Chapter 50 Antifungal Agents
| Abbreviations | |
|---|---|
| ABLC | Amphotericin B lipid complex |
| AIDS | Acquired immunodeficiency syndrome |
| CSF | Cerebrospinal fluid |
| GI | Gastrointestinal |
| IV | Intravenous |
Therapeutic Overview
| Therapeutic Overview |
|---|
| Cutaneous and Subcutaneous Mycoses |
| Treat with dermatological preparations, occasionally systemic agents |
| Epidermophyton species |
| Microspora species |
| Sporothrix species |
| Trichophyton species |
| Systemic Mycoses |
| Difficult to treat; available drugs often cause deleterious side effects; often need long-term therapy |
| Aspergillus species |
| Candida species |
| Blastomyces dermatitidis |
| Cryptococcus neoformans |
| Coccidioides immitis |
| Fusarium species |
| Histoplasma capsulatum |
| Paracoccidioides brasiliensis |
| Mucormycosis |
broad-spectrum antibacterial agent use). Many fungal infections are superficial and primarily annoying. Others are systemic and can be life-threatening, particularly in patients with compromised defenses, such as those receiving immunosuppressive drugs. The toxicity of many antifungal drugs limits their use, and unfortunately there are few agents useful in treating systemic fungal infections.
Mechanisms of Action
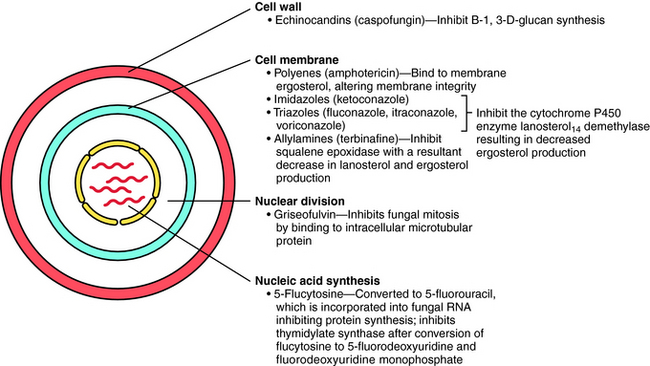
The principal antifungal drugs are the polyenes, azoles, allylamines, echinocandins, and others including flucytosine and griseofulvin. The sites of action of these agents are shown in Figure 50-1.
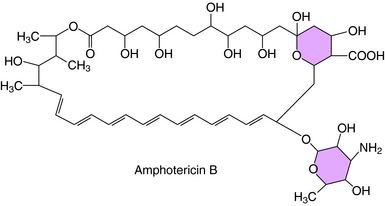
The polyene (i.e., multiple double bonds) drugs are macrocyclic lactones that contain a hydrophilic hydroxylated portion and a hydrophobic conjugated double bond portion. The structure of amphotericin B, the most widely used polyene antifungal drug, is shown in Figure 50-2.
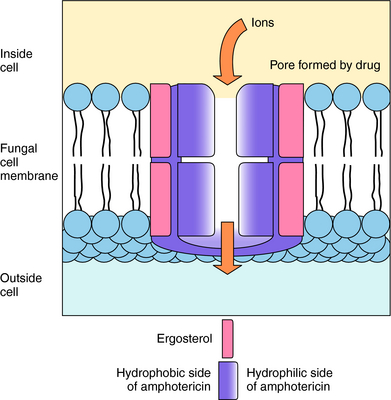
Polyenes act by binding to sterols in the cell membrane and forming channels, allowing K+ and Mg++ to leak out. The polyenes become integrated into the membrane to form a ring with a pore in the center approximately 0.8 nm in diameter. K+ leaks out through these pores, followed by Mg++, and with the loss of K+, cellular metabolism becomes deranged (Fig. 50-3). It is thought that derangement of the membrane alters activity of membrane enzymes. The principal sterol in fungal membranes, ergosterol, has a higher affinity for polyenes than does cholesterol, the principal sterol of mammalian cell membranes. Therefore the polyenes show greater activity against fungal cells than mammalian cells, and fungi that lack ergosterol are not susceptible to amphotericin B.
The structures of some of the principal azole antifungal agents are shown in Figure 50-4. Ketoconazole, miconazole, clotrimazole, and econazole are available, as are the newer agents fluconazole, itraconazole, and voriconazole.
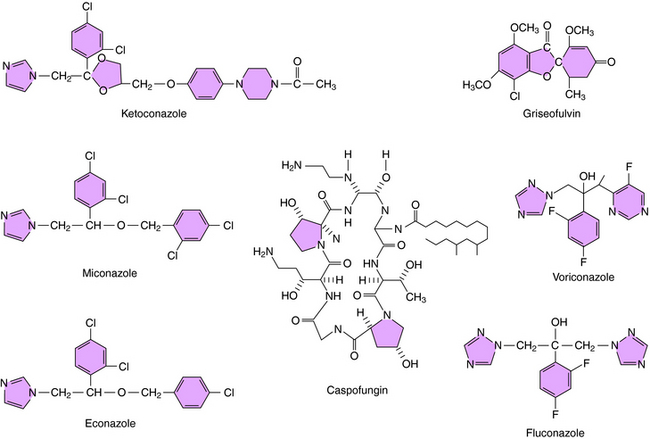
Caspofungin is the first echinocandin compound to gain approval for use in the United States. It blocks production of B-1,3-D-glucans, the major structural component of the fungal cell wall, by inhibition of glucan synthesis. Fungi have been shown to develop in vitro resistance to echinocandins through mutations in genes coding for the target enzymes.
Flucytosine is transported into susceptible fungi by a permease system for purines. The drug is then deaminated by cytosine deaminase to 5-fluorouracil. Because cytosine deaminase is not present in mammalian cells, the drug is not activated in humans. Fluorouracil in turn is converted by uridine phosphate pyrophosphorylase and other enzymes to 5-fluoro-2’-deoxyuridine 5’-monophosphate, which inhibits thymidylate synthase and interferes with deoxyribonucleic acid synthesis (Chapter 54).
Fungi can be resistant to flucytosine because they lack a permease, have a defective cytosine deaminase, or have a low concentration of the uridine monophosphate pyrophosphorylase. Whether the faulty ribonucleic acid produced by incorporation of fluorouracil contributes to its action is unclear.
Whether griseofulvin is fungicidal or fungistatic is not established. It enters susceptible fungi by an energy-dependent transport system and inhibits mitosis. It binds to the microtubules that form the mitotic spindle and blocks the polymerization of tubulin into microtubules. It also binds to a microtubule-associated protein, although the role of this protein is not known. The binding site for griseofulvin on tubulin differs from that of colchicine and the plant alkaloids. This effect on microtubule assembly probably explains the morphological changes, such as curling, that are observed in the fungi. Mechanisms of resistance are unknown, but may stem from decreased uptake of the drug. The structure of griseofulvin is shown in Figure 50-4.
Pharmacokinetics
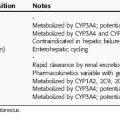
Pharmacokinetic parameters for the antifungal drugs are summarized in Table 50-1.
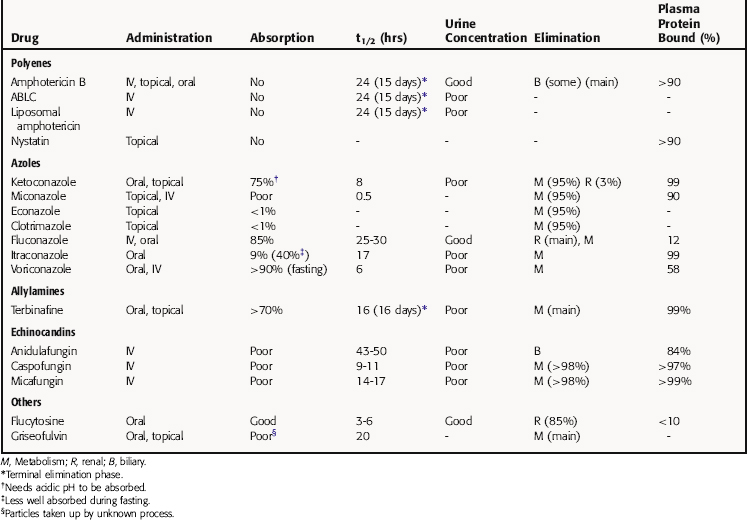
The principal pathway for amphotericin B elimination is not known. Some is excreted by the biliary route, and only 3% is eliminated in urine. Renal dysfunction does not affect plasma concentrations, and amphotericin B is not removed by hemodialysis.
Griseofulvin is widely distributed and becomes concentrated in fat, liver, and muscle. It is deposited in the keratin layer of the skin; becomes concentrated in keratin precursor cells in the stratum corneum of the skin, nails, and hair; and is secreted in perspiration. New keratin formed during treatment with griseofulvin is resistant to fungus, but griseofulvin does not destroy fungi in previously infected outer layers of skin. Thus a dermatophyte infection can be cured only when infected skin, nails, or hair is shed and the new keratin containing the griseofulvin replaces all the old keratin. Skin and hair infections require 4 to 6 weeks of therapy, fingernails require up to 6 months, and toenails require up to a year.
Relation of Mechanisms of Action to Clinical Response
Amphotericin was once the treatment of choice for most serious systemic mold infections and most endemic fungal infections because of its broad spectrum of activity, and it remains an important agent for most life-threatening mycoses, although newer, less toxic antifungals are beginning to be preferred over amphotericin. Amphotericin B inhibits most fungi listed in Table 50-2. Candida and Aspergillus are likely to be a cause of a systemic mycosis, as are Mucor, Rhizopus, and Absidia species, which are often present as opportunistic pathogens in debilitated patients. Amphotericin B also inhibits Sporothrix as well as some ameboflagellates and the freshwater ameba Acanthamoeba. It has variable activity against Trichosporon species, and treatment failures have been reported. A few fungal species, such as Pseudallescheria boydii and Fusarium species, show resistance.

Nystatin has a mode of action and antifungal spectrum of activity similar to those of amphotericin B. However, it is too toxic for parenteral administration and is only used topically to treat Candida infections of the skin and mucous membranes. It is effective for oral candidiasis, vaginal candidiasis, and Candida esophagitis. Although it is used prophylactically in neutropenic patients, it is ineffective except at very large daily doses.
Ketoconazole, the oldest of the azoles, inhibits most of the common dermatophytes and many of the fungi that cause the systemic mycoses listed in Table 50-2. Ketoconazole is effective for treatment of cutaneous mycoses and oral and esophageal candidiasis in immunocompromised patients. However, it is less effective than fluconazole and is not active against Aspergillus organisms or Phycomycetes, such as Mucor species. The membrane actions of ketoconazole also block the formation of branching hyphae, which may aid in white blood cell attack on the fungi. Because ketoconazole interferes with synthesis of ergosterol, it should not be used with amphotericin B because it would antagonize its effect. This has been demonstrated in vitro and in an animal model of Cryptococcus infection. As a result of its reduced selectivity for fungal P450 enzymes, ketoconazole can significantly inhibit the human P450 enzymes as well. Although the other azoles may also inhibit the cytochrome P450 system, they are not as potent as ketoconazole. For this reason, ketoconazole is rarely used systemically these days.
Griseofulvin inhibits dermatophytes of Microsporum, Trichophyton, and Epidermophyton species. It has no effect on filamentous fungi such as Aspergillus, yeasts such as Candida organisms, or dimorphoric species such as Histoplasma. It is used to treat only dermatophyte infections of the skin, nails, or hair. Mild infections can be effectively handled topically.
Pharmacovigilance: Side Effects, Clinical Problems, and Toxicity
Occasionally patients taking flucytosine experience nausea, vomiting, and diarrhea. Serious side effects are hematological and include anemia, leukopenia, and thrombocytopenia. Because this drug is usually coadministered with amphotericin B, there may be reduced renal clearance. Toxicity is attributable to the metabolite 5-fluorouracil. Some cases of transient hepatotoxicity have been reported.
| Drug | Adverse Effects |
|---|---|
| Polyenes | |
| Amphotericin B | Nephrotoxicity; fever, chills; phlebitis; hypokalemia; anemia; GI disturbance |
| Azoles | |
| Imidazoles | |
| Miconazole | Headache; pruritus; thrombophlebitis; hepatotoxicity; autoinduction of hepatic metabolizing enzymes |
| Ketoconazole | GI disturbance; hepatotoxicity, inhibition of hepatic P450 enzymes |
| Triazoles | |
| Itraconazole | GI disturbance; rare hepatotoxicity |
| Fluconazole | GI disturbance; rare hepatotoxicity; rare Stevens-Johnson syndrome |
| Voriconazole | Reversible photopsia, mild rash, Stevens-Johnson syndrome, toxic epidermal hepatotoxicity, necrolysis, visual hallucinations |
| Echinocandins | |
| Caspofungin, micafungin, anidulafungin | Fever, thrombophlebitis, rare hepatotoxicity |
| Others | |
| Flucytosine | Bone marrow suppression; hepatotoxicity; GI disturbance |
Johnson MD, Perfect JR. Caspofungin: First approved agent in a new class of antifungals. Expert Opin Pharmacother. 2003;4:1-17.
Menichetti F. Combining antifungal drugs: The future of antifungal therapy? Abstr Intersci Conf Antimicrob Agents Chemother. 2002;42:27-30. Sep
Steinbach WJ, Stevens DA. Review of newer antifungal and immunomodulatory strategies for invasive aspergillosis. Clin Infect Dis. 2003;37(suppl 3):S157-187.