Chapter 51 Antiviral Agents
| Abbreviations | |
|---|---|
| AIDS | Acquired immunodeficiency syndrome |
| AZT | Zidovudine (formerly known as azidothymidine) |
| CMV | Cytomegalovirus |
| CNS | Central nervous system |
| CSF | Cerebrospinal fluid |
| DNA | Deoxyribonucleic acid |
| GI | Gastrointestinal |
| HAART | Highly active antiretroviral therapy |
| HIV | Human immunodeficiency virus |
| HPV | Human papilloma virus |
| HSV | Herpes simplex virus |
| IM | Intramuscular |
| IV | Intravenous |
| NNRTIs | Non-nucleoside reverse transcriptase inhibitors |
| NRTIs | Nucleoside reverse transcriptase inhibitors |
| RNA | Ribonucleic acid |
| RSV | Respiratory syncytial virus |
| SC | Subcutaneous |
Therapeutic Overview
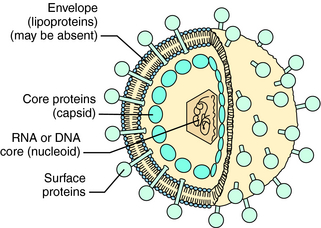
Viruses are responsible for significant morbidity and mortality in populations worldwide. These infectious agents consist of a core genome of nucleic acid (nucleoid) contained in a protein shell (capsid), which is sometimes surrounded by a lipoprotein membrane (envelope) (Fig. 51-1). Viruses cannot replicate independently. Rather, they must enter cells and use the energy-generating, deoxyribonucleic acid (DNA)-or ribonucleic acid (RNA)-replicating, and protein-synthesizing pathways of the host cell to replicate. Some viruses can integrate a copy of their genetic material into host chromosomes, achieving viral latency, in which clinical illness can recur without reexposure to the virus.
Some genera of viruses that cause human infections are listed in Table 51-1. Also listed is information about which genomic material—RNA or DNA—is present and examples of clinically important diseases attributed to each virus.
| Virus Genera or Groupings | Nucleic Acid | Clinical Examples of Illnesses |
|---|---|---|
| Adenovirus | DNA | Upper respiratory tract and eye infections |
| Hepadnaviridae | DNA | Hepatitis B, cancer |
| Herpesvirus | DNA | Genital herpes, varicella, meningoencephalitis, mononucleosis, retinitis |
| Papillomavirus | DNA | Papillomas (warts), cancer |
| Parvovirus | DNA | Erythema infectiosum |
| Arenavirus | RNA | Lymphocytic choriomeningitis |
| Bunyavirus | RNA | Encephalitis |
| Coronavirus | RNA | Upper respiratory tract infections |
| Influenzavirus | RNA | Influenza |
| Paramyxovirus | RNA | Measles, upper respiratory tract infections |
| Picornavirus | RNA | Poliomyelitis, diarrhea, upper respiratory tract infections |
| Retrovirus | RNA | Leukemia, AIDS |
| Rhabdovirus | RNA | Rabies |
| Togavirus | RNA | Rubella, yellow fever |
to allow detection of individual virus mutations, allowing physicians to predict viral susceptibility to many antiviral agents.
| Therapeutic Overview |
|---|
| Approaches to treatment of viral infections include: |
| Block viral attachment to cells |
| Block uncoating of virus |
| Inhibit viral DNA/RNA synthesis |
| Inhibit viral protein synthesis |
| Inhibit specific viral enzymes |
| Inhibit viral assembly |
| Inhibit viral release |
| Stimulate host immune system |
latency. Strains of viruses resistant to specific drugs can also develop.
Mechanisms of Action
Understanding the steps involved in virus infection and replication has led to the development of drugs that interfere with this process at various sites. This replication cycle and sites of action for the major classes of antiviral drugs are illustrated for the human immunodeficiency virus (HIV) in Figure 51-2.
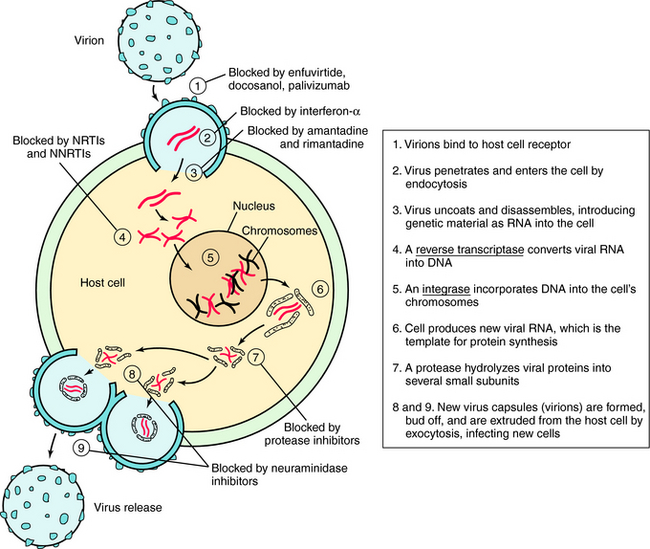
Inhibitors of Cell Penetration
HIV entry into cells is accomplished by a complex series of virus host interactions. Initially, virus approximates the CD4 cell by interactions of HIV surface protein and the host CD4 receptor. After approximation, host coreceptors interact with HIV surface proteins, resulting in folding of the HIV protein gp41. This folding results in fusion of the HIV membrane with the host cell membrane and insertion of the HIV nucleoid into the cell. Enfuvirtide prevents entry of HIV into cells by lying along the gp41 coils, causing steric hindrance of protein folding. Resistance occurs when mutations of gp41 occur that alter conformation and folding.
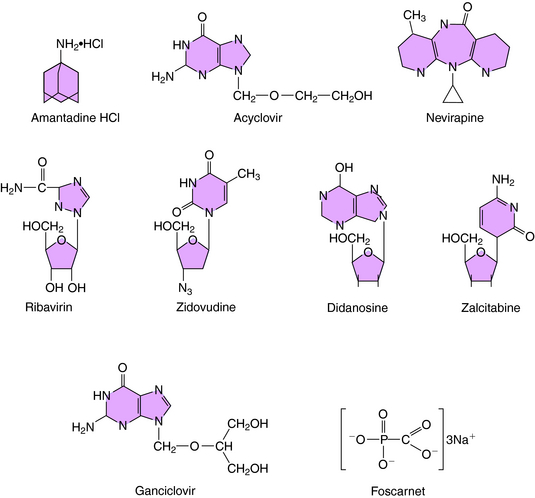
The structure of amantadine is shown in (Fig. 51-3). Its mechanism of action is not fully established but appears to involve blocking the ion channel activity of the M2 protein, thereby inhibiting late-stage uncoating of influenza A virions. This drug is not effective against influenza B, which lacks the M2 protein. A single amino acid change in the M2 protein results in amantadine resistance. Resistant virus is virulent and causes disease in exposed people. Rimantadine is a related compound with similar actions but an improved side effect profile.
Inhibitors of Viral DNA and RNA Synthesis
Acyclovir (see Fig. 51-3) is a synthetic guanosine analog and is the prototypical agent for this group of anti-HSV drugs. The group includes the related drugs valacyclovir and famciclovir, a prodrug for penciclovir. All of these drugs must be phosphorylated to be active and are initially monophosphorylated by viral thymidine kinase. Because the thymidine kinases of HSV types 1 and 2 are many times more active on acyclovir than host thymidine kinase, high concentrations of acyclovir monophosphate accumulate in infected cells. This is then further phosphorylated to the active compound acyclovir triphosphate. The triphosphate cannot cross cell membranes and accumulates further. The resulting concentration of acyclovir triphosphate is 50 to 100 times greater in infected cells than in uninfected cells.
The structure of ganciclovir is shown in Figure 51-3. It is also a synthetic guanosine analog active against many herpesviruses and must also be phosphorylated to be active. Infection-induced kinases, viral thymidine kinase, or deoxyguanosine kinase of various herpesviruses can catalyze this reaction. After monophosphorylation, cellular enzymes convert ganciclovir to the triphosphorylated form, and the triphosphate inhibits viral DNA polymerase rather than cellular DNA polymerase. Ganciclovir triphosphate competitively inhibits the incorporation of guanosine triphosphate into DNA. Because of its toxicity and the availability of acyclovir for treatment of many herpesvirus infections, its use is currently restricted to treatment of CMV retinitis.
Foscarnet (see Fig. 51-3) inhibits DNA polymerases, RNA polymerases, and reverse transcriptases. In vitro it is active against herpesviruses, influenza virus, and HIV. Foscarnet is used primarily in treatment of CMV retinitis. Viral resistance is attributable to structural alterations in CMV DNA polymerase. Foscarnet inhibits CMV herpesviruses that are resistant to acyclovir and ganciclovir.
Ribavirin is a synthetic purine nucleoside active against many viruses, including respiratory syncytial virus, Lassa fever virus, and influenza viruses (see Fig. 51-3). Ribavirin appears to be phosphorylated in host cells by host adenosine kinase. The 5’-monophosphate subsequently inhibits cellular inosine monophosphate formation, resulting in depletion of intracellular guanosine triphosphate. In some situations ribavirin triphosphate suppresses guanosine triphosphate-dependent capping of messenger RNA, thereby inhibiting viral protein synthesis. It also acts by suppressing the initiation or elongation of viral messenger RNA. Exogenous guanosine can reverse the antiviral effects of ribavirin with some viruses.
Nucleoside Reverse Transcriptase Inhibitors and Nucleotides
These drugs include zidovudine, didanosine, lamivudine, stavudine, and others (Box 51-1), and all work through a similar mechanism. Zidovudine (AZT) is the prototype for use in HIV infection. It is a thymidine analog that is phosphorylated to monophosphate, diphosphate, and triphosphate forms by cellular kinases in infected and uninfected cells. NRTIs have two primary methods of action. First, the triphosphate form acts as a competitive inhibitor of HIV reverse transcriptase. Second, after the nucleoside is incorporated into the elongating DNA chain, the forming sugar phosphate backbone of the DNA is blocked from further elongation by substitution at the 3 position. This results in chain termination. In the case of zidovudine, this substitution is an azido (N3) group. Zidovudine inhibits HIV reverse transcriptase at much lower concentrations than those needed to inhibit cellular DNA polymerases, leading to a more targeted effect against HIV.
Idoxuridine is an iodinated thymidine nucleoside analog that is incorporated into DNA in place of thymidine and blocks further DNA chain elongation. In vitro it inhibits many DNA viruses, and exogenous thymidine eliminates its antiviral effect. Idoxuridine affects mammalian cells, and its teratogenic, mutagenic, and immunosuppressive effects limit its use to topical preparations. It is used especially for the treatment of HSV infections of the cornea. Herpesviruses resistant to idoxuridine do occur and generally have decreased thymidine kinase activity.
Interferons are naturally occurring glycoproteins produced by lymphocytes, macrophages, fibroblasts, and other human cells (see Chapter 6). There are three distinct classes: α, β, and γ. They act as antiviral agents by inhibiting viral protein synthesis or assembly, or by stimulating the immune system. Interferons bind specific cell receptors and produce rapid changes in RNA. These effects may result in inhibition of viral penetration; uncoating, synthesis, or methylation of mRNA; translation of viral proteins; or assembly and release of virus. A 2’,5’-oligoadenylate synthetase and a protein kinase are usually produced that inhibit protein synthesis in the presence of double-stranded RNA. Interferons can also protect uninfected cells from infection by mechanisms that are as yet unclear. Interferons are used for the treatment of hepatitis B and C and papillomavirus infections.
Pharmacokinetics
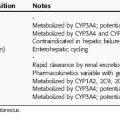
Because these compounds often interfere with human DNA or RNA synthesis, any antiviral agent should be used with the utmost caution in pregnancy and only when the potential benefits of treatment clearly outweigh the potential risks. The pharmacokinetic parameters for some antiviral drugs are listed in Table 51-2.
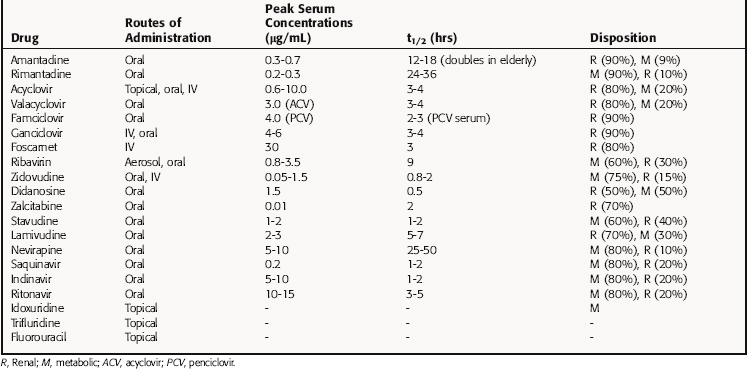
Amantadine is completely but slowly absorbed from the gastrointestinal (GI) tract, is 65% protein bound, and is distributed well throughout the body. Serum drug concentrations vary widely with the age of the patient, although peak drug concentrations occur 2 to 4 hours after ingestion. The plasma half-life of amantadine doubles in the elderly, necessitating dosage reductions. Amantadine is eliminated by kidney glomerular filtration and tubular secretion but is also metabolized to at least eight different compounds, although the biological activity of these metabolites is unknown.
Several protease inhibitors are now available for use. The absorption of these medications is usually improved with food; however, indinavir absorption is decreased with food. Peak serum concentrations of protease inhibitors are reached 1 to 3 hours after ingestion and are eliminated primarily by metabolism through cytochrome P4503A4. Inhibition of the P450 system can cause significant interactions with other medications. The elimination of protease inhibitors can be preferentially decreased by low-dose ritonavir, causing higher serum levels for longer periods of time. Lopinavir and ritonavir are available as a fixed-drug combination using this effect. Patients with liver disease who need protease inhibitor therapy need to be monitored carefully.
Because interferons are glycoproteins, their pharmacokinetics are difficult to assess. Simple detection of circulating interferon may not approximate clinical activity, because cellular binding is necessary, and an intact immune system is important to achieve a maximal response. Also, the biological activity may last days; even though the compound has been cleared from serum (see Chapter 6).
Relationship of Mechanisms of Action to Clinical Response
Systemic acyclovir is effective in reducing viral shedding, alleviating local symptoms, and decreasing the severity and duration of HSV infections. Recurrences after termination of therapy are common because of viral latency. Acyclovir decreases mortality in patients with herpes encephalitis to approximately 20%. Approximately 50% of acyclovir-treated patients return to normal life. High-dose acyclovir is needed to treat encephalitis to improve penetration across the blood-brain barrier. Acyclovir should be administered as soon as possible after encephalitis is diagnosed to lessen patient morbidity and mortality.
All three classes of human interferons (α, β, γ) are nonspecific immune stimulators that also have significant antiviral activity. As an antiviral agent, interferon is approved for the treatment of condyloma acuminata and chronic hepatitis B or C (often in combination with ribavirin). Adding polyethylene glycol to interferon has improved outcomes in chronic hepatitis C, presumably from greater sustained blood levels of interferon.
Some human immunoglobulins have high titers against specific viruses such as hepatitis B and rabies and are more efficacious against these viruses than nonspecific immunoglobulins. Some viral infections amenable to immunoglobulin therapy are listed in Box 51-2. Immunoglobulins are usually given IM, as close as possible to the time of exposure to the virus. In some circumstances an immunoglobulin should also be given very close to the lesion (as in rabies) to provide high concentrations to lymphatic tissues. In most situations IM injection provides systemic immunoglobulin concentrations adequate to prevent the development of clinical infection. However, because immunoglobulins do not confer long-term immunity, they must often be given in a series of injections, together with vaccine therapy.
Pharmacovigilance: Side Effects, Clinical Problems, and Toxicity
Amantadine also has anticholinergic properties that can cause urinary retention, ventricular arrhythmias, pupillary dilatation, and psychosis in some patients (see Chapter 28). The anticholinergic effects of amantadine are enhanced by antihistamines and anticholinergic drugs. Because its safety in pregnant and breast-feeding women is not established, caution should be exercised. Physostigmine given every 1 to 2 hours in adults may temporarily reverse serious neurological reactions. Overall, rimantadine is better tolerated than amantadine and does not appear to have significant anticholinergic effects.
Most clinical experience with ganciclovir has been gained in the treatment of CMV retinitis in AIDS patients, in whom the most common side effects are bone marrow suppression (up to 40%) and CNS abnormalities (up to 15%). Neutropenia and thrombocytopenia are the most common manifestations of bone marrow suppression, usually observed in the second week of therapy. Effects are usually reversible, but significant infections during granulocytopenia can occur. Concurrent use of NRTIs increases bone marrow toxicity. Approximately 33% of AIDS patients developed CNS or bone marrow toxicities significant enough to interrupt therapy. AIDS patients who have received long-term ganciclovir therapy have significant increases in their follicle-stimulating hormone, luteinizing hormone, and testosterone concentrations. Side effects of oral therapy are less frequent. Because of significant side effects, some patients with CMV retinitis receive intraocular ganciclovir implants. Ganciclovir has proved to be teratogenic and mutagenic in several different experimental systems.
Protease inhibitors are primarily metabolized by the P450 system and thus have the potential to alter the metabolism of other drugs. Ritonavir has the greatest capacity for drug-drug interactions, although this may occur with any protease inhibitor. Patients on protease inhibitor therapy need their medications routinely reviewed to check for significant drug-drug reactions. Lipodystrophy is common with all protease inhibitors, and glucose intolerance, dyslipidemia, may also occur. Patients on protease inhibitors have an increased risk of myocardial infarctions. Indinavir may precipitate in renal tubules and create clinically significant kidney stones. Adequate hydration is required for patients receiving indinavir. Other common side effects for protease inhibitors include GI upset and diarrhea.
| Amantadine | GI upset, CNS effects (nervousness, insomnia), anticholinergic effects |
| Rimantadine | GI upset, CNS effects |
| Acyclovir | CNS effects (nervousness) |
| Valacyclovir | Headache |
| Famciclovir | Decreased renal famciclovir function |
| Ganciclovir | Bone marrow suppression, CNS effects, rash, fever |
| Ribavirin | Headache, GI upset, dyspnea, teratogenic |
| Zidovudine | Bone marrow suppression, granulocytopenia, myositis |
| Didanosine | Pancreatitis, neuropathy |
| Zalcitabine | Neuropathy |
| Lamivudine | Bone marrow suppression, neuropathy, malaise |
| Stavudine | Neuropathy, GI upset |
| Nevirapine | Rash |
| Saquinavir | Drug-drug interactions |
| Indinavir | Renal stones, drug-drug interactions |
| Ritonavir | Drug-drug interactions |
New Horizons
in both HIV and other chronic viral infections such as hepatitis. The utility of using mismatched RNA to inhibit viral growth is also being evaluated (see Chapter 5). Management of CMV infections in the immunocompromised patient continues to be problematic, and agents more effective against this infection are being sought.
Anonymous. Drugs for HIV infection. Treat Guidel Med Lett. 2006;4:67-76.
Anonymous. Drugs for non-HIV viral infections. Treat Guidel Med Lett. 2007;5:59-70.
Hammer et al 2006 Hammer SM, Saag MS, Schechter M, et al. International AIDS Society, USA Panel. Treatment for adult HIV infection. 2006 Recommendations of the International Aids Society-USA Panel. http://www.iasusa.org/pub/arv_2006.pdf.
Herrick TM, Million RP. Tapping the potential of fixed dose combinations. Nat Rev Drug Discov. 2007;6:513-514.
Johnson et al 2007 Johnson VA, Brun-Vezinet F, Clotet B, et al. International AIDS Society USA Panel. Update of the Drug Resistance Mutations in HIV-1:2007. http://www.iasusa.org/resistance_mutations/mutations_figures.pdf