CHAPTER 53 Antiepileptic Medications
Principles of Clinical Use
This chapter presents a systematic review of the clinical pharmacology of each of the main established AEDs and the newer AEDs, including their pharmacokinetics, interactions, dosages, efficacy profile, and safety profile. The pharmacokinetic properties, as well as doses and therapeutic ranges of the AEDs to be discussed, are summarized in Table 53-1. The principles of drug treatment of epilepsy are discussed, such as the decision to initiate long-term prophylactic drug treatment, the sequence of drug choices for various seizure types or syndromes (Table 53-2), initiation and monitoring of antiepileptic therapy, and discontinuation of treatment.
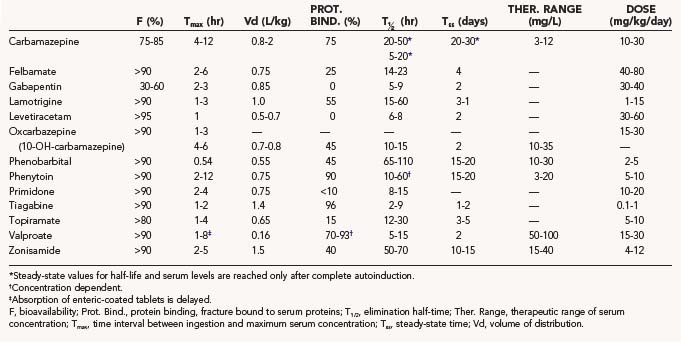
TABLE 53-2 Place of Newer Antiepileptic Drugs in the Treatment Sequence of Seizures and Epileptic Syndromes in Children
| Partial Seizures with or without Secondary Generalization | |
| First choice | Oxcarbazepine, carbamazepine, levetiracetam |
| Second choice | Lamotrigine, valproate, gabapentin |
| Third choice | Topiramate, zonisamide, phenytoin, phenobarbital, primidone |
| Consider | Pregabalin, tiagabine, benzodiazepine, acetazolamide |
| Generalized Tonic-Clonic Seizures | |
| First choice | Valproate, levetiracetam, lamotrigine |
| Second choice | (Ox)carbamazepine, topiramate, phenytoin |
| Third choice | Zonisamide, phenobarbital, primidone |
| Absence Seizures | |
| Before 10 years of age | |
| First choice | Ethosuximide, valproate |
| Second choice | Lamotrigine |
| Consider | Methsuximide, benzodiazepine, levetiracetam, topiramate, zonisamide, acetazolamide |
| After 10 years of age | |
| First choice | Valproate |
| Second choice | Lamotrigine |
| Third choice | Ethosuximide, methsuximide, levetiracetam, topiramate, zonisamide, benzodiazepine, acetazolamide |
| Juvenile Myoclonic Epilepsy | |
| First choice | Valproate |
| Second choice | Levetiracetam, lamotrigine, topiramate, clonazepam |
| Third choice | Zonisamide, phenobarbital, primidone |
| Lennox-Gastaut and Related Syndromes | |
| First choice | Topiramate, lamotrigine |
| Second choice | Valproate |
| Third choice | Ketogenic diet, felbamate, zonisamide, rufinamide, benzodiazepine, phenobarbital |
| Consider | Ethosuximide, methsuximide, levetiracetam, steroids |
| Infantile Spasms | |
| First choice | Adrenocorticotropic hormone, vigabatrin |
| Second choice | Valproate, topiramate, lamotrigine, zonisamide, benzodiazepine, ketogenic diet |
| Consider | Pyridoxine, levetiracetam, felbamate |
| Benign Epilepsy with Centrotemporal Spikes | |
| First choice | Sulthiame, gabapentin |
| Second choice | Valproate, levetiracetam |
| Consider | Lamotrigine, topiramate, zonisamide, pregabalin |
Clinical Pharmacology of Antiepileptic Drugs
Older Antiepileptic Drugs
Phenytoin
The use of phenytoin (PHT) has decreased, but it is still a widely used AED. In addition to its good efficacy against convulsive seizures and decades of experience with a good safety profile, PHT can be loaded rapidly without the need to titrate the dose slowly. This property is particularly valuable in neurosurgical practice. The volume of distribution of PHT is about 0.75 L/kg. A dose of 7.5 mg/kg given intravenously raises the level by 10 mg/L. Elimination of PHT is unique among AEDs because it is saturable at therapeutic concentrations1 in all age groups.2,3 This results in a nonlinear relationship between maintenance doses and steady-state concentrations. Especially in the upper therapeutic range, small increases in dosage can cause relatively large increases in levels. PHT does not have an elimination half-life because the time for the level to decrease by 50% becomes longer at higher levels. Steady-state levels are reached only after 2 to 3 weeks of a stable maintenance dose. To achieve average levels of about 15 mg/L, adult patients must usually take 5 to 6 mg/kg per day, which corresponds to 350 to 450 mg/day. The common dose of 300 mg/day often results in levels of 10 mg/L or less. PHT is a potent enzyme inducer and lowers the level of many other drugs. This affects other AEDs, such as carbamazepine, valproate, felbamate, lamotrigine, topiramate, zonisamide, and tiagabine, as well as many other drugs, including warfarin, oral contraceptives, and cyclosporine. PHT is highly protein bound and is displaced from serum proteins by valproate. Such displacement increases the free fraction of PHT and makes total serum levels unreliable.
The spectrum of activity of PHT includes partial seizures (simple or complex without or with secondary generalization), generalized convulsive seizures, status epilepticus,4 and neonatal seizures.5 Intravenous administration of PHT can cause bradyarrhythmia and hypotension, as well as skin necrosis.6 This local irritation in particular can be avoided by using the prodrug phosphenytoin instead of PHT for intravenous administration. The rate of administration of phosphenytoin is up to 150 mg/min (or 3 mg/kg per minute) instead of 50 mg/min (or 1 mg/kg per minute) for PHT.
The dose-related central nervous system side effects of PHT are nystagmus, ataxia, and lethargy. PHT can cause various forms of hypersensitivity reactions,7 as well as a hypersensitivity syndrome.8 Chronic or delayed adverse effects include gingival hyperplasia, hirsutism, peripheral neuropathy, and bone demineralization secondary to reduced vitamin D levels.
Carbamazepine
Carbamazepine (CBZ) is gradually being displaced from its exclusive place as the drug of first choice for partial and secondarily generalized seizures. The elimination kinetics of CBZ is linear.9 The characteristic feature of CBZ elimination is autoinduction of its metabolism,10 which results in an increase in CBZ clearance during the first weeks of treatment unless the patient already is taking another enzyme-inducing drug, such as phenytoin, phenobarbital, or primidone. Accordingly, the elimination half-life of CBZ decreases from about 36 hours to 10 to 20 hours.11 The practical consequence is that the dose of CBZ must be increased progressively during the first 3 to 4 weeks of treatment from 100 to 200 mg/day to 600 to 800 mg/day. Further increases in dosage may be needed and tolerated. Because of the relatively short half-life after induction, the regular CBZ preparations are best taken three times a day. Slow-release preparations are taken every 12 hours. CBZ is involved in many pharmacokinetic interactions. Similar to phenytoin, it is an enzyme inducer (see earlier). Other enzyme-inducing drugs, such as phenytoin, phenobarbital, and primidone, accelerate CBZ metabolism to a degree that exceeds CBZ autoinduction.
CBZ is effective against partial seizures without or with secondary generalization, as well as against generalized tonic-clonic seizures. Against partial and secondarily generalized seizures, phenytoin, CBZ, phenobarbital, and primidone are about equally effective in terms of seizure control.12 There is no parenteral preparation for CBZ. Dose-related central nervous system toxicity is the most common side effect. This toxicity may subside with time, can be minimized by careful titration, and is closely related to CBZ serum levels.13 Other side effects include neutropenia and rare, severe blood dyscrasias,14 hyponatremia, movement disorders, allergic rashes, and hypersensitivity syndrome.
Valproate
Valproate (VPA) is unique among the older AEDs because of its broad spectrum of activity against various seizure types. Absorption from enteric-coated VPA tablets can be delayed by several hours, but once it begins, it is rapid, as opposed to the slow release from extended-release tablets. Other oral preparations exhibit rapid and early absorption, except for enteric-coated sprinkles, which have an intermediate absorption pattern. VPA is highly bound to serum proteins and tends to displace other drugs, such as phenytoin. The elimination half-life of VPA varies as a function of comedication. In adults, the half-life is 13 to 16 hours in the absence of inducing drugs15 and 9 hours in induced patients.16 In addition to displacement from serum proteins, VPA is involved in two types of pharmacokinetic interactions: its metabolism is accelerated by inducing drugs such as phenytoin, carbamazepine, phenobarbital, and primidone, and VPA itself can prolong the elimination (and raise the levels) of other drugs, such as phenobarbital, ethosuximide, lamotrigine, and felbamate. The initial target dose of VPA is 15 mg/kg per day, which can be attained within a few days. Higher doses of 60 mg/kg per day or more may be necessary in certain patients, especially in children and those taking inducing drugs.
VPA has a broad spectrum of activity. In addition to being effective against partial seizures,17 VPA is highly effective against absence seizures, generalized tonic-clonic seizures, and myoclonic seizures. It is a drug of first choice in patients with primary (idiopathic) generalized epilepsies. It can be helpful in the treatment of infantile spasms18 and Lennox-Gastaut syndrome. VPA has several side effects that affect different systems and are of variable severity. Mild side effects include transient hair loss and dose-related tremor. VPA is not sedative, but drowsiness and lethargy may appear in some patients at levels around 100 mg/L, as well as idiosyncratic stuporous states at therapeutic levels.19 Gastrointestinal upset is less common with enteric-coated tablets. Fatal hepatotoxicity20 and pancreatitis21 are the most serious complications of VPA treatment. Thrombocytopenia, in conjunction with other VPA-mediated disturbances of hemostasis, such as impaired platelet function, fibrinogen depletion, and coagulation factor deficiencies,22 may cause excessive bleeding. The common practice of withdrawing VPA before elective surgery is recommended, although reports have found no objective evidence of excessive operative bleeding in neurosurgical patients maintained on VPA.23,24 In women of childbearing age, concerns associated with VPA treatment include not only an increased risk for neural tube defects in the fetus but also an increased risk for polycystic ovaries and metabolic and endocrine disturbances.25
Phenobarbital and Primidone
The use of phenobarbital (PB) and primidone (PRM) for the treatment of seizures has declined steadily because of their central nervous system side effects. PB and PRM produce more sedative and behavioral side effects than most other AEDs do, but they have relatively little systemic toxicity. PB has excellent pharmacokinetic properties, can be administered intravenously and intramuscularly, is effective in patients with status epilepticus, and is inexpensive. The volume of distribution of PB is 0.55 L/kg, with an elimination half-life averaging 80 to 100 hours in adults and newborns and shorter in infants and children. Maintenance doses range from 2 to 5 mg/kg per day. Although treatment with PRM results in the accumulation of significant levels of PB, PRM has independent pharmacologic activity and probably is not just a prodrug. PRM itself has a much shorter half-life than PB. Daily dosage requirements of PRM are about five times higher than those of PB. As an enzyme inducer, PB causes pharmacokinetic interactions that are shared by other enzyme-inducing drugs and by PRM because of the derived PB. Other enzyme-inducing drugs, in particular phenytoin,26 accelerate the conversion of PRM to PB, thereby increasing the PB-to-PRM serum level ratio.
PB and PRM are as effective against partial and secondarily generalized seizures as carbamazepine and phenytoin but were found to be associated with more treatment failures because of mostly early central nervous system side effects.12 PB can be used for the treatment of status epilepticus and neonatal seizures, as well as for the prophylaxis of febrile seizures. In addition to the well-known sedative and behavioral side effects, PB and PRM can cause allergic reactions. Use over many years may be associated with connective tissue disorders, such as Dupuytren’s contracture and frozen shoulder.
Newer Antiepileptic Drugs
Felbamate
Because of potentially serious side effects, felbamate (FBM) is currently used only in special circumstances. FBM is involved in multiple pharmacokinetic interactions; its levels are decreased by enzyme-inducing drugs, and it raises levels of phenytoin and valproate. The recommended initial dose of FBM is 1200 mg/day (15 mg/kg per day) during the first week. This dose can be doubled at the beginning of the second week and tripled at the beginning of the third week. It is prudent to reduce the dose of other AEDs by about a third when FBM is introduced. In double-blind studies, FBM was shown to be effective against partial onset seizures,27,28 as well as in the treatment of Lennox-Gastaut syndrome.29 Uncontrolled reports have suggested efficacy of FBM against absence seizures, juvenile myoclonic epilepsy, and infantile spasms.
The main common side effects of FBM have been nausea and vomiting, anorexia and weight loss, somnolence, and insomnia. Within 1 year after its marketing, it became evident that FBM was associated with a relatively high incidence of potentially fatal aplastic anemia30 and hepatic necrosis.31 Currently, the main indication for FBM is as a drug of third choice for the treatment of Lennox-Gastaut syndrome and focal onset seizures.
Gabapentin
Gabapentin (GBP) differs from previously used AEDs by the fact that it is eliminated entirely by the kidneys. As a consequence, it has no pharmacokinetic interactions. GBP absorption is saturable, and the daily dose should be divided into three or four fractions per day, especially with high doses.32 Initial target doses of GBP are about 1800 mg/day (30 mg/kg per day). Doses higher than 3600 mg/day (60 to 100 mg/kg per day) are often well tolerated and may be necessary to achieve the maximum benefit.
GBP has been shown in double-blind trials to be effective against focal onset seizures.33 It was found to be superior to placebo in a double-blind study in patients with benign epilepsy of childhood with centrotemporal spikes (rolandic epilepsy).34 Serious side effects of GBP appear to be exceedingly rare.35 However, excessive weight gain and behavioral problems in children are common.
Lamotrigine
Lamotrigine (LTG) has a relatively long half-life and can be administered two times a day. The pharmacokinetic interactions of LTG consist of a marked reduction of its levels by enzyme-inducing drugs and marked elevation of its levels by valproate.36 As a consequence of these interactions, dosage requirements for LTG vary from patient to patient. Patients taking valproate should receive 25 mg/day or less (0.2 mg/kg per day) during the first 2 weeks of treatment. Patients taking enzyme-inducing drugs without valproate may start with higher doses. Slow titration with increases in dosage at 2-week intervals is important because it seems to reduce the risk for the potentially severe rash, or even Stevens-Johnson syndrome, associated with LTG.37 Against focal onset seizures, LTG appears to be as effective as carbamazepine and phenytoin.38,39 LTG has also been found to be effective in the treatment of Lennox-Gastaut syndrome,40 absence seizures, generalized tonic-clonic seizures, and juvenile myoclonic epilepsy.
Topiramate
Topiramate (TPM) is also a broad-spectrum AED. It can be administered two times a day in most cases. The main pharmacokinetic interaction involving TPM consists of an approximately twofold increase in its clearance by enzyme-inducing AEDs.41 TPM can reduce the efficacy of oral contraceptives. To reduce the incidence of early side effects, the dose of TPM needs to be titrated slowly. The initial dosage should be 25 to 50 mg/day (0.5 to 1.0 mg/kg per day) with weekly increases by the same amount to achieve an initial target dose of 200 to 400 mg/day (5 to 6 mg/kg per day). The efficacy of TPM was found to be relatively high against focal onset seizures in an analysis comparing trials of several newer AEDs.42 TPM was also found to be effective in double-blind studies of patients with generalized tonic-clonic seizures43 and in patients with Lennox-Gastaut syndrome.44 The most common side effects of TPM include somnolence, impaired concentration, confusion, abnormal thinking, and impaired verbal memory. Other side effects include anorexia, weight loss, and nephrolithiasis, as well as metabolic acidosis and decreased sweating in children.
Tiagabine
Among the newer AEDs, tiagabine (TGB) has not found widespread use, mostly because of a narrow spectrum of activity and central nervous system side effects. TGB has a short half-life (see Table 53-1), which becomes shorter in the presence of enzyme-inducing drugs. There are no other pharmacokinetic interactions. When TGB is introduced, the dose should be titrated slowly with weekly increments.
Thus far, the known clinical efficacy of TGB is limited to partial onset seizures without or with secondary generalization.45 TGB does not appear to have severe or potentially life-threatening side effects. The main side effects include dizziness, tremor, difficulty with concentration, nervousness, and emotional lability.45
Oxcarbazepine
Oxcarbazepine (OXC) is a derivative of carbamazepine and shares most of the clinical characteristics of carbamazepine. OXC is a prodrug and is rapidly metabolized to the active compound monohydroxycarbamazepine, which is measured in serum for therapeutic monitoring.46,47 This metabolite has a half-life of 10 to 15 hours, and its level can be reduced by 30% to 40% in the presence of enzyme-inducing drugs such as phenytoin, carbamazepine, or phenobarbital. OXC is an enzyme inducer, but less so than phenytoin, phenobarbital, or carbamazepine. There is no parenteral preparation. The usual initial dose of OXC is 300 mg twice daily in adults and 5 to 10 mg/kg per day in children. The dose can be increased by that amount at weekly intervals to reach a target dose of 1200 to 1800 mg in adults and 20 to 30 mg/kg per day in children. A therapeutic range of 10 to 35 mg/L has been suggested.
OXC has the same narrow spectrum of efficacy as carbamazepine, with efficacy limited to partial onset and secondarily generalized seizures.48 The side effects of OXC are similar to those of carbamazepine, although they may be somewhat milder. They consist mostly of somnolence, dizziness, ataxia, diplopia, and blurred vision. An allergic rash can occur, and cross-reactivity with carbamazepine is at least 25%. Hyponatremia is more common with OXC than with carbamazepine and is more frequent in adults, especially in the elderly, than in children.49,50
Levetiracetam
Despite its relatively short half-life of 6 to 8 hours, levetiracetam (LEV) is usually administered twice daily. The pharmacokinetics are linear, protein binding is low, and no pharmacokinetic interactions caused by or involving LEV have been identified.51 The initial dose is 250 to 500 mg twice daily (500 to 1000 mg/day) in adults and 20 mg/kg per day in children. The maintenance dose is 1000 to 3000 mg/day in adults and 30 to 40 mg/kg per day in children. Higher doses may be needed and are tolerated. A therapeutic range of approximately 10 to 40 mg/L is probably appropriate in most patients, but levels of 40 to 80 mg/L may be beneficial and are well tolerated.
LEV has emerged as a broad-spectrum antiepileptic drug. It is approved for the treatment of partial and secondarily generalized seizures,52 primarily generalized tonic-clonic seizures in idiopathic general epilepsies,53 and myoclonic seizures in juvenile myoclonic epilepsy.54 LEV has also been used for absence seizures, severe myoclonic epilepsy in infancy, progressive myoclonic epilepsy (Unverricht-Lundborg), rolandic epilepsy,55 and posthypoxic and postencephalitic myoclonus. LEV has virtually no serious or life-threatening side effects. Its side effects may include somnolence, asthenia, dizziness, emotional lability, depression, and psychosis.56 Behavioral problems are particularly common in children and include agitation, hostility, oppositional behavior, anxiety, and aggression. Allergic reactions, liver failure, and bone marrow suppression are exceedingly rare.
Zonisamide
Zonisamide (ZNS) has a long half-life, about 60 hours in adults, and can be administered once or twice daily.57 Its pharmacokinetics is linear, and protein binding is 50% or less. Clearance of zonisamide is increased and levels of zonisamide are lowered by the addition of the following drugs (discontinuation of these drugs has an opposite effect): phenytoin, carbamazepine, phenobarbital, primidone, and valproic acid. ZNS has no known effect on the kinetics of other drugs. The initial dose is 100 mg/day in adults and 1.0 to 2.0 mg/kg per day in children. The initial target dose in adults is 100 to 600 mg/day (lower doses may be sufficient with monotherapy, and higher doses may be necessary with enzyme inducers). The initial target dose in children is 8 mg/kg per day with monotherapy and 12 mg/kg per day with enzyme inducers. Higher doses may be needed, particularly in infants and in patients comedicated with enzyme-inducing drugs.
ZNS is also a broad-spectrum drug.58 It can be effective against partial and secondarily generalized seizures,59 primarily generalized tonic-clonic seizures, Lennox-Gastaut syndrome, juvenile myoclonic epilepsy,60 absence seizures, infantile spasms,61 myoclonic astatic epilepsy (Doose’s syndrome), and progressive myoclonic epilepsy. Side effects of ZNS include drowsiness, fatigue, ataxia, psychomotor slowing, behavioral or psychiatric side effects, anorexia and weight loss, and allergic rash. Other possible side effects are metabolic acidosis (lowered serum bicarbonate or CO2, especially in children), hypohidrosis (decreased sweating, especially in children, may lead to hyperthermia),62 nephrolithiasis (1% to 2%), and paresthesias.
Pregabalin
Pregabalin (PGB) is very similar to gabapentin in most aspects, except for the fact that PGB has better bioavailability than gabapentin does.63 The elimination half-life of PGB is about 6 hours, which requires dosing twice or three times daily. PGB is not bound to serum proteins, is eliminated mostly unchanged in urine, and has no pharmacokinetic interactions. The usual dose in adults is 150 mg/day the first week, 300 mg/day the second week, 450 mg/day the third week, and 600 mg/day thereafter. A dosage schedule for children has not been established.
PGB, like gabapentin, has a narrow spectrum of activity that is limited to focal onset and secondarily generalized seizures.64 Its most common side effects are dizziness, somnolence, dry mouth, peripheral edema, blurred vision, excessive weight gain,65 and difficulty with concentration. PGB is a controlled substance because of a slight potential for recreational abuse and dependence.66
Principles of Treatment
Antiepileptic Drug Selection by Seizure Type or Epilepsy Syndrome
When a decision to treat has been made, the first step is to determine the drug of first choice for the patient. The choice of an AED is based first on the seizure type or on the epileptic syndrome. Among the drugs available for a particular seizure type or syndrome, the choice is based mainly on the adverse effect profile while taking into consideration the patient’s age and gender, as well preference. The place of any AED in the treatment sequence of epilepsy is not firmly established and not strictly scientifically determined because there have been no head-to-head comparisons of their efficacy against any given seizure type or epilepsy syndrome, especially for the newer drugs. With this concept in mind, Table 53-2 assigns places to AEDs in the treatment sequence of seizures. The listing of drugs as second and third choices applies only to patients whose seizures could not be controlled with a drug of first choice.
Discontinuation of Antiepileptic Drug Therapy
Similar to the initiation of therapy, discontinuation is based on a risk-versus-benefit analysis. Factors shown to increase the risk for seizure recurrence after stopping AED therapy include a known remote cause, seizure onset after the age of 12 years, a family history of epilepsy in patients with idiopathic epilepsy, focal or generalized slowing on EEG before discontinuation, a history of atypical febrile seizures, and an IQ of less than 50. The 2-year risk for recurrence after drug discontinuation may vary from about 10% in patients with none of these risk factors to about 80% in patients with remote symptomatic seizures and three risk factors. Contrary to widespread belief, seizure control can almost always be reestablished with medication if seizures recur after discontinuation. In general, the decision to discontinue an AED is made after 2 years without seizures. Because there is usually no urgency, the drug dosage should be tapered slowly over a period of at least 3 months. In patients who have become free of seizures after epilepsy surgery, it is a common practice to reduce the number of AEDs after 1 year and to discontinue all drugs after 2 years.67
Andermann F, Bourgeois BF, Leppik IE, et al. Postoperative pharmacotherapy and discontinuation of antiepileptic drugs. In: Engel J, editor. Surgical Treatment of the Epilepsies. New York: Raven Press; 1993:679-684.
Anderson GD, Lin YX, Berge C, et al. Absence of bleeding complications in patients undergoing cortical surgery while receiving valproate treatment. J Neurosurg. 1997;87:252-256.
Asconapé JJ, Penry JK, Dreifuss FE, et al. Valproate-associated pancreatitis. Epilepsia. 1993;34:177-183.
Ben-Menachem E. Pregabalin pharmacology and its relevance to clinical practice. Epilepsia. 2004;45(Suppl 6):13-18.
Berkovic SF, Knowlton RC, Leroy RF, et al. Placebo-controlled study of levetiracetam in idiopathic generalized epilepsy. for the Levetiracetam N01057 Study Group. Neurology. 2007;69;:1751-1760.
Brodie M, Richens A, Yuen A. Double-blind comparison of lamotrigine and carbamazepine in newly diagnosed epilepsy. Lancet. 1995;345:476-479.
Brodie MJ, Duncan R, Vespignani H, et al. Dose-dependent safety and efficacy of zonisamide: a randomized, double-blind, placebo-controlled study in patients with refractory partial seizures. Epilepsia. 2005;46:31-41.
Brodie MJ, Perucca E, Ryvlin P, et al. Comparison of levetiracetam and controlled-release carbamazepine in newly diagnosed epilepsy. for the Levetiracetam Monotherapy Study Group. Neurology. 2007;68;:402-408.
Bryant A, Dreifuss FE. Valproic acid hepatic fatalities: III. U.S. experience since 1986. Neurology. 1996;46:465-469.
Dam M, Ekberg R, Lyning Y, et al. A double-blind study comparing oxcarbazepine and carbamazepine in patients with newly diagnosed, previously untreated epilepsy. Epilepsy Res. 1989;3:70-76.
Guberman A, Besag F, Brodie M, et al. Lamotrigine-associated rash: risk/benefit considerations in adults and children. Epilepsia. 1998;40:985-996.
Leiderman D. Gabapentin as add-on therapy for refractory partial epilepsy: results of five placebo-controlled trials. Epilepsia. 1994;35:S74-S76.
Leppik IE. Practical prescribing and long-term efficacy and safety of zonisamide. Epilepsy Res. 2006;68S:S17-S24.
Marescaux C, Warter JM, Micheletti G, et al. Stuporous episodes during treatment with sodium valproate: report of seven cases. Epilepsia. 1982;23:297-305.
Mattson RH, Cramer JA, Collins JF. A comparison of valproate with carbamazepine for the treatment of complex partial seizures and secondarily generalized tonic-clonic seizures in adults. for the Department of VA Epilepsy Cooperative Study No. 264 Group. N Engl J Med. 1992;327;:765-771.
Mattson RH, Cramer JA, Collins JF, et al. Comparison of carbamazepine, phenobarbital, phenytoin and primidone in partial and secondarily generalized tonic-clonic seizures. N Engl J Med. 1985;313:145-151.
Nielsen OA, Johannessen AC, Bardrum B. Oxcarbazepine-induced hyponatremia, a cross-sectional study. Epilepsy Res. 1988;2:269-271.
O’Brien T, Cascino G, So E, et al. Incidence and clinical consequence of the purple glove syndrome in patients receiving intravenous phenytoin. Neurology. 1998;51:1034-1039.
Patsalos PN. Pharmacokinetic profile of levetiracetam: toward ideal characteristics. Pharmacol Ther. 2000;85:77-85.
Specchio LM, Gambardella A, Giallonardo AT, et al. Open label, long-term, pragmatic study on levetiracetam in the treatment of juvenile myoclonic epilepsy. Epilepsy Res. 2006;71:32-39.
Ward MM, Barbaro NM, Laxer KD, et al. Preoperative valproate administration does not increase blood loss during temporal lobectomy. Epilepsia. 1996;37:98-101.
1 Arnold K, Gerber N. The rate of decline of diphenylhydantoin in human plasma. Clin Pharmacol Ther. 1969;11:121-134.
2 Dodson WE. The nonlinear kinetics of phenytoin in children. Neurology. 1982;32:42-48.
3 Bourgeois BFD, Dodson WE. Phenytoin elimination in newborns. Neurology. 1983;33:173-178.
4 Cloyd J, Gumnit R, McLain W. Status epilepticus: the role of intravenous phenytoin. JAMA. 1980;244:1479-1481.
5 Painter MJ, Pippinger C, Wasterlain C, et al. Phenobarbital and phenytoin in neonatal seizures: metabolism and tissue distribution. Neurology. 1981;31:1107-1112.
6 O’Brien T, Cascino G, So E, et al. Incidence and clinical consequence of the purple glove syndrome in patients receiving intravenous phenytoin. Neurology. 1998;51:1034-1039.
7 Haruda F. Phenytoin hypersensitivity: 38 cases. Neurology. 1979;29:1480-1485.
8 Schlienger R, Shear N. Antiepileptic drug hypersensitivity syndrome. Epilepsia. 1998;39(Suppl 7):S3-S7.
9 Perucca E, Bittencourt P, Richens A. Effect of dose increments on serum carbamazepine concentration in epileptic patients. Clin Pharmacokinet. 1980;5:576-582.
10 Bertilsson L, Bengt H, Gunnel T, et al. Autoinduction of carbamazepine metabolism in children examined by a stable isotope technique. Clin Pharmacol Ther. 1980;27:83-88.
11 Eichelbaum M, Ekbom K, Bertilsson L, et al. Plasma kinetics of carbamazepine and its epoxide metabolite in man after single and multiple doses. Clin Pharmacol. 1975;8:337-341.
12 Mattson RH, Cramer JA, Collins JF, et al. Comparison of carbamazepine, phenobarbital, phenytoin and primidone in partial and secondarily generalized tonic-clonic seizures. N Engl J Med. 1985;313:145-151.
13 Hoppener R, Kuyer A, Meijer J, et al. Correlation between daily fluctuations of carbamazepine serum levels and intermittent side effects. Epilepsia. 1980;21:341-350.
14 Tohen M, Castillo J, Baldessarini R, et al. Blood dyscrasias with carbamazepine and valproate: A pharmacoepidemiological study of 2,228 patients at risk. Am J Psychiatry. 1995;152:413-418.
15 Gugler R, Schell A, Eichelbaum M, et al. Disposition of valproic acid in man. Eur J Clin Pharmacol. 1977;12:125-132.
16 Perucca E, Gatti G, Frigo GM, et al. Disposition of sodium valproate in epileptic patients. Br J Clin Pharmacol. 1978;5:495-499.
17 Mattson RH, Cramer JA, Collins JF. A comparison of valproate with carbamazepine for the treatment of complex partial seizures and secondarily generalized tonic-clonic seizures in adults. for the Department of VA Epilepsy Cooperative Study No. 264 Group. N Engl J Med. 1992;327;:765-771.
18 Bachman DS. Use of valproic acid in treatment of infantile spasms. Arch Neurol. 1982;39:49-52.
19 Marescaux C, Warter JM, Micheletti G, et al. Stuporous episodes during treatment with sodium valproate: report of seven cases. Epilepsia. 1982;23:297-305.
20 Bryant A, Dreifuss FE. Valproic acid hepatic fatalities: III. U.S. experience since 1986. Neurology. 1996;46:465-469.
21 Asconapé JJ, Penry JK, Dreifuss FE, et al. Valproate-associated pancreatitis. Epilepsia. 1993;34:177-183.
22 Gidal B, Spencer N, Maly M, et al. Valproate-mediated disturbances of hemostasis: relationship to dose and plasma concentration. Neurology. 1994;44:1418-1422.
23 Ward MM, Barbaro NM, Laxer KD, et al. Preoperative valproate administration does not increase blood loss during temporal lobectomy. Epilepsia. 1996;37:98-101.
24 Anderson GD, Lin YX, Berge C, et al. Absence of bleeding complications in patients undergoing cortical surgery while receiving valproate treatment. J Neurosurg. 1997;87:252-256.
25 Isojarvi JI, Laatikainen TJ, Knip M, et al. Obesity and endocrine disorders in women taking valproate for epilepsy. Ann Neurol. 1996;39:579-584.
26 Cloyd JC, Miller KW, Leppik IE. Primidone kinetics: effects of concurrent drugs and duration of therapy. Clin Pharmacol Ther. 1981;29:402-407.
27 Bourgeois BFD. Pharmacokinetics and pharmacodynamics in clinical practice. In: Wyllie E, editor. The Treatment of Epilepsy: Principles and Practice. Philadelphia: Lea & Febiger; 1993:726-734.
28 Faught E, Sachdeo RC, Remler MP, et al. Felbamate monotherapy for partial-onset seizures: an active-control trial. Neurology. 1993;43:688-692.
29 The Felbamate Study Group in Lennox-Gastaut Syndrome. Efficacy of felbamate in childhood epileptic encephalopathy (Lennox-Gastaut syndrome). N Engl J Med. 1993;328:29-33.
30 Kaufman D, Kelly J, Anderson T, et al. Evaluation of case reports of aplastic anemia among patients treated with felbamate. Epilepsia. 1997;38:1265-1269.
31 O’Neil M, Perdun C, Wilson M, et al. Felbamate-associated fatal acute hepatic necrosis. Neurology. 1996;46:1457-1459.
32 Gidal B, DeCerce J, Bockbrader H, et al. Gabapentin bioavailability: effect of dose and frequency of administration in adult patients with epilepsy. Epilepsy Res. 1998;31:91-99.
33 Leiderman D. Gabapentin as add-on therapy for refractory partial epilepsy: results of five placebo-controlled trials. Epilepsia. 1994;35:S74-S76.
34 Bourgeois B, Brown L, Pellock J, et al. Gabapentin (Neurontin) monotherapy in children with benign childhood epilepsy with centrotemporal spikes (BECTS): a 36-week, double-blind, placebo-controlled study. Epilepsia. 1998;39(Suppl 6):163.
35 McLean M, Morrell M, Willmore L, et al. Safety and tolerability of gabapentin as adjunctive therapy in a large, multicenter study. Epilepsia. 1998;40:965-972.
36 Vauzelle-Kervroedan F, Rey E, Cieuta C, et al. Influence of concurrent antiepileptic medication on the pharmacokinetics of lamotrigine as add-on therapy in epileptic children. Br J Clin Pharmacol. 1996;41:325-330.
37 Guberman A, Besag F, Brodie M, et al. Lamotrigine-associated rash: risk/benefit considerations in adults and children. Epilepsia. 1998;40:985-996.
38 Brodie M, Richens A, Yuen A. Double-blind comparison of lamotrigine and carbamazepine in newly diagnosed epilepsy. Lancet. 1995;345:476-479.
39 Steiner T, Dellaportas C, Findley L, et al. Lamotrigine monotherapy in newly diagnosed untreated epilepsy: a double-blind comparison with phenytoin. Epilepsia. 1999;40:601-607.
40 Motte J, Trevathan E, Arvidsson JF, et al. Lamotrigine for generalized seizures associated with the Lennox-Gastaut syndrome. N Engl J Med. 1997;337:1807-1812.
41 Bourgeois B. Drug interaction profile of topiramate. In: International League Against Epilepsy. Philadelphia: Lippincott-Raven; 1996.
42 Marson A, Kadir Z, Chadwick D. New antiepileptic drugs: a systematic review of their efficacy and tolerability. BMJ. 1996;313:1169-1174.
43 Biton V, Montouris G, Ritter F, et al. A randomized, placebo-controlled study of topiramate in primary generalized tonic-clonic seizures. Neurology. 1999;52:1330-1337.
44 Sachdeo R, Glauser T, Ritter F, et al. A double-blind, randomized trial of topiramate in Lennox-Gastaut syndrome. Neurology. 1999;52:1882-1887.
45 Uthman B, Rowan A, Ahmann P, et al. Tiagabine for complex partial seizures: A randomized, add-on, dose-response trial. Arch Neurol. 1998;55:56-62.
46 May TW, Korn-Merker E, Rambeck B. Clinical pharmacokinetics of oxcarbazepine. Clin Pharmacokinet. 2003;42:1023-1042.
47 127. Dickinson RG, Hooper WD, Dunstan PR, et al. First dose and steady-state pharmacokinetics of oxcarbazepine and its 10-hydroxy metabolite. Eur J Clin Pharmacol. 1989;37:69-74.
48 1Dam M, Ekberg R, Lyning Y, et al. A double-blind study comparing oxcarbazepine and carbamazepine in patients with newly diagnosed, previously untreated epilepsy. Epilepsy Res. 1989;3:70-76.
49 1Nielsen OA, Johannessen AC, Bardrum B. Oxcarbazepine-induced hyponatremia, a cross-sectional study. Epilepsy Res. 1988;2:269-271.
50 Borusiak P, Korn-Merker E, Holert N, et al. Hyponatremia induced by oxcarbazepine in children. Epilepsy Res. 1998;30:241-246.
51 Patsalos PN. Pharmacokinetic profile of levetiracetam: toward ideal characteristics. Pharmacol Ther. 2000;85:77-85.
52 Brodie MJ, Perucca E, Ryvlin P, et al. Comparison of levetiracetam and controlled-release carbamazepine in newly diagnosed epilepsy. for the Levetiracetam Monotherapy Study Group. Neurology. 2007;68;:402-408.
53 Berkovic SF, Knowlton RC, Leroy RF, et al. Placebo-controlled study of levetiracetam in idiopathic generalized epilepsy. for the Levetiracetam N01057 Study Group. Neurology. 2007;69;:1751-1760.
54 Specchio LM, Gambardella A, Giallonardo AT, et al. Open label, long-term, pragmatic study on levetiracetam in the treatment of juvenile myoclonic epilepsy. Epilepsy Res. 2006;71:32-39.
55 Verrotti A, Coppola G, Manco R, et al. Levetiracetam monotherapy for children and adolescents with benign rolandic seizures. Seizure. 2007;16:272-275.
56 Kossoff EH, Bergey GK, Freeman JM, et al. Levetiracetam psychosis in children with epilepsy. Epilepsia. 2001;42:1611-1613.
57 Mimaki T. Clinical pharmacology and therapeutic drug monitoring of zonisamide. Ther Drug Monitor. 1998;29:593-597.
58 Leppik IE. Practical prescribing and long-term efficacy and safety of zonisamide. Epilepsy Res. 2006;68S:S17-S24.
59 Brodie MJ, Duncan R, Vespignani H, et al. Dose-dependent safety and efficacy of zonisamide: a randomized, double-blind, placebo-controlled study in patients with refractory partial seizures. Epilepsia. 2005;46:31-41.
60 Kothare SV, Valencia I, Khurana DS, et al. Efficacy and tolerability of zonisamide in juvenile myoclonic epilepsy. Epileptic Disord. 2004;6:267-270.
61 Yanagaki S, Oguni H, Yoshii K, et al. Zonisamide for West syndrome: a comparison of clinical responses among different titration rate. Brain Dev. 2005;27:286-290.
62 Knudsen JF, Thambi LR, Kapcala LP, et al. Oligohidrosis and fever in pediatric patients treated with zonisamide. Pediatr Neurol. 2003;28:184-189.
63 Ben-Menachem E. Pregabalin pharmacology and its relevance to clinical practice. Epilepsia. 2004;45(Suppl 6):13-18.
64 Beydoun A, Nasreddine W, Atweh S. Efficacy and tolerability of pregabalin in partial epilepsy. Expert Rev Neurother. 2008;8:1013-1024.
65 Hoppe C, Rademacher M, Hoffmann JM, et al. Bodyweight gain under pregabalin therapy in epilepsy: mitigation by counseling patients? Seizure. 2008;17:327-332.
66 Drug Enforcement Administration, Department of Justice. Schedules of controlled substances: placement of pregabalin into schedule V. Final rule. Fed Reg. 2005;70:43633-43635.
67 Andermann F, Bourgeois BF, Leppik IE, et al. Postoperative pharmacotherapy and discontinuation of antiepileptic drugs. In: Engel J, editor. Surgical Treatment of the Epilepsies. New York: Raven Press; 1993:679-684.