[level-membership-for-basic-science-category]
Chapter 26 Antithrombotic Drugs
| Abbreviations | |
|---|---|
| APTT | Activated partial thromboplastin time |
| cAMP | Cyclic adenosine monophosphate |
| INR | International normalized ratio |
| IV | Intravenous |
| PT | Prothrombin time |
| t-PA | Tissue plasminogen activator |
| TXA2 | Thromboxane A2 |
| t-PA | Tissue plasminogen activator |
| u-PA | Urokinase plasminogen activator |
| vWF | Von Willebrand factor |
Therapeutic Overview
| Therapeutic Overview | |
|---|---|
| Anticoagulation | |
| Heparin and heparin derivatives, coumarins, directly acting thrombin inhibitors | Arterial thrombosis, atrial fibrillation, cardiomyopathy, cerebral emboli, hip surgery, vascular prostheses, heart valve disease, venous thromboembolism |
| Fibrinolysis | |
| Streptokinase, urokinase, tissue plasminogen activator and its derivatives | Acute myocardial infarction, deep venous thrombosis Pulmonary embolism |
| Platelet Aggregation Inhibition | |
| Aspirin | Cerebrovascular accident, stroke, after coronary artery bypass surgery, coronary angioplasty/stenting or thrombolysis, myocardial infarction, transient ischemic attack |
| Clopidogrel | Coronary artery disease, cerebrovascular accident, stroke, peripheral arterial disease |
| Glycoprotein IIb/IIIa inhibitors | Acute coronary syndromes, after coronary artery stenting |
contact with foreign materials, initiate coagulation and thrombus formation. In these settings prophylactic administration of anticoagulants diminishes unwanted thrombus formation. In situations where a thrombus has already formed, such as deep vein thrombosis, acute myocardial infarction, and pulmonary embolism, rapid activation of the fibrinolytic system to lyse the thrombus and initiation of anticoagulation therapy to minimize further clot formation are effective. In cardiovascular disease and stroke, clinical evidence supports the use of drugs that inhibit platelet function.
Mechanisms of Action
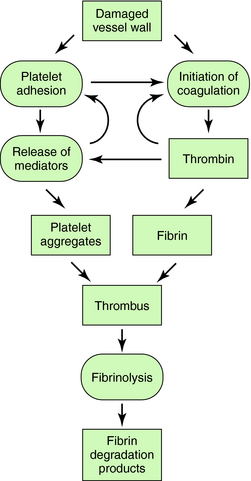
The interactions of the coagulation, fibrinolytic, and platelet systems are summarized in Figure 26-1. Endothelial cells in the blood vessel lumen normally present a nonthrombogenic surface. If the endothelium is damaged, blood comes into contact with thrombogenic substances within the subendothelium, such as collagen, which activates platelets, and tissue factor, which initiates blood coagulation. Foreign surfaces, such as prosthetic vascular grafts or mechanical cardiac valves, can also trigger clotting. Removal of thrombi by the fibrinolytic system depends on generation of plasmin from plasminogen by plasminogen activators.
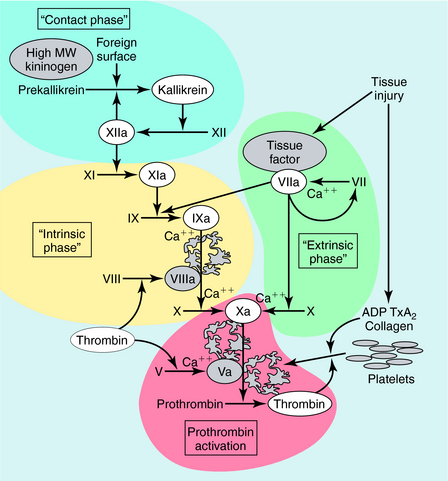
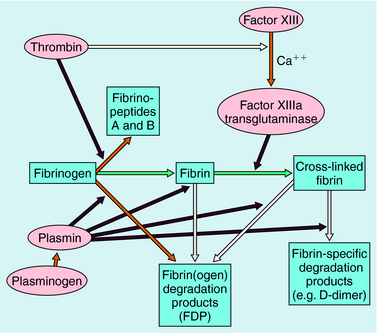
Blood coagulation occurs by sequential conversion of a series of inactive proteins into catalytically active proteases (Fig. 26-2). When the endothelium is damaged, blood comes into contact with cells that express tissue factor, a membrane-bound glycoprotein (the extrinsic pathway). A catalytically active complex of tissue factor and plasma factor VII is produced, which converts factor X to its enzymatically active form (Xa). In turn, factor Xa, in the presence of factor Va and a phospholipid surface (usually that of activated platelets), converts prothrombin to thrombin. Thrombin removes small peptides from fibrinogen (Fig. 26-3), converting it to fibrin monomer, which spontaneously polymerizes to form a clot. Fibrin is stabilized by factor XIIIa (transglutaminase), which introduces covalent bonds between fibrin molecules (see Fig. 26-3).
Most enzymes involved in coagulation are trypsin-like serine proteases with considerable homology. Plasma contains many inhibitors that regulate the coagulation cascade (Table 26-1). These proteins prevent inappropriate clotting and prevent appropriate, localized activation of the coagulation cascade from progressing to systemic coagulation.
TABLE 26–1 Plasma Protease Inhibitors That Regulate Blood Coagulation and Fibrinolysis
| Name | Principal Target |
|---|---|
| α1-Protease inhibitor | Elastase |
| α1-Antichymotrypsin | Cathepsin G1 |
| Antithrombin | Thrombin, Xa, IXa |
| α2-Macroglobulin | Plasmin, kallikrein, and other proteases |
| C1 inhibitor | Complement, XIIa |
| α2-Antiplasmin | Plasmin |
| Heparin cofactor II | Thrombin, Xa |
| Plasminogen activator inhibitor-1 (PAI-1) | t-PA, u-PA |
Parenteral Anticoagulant Drugs
Heparin is a linear polysaccharide with alternating residues of glucosamine and either glucuronic or iduronic acid (Fig. 26-4) derived from animal sources. The amino group of glucosamine is either acetylated or sulfated, and there is a variable degree of sulfation (≤40%) on the hydroxyl groups, rendering heparin a heterogeneous compound. Heparin acts by increasing the activity of antithrombin, a plasma glycoprotein that inhibits serine protease clotting enzymes. Heparin binds to antithrombin, causing a conformational change that renders the reactive site on antithrombin more accessible to serine proteases, inactivating thrombin and factors IXa and Xa; low molecular weight heparin inhibits mainly factor Xa. After the binding of antithrombin to thrombin, the heparin molecule is released and can bind to another antithrombin molecule. Although low doses of heparin act primarily by neutralizing factor Xa, at high doses it acts by preventing thrombin-induced platelet activation and prolongs bleeding time. Although the heparin-antithrombin complex is a very efficient inhibitor of free thrombin, clot-bound thrombin is resistant to inhibition.
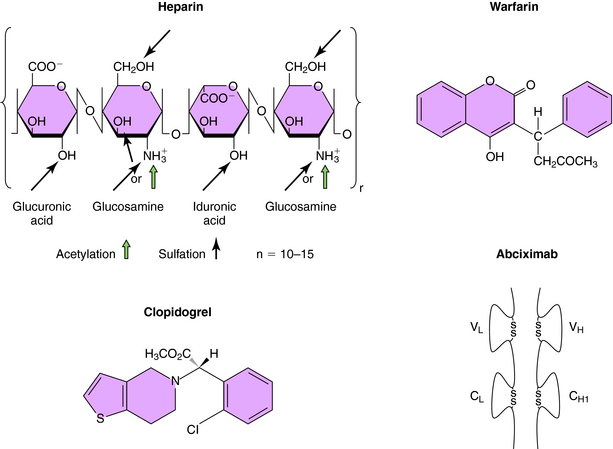
The oral anticoagulants, typified by warfarin and the coumarins (Fig. 26-4), represent a very important class of agents whose action involves their ability to inhibit vitamin K. A subset of blood coagulation factors (II, VII, IX, X) and anticoagulant proteins C and S are activated via the γ-carboxylation of several glutamic acid residues, which mediate their Ca++-dependent binding to phospholipid surfaces, critical for assembly of complexes necessary to generate thrombin. This activation requires vitamin K as a cofactor, and carboxylation of these vitamin K-dependent coagulation factors leads to the concomitant oxidation of vitamin K to its corresponding epoxide. The regeneration of vitamin K necessary to sustain the carboxylation reaction is mediated by vitamin K epoxide reductase, an enzyme inhibited by warfarin and the coumarins. Thus these compounds block recycling of the oxidized form of vitamin K to the reduced form required for cofactor function. Because these compounds inhibit the synthesis of clotting factors but have no direct effect on previously synthesized factors, plasma levels of preexisting vitamin K-dependent factors must decline before the anticoagulant effect of these agents becomes apparent, which requires several days. The first to decline is factor VII, followed by other factors with longer half-lives (see Table 26-2). The full anticoagulant effect of warfarin is typically reached within 4 to 7 days. Because of genetic variations in metabolism, drug interactions, and differences in vitamin K intake, significant variations between individuals exist in the time required for a maximal effect and in doses required for maintenance. Consequently, careful monitoring of prothrombin time (PT), a standard laboratory measure, is necessary.
TABLE 26–2 Rates of Disappearance (Half-Lives) of Vitamin K-dependent Proteins from Blood
| Protein | Time |
|---|---|
| Coagulation Factors | |
| Factor VII | 5 hours |
| Factor IX | 15 hours |
| Factor X | 1 day |
| Prothrombin | 2-3 days |
| Anticoagulant Proteins | |
| Protein C | 6 hours |
| Protein S | 10 hours |
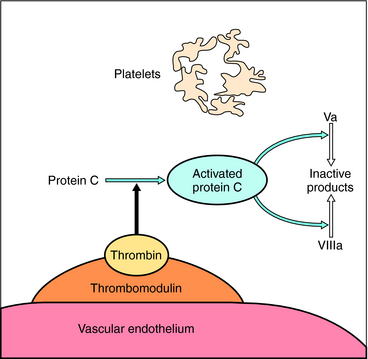
Proteins C and S, two other vitamin K-dependent factors, also inhibit excessive coagulation in the activated state. This mechanism involves the binding of thrombin to thrombomodulin, an endothelial cell surface protein, which results in a different proteolytic specificity than free thrombin. In this state thrombin does not cleave fibrinogen or activate platelets. Rather, it activates protein C, which in combination with protein S proteolytically inactivates clotting factors Va and VIIIa (Fig. 26-5), thereby providing a feedback inhibition to down regulate blood clotting after vascular injury. Genetic deficiency of protein C or protein S can cause thromboembolic disease.
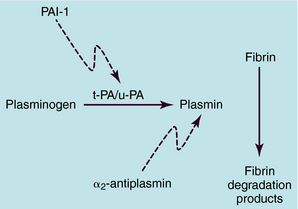
Fibrin clots are lysed mainly through the proteolytic action of plasmin, the enzyme produced by the proteolytic activation of plasminogen by plasminogen activators (Fig. 26-6). A minor aspect of clot lysis may result from release of proteolytic enzymes, such as elastase, from leukocytes. The two major classes of endogenous plasminogen activators are tissue plasminogen activator (t-PA) and urokinase plasminogen activator (u-PA). Recombinant forms of these proteins are used as clot-dissolving drugs.
The activation and subsequent aggregation of platelets is a major component of arterial thrombosis and may be involved in initiation of venous thrombosis. Interaction of platelets with vessel wall collagen appears to be a key step. Activation of platelets leads to formation and release of thromboxane A2 (TXA2) from arachidonic acid in platelet membranes (see Chapter 15). TXA2 is a potent aggregating agent and vasoconstrictor. Platelet activation also causes secretion of adenosine diphosphate from storage granules. Both TXA2 and adenosine diphosphate, which act through specific receptors, cause activation of integrin αIIbβIIIa receptors on the platelet surface for fibrinogen and for other adhesive proteins, including von Willebrand’s factor (vWF). Fibrinogen binding to its integrin receptor mediates aggregation, whereas vWF is involved primarily in adhesion of platelets to extracellular matrices in the vessel wall. Thrombin generated locally on the surface of activated platelets greatly amplifies the response by causing further activation and mediator secretion. Although TXA2, adenosine diphosphate, and thrombin all increase cytoplasmic Ca++, the mechanism by which thrombin activates its receptor is unique. Thrombin cleaves the N-terminal sequence of its platelet receptor (PAR1), forming a new N-terminus that serves as a “tethered ligand” and binds to and activates the receptor to induce transmembrane signaling (Fig. 26-7).
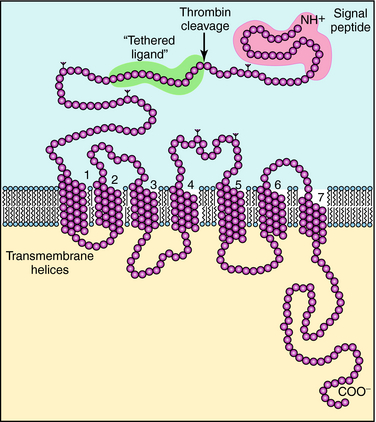
FIGURE 26–7 Predicted linear structure of the platelet thrombin receptor, protease activated receptor 1 (PAR1). It is thought to have a characteristic seven-transmembrane domain G-protein-coupled receptor structure (see Chapter 1). However, thrombin acts by cleaving the N-terminal extracellular tail of the receptor, releasing an activation peptide and leaving a new N-terminal sequence: Ser-Phe-Lys-Lys-Arg-. This sequence acts as a “tethered ligand,” which activates the receptor and is a powerful aggregating agent.
Platelet activation is inhibited by elevation of intracellular cyclic adenosine monophosphate (cAMP), and agents that increase this second messenger inhibit aggregation. The most active agent is prostacyclin, released by cells of the vessel wall (see Chapter 15). Other mediators from endothelial cells may also contribute.
The major therapeutic approach to reducing platelet aggregation is through inhibition of cyclooxygenase. Aspirin irreversibly inhibits platelet cyclooxygenase by acetylating a serine residue near the active site of the enzyme, thereby blocking TXA2 formation (see Chapter 36). Aspirin also blocks the synthesis of the endogenous vasodilator and platelet inhibitor prostacyclin, although at standard doses this prothrombotic effect is insignificant.
Clopidogrel (see Fig. 26-4) irreversibly blocks activation of the platelet P2Y receptor by adenosine diphosphate. Clopidogrel is a prodrug that is metabolized by cytochrome CYP3A to an active metabolite. Clopidogrel has become a mainstay in the treatment of patients with coronary artery disease, and it is also commonly administered to patients with peripheral arterial occlusive disease and cerebrovascular disease. It is routinely administered with aspirin, particularly to patients who have received coronary artery stents.
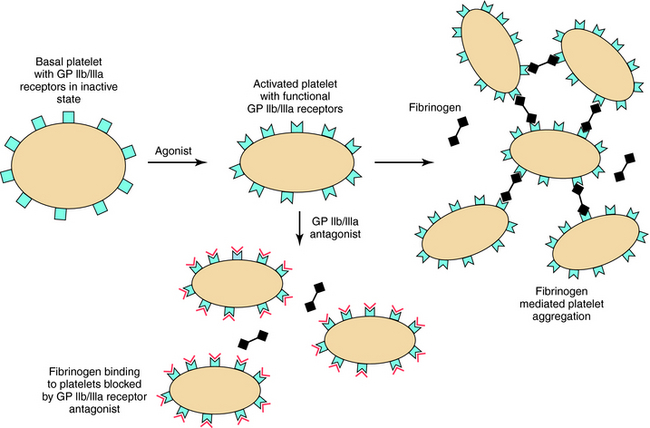
Inhibitors of glycoprotein IIb/IIIa (Fig. 26-8) bind to αIIbβIIIa, or glycoprotein IIb/IIIa, the platelet integrin receptor for fibrinogen, vWF, and other adhesive ligands. These inhibitors prevent fibrinogen cross-linking of platelets, which is the final common pathway of aggregation. These agents are administered by intravenous infusion, primarily to prevent platelet-dependent thrombosis during treatment of acute coronary artery syndromes and after implantation of intracoronary stents. Abciximab is the Fab fragment of a chimeric human-murine monoclonal antibody that binds to the glycoprotein IIb/IIIa receptor of human platelets (see Fig. 26-4). Abciximab also binds to the vitronectin receptor (αVβ3) present on platelets, vascular endothelial cells, and vascular smooth muscle cells. Platelet function gradually recovers after abciximab infusion is stopped. Plasma drug levels fall quickly, though platelet-bound abciximab can be detected for up to 15 days.
Pharmacokinetics
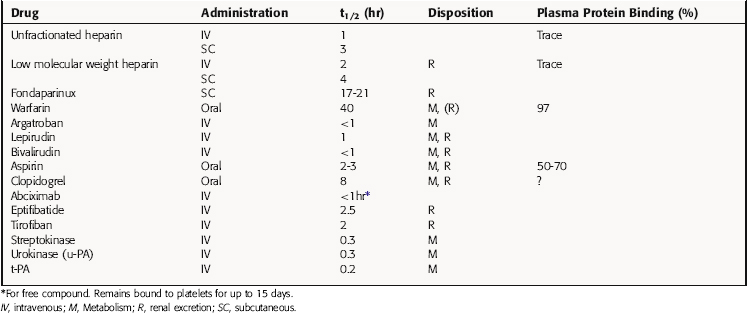
The principal pharmacokinetic parameters of the anticoagulants, fibrinolytics, and antiplatelet drugs are given in Table 26-3. Heparin is administered by IV infusion or subcutaneously. Because of different mechanisms of action, the anticoagulant effect of heparin is immediate, whereas that of warfarin typically occurs within 3 to 7 days. Heparin is not bound to plasma albumin, and the t1/2 of low molecular weight heparins is longer than that of the naturally occurring, unfractionated compound. Thus low molecular weight heparins are effective when administered by subcutaneous injection once or twice daily.
Aspirin is hydrolyzed in plasma to salicylic acid, with a t1/2 of 15 to 20 minutes, as discussed in Chapter 36. The antithrombotic effect of aspirin, however, persists for at least 2 days, because circulating platelets cannot synthesize cyclooxygenase. New platelets must be produced to restore TXA2 concentrations.
Relationship of Mechanisms of Action to Clinical Response
If a patient has an acute thrombus or is at high risk of forming one within the next few days, heparin is used, because its antithrombotic effect is immediate, whereas that of warfarin is delayed. If long-term anticoagulation is necessary, warfarin can be started as soon as therapeutic anticoagulation with heparin is achieved. After therapeutic anticoagulation with warfarin is achieved, heparin is often continued for 1 to 2 days. This overlap is commonly used because the antithrombotic effect of warfarin can lag behind laboratory measurements of warfarin anticoagulation, which is largely affected by a reduction in the level of factor VII, which has a short t1/2 (Table 26-3). If warfarin is started for prophylaxis of thrombosis, and the short-term risk is not high (e.g., a patient with atrial fibrillation being started on warfarin to lower stroke risk), heparin may be unnecessary. Most experts recommend that loading doses of warfarin should not be used, that is, the patient be started on the anticipated maintenance dose. During the first week of warfarin therapy, the coagulation response should be checked at least twice. Depending on the rapidity and stability of this measure, the time interval between subsequent determinations is gradually increased.
Clinical trials have shown that aspirin reduces the incidence of myocardial infarction and death from cardiac causes by 30% to 50% in patients with unstable angina. Aspirin also significantly reduces the incidence of a first myocardial infarction in men with stable angina and is effective as an antithrombotic agent after coronary angioplasty/stenting or bypass grafting. Aspirin is effective in secondary prevention of myocardial infarction. Aspirin is also recommended for use in patients with transient ischemic attacks. There are, of course, adverse effects of long-term aspirin therapy (see Chapter 36).
Pharmacovigilance: Side Effects, Clinical Problems, and Toxicity
incidence ranges from 0.3% to 3% in patients exposed to unfractionated heparin for greater than 4 days and is less common with low molecular weight heparins than with unfractionated heparin. It is associated with arterial or venous thrombosis in a small but significant subset of patients and occasionally can be extremely serious and even fatal. Platelet counts should be monitored at regular intervals in patients receiving heparin for prolonged periods. When heparin is discontinued, the platelet count usually returns to normal within 4 days.
Because warfarin treatment often extends over months or years, the possibility of drug-drug interactions is high because of a high degree of plasma protein binding and renal elimination. Common warfarin-drug interactions are listed in Box 26-1. Hereditary resistance to warfarin is rare but has been described; those affected require 5 to 20 times the average normal dose.
BOX 26–1 Drug Interactions with Warfarin
Decreased Anticoagulation
Martínez-Sánchez et al. 2007 Martínez-Sánchez P, Díez-Tejedor E, Fuentes B, et al. Systemic reperfusion therapy in acute ischemic stroke. Cerebrovasc Dis. 2007;24:143-152.
Mukherjee D, Eagle KA. The use of antithrombotics for acute coronary syndromes in the emergency department: Considerations and impact. Prog Cardiovasc Dis. 2007;50:167-180.
McRae SJ, Ginsberg JS. New anticoagulants for venous thromboembolic disease. Curr Opin Cardiol. 2005;20:502-508.
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
Chapter 26 Antithrombotic Drugs
| Abbreviations | |
|---|---|
| APTT | Activated partial thromboplastin time |
| cAMP | Cyclic adenosine monophosphate |
| INR | International normalized ratio |
| IV | Intravenous |
| PT | Prothrombin time |
| t-PA | Tissue plasminogen activator |
| TXA2 | Thromboxane A2 |
| t-PA | Tissue plasminogen activator |
| u-PA | Urokinase plasminogen activator |
| vWF | Von Willebrand factor |
Therapeutic Overview
| Therapeutic Overview | |
|---|---|
| Anticoagulation | |
| Heparin and heparin derivatives, coumarins, directly acting thrombin inhibitors | Arterial thrombosis, atrial fibrillation, cardiomyopathy, cerebral emboli, hip surgery, vascular prostheses, heart valve disease, venous thromboembolism |
| Fibrinolysis | |
| Streptokinase, urokinase, tissue plasminogen activator and its derivatives | Acute myocardial infarction, deep venous thrombosis Pulmonary embolism |
| Platelet Aggregation Inhibition | |
| Aspirin | Cerebrovascular accident, stroke, after coronary artery bypass surgery, coronary angioplasty/stenting or thrombolysis, myocardial infarction, transient ischemic attack |
| Clopidogrel | Coronary artery disease, cerebrovascular accident, stroke, peripheral arterial disease |
| Glycoprotein IIb/IIIa inhibitors | Acute coronary syndromes, after coronary artery stenting |
contact with foreign materials, initiate coagulation and thrombus formation. In these settings prophylactic administration of anticoagulants diminishes unwanted thrombus formation. In situations where a thrombus has already formed, such as deep vein thrombosis, acute myocardial infarction, and pulmonary embolism, rapid activation of the fibrinolytic system to lyse the thrombus and initiation of anticoagulation therapy to minimize further clot formation are effective. In cardiovascular disease and stroke, clinical evidence supports the use of drugs that inhibit platelet function.
Mechanisms of Action
The interactions of the coagulation, fibrinolytic, and platelet systems are summarized in Figure 26-1. Endothelial cells in the blood vessel lumen normally present a nonthrombogenic surface. If the endothelium is damaged, blood comes into contact with thrombogenic substances within the subendothelium, such as collagen, which activates platelets, and tissue factor, which initiates blood coagulation. Foreign surfaces, such as prosthetic vascular grafts or mechanical cardiac valves, can also trigger clotting. Removal of thrombi by the fibrinolytic system depends on generation of plasmin from plasminogen by plasminogen activators.
Blood coagulation occurs by sequential conversion of a series of inactive proteins into catalytically active proteases (Fig. 26-2). When the endothelium is damaged, blood comes into contact with cells that express tissue factor, a membrane-bound glycoprotein (the extrinsic pathway). A catalytically active complex of tissue factor and plasma factor VII is produced, which converts factor X to its enzymatically active form (Xa). In turn, factor Xa, in the presence of factor Va and a phospholipid surface (usually that of activated platelets), converts prothrombin to thrombin. Thrombin removes small peptides from fibrinogen (Fig. 26-3), converting it to fibrin monomer, which spontaneously polymerizes to form a clot. Fibrin is stabilized by factor XIIIa (transglutaminase), which introduces covalent bonds between fibrin molecules (see Fig. 26-3).
Most enzymes involved in coagulation are trypsin-like serine proteases with considerable homology. Plasma contains many inhibitors that regulate the coagulation cascade (Table 26-1). These proteins prevent inappropriate clotting and prevent appropriate, localized activation of the coagulation cascade from progressing to systemic coagulation.
TABLE 26–1 Plasma Protease Inhibitors That Regulate Blood Coagulation and Fibrinolysis
| Name | Principal Target |
|---|---|
| α1-Protease inhibitor | Elastase |
| α1-Antichymotrypsin | Cathepsin G1 |
| Antithrombin | Thrombin, Xa, IXa |
| α2-Macroglobulin | Plasmin, kallikrein, and other proteases |
| C1 inhibitor | Complement, XIIa |
| α2-Antiplasmin | Plasmin |
| Heparin cofactor II | Thrombin, Xa |
| Plasminogen activator inhibitor-1 (PAI-1) | t-PA, u-PA |
Parenteral Anticoagulant Drugs
Heparin is a linear polysaccharide with alternating residues of glucosamine and either glucuronic or iduronic acid (Fig. 26-4) derived from animal sources. The amino group of glucosamine is either acetylated or sulfated, and there is a variable degree of sulfation (≤40%) on the hydroxyl groups, rendering heparin a heterogeneous compound. Heparin acts by increasing the activity of antithrombin, a plasma glycoprotein that inhibits serine protease clotting enzymes. Heparin binds to antithrombin, causing a conformational change that renders the reactive site on antithrombin more accessible to serine proteases, inactivating thrombin and factors IXa and Xa; low molecular weight heparin inhibits mainly factor Xa. After the binding of antithrombin to thrombin, the heparin molecule is released and can bind to another antithrombin molecule. Although low doses of heparin act primarily by neutralizing factor Xa, at high doses it acts by preventing thrombin-induced platelet activation and prolongs bleeding time. Although the heparin-antithrombin complex is a very efficient inhibitor of free thrombin, clot-bound thrombin is resistant to inhibition.
The oral anticoagulants, typified by warfarin and the coumarins (Fig. 26-4), represent a very important class of agents whose action involves their ability to inhibit vitamin K. A subset of blood coagulation factors (II, VII, IX, X) and anticoagulant proteins C and S are activated via the γ-carboxylation of several glutamic acid residues, which mediate their Ca++-dependent binding to phospholipid surfaces, critical for assembly of complexes necessary to generate thrombin. This activation requires vitamin K as a cofactor, and carboxylation of these vitamin K-dependent coagulation factors leads to the concomitant oxidation of vitamin K to its corresponding epoxide. The regeneration of vitamin K necessary to sustain the carboxylation reaction is mediated by vitamin K epoxide reductase, an enzyme inhibited by warfarin and the coumarins. Thus these compounds block recycling of the oxidized form of vitamin K to the reduced form required for cofactor function. Because these compounds inhibit the synthesis of clotting factors but have no direct effect on previously synthesized factors, plasma levels of preexisting vitamin K-dependent factors must decline before the anticoagulant effect of these agents becomes apparent, which requires several days. The first to decline is factor VII, followed by other factors with longer half-lives (see Table 26-2). The full anticoagulant effect of warfarin is typically reached within 4 to 7 days. Because of genetic variations in metabolism, drug interactions, and differences in vitamin K intake, significant variations between individuals exist in the time required for a maximal effect and in doses required for maintenance. Consequently, careful monitoring of prothrombin time (PT), a standard laboratory measure, is necessary.
TABLE 26–2 Rates of Disappearance (Half-Lives) of Vitamin K-dependent Proteins from Blood
| Protein | Time |
|---|---|
| Coagulation Factors | |
| Factor VII | 5 hours |
