sterility during the addition of protein. After any media preparation or addition, it should be filtered for assurance of sterility.
Oocyte Collection and Evaluation
Oocyte Assessment
Oocyte–cumulus complexes are scored before incubation. Accurate assessment of oocyte maturity at the time of the retrieval may be informative for timing and success of fertilization and may permit identification of some oocyte abnormalities. Initial oocyte grading relies on direct optical analysis of oocyte– cumulus complex morphology. The grade is based on characteristics of the cumulus oophorus and corona radiata. This first observation should be made quickly while searching through the follicular fluid. In cases of IVF by conventional insemination, the cumulus is not removed before insemination, so observation is somewhat limited.
Immature oocytes are defined as being at a stage of meiosis prior to metaphase of meiosis II. This includes oocytes in prophase of meiosis I, which are identified by the presence of a germinal vesicle or nuclear envelope in the cytoplasm, without any polar body present in the perivitelline space (Fig. 39-1). If present, cumulus and corona are commonly very tightly condensed. As prophase I resumes, the oocyte enters into metaphase of meiosis I. This intermediate stage of maturation is recognized by the disappearance of the germinal vesicle and the absence of the first polar body (Fig. 39-2). For meiosis I oocytes the cumulus may be expanded, but the corona can still be compact. The extrusion of the first polar body marks the transition to a mature oocyte, which is now considered to be at meiosis II (Fig. 39-3). Metaphase II oocytes will usually have a fully expanded cumulus. Under normal circumstances the oocyte will remain at meiosis II until fertilization, when meiosis II resumes, the second polar body is extruded, and the male and female pronuclei form (Fig. 39-4).
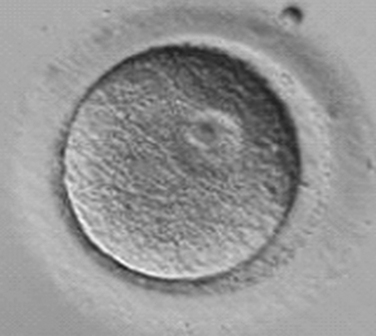
Figure 39-1 Germinal vesicle intact or immature human oocyte after cumulus removal.
(Courtesy of Kathleen A. Miller, Reproductive Medicine Associates of New Jersey, Morriston, N.J.)
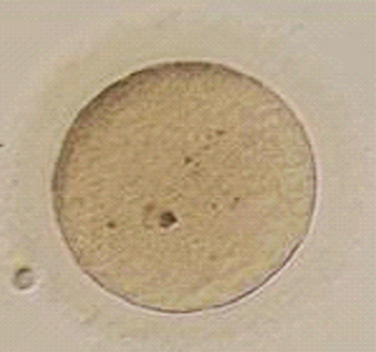
Figure 39-2 Metaphase I human oocyte without a polar body after cumulus cell removal.
(Courtesy of Kathleen A. Miller, Reproductive Medicine Associates of New Jersey, Morriston, N.J.)
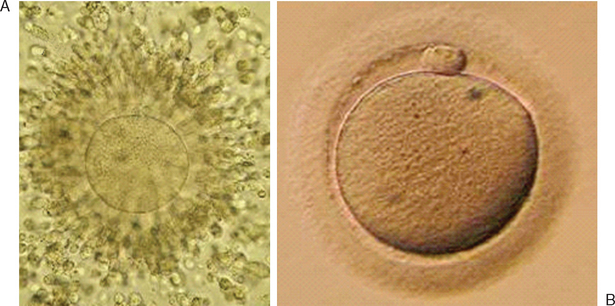
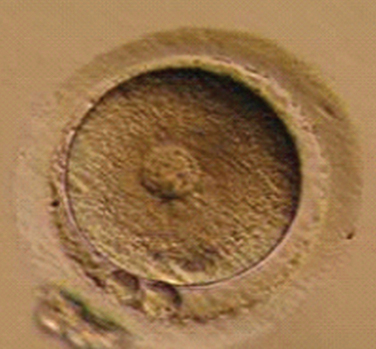
Figure 39-4 Normal zygote showing presence of two polar bodies and two pronuclei.
(Courtesy of Kathleen A. Miller, Reproductive Medicine Associates of New Jersey, Morriston, N.J.)
Assessment of oocyte morphology after cumulus cell removal may reveal its true morphologic quality. The appearance of cytoplasm, zona pellucida, and polar body should be considered. A homogeneous cytoplasm, with uniform color and granularity and occupying most of the space inside the zona pellucida, represents a good quality oocyte. Vacuoles, a dark color, shrunken shape, or granularity are considered negative signs in the morphological quality evaluation of oocytes. It may be important to observe shape and thickness of the zona pellucida; it should not be cracked and should have an appropriate thickness for its developmental stage.
IN VITRO MATURATION OF OOCYTES
In vitro maturation of human oocytes was first demonstrated by Dr. Edwards in 1965. Although this technique was viewed as having great potential in providing oocytes for early IVF work, the use of exogenous gonadotropin administration proved more efficient and became the gold standard in infertility treatment. Because of the numerous perceived advantages of in vitro maturation, continued interest and investigation have persisted in the area of human oocyte in vitro maturation. Pregnancies have been reported using in vitro maturation of immature oocytes that were retrieved after conventional follicle-stimulating hormone (FSH) stimulation and hCG administration.28–30 However, such an approach minimizes the advantages that warrant in vitro maturation. In addition, it could be argued that such an approach represents pseudo in vitro maturation of oocytes because the exogenous gonadotropins and resulting follicular development most likely progressed oocytes’ cytoplasmic maturation while nuclear maturation lagged.
Recent reports have utilized a single dose of hCG (no exogenous FSH administration) on day 10 to 14 of the cycle with oocyte isolation 36 hours after hCG administration.31,32 These studies were performed in patients with polycystic ovary syndrome (PCOS). In this subpopulation the use of single-dose hCG administration coupled with in vitro maturation and ICSI appears to be an efficient means of treating infertility while reducing the cost and concerns of hyperstimulation syndrome. Although the numbers in the study are small, the resulting clinical pregnancy rate is encouraging, at approximately 39%.32 Whether such a treatment regimen will work in non-PCOS patients remains to be determined.
At the other extreme, true in vitro maturation entails collection of oocytes from ovaries without exogenous gonadotropin stimulation. Such an approach will, when it becomes efficient, allow realization of all the perceived advantages of in vitro maturation. Clinical pregnancies using in vitro maturation without exogenous gonadotropin stimulation have been reported.33–35 These results suggest that using an approach of in vitro maturation of human oocytes, coupled with IVF, is a means of obtaining live births. However, this technique requires extensive evaluation because the cumulative success in all of these studies, as determined by live births, is less than 5%. Numerous factors individually, or in combination, may be contributing to the poor success of IVF after in vitro maturation. These include oocyte quality, culture conditions, and endometrial receptivity.
IN VITRO FERTILIZATION
Conventional Insemination
If spermatozoa are of sufficient quality and quantity, IVF can be attempted with conventional insemination. Although the terms of sufficient quality and quantity are indeed subjective, each laboratory will have its own criteria for when a couple uses conventional insemination or ICSI to achieve fertilization. Spermatozoa can be processed in numerous manners or with a combination of techniques. The primary goal is to remove seminal plasma and isolate motile spermatozoa of normal morphology with as little nongamete cellular contamination as possible. The processes used include semen washing, density gradient separation, and swim-up procedures.
Intracytoplasmatic Sperm Injection
Technique
The equipment for ICSI, as in any other micromanipulation procedure, consists of an inverted phase contrast microscope with micromanipulators and injectors, which permit the manipulation and cellular surgery. A warming stage may be recommended on the microscope for maintenance of a consistent temperature, although some groups show quite acceptable results without using a warming stage for micromanipulation.36 The hydraulic system in a micromanipulator should work with maximal precision, and the microscope should be placed in a very stable location.
The spermatozoon is then brought to the tip of the pipette, which is pushed against the zona pellucida. The needle is pushed forward through the zona pellucida and into the oolemma. Gentle suction is applied when the tip is believed to have penetrated the oolemma and a small amount of ooplasm is aspirated to ensure that the membrane has been breached. The spermatozoa is then released into the ooplasm and the pipette is removed. Injection of any extra medium should be avoided. A timeline schematic of the entire ICSI process is depicted in Figure 39-5.
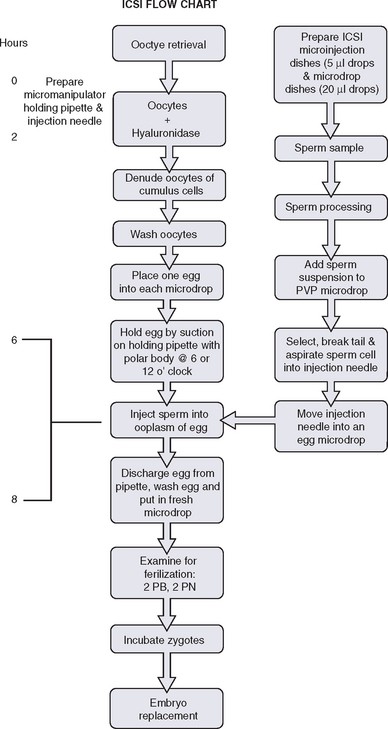
PREPARATION AND EVALUATION OF ZYGOTES AND EMBRYOS
Embryo Assessment
Subtract from the grade for irregularities as follows:
Embryo Culture
With the improvement of culture systems and extended culture media, it is now possible to culture embryos to the blastocyst stage (day 5 or 6). Typically patients who are considered candidates for blastocyst transfer are young women (less than 36 years old) with good ovarian reserve or older patients with a high number of embryos in a cycle (more than 5). One of the most important factors influencing blastocyst culture and transfer success is the quality and quantity of embryos on day 3. An effective blastocyst grading system using both number and letter assignment based on degree of development and assessment of both the trophectoderm and the inner cell mass has been proposed.37 The program should consider advantages and disadvantages in each individual patient and cycle before considering the option of blastocyst culture.
Assisted Hatching
There are two common methods of assisted hatching used clinically for embryos at the six- to eight-cell stage: acid Tyrode’s solution and laser assisted hatching. Acid Tyrode’s solution can be prepared in the laboratory, following a standard protocol, and the pH adjusted to 2.5,38 or it can be commercially purchased. It is important to appreciate that use of this acid can be detrimental to the blastomeres closest to the point where the acid is applied. In cases of erroneous manipulation, it can compromise the viability of the embryo. In this procedure, an assisted hatching micropipette expels the acid close to the zona pellucida of the embryo, which is secured by a holding pipette. As soon as a hole is observed, rapid suction is applied to avoid excess acid in the perivitelline space and in contact with blastomeres. After the procedure the embryos must be washed several times before further culture. Use of a laser for assisted hatching may be considered by some to be less dangerous than use of acid Tyrode’s solution assisted hatching, but its application, if not precise, may cause thermal or mutagenic effects in the embryo. Both techniques have risks and mandate fine proficiency.
Preimplantation Genetic Screening
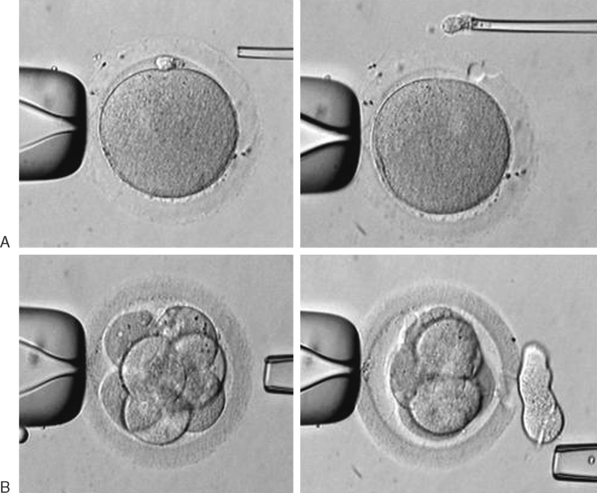
Preimplantation genetic screening (PGS) was originally seen as a means to detect and avoid congenital diseases by evaluation of individual blastomeres of preimplantation embryos. This has value for couples with probability of having a child affected by some genetic disturbance. Initially, molecular PGS was employed for embryo sexing in couples at risk for X-linked diseases.39 Technically, PGS consists of two steps, including micromanipulation and biopsy, and DNA analysis of gametes or embryos.
Micromanipulation for the biopsy includes the mechanical opening of the zona pellucida and retrieval of one or two polar bodies, in the case of diagnosis performed on oocytes or zygotes, or the retrieval of a blastomere in cases of biopsy of embryos. This procedure is briefly illustrated in Figure 39-6. For DNA analysis, there are two main techniques currently employed, polymerase chain reaction (PCR) for single-gene disease detection and fluorescence in situ hybridization (FISH) for aneuploidy.
Single-Gene Defects
Even with the high number of copies generated with PCR cycle amplification, a single PCR procedure may yield insufficient genetic material for gene detection. Efficiency and specificity of the DNA amplification can be improved by using a nested PCR method that consists of a two-step procedure.40
It is of extreme importance to realize that many factors can influence the reliability of PCR analysis. It is essential that numerous steps in quality assurance be implemented to protect against erroneous results and to ensure confidence in the final results.
Fluorescent In Situ Hybridization
Another way to analyze chromatin for PGS is FISH. This method utilizes direct-labeled probes, with the specific annealing of fluorescent nucleic acid probes to complementary sequences of DNA in a fixed specimen in which chromatin or chromosomes have been isolated.41–44 Chromatin from polar bodies or blastomeres can be treated and fixed for FISH. Cells in this procedure are fixed on a slide in a manner that ruptures the cell, removes the cytoplasmic material, and spreads and adheres chromosomes. These chromosomes are then hybridized or annealed to the chromosome-specific fluorescent probes. Using a fluorescent microscope one can count specific chromosomes and thus identify normal or abnormal chromosome numbers (Fig. 39-7).
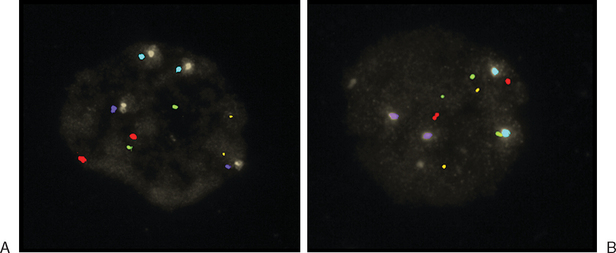
Figure 39-7 Examples of FISH analysis micrographs. A, A normal female. B, Trisomy 21 (in green).
(Courtesy of Kathleen A. Miller, Reproductive Medicine Associates, New Jersey, Morriston, N.J.)
Currently, the most utilized FISH probes determine number of chromosomes 13, 16, 18, 21, 22, X, and Y in the cells.45 As is the case in PCR-based PGS, numerous controls and implementation of stringent quality assurance programs are essential for the laboratory performing FISH-based PGS.
CRYOPRESERVATION
Cryopreservation of gametes and embryos maximizes the success of conception in any IVF cycle and prevents waste of specimens. Currently, freezing of embryos is accepted and is considered an indispensable technology in most ART laboratories. Such importance has even been confirmed by the ethics committee of the American Society for Reproductive Medicine.46 It has been stated that an effective cryopreservation program can decrease multiple pregnancies, costs, and need for hormonal stimulation of additional cycles and prevention of embryo waste. However, it is important to realize that there are many ethical, religious, legal, and social implications to embryo storage. Some countries, such as Italy, Germany, Austria, Switzerland, Denmark, and Sweden, have restricted or forbidden cryopreservation of embryos.47
Vitrification
Vitrification is a form of rapid cooling that utilizes very high concentrations of cryoprotectant that solidify without forming ice crystals. Ice crystals are a major cause of intracellular cryodamage. The term vitrification is derived from the Latin word vitreous, which means glassy or resembling glass. Vitrification can be considered a nonequilibrium approach to cryopreservation originally developed for cryopreservation of mammalian embryos.48
The vitrified solids therefore contain the normal molecular and ionic distributions of the original liquid state and can be considered an extremely viscous, supercooled liquid. In this technique oocytes or embryos are dehydrated by brief exposure to a concentrated solution of cryoprotectant before the samples are plunged directly into liquid nitrogen. Application of vitrification for both oocytes and embryos is a current area of focus for many clinical, rodent, and domestic animal production laboratories. An excellent review of the history of vitrification and potential advantages is available.49
1 Mortimer D, Templeton AA, Lenton EA, Coleman RA. Influence of abstinence and ejaculation-to-analysis delay on semen analysis parameters of suspected infertile men. Arch Androl. 1982;8:251-256.
2 Dickerman Z, Sagiv M, Savion M, et al. Andrological parameters in human semen of high (≥6ml) and low (≤1 ml) volume. Andropologia. 1989;21:353-362.
3 World Health Organization. The WHO Laboratory Manual for the Examination of Human Semen and Sperm–Cervical Mucus Interaction, 3rd ed. Cambridge: Cambridge University Press, 1992.
4 Lilja H, Oldbring J, Rannevik G, Laurell CB. Seminal-secreted proteins and their reactions during gelation and liquefaction of human semen. J Clin Invest. 1987;80:281-285.
5 Propping D, Tauber PF, Zaneveld LJD, Schumacher GFB. Purification and characterization of two plasminogen activators from human seminal plasma. Fed Proc. 1974;33:289-293.
6 Keel BA, Webster BW. The Handbook of Laboratory Diagnosis and Treatment of Infertility. Boca Raton, Fla.: CRC Press, 1990.
7 World Health Organization. The WHO Laboratory Manual for the Examination of Human Semen and Sperm–Cervical Mucus Interaction, 4th ed. Cambridge: Cambridge University Press, 1999.
8 Kruger TF, Menkveld R, Stander FS, et al. Sperm morphologic features as a prognostic factor in in vitro fertilization. Fertil Steril. 1986;46:1118-1123.
9 Jeyendran RS, Van der Van HH, Perez-Pelaez M, et al. Development of an assay to assess the functional integrity of the human spermatozoa membrane and its relationship to other semen characteristics. J Reprod Fertil. 1984;70:219-228.
10 Burkman LJ, Coddington CC, Fraken DR, et al. The hemizona assay (HZA): Development of a diagnostic test for the binding of human spermatozoa to the human hemizona pellucida to predict fertilization potential. Fertil Steril. 1988;49:688-697.
11 Yanagamachi R, Yanagamachi H, Rogers BJ. The use of zona-free animal ova as a test system for the assessment of fertilizing capacity of human spermatozoa. Biol Reprod. 1976;15:471-476.
12 Rogers BJ, Van Campen H, Ueno M, et al. Analysis of human spermatozoa fertilizing ability using zona-free ova. Fertil Steril. 1979;32:664-670.
13 Evenson DP, Jost LK, Baer RK, et al. Individuality of DNA denaturation patterns in human sperm as measured by the sperm chromatin structure assay. Reprod Toxicol. 1991;5:115-125.
14 Hayes WA, editor. Principles and Methods of Toxicology. New York: Raven Press, 1994.
15 Aitken RJ, Clarkson JS. Cellular basis of defective sperm function and its association with the genesis of reactive oxygen species by human spermatozoa. J Reprod Fertil. 1987;91:459-469.
16 Mahadevan M, Leeton JF, Trounson AO. Noninvasive method of semen collection for successful artificial insemination in a case of retrograde ejaculation. Fertil Steril. 1981;36:243-247.
17 Ohl DA, Menge AC, Sonksen J. Penile vibratory stimulation in spinal cord injured men: Optimized vibration parameters and prognostic factors. Arch Phys Med Rehabil. 1996;77:903-905.
18 Brindley GE. Electroejaculation: Its technique, neurological application implications and uses. J Neurol Neurosurg Psych. 1981;44:9-18.
19 Ohl DA, Bennett CJ, McCabe M. Predictors of success in electroejaculation of spinal cord injured men. J Urol. 1989;142:1483-1486.
20 Ohl DA, Menge AC, Jarow JP. Seminal vesicle aspiration in spinal cord injured men: Insight into poor sperm quality. J Urol. 1999;162:2048-2051.
21 Sheynkin YR, Ye Z, Menendez S, et al. Controlled comparison of percutaneous and microsurgical sperm retrieval in men with obstructive azoospermia. Hum Reprod. 1998;13:3086-3089.
22 Schlegel PN, Palermo GD, Goldstein M, et al. Testicular sperm extraction with intracytoplasmic sperm injection for nonobstructive azoospermia. Urol. 1997;49:435-440.
23 Schlegel PN. Testicular sperm extraction: Microdissection improves sperm yield with minimal tissue excision. Hum Reprod. 1999;14:131-135.
24 Manning M, Junemann KP, Alken P. Decrease in testosterone blood concentrations after testicular sperm extraction for intracytoplasmic sperm injection in azoospermic men. Lancet. 1988;352:37.
25 Zhu JJ, Tsirigotis M, Pelekanos M, Craft IL. In vitro maturation of human testicular spermatozoa. Hum Reprod. 1996;111:231-232.
26 Sherman JK. Freezing and freeze-drying of human spermatozoa. Fertil Steril. 1953;14:49-64.
27 Tournaye H, Merdad T, Silber S. No differences in outcome after intracytoplasmic sperm injection with fresh or with frozen–thawed epididymal spermatozoa. Hum Reprod. 1999;14:90-95.
28 Veeck LL, Wortham JW, Witmyer J, et al. Maturation and fertilization of morphologically immature human oocytes in a program of in vitro fertilization. Fert Steril. 1983;39:594-602.
29 Prins GS, Wagner C, Weidel L, et al. Gonadotropins augment maturation and fertilization of human immature oocytes cultured in vitro. Fertil Steril. 1987;47:1035-1037.
30 Nagy ZP, Cecile J, Liu J, et al. Pregnancy and birth after intracytoplasmic sperm injection of in vitro matured germinal-vesicle stage oocytes: Case report. Fertil Steril. 1996;65:1047-1050.
31 Chian RC, Buckett WM, Too LL, Tan SL. Pregnancies resulting from in vitro matured oocytes retrieved from patients with polycystic ovary syndrome after priming with human chorionic gonadotropin. Fertil Steril. 1999;72:639-642.
32 Chian RC, Buckett WM, Tulandi T, Tan SL. Prospective randomized study of human chorionic gonadotrophin priming before immature oocyte retrieval from unstimulated women with polycystic ovarian syndrome. Hum Reprod. 2000;15:165-170.
33 Cha KY, Koo JJ, Ko JJ, et al. Pregnancy after in vitro fertilization of human follicular oocytes collected from nonstimulated cycles, their culture in vitro and their transfer in a donor oocyte program. Fertil Steril. 1991;55:109-113.
34 Trounson A, Wood C, Kausche A. In vitro maturation and the fertilization and developmental competence of oocytes recovered from untreated polycystic ovarian patients. Fertil Steril. 1994;62:353-362.
35 Russell JB, Knezevich KM, Fabian KF, Dickson JA. Unstimulated immature oocyte retrieval: Early versus midfollicular endometrial priming. Fertil Steril. 1997;67:616-620.
36 Atiee SH, Pool TB, Martin JE. A simple approach to intracytoplasmatic sperm injection. Fertil Steril. 1995;63:652-655.
37 Gardner DK, Lane M. Blastocyst transfer. Clin Obstet Gynecol. 2003;46:231-238.
38 Hogan B, Constantini F, Lacy E. Manipulating the mouse embryo: A laboratory manual. New York: Cold Spring Harbor Laboratory Press, 1986.
39 Yaron Y, Gamzu R, Malcov M. Genetic analysis of the embryo. In: Gardner DK, Weissman A, Howles CM, Shoham Z, editors. Textbook of Assisted Reproductive Techniques: Laboratory and Clinical Perspectives. New York: Taylor & Francis; 2004:319-332.
40 Wells D, Delhanty JD. Preimplantation genetic diagnosis: Applications for molecular medicine. Trends Mol Med. 2001;7:23-30.
41 Dyban A, Freidine M, Severova E, et al. Detection of aneuploidy in human oocytes and corresponding first polar bodies by FISH. J Assist Reprod Genet. 1996;13:73-78.
42 Verlinsky Y, Cieslak J, Lifches A, et al. Birth of healthy children following preimplantation diagnosis of common aneuploidies by polar FISH analysis. Fertil Steril. 1996;65:358-360.
43 Harper JC, Dawson K, Delhanty JDA, Winston RML. Use of fluorescent in situ hybridization (FISH) for the analysis of in vitro fertilization embryos: A diagnostic tool for the infertile couple. Hum Reprod. 1995;10:3255-3258.
44 Wilkinson DG. The theory and practice of in situ hybridization. In: Wilkinson DG, editor. In Situ Hybridization: A Practical Approach. New York: Oxford University Press; 1992:1-13.
45 Verlinsky Y, Kuliev A. Preimplantation diagnosis of common aneuploidies in infertile couples of advanced maternal age. Hum Reprod. 1996;10:2076-2077.
46 The Ethics Committee of the American Fertility Society. Ethical considerations of assisted reproductive technologies. The cryopreservation of pre-embryos. Fertil Steril. 1994;62:56s-59s.
47 Porcu E. Oocyte cryopreservation. In: Gardner DK, Weissman A, Howles CM, Shoham Z, editors. Textbook of Assisted Reproductive Techniques: Laboratory and Clinical Perspectives. 2nd ed. New York: Taylor & Francis; 2004:233-242.
48 Rall W, Fahy G. Ice-free cryopreservation of mouse embryos at −196°C by vitrification. Nature. 1985;313:573-575.
49 Liebermann J, Tucker MJ, Graham JR, et al. Blastocyst development after vitrification of multipronuclear zygotes using flexipet denuding pipette. Reprod Biomed Online. 2002;4:146-150.
[/level-membership-for-surgery-category][not-level-membership-for-surgery-category]
Chapter 39 Assisted Reproductive Technology: Laboratory Aspects
HUMAN SEMEN: PREPARATION AND EVALUATION
Semen Collection
Many clinicians limit male infertility evaluation to a single semen analysis if the initial sample is found to be normal. However, a thorough infertility evaluation requires analysis of two ejaculates collected as least 1 month apart because semen quality varies over time.1 If either one of the two ejaculates are abnormal, additional samples should be evaluated. Some clinicians recommend that all fertility patients undergo semen analysis once or twice yearly during the duration of their treatment. More frequent analysis may be indicated in postsurgical patients as a part of routine follow-up.
It is preferable for samples to be collected near the andrology assessment area. Rooms designated for production of semen samples should be located in a quiet part of the facility and doors should be labeled with DO NOT DISTURB signs. Reading materials and/or video aids should be available for those men requiring them.
Semen Parameters
Semen Volume
The suffix -spermia refers to the volume of seminal fluid. A man is aspermic if he produces no semen after orgasm, hypospermic if he produces less than 0.55 mL of ejaculate, and hyperspermic if ejaculate volume is above 6.0 mL. The most common cause of low ejaculate volume is incomplete collection. Any abnormalities or interruptions during collection should be noted. Sample loss often compounds interpretation because distinct sperm-rich and sperm-poor fractions can be emitted. Short duration of abstinence from ejaculation may also lead to low semen volume. Hyperspermia is a rare clinical entity with unknown significance. Although spermatozoa density may be decreased, spermatozoa motility is unchanged.2
Semen pH
The semen pH should be recorded because it may be a useful parameter for diagnosis of some conditions affecting semen quality. Normal semen is alkaline, with a pH between 7.2 and 8.0.3 Values up to 8.5 have been noted in otherwise normal samples. Measurement is most easily performed with pH paper.
Semen Liquefaction
Fresh semen quickly coagulates into a gelatinous mass after ejaculation before liquefying over 5 to 25 minutes at room temperature. Coagulation is due to coagulating proteins contributed by the seminal vesicles in the final portion of the ejaculate; secretions from the bulbourethral glands of Cowper and prostate present in the initial fraction of the ejaculate are responsible for liquefaction.4
An absence of coagulation may indicate absent or hypoplastic seminal vesicles, or ejaculatory duct obstruction. Conversely, prolonged liquefaction time may be due to a lack of prostate-supplied, liquefying proteases such as prostate-specific antigen, amylase, and plasminogen activators.5 Absence of liquefaction may cause infertility, although impaired liquefaction is frequently associated with normal fertility.
Spermatozoa Concentration
The Makler counting chamber (Sefi Medical Instruments, Haifa, Israel) is a specialized instrument designed for rapid semen analysis. Since its inception in 1980, the Makler counting chamber has gained wide acceptance and is used in many laboratories. The main advantage of the Makler counting chamber over the Neubauer hemocytometer is that except in cases of extremely high spermatozoa concentration, no dilution of the semen specimen is needed. Additionally, motility and forward progression may be assessed at the time of spermatozoa concentration determination. Although the Makler Chamber simplifies determination of spermatozoa concentration in semen analysis, the method lacks the accuracy of traditional hemocytometry and may be prone to errors.6
Spermatozoa Motility and Forward Progression
The World Health Organization (WHO) 1999 progression system classifies spermatozoa into one of four categories: A represents spermatozoa with rapid progressive motility; B, slow or sluggish progressive motility; C, nonprogressive motility; and D, no motility.7 The percentage of spermatozoa in each category is reported. A normal semen specimen contains at least 50% motile spermatozoa with the majority exhibiting good (modal progression class >2, WHO class A and B) forward progression. Athenozoospermia is defined as less than 50% of spermatozoa with good motility.
Spermatozoa Morphology
In 1986, Kruger and associates proposed a strict criteria for classification of normal and abnormal spermatozoa morphology.8 Objective measurements of individual spermatozoa components are included in this system, also known as the Tygerberg strict criteria classification system. According to this system, a spermatozoon is considered normal only if: (1) the head measures 5 to 6 microns in length and 2.5 to 3.5 microns in width; (2) acrosomal staining covers 40% to 70% of the anterior aspect of the sperm head; (3) the midpiece is 1.5 times as long as the head length and less than 1 micron in width; (4) the tail is approximately 45 microns long, uniform, uncoiled, and free from kinks; and (5) cytoplasmic droplets are no more than half the size of the spermatozoa head and occupy the midpiece only. Borderline forms are considered abnormal in this classification scheme.
In men with spermatozoa counts greater than 20 million/mL and motility greater than 30%, fertilization rates during IVF cycles were 91% for men with greater than 14% normal spermatozoa by Tygerberg strict criteria and 37% for men with less than 14% normal forms.8 This study established the reference value for normal morphology as 15% or greater by strict criteria.
In 1992, the WHO adopted a grading system based on the Tygerberg strict characterization of distinct spermatozoon components.3 Under these guidelines, a spermatozoon is considered normal if: (1) the head measures 4 to 5.5 microns in length and 2.5 to 3.5 microns in width; (2) acrosomal staining covers 40% to 70% of the anterior aspect of the sperm head; (3) there are no neck, midpiece, or tail defects; and (4) cytoplasmic droplets are less than one third the size of the head. Although these parameters appear quite similar to Tygerberg criteria, slightly looser standards increase the reference value for normal morphology to 30%. Use of both grading schemes varies from laboratory to laboratory.
Spermatozoa Viability
Another method for assessing spermatozoa viability is the hypo-osmotic swelling test. The test relies on the ability of live spermatozoa to tolerate moderate hypo-osmotic stress as evidenced by controlled swelling. Dead spermatozoa do not show discernible swelling when exposed to hypo-osmotic conditions, whereas viable spermatozoa exhibit a characteristic coiling pattern in the tail.9
Functional Spermatozoa Tests
Zona Pellucida Binding Assay
The zona pellucida binding assay is another adjunctive test of spermatozoa function. This test assesses the capacity of spermatozoa to bind to the outer surface of the zona pellucida, a prerequisite for binding and penetrating the egg. Microscopically, the zona pellucida is divided and each half is mixed with a sample of the patient’s spermatozoa or a sample of a fertile donor. Spermatozoa bound to the zona pellucida outer surface are counted. A hemizona index is calculated by dividing the total number of patient spermatozoa bound by the total number of donor spermatozoa bound. Successful IVF is associated with an index greater than 60%.10
Hamster Egg Penetration Assay
The zona pellucida, the acellular glycoprotein layer surrounding the ovum, regulates species-specific fertilization. Removal of this layer allows evaluation of interspecies, spermatozoa–oocyte interaction using the sperm penetration assay (SPA). This procedure assesses the ability of human spermatozoa to penetrate a hamster oocyte as a test of the fertilizing capability of the human spermatozoa.11 Human spermatozoa penetration of zona-free hamster oocytes has been shown to correlate with human spermatozoa penetration of human ova.12
Sperm Chromatin Structure Assay/Single-cell Gel Electrophoresis
Assays utilize direct staining and evaluation of spermatozoa chromatin to assess the integrity of the genetic material. The tests provide useful information on the presence of genetic damage to spermatozoa DNA. Briefly, both procedures evaluate shifts from intact, double-stranded DNA to denatured, single-stranded DNA.13 These assays can be used to assess male spermatozoa DNA integrity as related to fertility potential and embryo development as well as effects of reproductive toxins.14
Collection-Specific Spermatozoa Evaluation and Processing
Processing of Semen Collected by Masturbation
Spermatozoa Migration Techniques (Swim-up Method)
Spermatozoa washing of a sample before enrichment for motile spermatozoa (swim-up from pellet) has been criticized because dead and abnormal spermatozoa present in the original sample remain in the pellet. Aitken and Clarkson showed that the functional fertilizing capacity of motile spermatozoa isolated from such a pellet can be impaired when compared to that of motile spermatozoa isolated before spermatozoa washing and pelleting.15 The authors propose that the production of oxygen free radicals by abnormal spermatozoa in the pellet may result in cellular damage in motile spermatozoa, leading to abnormal spermatozoa function. Additionally, the centrifuged pellet has a relatively small interface with the overlying layer, making it difficult for motile spermatozoa in the bottom of the pellet to migrate into the media.
Processing of Spermatozoa Collected Following Retrograde Ejaculation
Viable spermatozoa may be retrieved from the urine of patients failing medical therapy.16 Because urine’s acidity makes it naturally toxic to spermatozoa, it is first suggested to alter the chemical milieu of the bladder to minimize its deleterious effects on spermatozoa quality. Alkalinization of the urine above a pH of 7.5 can be achieved by instructing the patient to ingest sodium bicarbonate for several days prior to collection. The patient empties the bladder before masturbation. Immediately after orgasm, the patient urinates into a sterile container, which is taken to the laboratory for spermatozoa extraction. Alternatively, for men unable to volitionally void, the bladder may be catheterized and washed with fresh, pH-adjusted, spermatozoa processing medium. After irrigation, a small volume of medium is left in the bladder. The patient masturbates and ejaculates, and the bladder is catheterized again to obtain fluid containing retrograde ejaculate, which is collected and transported to the laboratory for processing. Laboratory procedures begin with centrifugation and removal of urine from the spermatozoa. The resuspended pellet can then be processed as previously described for samples produced from masturbation.
Processing of Spermatozoa Collected Following Vibratory Stimulation/Electroejaculation
Failure of emission is termed anejaculation. Causes of anejaculation are similar to those of retrograde ejaculation, although spinal cord injury is a much more common cause. Because most of these patients do not respond to sympathomimetic therapy, they may require the use of external stimulatory devices to achieve emission. The most common methods of stimulation are penile vibratory stimulation and rectal electroejaculation. Penile vibratory stimulation entails the application of a penile vibrator to the glans penis, leading to a reflexive ejaculation. This technique induces ejaculation in approximately 70% of anejaculatory men with spinal cord injuries.17
Men with lesions above the level of T10 are more likely to respond than those with lesions below that level. Electroejaculation may be used in men who do not respond to vibratory stimulation. This procedure involves carefully inserting a probe into the rectum, followed by the intermittent application of electrical current to the periprostatic nerve plexus. The current stimulates the muscular tissue of the prostate, seminal vesicle, vas deferens, and epididymis to contract and propel semen into the urethra. It is successful in approximately 75% of patients.18,19 If antegrade ejaculation ensues, it may be collected from the urethra. If retrograde ejaculation occurs, then the urine is alkalinized beforehand and the bladder irrigated with medium and catheterized to remove any spermatozoa-bearing fluid.
Risks of electroejaculation include rectal thermal injury or perforation. The rectum should be examined with proctoscopy before and after the procedure. Life-threatening autonomic dysreflexia, or massive, uncoordinated sympathetic release, may occur in patients with spinal cord injuries above the level of T6. Blood pressure monitoring is imperative in this patient population. Semen obtained by vibratory stimulation and electroejaculation frequently has low volume, high spermatozoa concentration, and poor motility, likely owing to stasis within the genital tract.20 Again, processing of samples produced with vibratory stimulation or electroejaculation continues as in samples produced with masturbation.
