[level-membership-for-basic-science-category]
Chapter 20 Antihypertensive Drugs
| Abbreviations | |
|---|---|
| ACE | Angiotensin-converting enzyme |
| ARB | Angiotensin receptor blocker |
| AV | Atrioventricular |
| CNS | Central nervous system |
| CO | Cardiac output |
| Epi | Epinephrine |
| NE | Norepinephrine |
| TPR | Total peripheral resistance |
Therapeutic Overview
upper end of the normal distribution of blood pressure values for their population group. This most common form of hypertension, with no readily identifiable cause, is called essential hypertension. It is usually first diagnosed in middle-aged people but can also be found in children and young adults. Because of its prevalence, it is the disease most often treated with antihypertensive drugs.
Adoption of healthy lifestyles may lower blood pressure as much as some drugs. It may also prevent the onset or progression of hypertension. Patients differ in their sensitivity to these techniques. For example, maintenance of normal body weight and increased physical activity lowers blood pressure in most sedentary and overweight hypertensive individuals, whereas Na+ restriction lowers
| Therapeutic Overview |
|---|
| Hypertension is defined as: |
| Systolic pressure >140 mm Hg and/or diastolic pressure >90 mm Hg |
| Hypertension is a major risk factor for: |
| Atherosclerosis |
| Coronary artery disease |
| Congestive heart failure |
| Diabetes |
| Insulin resistance |
| Stroke |
| Renal disease |
| Retinal disease |
| Hypertension therapy |
| Nonpharmacological |
| Weight reduction, dietary (reduce salt and saturated fat, increase fruits and vegetables, use low-fat dairy products), exercise, smoking cessation, decrease excessive (>30 mL/day) alcohol intake |
| Pharmacological |
| Diuretics, renin-angiotensin inhibitors, sympatholytics, Ca++ channel blockers, direct vasodilators |
blood pressure mainly in hypertensive people categorized as “salt-sensitive.” The major advantage of nonpharmacological therapies is relative safety, as compared with drug therapy. Their principal limitation is the lack of compliance by most people. For most hypertensive patients control of hypertension requires drug treatment to achieve an adequate, sustained blood pressure reduction. Nevertheless, lifestyle modification plays a valuable and important role in management.
Mechanisms of Action
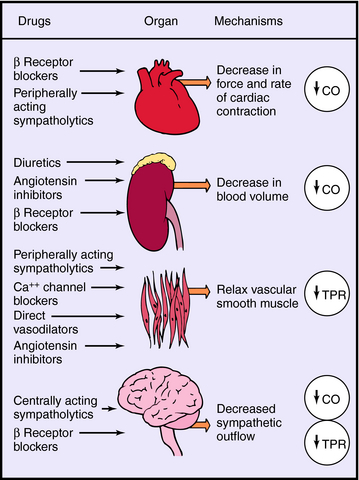
Systemic blood pressure is regulated redundantly by several physiological control systems to ensure optimal tissue perfusion throughout the body. When blood pressure decreases by any means, including antihypertensive drug therapy, one or more of these regulatory mechanisms are activated to compensate for decreases in arterial blood pressure (Fig. 20-1).
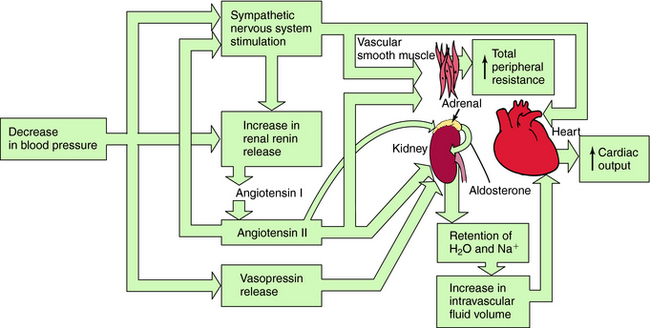
The Sympathetic Nervous System
A decrease in blood pressure activates the baroreceptor reflex (Chapter 19), producing increased sympathetic activity, leading to:
Renin-Angiotensin-Aldosterone System
A decrease in arterial pressure produces a decrease in renal perfusion pressure and baroreflex-mediated sympathetic activation of renal β1 adrenergic receptors, inducing the release of renin from the juxtaglomerular cells of the kidney into the blood. Renin cleaves the decapeptide angiotensin I from a circulating glycoprotein, angiotensinogen, which is synthesized mainly in liver. Angiotensin I is converted to the octapeptide angiotensin II by angiotensin–converting enzyme (ACE) present in endothelial cell membranes, especially in the lung. Angiotensin II constricts blood vessels, enhances sympathetic nervous system activity, and causes renal Na+ and H2O retention by direct intrarenal actions and by stimulating the adrenal cortex to release aldosterone (see Fig. 20-1).
Diuretics cause Na+ excretion and reduce fluid volume by inhibiting electrolyte transport in the renal tubules. The diuretics can be classified into three broad categories related to their sites and mechanisms of action (see Chapter 21). Thiazide diuretics inhibit the Na+/Cl− cotransporter principally in the distal convoluted tubules and produce a relatively sustained diuresis, natriuresis, and kaliuresis. These diuretics are most effective in patients with adequate renal function.
Drugs Affecting the Sympathetic Nervous System
Adrenergic Receptor Antagonists
There is wide diversity in the pharmacological profile of adrenergic β receptor antagonists (see Chapter 11). Some of these compounds, such as propranolol and pindolol, are nonselective and antagonize both β1 and β2 receptors, whereas others, like atenolol, are selective for the β1 receptor subtype. In addition, some β receptor blockers such as pindolol have modest intrinsic sympathomimetic activity, while others including labetalol and carvedilol are competitive antagonists at α1, β1, and β2 adrenergic receptors. Despite these differences, all β receptor blockers used for the treatment of hypertension share the common characteristic of competitively antagonizing the effects of norepinephrine (NE) and epinephrine (Epi) on β1 adrenergic receptors in the heart and renin-secreting cells of the kidney. Furthermore, clinically useful α1 adrenergic receptor antagonists lower blood pressure by blocking α1 receptors on vascular smooth muscle.
Centrally Acting Sympatholytics
The antihypertensive effects of the prodrug α-methyldopa are attributed to its conversion in the brain by l-aromatic amino acid decarboxylase and dopamine-β-hydroxylase to α-methyl-NE, which is a preferential agonist at α2 adrenergic receptors (see Chapter 11, Fig. 11-12). Clonidine, guanfacine, and guanabenz, which readily enter the brain after systemic administration, are selective agonists at central α2 receptors (see Chapter 11). In addition, because these drugs are taken orally for the treatment of hypertension, activation of presynaptic α2 adrenergic receptors on peripheral sympathetic nerve terminals may inhibit the release of NE and potentially contribute to their antihypertensive action.
The central site(s) where α2 receptor agonists act to lower blood pressure have not been completely identified and characterized but may include the nucleus of the solitary tract and the C1 neurons of the rostral ventrolateral medulla (see Chapter 19).
Voltage-dependent Ca++ channels play important roles in the excitation-contraction-relaxation cycle (see Chapter 22 and Chapter 24). Under resting conditions, when intracellular Ca++ is low, regulatory proteins prevent actin and myosin filaments from interacting with each other, and muscle is relaxed. When intracellular Ca++ concentrations increase by influx or release from internal stores, Ca++ occupies binding sites on Ca++-binding regulatory proteins, such as troponin C (in cardiac and skeletal muscle) and calmodulin (in vascular smooth muscle). These proteins then interact with other proteins and enzymes (e.g., troponin I in cardiac and skeletal muscle and myosin light-chain kinase in smooth muscle), facilitating cross-bridge formation between actin and myosin, which underlies contraction. When Ca++ channels inactivate, Ca++ is pumped out of the cell, activation of contractile proteins is reversed, actin dissociates from myosin, and the muscle relaxes.
The direct-acting vasodilators are among the most powerful drugs used to lower blood pressure and include hydralazine, minoxidil, diazoxide, nitroprusside, and fenoldopam. As indicated in Chapter 24, several purported mechanisms have been proposed to mediate the ability of hydralazine to dilate arterioles, whereas minoxidil and pinacidil bind to ATP-sensitive K+ channels, causing them to open. This allows K+ to partially equilibrate along its concentration gradient, which shifts the membrane potential toward the K+ hyperpolarizing reversal potential. The net effect is to reduce the probability of arterial smooth muscle depolarization and resultant contraction. Diazoxide produces arteriolar vasodilation and decreases peripheral vascular resistance through an action that may also involve K+ channels. Nitroprusside rapidly decomposes to release nitric oxide that activates guanylyl cyclase, which increases intracellular cGMP concentrations, particularly in veins (see Chapter 24). Fenoldopam is an agonist at D1 dopamine receptors with an action on renal arterioles, and trimethaphan has its vasodilatory effect by blocking autonomic ganglia.
The direct-acting vasodilators may produce marked compensatory reactions (see Fig. 20-1), including fluid retention and reflex-mediated increases in renin release, heart rate, and contractility. Because of their pronounced antihypertensive action and potential for producing these and other side effects (see Chapter 24), the direct-acting vasodilators are usually reserved for the treatment of hypertension that is severe or refractory to other drugs.
Pharmacokinetics
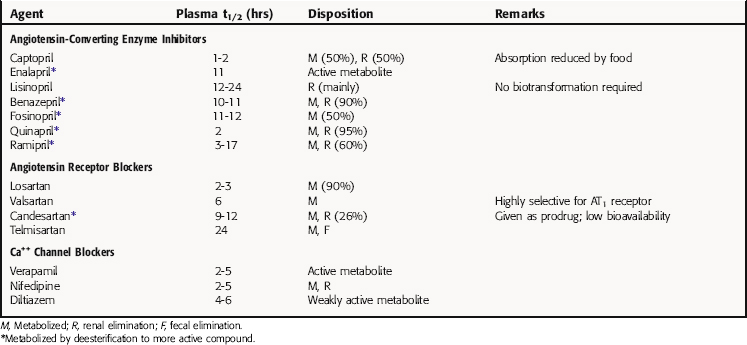
Pharmacokinetic parameters for ACE inhibitors, ARBs, and Ca++ channel blockers are summarized in Table 20-1. Pharmacokinetic parameters for the β receptor blocking drugs and sympatholytics are discussed in Chapter 11, for diuretics in Chapter 21, and for direct-acting vasodilators in Chapter 24.
Of the direct-acting vasodilator drugs used for the emergency reduction of hypertension, both nitroprusside and fenoldopam are administered by continuous intravenous infusion. Nitroprusside achieves full effects in seconds, and recovery takes place within a few minutes of terminating the infusion. Fenoldopam produces steady-state plasma levels proportionate to its rate of infusion, with an elimination t1/2 of approximately 5 minutes. Diazoxide is usually given in repeated low-dose intravenous injections, with the desired reduction in blood pressure occurring 1 to 5 minutes after dosing. The duration of effect varies from hours to a day. Trimethaphan must be administered by continuous intravenous infusion; a full response occurs in seconds and is greater when the patient is upright. It takes 10 to 60 minutes for blood pressure to recover after trimethaphan infusion.
Relationship of Mechanisms of Action to Clinical Response
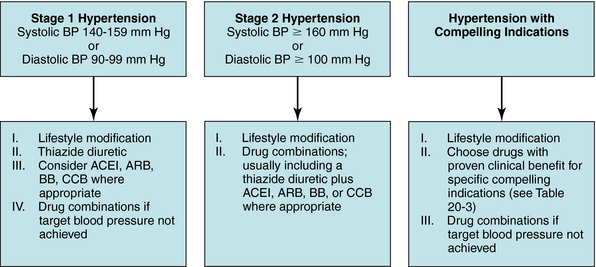
The seven classes of antihypertensives produce different physiological responses (Table 20-2) and have proven to be of clinical benefit, either alone or in combination, for several indications (Table 20-3). The choice of therapy for a patient with essential hypertension depends on the initial blood pressure of the individual, as well as age, race, family history of cardiovascular disease, and other risk factors such as smoking, obesity, and sedentary lifestyle. Most important is the presence of other conditions, such as kidney disease, ischemic heart disease, heart failure, previous myocardial infarction or stroke, or diabetes. Each of these must be given due consideration to determine an appropriate treatment plan.
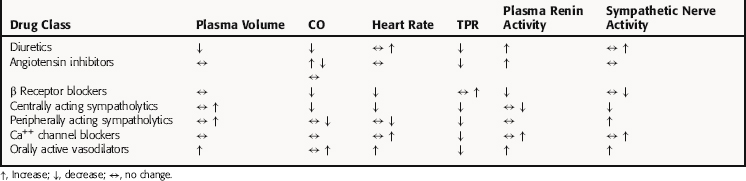
TABLE 20–3 Compelling Indications for Use of Individual Drug Classes Rights were not granted to include this table in electronic media. Please refer to the printed book.

Indications for which specific classes of antihypertensive drugs have proven clinical benefit. These drugs may be used alone or in combination with a thiazide diuretic. BB, β receptor blocker; ACEI, angiotensin-converting enzyme inhibitor; ARB, angiotensin receptor blocker; CCB, Ca++ channel blocker; Aldo ANT, aldosterone antagonist (see Chapter 23).
Modified from Chobanian AV, Bakris GL, Black HR, et al. Hypertension 2003; 42:1206-1252.
In general, more than 50% of patients require more than one drug to control their hypertension (Fig. 20-2). If two or more drugs are used, each should target a different physiological mechanism (Fig. 20-3). For example, it would be more beneficial to combine a diuretic with a vasodilator than to use two drugs that both reduce smooth muscle contraction. In most instances a diuretic should be included in any regimen using two or more antihypertensive drugs.
Although the renal targets of the diuretics are well known (see Chapter 21), the precise mechanism(s) responsible for their antihypertensive action are not as well understood. The decline in TPR may initially involve autoregulatory vascular adjustments in response to decreased perfusion, but this would not be expected to remain operative after CO is normalized. Other possible mechanisms include a decreased vascular reactivity to NE and other endogenous pressor substances, and a decreased “structural” vascular resistance caused by the removal of Na+ and H2O from the blood vessel wall. These changes could result directly from the actions of diuretic drugs or indirectly from the generalized loss of Na+ and H2O. The latter seems probable, because diuretics fail to lower blood pressure in patients who do not exhibit salt and H2O loss (i.e., nephrectomized patients on hemodialysis). However, the antihypertensive actions of diuretics do not parallel their efficacy in causing fluid loss, except in patients with renal insufficiency.
In vascular smooth muscle, there is some evidence that inhibition of vascular ACE activity correlates temporally with the hypotensive response to ACE inhibitors. In the kidney, angiotensin II can be produced locally by intrarenal renin and may exert an antinatriuretic and antidiuretic effect. Inhibition of intrarenal angiotensin II formation by ACE inhibitors could lower blood pressure by promoting salt and H2O excretion in a manner similar to that of diuretics. A third potential site is angiotensin II formed in brain. In experimental animals CNS administration of angiotensin II increases sympathetic nervous system activity and blood pressure. ACE inhibitors could decrease blood pressure in hypertensive individuals by reducing sympathetic nervous system activity in a manner similar to that of the centrally acting sympatholytics. ACE inhibitors are particularly useful for treating hypertension associated with other risk factors such as heart failure, postmyocardial infarction, diabetes, kidney disease, and stroke (see Table 20-3).
Verapamil was the first selective Ca++ channel inhibitor available for treatment of cardiovascular disorders, including hypertension. Like the dihydropyridines, it relaxes both coronary and peripheral arterioles. However, it has significantly more potent negative inotropic effects than the dihydropyridines or diltiazem. Verapamil can also depress AV nodal rate and conduction, and for this reason it can be used for the treatment of supraventricular tachycardia (see Chapter 22). It is also effective for the treatment of angina pectoris and hypertension. The reflex increase in adrenergic tone caused by a sudden decrease in blood pressure mitigates but does not overcome its strong direct negative inotropic and chronotropic effects. Because of its cardiodepressant effects, verapamil is generally contraindicated for the treatment of increased peripheral resistance associated with heart failure.
Dihydropyridines such as nifedipine are relatively selective arteriolar dilators. These drugs reduce peripheral resistance, arterial pressure, and afterload on the heart. These effects are larger in hypertensive than in normotensive individuals. However, if the drug has a relatively sudden onset of action, the decrease in blood pressure can produce reflex sympathoexcitation, tachycardia, and augmented cardiac contractility. These effects may be counteracted by the cardiodepressant action of some Ca++ channel blockers such as verapamil, but usually not by dihydropyridines, at doses typically used for treatment of hypertension. Nevertheless, because of the potential to exacerbate cardiac disease, slow-release, sustained-action formulations of dihydropyridine-type drugs are preferred for chronic therapeutic applications, such as the treatment of hypertension.
The orally active direct vasodilators, hydralazine and minoxidil, lower blood pressure by directly and preferentially relaxing arterial smooth muscle. Their selectivity for arterioles is greater than that of the Ca++ channel blockers (see Chapter 24).
Under some clinical circumstances, blood pressure must be reduced rapidly for a relatively short period of time (Box 20-1). Although several of the antihypertensive agents discussed can be administered parenterally for this purpose, the direct-acting vasodilators nitroprusside and diazoxide and the short-acting ganglionic blocker trimethaphan are used exclusively to rapidly reduce blood pressure. Diazoxide is used only for short-term treatment of severe hypertension because of its hyperglycemic properties.
Lower doses of diuretics are used to treat hypertension than to treat edema. Larger doses do not produce greater blood pressure reduction, but they significantly increase the incidence and severity of side effects, particularly decreased plasma K+ and increased uric acid concentrations. There is some concern that diuretic-induced hypokalemia may increase the incidence of sudden cardiac death. Diuretics may also impair glucose tolerance and increase serum lipid concentrations. Monitoring serum K+, using relatively low doses of thiazide diuretics and including a K+-sparing diuretic or adding K+ supplements in the drug regimen, should all be considered when thiazide or loop diuretics are used. In addition, K+ depletion can be reduced when ACE inhibitors or ARBs are used in combination with diuretics. Additional details on the side effects of diuretics are presented in Chapter 21.
Both ACE inhibitors and ARBs can produce hyperkalemia, particularly in patients with impaired renal function, or if used with K+-sparing diuretics or nonsteroidal anti-inflammatory drugs; hyperkalemia is uncommon with aliskiren. Glomerular filtration in some patients with renal artery stenosis may be dependent on the angiotensin-mediated constriction of the efferent glomerular arterioles. Inhibition of angiotensin function in these patients may precipitate renal failure.
Drugs Affecting the Sympathetic Nervous System
Side effects of β receptor blocker therapy are discussed in Chapter 11. It is preferable to use selective β1 receptor antagonists in patients with asthma or diabetes, but any β receptor antagonist should be used with some caution. Because β1 receptor blockade impairs sympathetic stimulation of the heart, β receptor blockers can impair exercise tolerance. Abrupt cessation of β receptor antagonists has been associated with tachycardia, angina pectoris, and (rarely) myocardial infarction.
Centrally acting sympatholytics are notable for causing less orthostatic hypotension than many other antihypertensives. They also do not impair renal function and thus are suitable for patients with renal insufficiency. The preferred drug for treatment of hypertension during pregnancy is α-methyldopa (see Chapter 11). A special problem associated with clonidine and other centrally acting α2 receptor agonists is a dramatic sympathetic hypertensive response that occurs in some patients after abrupt withdrawal of therapy. For this reason termination of these drugs should be done gradually, and clonidine-like drugs should be used cautiously, or not at all, in potentially noncompliant patients.
Because of the marked lowering of blood pressure produced by these drugs, fluid retention and reflex tachycardia are common. These compensatory reactions may be so pronounced that they mask part of the antihypertensive action of the direct vasodilators. For this reason a diuretic and a β receptor blocker are usually given with a vasodilator to offset these compensatory responses. Other side effects of the vasodilator drugs are discussed in Chapter 24.
The side effects of drugs used for hypertensive emergencies can be significant. The usual side effects of diazoxide are fluid retention, tachycardia, and hyperglycemia. Nitroprusside reacts with blood and tissue to release cyanide ions, which are converted to thiocyanate by the liver. Other side effects of nitroprusside are discussed in Chapter 24. Side effects of trimethaphan are those expected of ganglionic blockade and include mydriasis, cycloplegia, constipation, and urinary retention (see Chapter 10). Tachyphylaxis occurs a day or two after the hypotensive action of trimethaphan develops.
Although the goal of antihypertensive therapy is to reduce end-organ damage associated with chronically elevated blood pressure, the effects of therapy on other cardiovascular risk factors must also be considered. End-organ damage is not related exclusively to blood pressure. If an antihypertensive drug effectively lowers blood pressure but increases the influence of other risk factors for cardiovascular disease, the benefit of therapy may be reduced. In some studies thiazide diuretics did not decrease the incidence of coronary artery disease, despite their ability to significantly reduce blood pressure. This may be related to the modest elevation of low-density lipoprotein and total triglycerides produced by K+-losing diuretics, although the causative link or potential clinical significance of this finding has not been established. Other risk factors that can be affected by antihypertensive drugs include alterations in plasma glucose, K+, and uric acid concentrations. In particular, insulin resistance is now recognized to be prevalent in patients with hypertension. Elevated insulin is a risk factor for coronary artery disease. Thus it is noteworthy that thiazide diuretics and β receptor blockers increase, ACE inhibitors and prazosin decrease, and Ca++ channel blockers have no effect on insulin resistance. Because of wide interpatient variability in risk factors and disease, therapeutic generalizations are difficult, and antihypertensive drug therapy must be tailored to each patient.
(In addition to generic and fixed-combination preparations, the following trade-named materials are some of the important compounds available in the United States.*)
* With the exception of the combination products listed, the trade-named materials available for diuretics are presented in Chapter 21, β receptor blockers and sympatholytics in Chapter 11, angiotensin receptor blockers and aldosterone antagonists in Chapter 23, and vasodilators in Chapter 24
ALLHAT Collaborative Research Group. Major outcomes in high-risk hypertensive patients randomized to angiotensin-converting enzyme inhibitor or calcium channel blocker vs diuretic. The antihypertensive and lipid-lowering treatment to prevent heart attack trial (ALLHAT). JAMA. 2002;288:2981-2997.
Anonymous. Drugs for hypertension. Treat Guidel Med Lett. 2005;3:39-48.
Chobanian et al. 2003 Chobanian AV, Bakris GL, Black HR, et al. Seventh report of the Joint National Committee on prevention, detection, evaluation, and treatment of high blood pressure. Hypertension. 2003;42:1206-1252.
Kaplan NM. Kaplan’s clinical hypertension, 8. New York: Lippincott Williams & Wilkins, 2002.
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
Chapter 20 Antihypertensive Drugs
| Abbreviations | |
|---|---|
| ACE | Angiotensin-converting enzyme |
| ARB | Angiotensin receptor blocker |
| AV | Atrioventricular |
| CNS | Central nervous system |
| CO | Cardiac output |
| Epi | Epinephrine |
| NE | Norepinephrine |
| TPR | Total peripheral resistance |
Therapeutic Overview
upper end of the normal distribution of blood pressure values for their population group. This most common form of hypertension, with no readily identifiable cause, is called essential hypertension. It is usually first diagnosed in middle-aged people but can also be found in children and young adults. Because of its prevalence, it is the disease most often treated with antihypertensive drugs.
Adoption of healthy lifestyles may lower blood pressure as much as some drugs. It may also prevent the onset or progression of hypertension. Patients differ in their sensitivity to these techniques. For example, maintenance of normal body weight and increased physical activity lowers blood pressure in most sedentary and overweight hypertensive individuals, whereas Na+ restriction lowers
| Therapeutic Overview |
|---|
| Hypertension is defined as: |
| Systolic pressure >140 mm Hg and/or diastolic pressure >90 mm Hg |
| Hypertension is a major risk factor for: |
| Atherosclerosis |
| Coronary artery disease |
| Congestive heart failure |
| Diabetes |
| Insulin resistance |
| Stroke |
| Renal disease |
| Retinal disease |
| Hypertension therapy |
| Nonpharmacological |
| Weight reduction, dietary (reduce salt and saturated fat, increase fruits and vegetables, use low-fat dairy products), exercise, smoking cessation, decrease excessive (>30 mL/day) alcohol intake |
| Pharmacological |
| Diuretics, renin-angiotensin inhibitors, sympatholytics, Ca++ channel blockers, direct vasodilators |
blood pressure mainly in hypertensive people categorized as “salt-sensitive.” The major advantage of nonpharmacological therapies is relative safety, as compared with drug therapy. Their principal limitation is the lack of compliance by most people. For most hypertensive patients control of hypertension requires drug treatment to achieve an adequate, sustained blood pressure reduction. Nevertheless, lifestyle modification plays a valuable and important role in management.
Mechanisms of Action
Systemic blood pressure is regulated redundantly by several physiological control systems to ensure optimal tissue perfusion throughout the body. When blood pressure decreases by any means, including antihypertensive drug therapy, one or more of these regulatory mechanisms are activated to compensate for decreases in arterial blood pressure (Fig. 20-1).
The Sympathetic Nervous System
A decrease in blood pressure activates the baroreceptor reflex (Chapter 19), producing increased sympathetic activity, leading to:
Renin-Angiotensin-Aldosterone System
A decrease in arterial pressure produces a decrease in renal perfusion pressure and baroreflex-mediated sympathetic activation of renal β1 adrenergic receptors, inducing the release of renin from the juxtaglomerular cells of the kidney into the blood. Renin cleaves the decapeptide angiotensin I from a circulating glycoprotein, angiotensinogen, which is synthesized mainly in liver. Angiotensin I is converted to the octapeptide angiotensin II by angiotensin–converting enzyme (ACE) present in endothelial cell membranes, especially in the lung. Angiotensin II constricts blood vessels, enhances sympathetic nervous system activity, and causes renal Na+ and H2O retention by direct intrarenal actions and by stimulating the adrenal cortex to release aldosterone (see Fig. 20-1).
Diuretics cause Na+ excretion and reduce fluid volume by inhibiting electrolyte transport in the renal tubules. The diuretics can be classified into three broad categories related to their sites and mechanisms of action (see Chapter 21). Thiazide diuretics inhibit the Na+/Cl− cotransporter principally in the distal convoluted tubules and produce a relatively sustained diuresis, natriuresis, and kaliuresis. These diuretics are most effective in patients with adequate renal function.
Drugs Affecting the Sympathetic Nervous System
Adrenergic Receptor Antagonists
There is wide diversity in the pharmacological profile of adrenergic β receptor antagonists (see Chapter 11). Some of these compounds, such as propranolol and pindolol, are nonselective and antagonize both β1 and β2 receptors, whereas others, like atenolol, are selective for the β1 receptor subtype. In addition, some β receptor blockers such as pindolol have modest intrinsic sympathomimetic activity, while others including labetalol and carvedilol are competitive antagonists at α1, β1, and β2 adrenergic receptors. Despite these differences, all β receptor blockers used for the treatment of hypertension share the common characteristic of competitively antagonizing the effects of norepinephrine (NE) and epinephrine (Epi) on β1 adrenergic receptors in the heart and renin-secreting cells of the kidney. Furthermore, clinically useful α1 adrenergic receptor antagonists lower blood pressure by blocking α1 receptors on vascular smooth muscle.
Centrally Acting Sympatholytics
The antihypertensive effects of the prodrug α-methyldopa are attributed to its conversion in the brain by l-aromatic amino acid decarboxylase and dopamine-β-hydroxylase to α-methyl-NE, which is a preferential agonist at α2 adrenergic receptors (see Chapter 11, Fig. 11-12). Clonidine, guanfacine, and guanabenz, which readily enter the brain after systemic administration, are selective agonists at central α2 receptors (see Chapter 11). In addition, because these drugs are taken orally for the treatment of hypertension, activation of presynaptic α2 adrenergic receptors on peripheral sympathetic nerve terminals may inhibit the release of NE and potentially contribute to their antihypertensive action.
The central site(s) where α2 receptor agonists act to lower blood pressure have not been completely identified and characterized but may include the nucleus of the solitary tract and the C1 neurons of the rostral ventrolateral medulla (see Chapter 19).
Voltage-dependent Ca++ channels play important roles in the excitation-contraction-relaxation cycle (see Chapter 22 and Chapter 24). Under resting conditions, when intracellular Ca++ is low, regulatory proteins prevent actin and myosin filaments from interacting with each other, and muscle is relaxed. When intracellular Ca++ concentrations increase by influx or release from internal stores, Ca++ occupies binding sites on Ca++-binding regulatory proteins, such as troponin C (in cardiac and skeletal muscle) and calmodulin (in vascular smooth muscle). These proteins then interact with other proteins and enzymes (e.g., troponin I in cardiac and skeletal muscle and myosin light-chain kinase in smooth muscle), facilitating cross-bridge formation between actin and myosin, which underlies contraction. When Ca++ channels inactivate, Ca++ is pumped out of the cell, activation of contractile proteins is reversed, actin dissociates from myosin, and the muscle relaxes.
The direct-acting vasodilators are among the most powerful drugs used to lower blood pressure and include hydralazine, minoxidil, diazoxide, nitroprusside, and fenoldopam. As indicated in Chapter 24, several purported mechanisms have been proposed to mediate the ability of hydralazine to dilate arterioles, whereas minoxidil and pinacidil bind to ATP-sensitive K+ channels, causing them to open. This allows K+ to partially equilibrate along its concentration gradient, which shifts the membrane potential toward the K+
[/not-level-membership-for-basic-science-category]
