[level-membership-for-endocrinology-diabetes-and-metabolism-category]Chapter 4
Anorexia Nervosa, Bulimia Nervosa, and Other Eating Disorders
“A young woman thus afflicted, her clothes scarcely hanging together on her anatomy, her pulse slow and slack, her temperature two degrees below the normal mean, her bowels closed, her hair like that of a corpse—dry and lustreless, her face and limbs ashy and cold, her hollow eyes the only vivid thing about her—this wan creature whose daily food might lie on a crown piece, will be busy with mother’s meetings, with little sister’s frocks, with university extension and with what you please else of unselfish effort, yet on what funds God only knows.”1
Anorexia nervosa (AN) and bulimia nervosa (BN) are eating disorders that have been recognized for hundreds of years, yet their etiologies remain poorly understood.2–9 Both illnesses carry significant physical and psychological morbidity, and in the case of AN, death can occur in severe, untreated cases. Early recognition and aggressive treatment thus are particularly important. Because the physical manifestations of these illnesses are so prominent, most patients have their first encounters with nonpsychiatric physicians. Lengthy diagnostic workups for underlying physical illness may be conducted, and a psychiatric disturbance may be considered only when the results of the workup do not fit a known physical illness. The careful application of psychiatric diagnostic criteria for AN and BN permits these illnesses to be suspected early in the medical workup and facilitates referral to the psychiatric specialist in a manner acceptable to the patient and family.
History and Epidemiology
AN and BN are diseases primarily of adolescent girls; approximately 95% of AN cases are female.10 Although there is documentary evidence of AN occurring in the Middle Ages, the first medical accounts appeared in the 17th century as A Discourse upon Prodigious Abstinence by John Reynolds in 1669 and Phthisiologia; or, a Treatise of Consumptions by Richard Morton in 1689.3,11,12 AN as a modern clinical entity derives from publications in 1873 by Charles Lasègue and William Gull, who referred to the illness as hysterical anorexia,13,14 and Gull’s publication in 1874 entitled Anorexia Nervosa.15 Bliss and Branch3 commented, “It is revealing to note the extraordinary differences in the description of the same condition by the two men. While Gull’s comments were as direct and precise as a pathologic report, Lasègue conveyed a sense of the spirit and feeling of these people, the nuances of their disturbed relationships, and the subtleties of their intrapsychic turmoil” (p. 14). The early 20th-century history of AN primarily involves its distinction from physical illnesses such as Simmonds’ cachexia.16
Although binge eating (bulimia) has long been recognized as part of the symptomatology of AN, BN as a distinct syndrome was first proposed by Russell in 1979.17 He elaborated two criteria for BN: an irresistible urge to overeat followed by self-induced vomiting or purging, and a morbid fear of becoming fat. In common with AN, such patients kept their body weight somewhat below normal but not to the same extent as AN patients. They also tended not to develop amenorrhea and were more active sexually. Russell considered BN to be an “ominous variant” of AN, in that comorbid depressive symptoms were often severe and distressing, leading to a high risk of suicide.
In the 1990s, there were opposing views about whether to treat binge eating disorder (BED) as a separate entity. Some argued that too little was known about it and other recurrent forms of overeating, and inclusion would be a source of diagnostic confusion. Others argued that there was a relatively pure group of individuals with Eating Disorder Not Otherwise Specified (EDNOS) who met criteria for BED and not BN, and that recognizing BED as a new disorder would stimulate research and clinical programs for these patients. Since 1994, BED became a provisional eating-disorder diagnosis and was included in the appendix of the fourth edition text revision of the Diagnostic and Statistical Manual of Mental Disorders (DSM-IV-TR).18
The reported incidence of AN has varied between about 0.5 and 15 cases per 100,000 population per year.19–21 The large variation is related to the diagnostic criteria used, the methods of case ascertainment, and the population studied (e.g., clinic versus community). The prevalence of AN ranges from 0.5% to 1.0% among females, with males being affected about one tenth as frequently, although the prevalence has been considerably higher in some studies.20,21 A meta-analysis of 42 studies revealed the crude mortality rate in AN (due to all causes of death) to be 5.9%, which is substantially greater than that for female inpatients and for the general population.22
The prevalence of BN varies between 2% and 4%.20,21,23,24 Both AN and BN, as well as subsyndromal anorexia and bulimia, are far more common in certain groups of young women, notably athletes and ballet dancers, where occupational demands place a premium on thinness. Primary and secondary amenorrheas in these individuals are extraordinarily common. Overweight and obesity are common comorbidities of BED, which has an estimated prevalence rate between 0.7% and 3%.25
Since the 1980s, large-scale twin and family studies have suggested that eating disorders are familial.26,27 There is cross-transmission between AN and BN, and they appear to share a common vulnerability, but the transmissible elements remain elusive. Twin and family studies suggest that major depressive disorder and substance dependence most likely do not share a common etiology with the eating disorders. The genetic contribution is likely predisposing rather than determining, in that sociocultural circumstances, as well as personal psychological stressors, are risk factors.28,29 The risk of developing AN is higher in female twins than in male twins, but male members of opposite-sex pairs have almost the same risk as in female twins.30 This suggests that an intrauterine factor, such as sex steroid hormones, also influences the development of AN.
Diagnosis
The DSM-IV-TR18 provides the current clinical criteria for AN (Table 4-1) and BN (Table 4-2) and EDNOS, which includes the provisional diagnostic category of BED. Since the DSM became a criterion-based system with DSM-III in 1980, it has undergone two revisions, and several diagnostic categories have undergone considerable change across these revisions. In contrast, the criteria for AN have remained relatively stable, although future revision might be useful, such as the elimination of amenorrhea as a criterion.31 The criteria for bulimia are in greater flux. For example, BED, characterized as recurrent episodes of binge eating and associated distress without inappropriate compensatory purging or other behaviors, has been included in the DSM-IV-TR Appendix as a set of criteria for further study.18 There have been recommendations to include BED as a distinct eating disorder in the upcoming DSM-V.32,33 Although there are far fewer men with eating disorders than women, the clinical features are similar.34
Table 4-1
DSM-IV-TR Diagnostic Criteria for Anorexia Nervosa
A Refusal to maintain body weight at or above a minimally normal weight for age and height (e.g., weight loss leading to maintenance of body weight less than 85% of that expected; or failure to make expected weight gain during period of growth, leading to body weight less than 85% of that expected).
B Intense fear of gaining weight or becoming fat, even though underweight.
C Disturbance in the way in which one’s body weight or shape is experienced, undue influence of body weight or shape on self-evaluation, or denial of the seriousness of the current low body weight.
D In postmenarcheal females, amenorrhea—i.e., the absence of at least three consecutive menstrual cycles. (A woman is considered to have amenorrhea if her periods occur only following hormone [e.g., estrogen] administration.)
Type
Restricting type: During the current episode … the person has not regularly engaged in binge-eating or purging behavior (i.e., self-induced vomiting or the misuse of laxatives, diuretics, or enemas).
Binge-eating/purging type: During the current episode … the person has regularly engaged in binge-eating or purging behavior (i.e., self-induced vomiting or the misuse of laxatives, diuretics, or enemas).
Reprinted with permission from American Psychiatric Association: Diagnostic and Statistical Manual of Mental Disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association, 2000.
Table 4-2
DSM-IV-TR Diagnostic Criteria for Bulimia Nervosa
A Recurrent episodes of binge eating. An episode of binge eating is characterized by both of the following:
(1) Eating, in a discrete period of time (e.g., within any 2-hour period), an amount of food that is definitely larger than most people would eat during a similar period of time and under similar circumstances.
(2) A sense of lack of control over eating during the episode (e.g., a feeling that one cannot stop eating or control what or how much one is eating).
B Recurrent inappropriate compensatory behavior in order to prevent weight gain, such as self-induced vomiting; misuse of laxatives, diuretics, enemas, or other medications; fasting; or excessive exercise.
C The binge eating and inappropriate compensatory behaviors both occur, on average, at least twice a week for 3 months.
D Self-evaluation is unduly influenced by body shape and weight.
E The disturbance does not occur exclusively during episodes of anorexia nervosa.
Type
Purging type: During the current episode … the person has regularly engaged in self-induced vomiting or the misuse of laxatives, diuretics, or enemas.
Nonpurging type: During the current episode … the person has used other inappropriate compensatory behaviors, such as fasting or excessive exercise, but has not regularly engaged in self-induced vomiting or the misuse of laxatives, diuretics, or enemas.
Reprinted with permission from American Psychiatric Association: Diagnostic and Statistical Manual of Mental Disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association, 2000.
The DSM-IV-TR criteria for AN, BN, and BED highlight the many identifiable behavioral and psychological aspects of these illnesses.35 If the patient and her family are queried about these aspects, considerable information pointing toward the diagnosis can be gleaned, psychiatric consultation can be considered early in the diagnostic process, and specialized and expensive laboratory testing often can be avoided.
For AN, a key feature of the weight loss is the patient’s refusal to maintain body weight, which often manifests as an active resistance to increasing caloric intake. As illustrated in Fig. 4-1, the cachexia can be severe, and as mentioned, self-starvation can lead to death. If the onset is in childhood or early adolescence, there may be a failure to make the expected weight gain during the active growth phase. This refusal to gain or maintain body weight is rooted psychologically in the second criterion, an intense fear of gaining weight or of becoming fat, even though the patient is underweight. There is a subjective distortion of body image such that the emaciated individual appears to herself to be either at an acceptable weight or even fat. Indeed, initiation of treatment by insisting on the patient’s eating can lead to purging behavior that did not occur prior to treatment. How inflexible a given patient may be in refusing to gain weight, however, can vary considerably.36
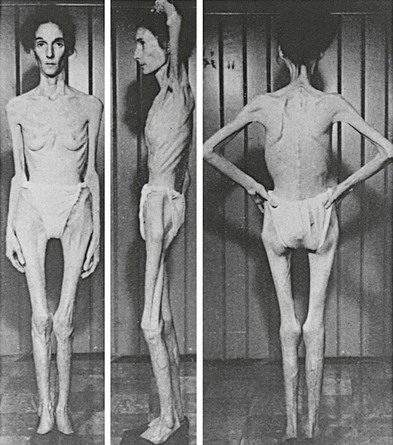
FIGURE 4-1 Extreme cachexia in an anorexia nervosa patient. (From Bliss EL, Branch CHH: Anorexia Nervosa: Its History, Psychology, and Biology. New York: Paul B. Hoeber, 1960.)
A corollary of this is the third criterion, disturbance of the experience of one’s body weight or shape (e.g., undue influence of body weight or shape on self-evaluation/self-esteem and/or denial of the seriousness of the weight loss), even though there may be clear adverse physical sequelae in addition to the current fourth criterion, secondary amenorrhea. For the endocrinologist, questions that should be asked of the patient and her family to address these psychological and behavioral aspects of AN include the following (adapted from the Structured Clinical Interview for DSM-IV37):
Why do you limit yourself to those foods?
Are you concerned that you might become fat if you ate more?
Do you weigh less than other people think you should weigh?
Do you need to be very thin in order to feel good about yourself?
Has anyone told you it can be dangerous to be as thin as you are?
What kinds of things do you do to keep from gaining weight?
Have you ever made yourself vomit or take laxatives, enemas, or water pills? How often?
Before now, were you having periods? Were they regular? When did they stop?
Rather than rigidly adhering to the DSM-IV-TR algorithm for diagnosing AN—that is, the reduction of body weight to less than 85% of expected and complete amenorrhea for at least 3 months—the clinician should consider both the patient’s core symptoms and current medical impairment; such impairment may require treatment even though body weight may be greater than 85% of expected and/or menses still may occur sporadically.31
For BN, the distinguishing features are uncontrollable binge eating of a definitely larger-than-normal amount of food (first criterion) that is clearly repetitive (third criterion). Here too, one’s sense of self is unduly influenced by weight and body shape (fourth criterion). Compensatory behaviors such as self-induced vomiting, purging, fasting between binges, and exercise are invoked to prevent weight gain (second criterion). The frequency and chronicity of the bingeing, and especially the compensatory behaviors, help distinguish BN from overeating in general. And because binge eating and purging can occur as part of AN as a subtype, a fifth criterion for BN is that it does not occur exclusively during episodes of AN. The validity of the DSM-IV-TR eating disorders classification has been questioned, owing to the high rates of diagnostic crossover among the AN subtypes and BN. Recent findings support the longitudinal distinction between AN and BN, but they may not support the AN subtyping schema.38,39 BED also is being distinguished from BN18,40 because obesity rather than inanition often results, and the endocrinologic and other metabolic changes are different from those of AN and BN.
For the endocrinologist, questions that should be asked of the patient and her family to address the criteria for BN include the following (adapted from the Structured Clinical Interview for DSM-IV37):
Do you have times when your eating gets out of control? Tell me about these times.
During these times, do you often eat within a 2-hour period what most people would regard as an unusual amount of food?
Can you give me an example of the kinds and amounts of food that you might eat during one of these times?
How often do these times occur?
Do you do anything to counteract the effects of eating that much—like making yourself vomit; taking laxatives, enemas, or water pills; strict fasting between periods of eating a lot of food; or exercising a lot?
How important is your body shape and size in how you feel about yourself?
These questions and those regarding AN should be phrased in a way that is comfortable to both the health care professional and the patient. Depending on the patient, questions about both bulimic and anorexic behaviors may need to be asked; the patient’s history and presenting symptoms should dictate the emphasis of the interview.41 The sample questions are given to indicate that one can (and should) ask about eating behavior forthrightly. If the topics of the questions are followed, information about all the diagnostic criteria for both AN and BN will be gleaned. The patient should also be asked if this is the first time these behaviors have occurred or if there have been past episodes; in the case of the latter, a careful history taking about each episode, its severity and duration, and treatments and their success should be done. This information may yield important clues as to how to approach the patient therapeutically during the current episode.
Comorbid symptoms of depression and anxiety occur with some frequency in both AN and BN and should be assessed, because their persistence during treatment may portend poorer outcomes.42–48 Symptoms of depression and anxiety are exaggerated by malnutrition and improved with nutrition. AN patients with obsessive-compulsive disorder tend to have an obsessional need for symmetry/exactness and compulsions toward ordering and arranging.49 Mild to moderate negative mood states and obsessive symptoms can persist after recovery in both anorexic and bulimic individuals,50,51 suggesting that these traits may contribute to the pathogenesis of eating disorders.52 As well, comorbid impulse control disorders (ICD), the most common being compulsive buying disorder and kleptomania, occur more frequently in individuals with binge eating subtypes and are associated with greater use of laxatives, diuretics, appetite suppressants, and fasting.53
There also is a high incidence of comorbid obsessive-compulsive disorder (OCD) in anorexic and bulimic women and their families, as well as increased rates of AN and BN in persons with primary OCD. It may be that the core eating disorder symptoms (e.g., fear of fatness, pursuit of thinness) are a specific type of obsession. Symmetry, ordering, and perfectionism are the most common target symptoms in women with AN and BN and often persist after recovery. Leckman and others54 delineated four symptom dimensions of OCD, one of which was symmetry and ordering; these occurred most commonly in men. OCD in men and eating disorders in women may be gender-specific expressions of a common psychobiology related to obsessions with symmetry and ordering compulsions.
There are primary central nervous system (CNS) conditions for which anorexia or bulimia may be an associated symptom; these include both organic pathology55 and “functional” illnesses. As an example of the latter, one of the authors was asked to consult on a hospitalized adolescent girl with electrolyte disturbances suggestive of Bartter’s syndrome. With no evidence of somatic causes of the patient’s cachexia, and with nurses’ observations of bizarre eating habits, a diagnosis of AN was entertained, and a psychiatric interview of the patient during endocrine rounds was requested. After introductions as to who the psychiatrist was and why he was there, questioning began with what the patient ate (lettuce and carrots only) and why she ate only those two items (the computer in the hospital basement was giving her instructions to do so). Elucidation of additional symptoms of psychosis led to a presumptive diagnosis of schizophrenia with secondary anorexia and referral for inpatient psychiatric care.
Genetics
Eating disorders in both women and men are familial, as shown by both family and twin studies.56–61 The heritability of both AN and BN has been estimated between 33% and 84%.62 Recent findings corroborate previous research implicating the transition from early to mid-adolescence as a critical time for the emergence of a genetic diathesis for disordered eating.63 Studies of the genetics of AN and BN have been primarily by linkage analysis and association studies. In linkage analysis, correlations are determined between the occurrence of a disease in families and the inheritance of specific chromosomal regions in those families. In association studies, nucleotide polymorphisms are searched for in specific candidate genes suggested by chromosomal linkage studies and/or the known functions of the gene product.
Linkage studies in AN and BN families have suggested susceptibility genes on several chromosomes, but the strength of the associations has depended on the diagnostic stringency of the cases. For example, in 192 families with at least one affected relative pair with AN, BN, and related eating disorders, modest linkage was found with a marker on chromosome 4.64 However, when the analysis was restricted to 37 families in which at least two relatives had a diagnosis of the restricting subtype of AN (without bingeing and purging), there was a much stronger linkage to a marker on chromosome 1p. With reference to specific behavioral traits, two variables were identified in a sample of 196 families with an AN proband: drive for thinness and obsessionality.65 When these variables were incorporated into a linkage analysis, there again was highly significant linkage to a region on chromosome 1, as well as linkages of lesser significance to regions on chromosomes 2 and 13. In contrast, linkage analysis of 308 families with a BN proband yielded a high linkage score to chromosome 10.66 In a subset of 133 families in which two or more BN members reported self-induced vomiting, an even higher linkage was found to a region on chromosome 10p. Another region on chromosome 14q was suggestive of linkage. These studies can serve to suggest specific chromosomal regions for association studies.
In association studies, researchers have targeted serotonin, dopamine, and norepinephrine-related genes. The most promising results have been with the serotonergic system, because serotonin (5-HT) neurotransmission inhibits feeding behavior.56,67 Several studies have suggested an association between polymorphisms in the promoter region of the 5-HT2A receptor gene and both AN and BN, but the results have been inconsistent, the association has been rather weak, and the functionality of the 5-HT2A polymorphism is unknown.68,69 Studies reporting an association between a polymorphism in the promoter region of the 5-HT transporter gene and eating disorders have been inconsistent as well. In one study, a relationship was suggested between a polymorphism in the 5-HT1B receptor gene and BN; in another study, a relationship was suggested between a polymorphism in the 5-HT2C receptor gene and vulnerability to rapid weight loss following reduced food intake in AN. On the other hand, studies of the 5-HT receptor genes 2C, 7, and 1Dβ, as well as the tryptophan hydroxylase gene (the rate-limiting enzyme in the biosynthesis of 5-HT), have been negative. Phenotypically, reduced serotonergic activity, as measured by platelet 3H-paroxetine binding, has been found in both AN and BN and was unrelated to diagnostic subtype or ancillary dimensions such as impulsive behavior or depression.70 The use of serotonin uptake inhibiting antidepressants in the treatment of AN and BN will be discussed later.
The dopaminergic system also has received attention because dopamine (DA) neurotransmission has been implicated in feeding behavior and in motor activity, amenorrhea, and distortion of body image.56,57 One study each of D3 and D4 receptor polymorphisms and food restriction in AN have been negative. One of three studies of polymorphisms in the catechol-O-methyl transferase gene (an enzyme in DA metabolism) in AN was positive, but the other two were negative.
Other candidate genes controlling hormones and proteins putatively related to food intake that have been studied in eating disorders include the estrogen receptor, the uncoupling proteins UCP-2 and UCP-3, pro-opiomelanocortin, the melanocortin-4 receptor, leptin, agouti-related protein, neuropeptide Y, cholecystokinin (CCK), the β3-adrenergic receptor, and tumor necrosis factor. All but one have been negative.56,71 Thus, although the familial clustering of both AN and BN is clear, there is no solid evidence that single nucleotide polymorphisms in any of the aforementioned genes are other than mildly related to either of these eating disorders.
General Physical and Laboratory Findings
The general physical and laboratory findings in AN and BN are presented in Tables 4-3 and 4-4.72,73 Metabolic changes and medical complications occur secondary to chronic starvation and malnutrition and to bingeing and purging.7,73–75 Malnutrition-associated disturbances as severe as pulmonary bronchiectasis and emphysema have been reported.76 Cardiac complications are also well recognized in patients with eating disorders, including the more common abnormalities of bradycardia, orthostatic hypotension, and (although less common) the risk for more serious abnormalities, including prolongation of the QT interval and silent pericardial effusion.77
Table 4-3
Physical and Laboratory Findings in Anorexia Nervosa
Physical
Cachexia, emaciation, dehydration, shock or impending shock
Covert infectious processes, immunologic problems late in process
Xerosis (dry, scaly skin), desquamation, yellowish palms and soles
Scalp and pubic hair loss, lanugo, increased pigmented body hair
Hypothermia, decreased metabolic rate, bradycardia, hypotension
Acrocyanosis (circulatory changes resulting in cold, blue, and occasionally sweaty hands and feet)
Bradypnea (respiratory compensation for alkalosis)
Edema of lower extremities, heart murmur (infrequent)
Signs of estrogen deficiency (dry skin, osteoporosis, small uterus and cervix, dry vaginal mucosa) and androgen deficiency (no acne or oily skin)
Chemical
Normal laboratory results early in process
Elevated BUN secondary to dehydration
Elevated serum cholesterol early in process; may decrease later
Decreased plasma transferrin, complement, fibrinogen, prealbumin; usually normal protein and albumin:globulin ratio
Elevated serum lactic dehydrogenase and alkaline phosphatase
Depressed serum magnesium, calcium, phosphorus, the last a late and ominous sign
Hematologic
Panleukopenia with relative lymphocytosis
Thrombocytopenia (causing purpura)
Very low erythrocyte sedimentation rate
Mild anemia (folate or iron deficiency); severe anemia (rare and late in process), especially with rehydration
Adapted from Comerci GD: Medical complications of anorexia nervosa and bulimia nervosa. Med Clin North Am 74:1293–1310, 1990; and Mitchell JE, Crow S: Medical complications in adolescents with anorexia nervosa: a review of the literature. Curr Opin Psychiatry 19:438–443, 2006.
Table 4-4
Physical and Laboratory Findings in Bulimia Nervosa
Physical
Usually well groomed with good hygiene
Usually normal weight or mild to moderate obesity
Generalized or localized edema of lower extremities
Swelling of parotid and other salivary glands
Bruises and lacerations of posterior pharynx, scar/callus formation over dorsum of hands (Russell’s sign) secondary to induced vomiting
Dental enamel discoloration and dysplasia secondary to vomiting of gastric acid
Pyorrhea and other gum disorders
Diminished reflexes, muscle weakness, paralysis, infrequently peripheral neuropathy
Muscle cramping (with induced hypoxia or positive Trousseau’s sign)
Signs of hypokalemia (hypotension, weak pulse, arrhythmias, decreased cardiac output, poor-quality heart sounds; shortness of breath; ileus, abdominal distension, acute gastric dilatation; depression, mental clouding)
Additional physical features of anorexia nervosa, if food restriction is part of syndrome
Chemical
No abnormalities reported; possible abnormal glucose metabolism
Metabolic alkalosis (hypochloremia, elevated serum bicarbonate)
Hypokalemia (secondary to metabolic alkalosis)
Hypovolemia with secondary hyperaldosteronism (also contributes to hypokalemia), pseudo-Bartter’s syndrome
Adapted from Comerci GD: Medical complications of anorexia nervosa and bulimia nervosa. Med Clin North Am 74:1293–1310, 1990.
Gastrointestinal (GI) disturbances occur in both AN and BN.78 Delayed gastric emptying and delayed colonic transit time resulting in constipation are common in AN patients and are associated with complaints of early satiety, bloating, and abdominal distension, leading the patient to feel fat and avoid eating. GI disturbances in BN include increased gastric capacity, decreased gastric relaxation, delayed gastric emptying, and abnormal function of the enteric autonomic nervous system. Abnormalities of GI function in AN and BN are reversible to various degrees with improvement in the underlying disorders. Treatments targeted at these abnormalities, such as agents to improve gastric emptying in AN and neurotransmitter modulators to reduce bingeing in BN, have met with varying success and require further study.
Increasing recognition is being given to osteopenia as an early and serious complication of AN.79–82 Adolescence is a time when optimal bone mineralization supporting physical growth is critical, and adolescent girls with AN have relatively poor bone mineral accrual. Several factors contribute to osteopenia in AN, including estrogen, androgen, and insulin-like growth factor deficiency; elevated cortisol, ghrelin, and peptide YY; excessive exercise; and nutritional deficiencies such as calcium and vitamin D. Multifaceted treatment of AN is required to restore bone mineral density to the normal range.
The electrolyte disturbances of AN and BN depend on whether purging is primarily by vomiting or by abuse of laxatives or diuretics (or both) and can be life threatening. The medical management of these cases can be complex and must be individualized. Refeeding syndrome may occur in severely malnourished patients as a result of severe shifts in fluids and electrolytes, in particular phosphate, from extracellular to intracellular spaces. These electrolyte shifts can lead to cardiovascular, neurologic, and hematologic complications and can be associated with significant morbidity and mortality.83 Comerci72 presented detailed case histories of an anorexic and a bulimic patient, including critiques of their treatment, which highlight the intricacies of successful medical management of these disorders.
Neuroimaging
This is a growing area of eating-disorders research supported by major improvements in imaging technology.84,85 In AN, enlargement of cortical sulci and subarachnoid spaces suggestive of cerebral, and on occasion cerebellar, atrophy have been reported, the changes being positively correlated with poor neuropsychological test performance and being reversible to varying degrees with weight gain.86–88 Reduced gray matter volume in the anterior cingulate cortex has been reported in AN compared to healthy controls, the amount of the decrease being correlated with illness severity.89 Studies in recovered AN and BN patients have differed, showing either persistent alterations or normalization of gray- and white-matter volumes.90,91
With regard to CNS metabolites, proton magnetic resonance spectroscopy (1H-MRS) indicates higher ratios of choline-containing compounds to creatine and N-acetylaspartate, as well as reduced myoinositol, in the frontal white matter and occipital gray matter in association with decreased body mass index (BMI), suggesting starvation-associated increased cell membrane turnover.92 Consistent with these findings, 31P-MRS has shown altered phosphodiester and phosphomonoester peaks in malnourished anorexics, suggesting that reduced body mass alters CNS cellular membrane phospholipid metabolism.93 Reduced blood flow in frontal, parietal, and frontotemporal cortex, as shown by single photon emission computed tomography (SPECT), has been reported in AN, which reverted to normal perfusion with clinical remission.94 Another SPECT study suggested reduced blood flow in the anterior cingulate in patients with AN compared to controls, both before and after weight restoration.95
Functional neuroimaging (fMRI) also is delineating activation of limbic and paralimbic areas that may be involved with calorie fear, including exaggerated activation of the caudate and reduced activation of the insula to taste stimuli in women recovered from AN compared to normal women.96,97 These areas also have been implicated as neural substrates of obsessive-compulsive and depressive symptoms.98 Decreased food-stimulated activation of several cortical areas has also been shown by fMRI in chronically ill anorexics, compared to those exhibiting long-term recovery.99 Positron emission tomography (PET) imaging has demonstrated reduced 5-HT1A and 5-HT2A receptor binding in frontal, parietal, and occipital cortex in both AN and BN patients, which may persist after recovery in both conditions.8,100,101 This may tie in with the candidate gene studies of these receptors discussed earlier; correlative studies have yet to be undertaken.
Neuroendocrinology
Abnormal hormone profiles and responses to challenge are closely related to the “starvation” status of AN and BN patients.102–106 Hormone abnormalities also may be present (but to a lesser extent) in normal-weight women with BN. The presence of starvation in AN is evident from the weight loss, but it may not be recognized in normal-weight BN: Although bulimic women often maintain a normal weight, they do so by restricting food intake when not bingeing and purging, and they often have poorly balanced meals. Starvation-induced depletion of hepatic glycogen stores results in free fatty acids and ketone bodies replacing glucose as the primary energy source. This shift from glycogenolysis to lipolysis and ketogenesis is associated with an increase in free fatty acids and their metabolites. β-Hydroxybutyric acid levels are elevated in both AN and BN,107 indicating that bulimic patients are nutritionally depleted despite their normal body weight.108
The relationship of starvation and eating disorders to neuroendocrine function is most clearly seen for the pituitary-gonadal axis. As mentioned, secondary amenorrhea is one of the criteria for AN in postmenarcheal women,74 and oligomenorrhea occurs in about 50% of bulimics. Table 4-5 lists the major endocrine disturbances that occur in AN and BN.103,109–112 The secondary amenorrhea is a direct result of altered gonadotropin secretion. Serum sex hormone–binding globulin may be increased, and both estrogen and testosterone are decreased.113 As indicated in Table 4-3, there are physical signs of severe estrogen deficiency. The luteinizing hormone (LH) response to LH-releasing hormone stimulation is blunted, but the follicle-stimulating hormone response is usually normal.
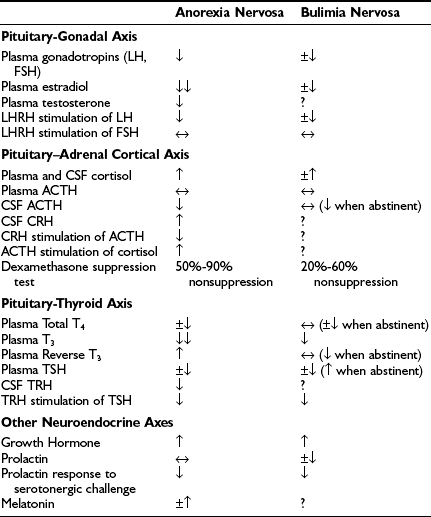
Fig. 4-2 illustrates circadian serum LH profiles of girls at different pubertal stages and in a 21-year-old woman with AN of 14 months’ duration whose body weight was 60% of ideal weight.109 This patient’s menarche was at age 14, and during the period of weight loss, her LH profile was that of a prepubertal girl. BN patients also may have decreased gonadotropin secretion if they have lost 15% or more of their body weight, but they usually have normal circulating gonadotropins and continue their menses.
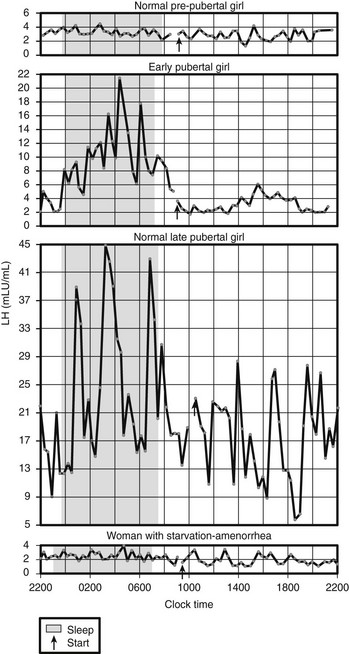
FIGURE 4-2 Circadian serum luteinizing hormone (LH) profiles in girls at different pubertal stages and in a 21-year-old woman with anorexia nervosa of 14 months’ duration whose body weight was 60% of ideal weight. Menarche was at age 14 years, and her LH profile is that of a prepubertal girl. (From Vande Wiele RL: Anorexia nervosa and the hypothalamus. Hosp Pract 12:45–51, 1977. © 1977, The McGraw-Hill Companies Inc. Illustration by Albert Miller.)
With reference to the hypothalamic-pituitary–adrenal cortical (HPA) axis (see Table 4-5), the abnormalities in AN and in reduced-weight BN111,114,115 are strikingly similar to those occurring in 30% to 50% of patients with major depression.111,116,117 Circulating cortisol is increased at all times of the day and night, but the amplitude and timing of its circadian rhythm are preserved. Circulating adrenocorticotropic hormone (ACTH) is usually normal when determined by radioimmunoassay, as it is in major depression; the more specific immunoradiometric assay for ACTH1-39 has shown decreased ACTH in major depression,118 which also could be the case for ACTH1-39 in these eating disorders. Cerebrospinal fluid (CSF) ACTH concentrations appear to be decreased, but CSF corticotropin-releasing hormone (CRH) concentrations may be increased,111,115 as may be CSF vasopressin,111,119 a secretagogue for ACTH in addition to CRH,120 which appears to play a greater role in stress states than under normal conditions.121
Stimulation and suppression tests of the HPA axis have been conducted mainly in AN, and they are in accord with the baseline hormone findings. The ACTH response to CRH administration is reduced, undoubtedly secondary to enhanced negative feedback on the pituitary corticotrophs exerted by elevated circulating cortisol. The cortisol response to ACTH administration is increased, suggesting increased secretory capacity of the adrenal cortex. The low-dose dexamethasone (DEX) suppression test is abnormal in 50% to 90% of anorexics and in 20% to 60% of bulimics, depending on weight loss. Because DEX acts primarily at the pituitary, ACTH and cortisol escape from DEX suppression suggest increased suprapituitary stimulation of corticotrophs by CRH and vasopressin. CRH may be more influential than AVP in stimulating the HPA axis in AN, because the cortisol response to exogenous AVP administration is blunted.122 Taken together, the pituitary-adrenocortical findings indicate a mild to moderate activation of this hormone axis in AN and BN. As well, plasma neuroactive steroids are reported to be elevated in untreated women with both AN and BN.123
With reference to the pituitary-thyroid axis, starvation leads to considerably decreased plasma free triiodothyronine (T3) concentrations, along with somewhat decreased plasma free thyroxine (T4) and increased plasma reverse T3 concentrations. This represents the “euthyroid sick syndrome” hormone profile.124–129 The decreased circulating T3 helps reduce energy expenditure and minimizes muscle protein catabolism into amino acids for gluconeogenesis. CSF thyrotropin-releasing hormone (TSH) also appears to be reduced in AN.130 When bingeing, bulimic patients generally have normal thyroid indices with perhaps reduced T3 and TSH concentrations; however, when they stop bingeing, their pituitary-thyroid axis function resembles that of anorexic patients.131–133
Insulin-like growth factor type 1 (IGF-1) concentrations are low in both AN and BN, and circulating growth hormone (GH) is increased, likely owing to both diminished feedback of IGF-1 on GH secretion and primary hypothalamic dysfunction.134,135 Circulating prolactin is usually unchanged in AN and may be reduced in BN. Prolactin responses to serotonergic challenges are diminished in both AN and BN,136–142 suggesting decreased CNS serotonergic neurotransmitter function.101 (Serotonin uptake–inhibiting antidepressants have shown promise in the treatment of these eating disorders, as discussed later.) Circulating melatonin has been reported as both unchanged and increased in AN and as unchanged in BN.143–146
The effect of reduced caloric intake on these endocrine systems has been studied in healthy individuals.147,148 When healthy women were starved, plasma gonadotropin concentrations declined. The women also experienced increased circulating concentrations of cortisol and GH and decreased plasma T3 concentrations despite normal plasma T4—the “euthyroid sick syndrome” hormone profile. The endocrine abnormalities associated with starvation in the healthy female subjects reversed with the resumption of normal eating.
The endocrine changes in both AN and BN revert to normal with successful treatment of these illnesses, indicating that the endocrine changes are “state” markers of the metabolic stress of starvation and malnutrition. It should be emphasized that in addition to its effects on hormone secretion, starvation can lead to abnormal psychological states. Semistarvation of male conscientious objectors to military service was associated with increased irritability, labile mood, depression, decreased concentration, decreased libido, and decreased motor activity,149 and starvation and malnutrition can exaggerate the comorbid psychiatric symptoms of AN and BN. These changes reinforce the concept of starvation-related “state” changes influencing both the behavioral and the endocrine aspects of eating disorders.
Central Nervous System–Related Neuropeptides
Since the 1980s, the realization has evolved that the peripheral hormonal disturbances in AN and BN are a consequence of the malnutrition associated with starvation and bingeing, rather than being etiologic. Contemporaneously, an understanding of how CNS neuronal pathways contribute to starvation-induced alterations in peripheral hormonal secretion has developed. The mechanisms for controlling food intake and energy homeostasis involve a complex interplay among peripheral (taste, local autonomic influences on GI/neuropeptides, vagal afferent nerves) and CNS neurotransmitters and neuromodulators, including monoamines and neuropeptides that influence hunger and satiety.8,9,115,150 Compounds such as norepinephrine, serotonin, insulin, opioids, neuropeptide Y and YY, leptin, CRH, vasopressin, and the orexins/hypocretins contribute to regulating the rate, duration, and size of meals, as well as the selection of carbohydrates and protein.106,151–153
Neuropeptides were initially determined to be regulators primarily of hypothalamic functions such as food and water consumption and metabolism, sexuality, sleep, body temperature, pain sensation, and autonomic function. These compounds also have been localized to other areas of the CNS besides the hypothalamus and pituitary, where they appear to regulate complex human mental functions such as mood, obsessionality, attachment formation, and risk-taking and addictive behaviors. Some of the behavioral disturbances occurring during starvation therefore may be related to alterations in function of neuropeptides throughout the CNS. These peptide systems work together multiply in overlapping CNS pathways that influence the spectrum of energy balance states. Table 4-6 lists the major neuropeptide disturbances that occur in AN and BN.8,9,104,115,119,154–157 These changes represent an adaptive profile to weight loss specific to AN and are not present in constitutionally lean women.158
Table 4-6
Central Nervous System Neuropeptide Disturbances in Anorexia Nervosa and Bulimia Nervosa
| Anorexia Nervosa | Bulimia Nervosa | |
| CSF neuropeptide Y | ↑ | ↔ |
| CSF peptide YY | ↔ | ↔ ↑ During abstinence |
| Plasma ghrelin | ↑ | ↑ |
| Plasma leptin | ↓ | ↓ |
| CSF leptin | ↓ | ? |
| Plasma adiponectin | ↑ | ↑ |
| Plasma resistin | ↓ | ? |
| Plasma cholecystokinin | ? | ↓ |
| CSF cholecystokinin | ? | ↓ |
| CSF β-endorphin | ↓ | ↓ |
| CSF dynorphin | ↔ | ↔ |
| CSF vasopressin | ↑ | ↑ |
| CSF oxytocin | ↓ | ↔ |
↑, Increased; ↓, decreased; ↔, unchanged; ?, insufficient or conflicting data.
Neuropeptide Y and Peptide YY
These phylogenetically and structurally related peptides share the same receptor family (Y1, Y2, Y4, Y5) and are among the most potent stimulants of feeding behavior in animals, particularly for carbohydrate-rich foods.115 Neuropeptide Y (NPY) occurs in high concentrations in limbic structures, including the hypothalamus, and is present throughout the cerebral cortex.159 NPY-producing cells in the arcuate nucleus of the hypothalamus co-express agouti-related protein and are inhibited by leptin. NPY increases during hunger, falls during meals, and acts on the paraventricular nucleus to mediate increased eating and reduce energy expenditure. Its effect is counteracted by CRH, and a dynamic equilibrium between NPY and CRH neuronal activity appears to be an important regulator of food intake. The Y1, Y2, and Y5 subtypes of NPY receptors have been implicated in the regulation of feeding.159
In contrast, peptide YY (PYY) occurs in lower concentrations in the CNS, caudal brainstem, and spinal cord. PYY occurs in two forms and is primarily located in endocrine cells of the lower GI tract, where it helps mediate motility and function. PYY1-36 is strongly orexigenic. PYY3-36, on the other hand, has particular affinity for the Y2 receptor, it increases in response to meals, and its actions are to decrease appetite and reduce food intake.160
Intracerebroventricular (ICV) NPY administration in animals produces many of the physiologic and behavioral changes associated with AN, including gonadal steroid-dependent effects on LH secretion, suppression of sexual activity, increased CRH in the hypothalamus, and hypotension.115 Underweight AN patients have elevated CSF NPY, likely a result of their malnourished status. In contrast, such patients, whether underweight or recovered, have normal CSF PYY concentrations. CSF NPY returns to normal with recovery, although patients with amenorrhea may continue to have higher CSF NPY concentrations. Elevated CSF NPY is not an effective stimulant of feeding in underweight anorexics, as evidenced by their resistance to eating and weight gain. Anorexics typically display an obsessive and paradoxical interest in dietary intake and food preparation, and it may be that increased NPY activity in extrahypothalamic areas of the CNS contributes to these cognitions and behaviors.
ICV PYY administration in rats causes massive food ingestion to which tolerance does not develop, which prompted speculation that increased CNS PYY activity may contribute to bulimia. CSF PYY concentrations in normal-weight bulimic women when bingeing and vomiting were similar to those of controls,115 whereas CSF PYY was significantly elevated in bulimic women after a month of abstinence from bingeing and vomiting, compared to healthy volunteer women and AN patients. CSF NPY concentrations were normal in the bulimic women, in contrast to the elevated CSF NPY concentrations in the anorexic women. Plasma NPY and PYY concentrations in AN and BN patients, however, do not necessarily accord with CSF concentrations of these peptides and may have peripheral effects unrelated to the CNS.9 For example, AN patients may have normal CSF PYY concentrations, as noted earlier, but they also may have elevated plasma PYY concentrations, which can contribute to the osteoporosis noted in these individuals.79–82
Leptin
Leptin is secreted predominantly by adipose cells and acts in the CNS to decrease food intake.161 Leptin activates OB receptors primarily in the hypothalamus, where it may inhibit NPY secretion, and in extraneural tissues to increase energy expenditure.162 In rodents, defects in the leptin gene coding sequence, resulting in leptin deficiency, and defects in leptin receptors are associated with obesity. Treatment with recombinant leptin reduces fat mass in both obese and normal-weight animals in a dose-dependent manner. In humans, serum leptin concentrations are positively correlated with fat mass in individuals in all weight ranges. Women tend to have higher serum leptin than men of the same weight, presumably because of their higher proportion of body fat.163 Obesity in humans is not thought to be a result of leptin deficiency per se, but obesity may be associated with leptin resistance.164
Malnourished and underweight AN patients have significantly reduced plasma and CSF leptin concentrations compared with normal-weight controls,155,165–167 implying a normal physiologic response to starvation. Reduced plasma/CSF leptin ratio has been found in anorexics compared with controls, suggesting that the proportional decrease in leptin with weight loss is greater in plasma than in CSF. As in normal control women, plasma leptin concentrations in anorexics are positively correlated with body weight and fat mass, and they are negatively correlated with physical activity.168 During refeeding in AN patients, CSF leptin concentrations increase to normal values before full weight restoration,155,169 possibly as a consequence of the relatively rapid and disproportionate accumulation of fat during refeeding. Preliminary evidence suggests that hyperleptinemia is associated with an increased risk of recurrent weight loss170; however, further research studies are warranted to delineate whether serum leptin levels at the time of hospital discharge are predictive of posthospital clinical course in patients with AN.170
Patients with BN appear to have significantly decreased serum leptin following an overnight fast.155 During sustained recovery from BN, serum leptin remains decreased compared to controls matched on amount of body fat. This finding, along with persistently decreased thyroid activity and elevated ghrelin in recovered BN patients, may result in decreased metabolic rate and a tendency toward weight gain contributing to the preoccupation with body weight that is characteristic of BN.
Leptin also appears to modulate fertility162,164,171,172 and may be the metabolic signal that mediates impaired reproductive ability in extreme overweight and underweight conditions.162 Leptin treatment of patients with hypothalamic amenorrhea has been found to restore reproductive function, but it is uncertain whether these patients had a history of eating disorders before onset of amenorrhea.173 In some anorexic patients, amenorrhea may occur before significant weight loss, and leptin may be the mediating hormone. Weight loss generally causes circulating leptin concentrations to fall in proportion to the loss of body fat mass, but acute, fasting-induced weight loss can provoke a fall in leptin disproportionately greater than would be expected from the amount of fat lost. This suggests that under conditions of intense food deprivation, leptin may instigate metabolic responses before a significant weight or fat loss has occurred. Reduced leptin concentrations appear to be a critical signal that initiates the neuroendocrine response to starvation, including limiting procreation, decreasing thyroid thermogenesis, and increasing secretion of stress steroids. Leptin administration during fasting partially restores LH and testosterone concentrations, blunts falling T4 concentrations, and attenuates the rise in ACTH and glucocorticoids, without affecting plasma concentrations of insulin, glucose, or ketone bodies. Studies in rat models and more recently in patients with AN174 have suggested that hypoleptinemia can trigger semistarvation-induced hyperactivity, supporting the notion that administration of leptin to severely hyperactive patients with AN might prove beneficial.167
As mentioned earlier, during starvation, circulating concentrations of NPY increase, inhibiting gonadotropin release and activating the HPA axis. Leptin inhibits starvation-induced elevations in NPY,175 and it may also reduce food intake and body weight by increasing the metabolic rate through activation of β-adrenergic receptors and possibly through its own anorexigenic properties.154
Adiponectin
Adiponectin also is secreted by adipose cells.156 Circulating levels are two to three times higher in women than in men. It exists in three forms made up of different numbers of trimers. The globular form of adiponectin appears to increase muscle sensitivity to insulin by increasing fatty acid oxidation, similar to the effect of leptin. Unlike leptin, however, circulating adiponectin is inversely correlated with fat mass, being decreased in obesity.176–178 There does not appear to be a direct relationship between circulating leptin and adiponectin, in that decreasing serum leptin by acute fasting or increasing it by administration of physiologic or pharmacologic doses had no effect on serum adiponectin.179 On the other hand, ghrelin impairs adiponectin expression by adipocytes.180
Despite increased circulating adiponectin in patients with AN, in contrast to reduced circulating leptin,165,181,182 studies on the effect of long-term malnutrition of these patients on plasma adiponectin levels show contradictory results, with plasma levels variably increased, decreased, or unchanged.183,184 Nonoxidative glucose metabolism is reduced as well. Patients with BN also were found to have increased circulating adiponectin concentrations, which were positively correlated with the frequency of binge/vomiting episodes.185 In contrast, women with binge eating disorder (bingeing without compensatory behaviors—vomiting and/or purging) had reduced circulating adiponectin along with increased glucose, cholesterol, and triglycerides. In these patients, adiponectin concentrations were not significantly related to the frequency of their bingeing episodes.
Other adipocytokines include resistin and acylation-stimulating protein.156,177 Resistin may play a role in obesity- and diabetes-associated insulin resistance, although this effect in humans is still unclear. However, neither insulin sensitivity nor body size and adiposity are major determinants of plasma resistin levels in patients with AN.186 Although decreased plasma resistin has been suggested to occur in AN, this is not a consistent finding.165,184
Ghrelin
Ghrelin, so named because it stimulates GH secretion, is released mainly from endocrine cells in the stomach and GI tract.154,187–189 In turn, its secretion is inhibited by GH. There are two forms of ghrelin: N-octanoyl-modified ghrelin, the active form, and des-acyl ghrelin.189 Fasting increases plasma concentrations of both forms. In contrast, hyperphagia and obesity lead to decreased des-acyl ghrelin but no change in concentrations of the active form.
Ghrelin antagonizes the action of leptin, strongly stimulates feeding and weight gain by promoting NPY and agouti-related protein, and has a number of other central and peripheral neuroendocrine and metabolic actions. In humans, fasting plasma ghrelin is inversely related to body mass index, percent body fat, and fasting plasma leptin and insulin concentrations. There is a normal preprandial rise and postprandial fall in plasma ghrelin in humans. Plasma ghrelin, including the active, octanoyl form, is elevated in AN patients, particularly in the bingeing and purging subtype,155,190–193 and it does not decrease normally after a standardized meal or fiber intake.194 On the other hand, ghrelin is not elevated in constitutionally thin women with similar body mass index as AN patients, and it returns to normal values in AN patients after weight recovery.190 Fasting plasma ghrelin also has been reported as elevated in BN patients compared to controls with similar body mass index, suggesting that the abnormal eating behavior (bingeing and purging) influences circulating ghrelin, as it can in the bingeing and purging subtype of AN.191,192 Ghrelin administration to medically ill patients led to improved appetite, body composition, and muscle wasting, but its usefulness as an appetite stimulant in AN remains to be clearly demonstrated.195
Cholecystokinin
Cholecystokinin, secreted by the GI tract in response to food intake,155 is known as the satiety hormone196 because it is thought to signal satiety to the CNS via vagal afferents. It is the most abundant peptide in the human brain.197 Exogenously administered CCK reduces food intake in humans. High postprandial levels of CCK have been observed in girls with AN and may aggravate the course of this disease by intensifying nausea and vomiting.198 To date, however, studies of CCK in patients with AN have been inconsistent in their findings. In contrast, patients with BN appear to have reduced CSF and lymphocyte CCK concentrations and diminished CCK secretion following a test meal.199–201 The blunted postprandial CCK response may contribute to the diminished post-ingestive satiety that BN patients experience.
Melanocortin and Corticotropin-Releasing Hormone
The central melanocortin system is important in the regulation of energy balance.202,203 The key melanocortin receptor agonist is α-melanocyte-stimulating hormone (α-MSH), and the key receptor antagonist is agouti-related peptide; these have anorexigenic and orexigenic activity, respectively.204 Plasma agouti-related peptide has been reported as increased in AN, but plasma α-MSH has not been different from control values.205 An amino-acid substitution polymorphism (Ala67Thr) in agouti-related peptide has been described in some AN subjects but not in others.202,206 The substitution has not been shown to alter the potency of the peptide to stimulate the melanocortin 4 receptor, so this polymorphism does not appear to play a role in the pathogenesis of AN.206
The hypercortisolism in AN and BN is most likely due to hypersecretion of CRH, which is most probably a response to weight loss per se; the change in CRH is likely mediated through the central melanocortin system.207 CRH also has central effects beyond the HPA axis; ICV administration of CRH to animals produces physiologic and behavioral changes associated with AN, including hypothalamic hypogonadism, decreased sexual activity, decreased feeding behavior, and hyperactivity.115,155 The anorexigenic action of leptin may be mediated at least partially through CRH.208 The anorectic activity of serotonergic drugs appears to be through activation of pro-opiomelanocortin neurons in the arcuate nucleus, a circuit which is tonically regulated by leptin.209,210
Vasopressin and Oxytocin
In addition to the effects of vasopressin on HPA axis regulation and free-water clearance by the kidney and the effects of oxytocin during the puerperium, these structurally related neuropeptides are distributed throughout the CNS and function as long-acting neuromodulators of complex behaviors. The effects of vasopressin appear to be reciprocal to those of oxytocin: Central administration of vasopressin to rats enhances memory consolidation and retrieval, whereas administration of oxytocin disrupts memory.115
In addition to abnormally high CSF vasopressin concentrations111 and impaired osmoregulation of plasma vasopressin,211 AN patients have reduced CSF oxytocin concentrations and impaired plasma oxytocin responses to stimulation.115 Underweight anorexics also have an impaired plasma oxytocin response to challenging stimuli.115 These abnormalities tend to normalize after weight restoration, suggesting they are secondary to malnutrition or abnormal fluid balance, or both. In underweight anorexics, low CNS oxytocin might interact with high CNS vasopressin to enhance the retention of cognitive distortions of the aversive consequences of eating, thereby reinforcing these patients’ perseverative preoccupation with the adverse consequences of food intake.
Patients with normal-weight BN were found to have elevated CSF vasopressin concentrations but normal CSF oxytocin, both on admission and after 1 month of nutritional stabilization and abstinence from bingeing and purging.111 In these patients as well, high CNS vasopressin might contribute to their obsessional preoccupation with the aversive consequences of weight gain. Some recovered bulimics may have continued elevation of CSF vasopressin, perhaps related to a lifetime history of major depression.212
Opioid Peptides
CNS opioid agonists increase food intake, and opioid antagonists decrease food intake,115 suggesting that these compounds may mediate some aspects of AN.115,155 Although assessment of brain opioid activity in vivo in humans is problematic because of the many CNS neuropeptides with opioid activity and the multiplicity of CNS opioid receptors, the CSF concentrations of some opioid peptides have been determined in anorexic patients. Underweight anorexics were found to have significantly reduced CSF β-endorphin concentrations compared with healthy volunteers.115 CSF β-endorphin concentrations remained significantly below normal after short-term weight restoration but returned to normal after long-term weight restoration. A down-regulation in opioid circuits or a hyposensitivity at receptor sites could lead to a loss of appetite involving either a passive or active resistance to eating. Evidence suggests that polymorphisms in opioid delta 1 (OPRD1) receptor genes show a significant association with restricting AN.213,214 However, further work is required to understand the processes that may be mediated by these genes. CSF β-endorphin concentrations also have been shown to be reduced in women with BN.115 CSF dynorphin concentrations have been reported as normal in all stages of AN and BN.115,155
Open trials of high doses of the opiate antagonist naltrexone have been reported to reduce binge frequency in BN,215,216 but double-blind, controlled trials with lower naltrexone doses have shown no effect on binge frequency or macronutrient intake.217,218 Relatively high-dose naltrexone treatment reduced binge and purge frequency and total daily food intake of bulimics, but it did not affect their ability to resist the desire to binge or purge.219 Whether high-dose opioid antagonist treatment has a role in the treatment of BN is still unclear.
A disturbance in CNS opioid function also may contribute to the neuroendocrine abnormalities in AN and BN (e.g., disturbances in HPA and pituitary-gonadal axis function).115,155 Brain opioid pathways inhibit ACTH and cortisol release in humans, and they suppress pulsatile gonadotropin secretion in rats and in sexually mature humans. Underweight anorexics frequently have a blunted response of LH secretion to opiate antagonists, and weight restoration tends to normalize this response.115 The failure of opioid antagonists to increase LH secretion in underweight anorexics suggests that another neurotransmitter system (or systems) may be responsible for this neuroendocrine disturbance.
Long-Term Effects of Multiple Neuropeptide Disturbances
Along with the peripheral endocrine changes in pituitary and target-gland hormones in AN and BN, multiple neuropeptide disturbances occur when patients engage in pathologic eating behaviors and become malnourished, as reviewed earlier. The list of neuropeptides involved continues to increase. A recent finding has been reduced circulating brain-derived neurotrophic factor (BDNF) in both AN and BN, suggesting its CNS satiety-inducing effect might be reduced.106 As with the peripheral endocrine markers, many of these peptide systems tend to become normal with long-term recovery. The slow correction of these neuropeptide disturbances with weight restoration in AN and BN implies that the disturbances are secondary to malnutrition or weight loss, or both, and are not etiologic, although once established, the disturbances may perpetuate some of the symptomatology, providing a biological dimension to the question why many anorexics and bulimics cannot easily reverse their illness. Secondary symptoms such as dysphoric mood, obsessions, and compulsions related to behaviors other than eating may be exaggerated by CNS neuropeptide alterations and cause the primary illness to be more refractory to treatment. That the neuropeptide disturbances eventually become normal during long-term recovery suggests that treatment of AN and BN must be sustained for months after weight normalization to rectify the many physiologic disturbances.
Treatment
Treatment of the eating disorders continues to be complex and difficult. In a 20-year follow-up of AN patients, about one third rated their outcomes as good, one third as intermediate, and one third as poor.220 Cognitive-behavioral, educational, psychodynamic, and psychopharmacologic treatments have been used with varying degrees of long-term success.221–223 As is often the case in the management of psychiatric patients, a combination of therapies is used under the rationale that moderate successes with individual treatments might be at least additive, if not synergistic in their overall effectiveness. The American Psychiatric Association practice guideline on the treatment of eating disorders discusses the spectrum of options and emphasizes developing individualized treatment plans for weight restoration in the least restrictive setting that is likely to be effective.222
There have been more large-scale, randomized, controlled clinical treatment trials in BN than in AN or BED.25,224–227 A prominent focus has been on interventions previously tested on comorbid psychiatric disorders such as depression and substance abuse. However, eating disorders and these comorbid syndromes appear to be independently transmitted familial liabilities,228 so that treatments also need to be targeted to the eating disorders themselves.
Controlled treatment studies of AN have shown the efficacy of various psychological therapies in promoting weight gain in acutely ill patients229–232 and in preventing relapse following restoration of normal body weight.222,233 Substantial improvement in body mass and psychosocial adjustment can be achieved in many anorexic subjects through cognitive-behavioral, psychoeducational, and family therapy techniques, with or without dietary counseling. Therapeutic gains have not been as robust in patients with more chronic disability. Specialized eating disorders hospital units offer combinations of enforced weight-gain regimens along with a range of psychosocial treatments. Compulsory in-hospital treatment often is effective over the short term and can achieve a weight gain of 2 to 4 pounds per week.234–237
Serotonin uptake–inhibiting antidepressant drugs (SUIs) also show some promise. As noted earlier, serotonergic neurotransmission appears to be compromised in eating disorders, including a possible underlying hypersensitivity in AN that may be partially ameliorated by self-starvation and a subsensitivity in BN that is responsive to SUI-mediated increases in serotonergic activity.85,225,238 SUIs are relatively ineffective in weight-reduced AN patients,239 possibly because of reduced intake of tryptophan, the amino-acid precursor of serotonin, with consequently diminished serotonin production. After weight restoration in AN and improved serotonin production, SUIs such as fluoxetine possibly can reduce obsessionality and the incidence of relapse, although a large, well-controlled study comparing fluoxetine and placebo in weight-restored patients found no significant benefit to medication during the year following nutritional rehabilitation.240 Atypical neuroleptics also may have some utility, not only to reduce anxiety and delusional thinking, but also because they promote weight gain.241–243 The bingeing and purging subtype of AN may have a poorer prognosis that the restricting subtype, and weight restoration alone is unlikely to be effective in the bulimic subtype of AN.244 Improved treatment of AN remains of great clinical and public health importance, in that it is a chronic, relapsing illness with substantial and costly medical morbidity. Other types of treatments continue to be studied (e.g., the use of the anabolic steroid, dehydroepiandrosterone, to increase bone density and improve psychological well-being).245 Yet overall, no pharmacologic intervention for AN thus far has had a significant impact on weight gain or its psychological features. Based on current evidence, managing AN patients only with medications is inappropriate and often associated with high dropout rates.227
Controlled treatment studies of BN have shown the efficacy of both antidepressant medications225,226 and psychological therapies, especially cognitive-behavioral therapy (CBT), in reducing both the frequency of bingeing and purging and the severity of body dissatisfaction, pursuit of thinness, and perfectionism.225,246–248 However, medication alone rarely produces full remission; many patients require multiple medication trials before achieving clinically significant improvement. There have been significant dropout rates in clinical trials, and relapse during continuation therapy is high.
CBT alone produces higher rates of full remission in BN than does antidepressant monotherapy.225 Even so, 40% to 60% of patients receiving CBT remain symptomatic to some degree after acute treatment.224,225 Interpersonal therapy (IPT) that strictly avoids direct reference to abnormal eating attitudes or dietary behaviors may achieve long-term benefits in controlling binge eating and purging equal to those obtained with CBT.249
How long to continue treatment in BN once binge eating abates, in order to minimize relapse, remains undetermined. With antidepressant continuation therapy, the risk of relapse may be higher than in patients with unipolar depression.225 Low serum T4 may predict poor treatment outcome.250 Combined treatment may have an advantage over CBT alone in reducing binge eating and purging, but the incremental benefit is modest. Remaining questions include the length of continuation of psychosocial and antidepressant therapies needed to sustain gains achieved during acute treatment, the mechanisms underlying the possibly synergistic effects of combined treatment, the predictors of differential treatment outcomes, the reasons for a more rapid decay of acute antidepressant treatment effects in BN compared to major depression, and the anticipated effects of crossing over to alternative modalities of treatment when initial treatment fails.225
Pharmacotherapies for BED, as distinct from BN, also are being investigated: Antiepileptic drugs such as topiramate and zonisamide, serotonin/norepinephrine uptake inhibitors such as sibutramine, and antiobesity agents such as the lipase inhibitor orlistat have shown promise in regard to weight reduction and reduction in binge eating.25,251–256 Novel pharmacologic interventions for eating disorders also are being evaluated, such as peptide hormones, ghrelin agonists, neuropeptide Y-1 and Y-5 antagonists, orexin receptor antagonists, CRH-2 receptor antagonists, histamine-3 receptor antagonists, melanocortin-4 receptor antagonists, β3-adrenoceptor agonists, serotonin-2A receptor antagonists, and growth hormone agonists.255
References
1. Allbutt, TC, Rolleston, HD. A System of Medicine. London: Macmillan; 1908.
2. Berkman, JM. Anorexia nervosa, anorexia, inanition, and low basal metabolic rate. Am J Med Sci. 1930;180:411–424.
3. Bliss, EL, Branch, CHH. Anorexia Nervosa: Its History, Psychology, and Biology. New York: Paul B. Hoeber; 1960.
4. Bruch, H. Eating Disorders: Obesity, Anorexia Nervosa and the Person Within. New York: Basic Books; 1973.
5. Vigersky RA, ed. Anorexia Nervosa. New York: Raven Press, 1977.
6. Halmi KA, ed. Psychobiology and Treatment of Anorexia Nervosa and Bulimia Nervosa. Washington, DC: American Psychiatric Press, 1992.
7. Kaplan, AS, Garfinkel, PE. Medical Issues and the Eating Disorders: The Interface. New York: Brunner/Mazel; 1993.
8. Kaye, W. Neurobiology of anorexia and bulimia nervosa. Physiol Behav. 2008;94:121–135.
9. Kaye, WH, Strober, M, Jimerson, D. The neurobiology of eating disorders. In Charney DS, Nestler RJ, eds.: The Neurobiology of Mental Illness, ed 3, New York: Oxford University Press, 2008.
10. Hsu, LK. Epidemiology of the eating disorders. Psychiatr Clin North Am. 1996;19:681–700.
11. Morton, R. Phthisiologia, seu exercitationes de phthisi tribus libris comprehensæ. Totumque opus variis historiis illustratum. London: Smith & Walford; 1689.
12. Reynolds, J. A Discourse upon prodigious abstinence: occasioned by the twelve months fasting of Martha Taylor, the famed Derbyshire damosell: proving that without any miracle, the texture of humane bodies may be so altered, that life may be long continued without the supplies of meat and drink. With an account of the heart, and how far it is interessed in the business of fermentation. London: Royall Society; 1669.
13. Lasègue, C. De l’anorexie hysterique. Arch Gén Méd. 1873;1:385.
14. Gull, WW. Anorexia hysterica (apepsia hysterica). Brit Med J. 1873;2:527.
15. Gull, WW. Anorexia nervosa (apepsia hysterica, anorexia hysterica). Trans Clin Soc Lond. 1874;7:22–31.
16. Simmonds, M. Über Hypophysisschwund mit tödlichem Ausgang. Deutsche Med Wochenschr. 1914;40:322.
17. Russell, GFM. Bulimia nervosa: An ominous variant of anorexia nervosa. Psychol Med. 1979;9:429–448.
18. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, ed 4. Washington, DC: American Psychiatric Association; 2000.
19. Vandereycken, W, Hoek, HW. Are eating disorders culture-bound syndromes? In: Halmi KA, ed. Psychobiology and Treatment of Anorexia Nervosa and Bulimia Nervosa. Washington, DC: American Psychiatric Press; 1992:19–36.
20. Wakeling, A. Epidemiology of anorexia nervosa. Psychiatry Res. 1996;62:3–9.
21. Hudson, JI, Hiripi, E, Pope, HG, Jr., et al. The prevalence and correlates of eating disorders in the National Comorbidity Survey replication. Biol Psychiatry. 2007;61:348–358.
22. Sullivan, PF. Mortality in anorexia nervosa. Am J Psychiatry. 1995;152:1073–1074.
23. Ben-Tovim, DI, Subbiah, N, Scheutz, B, et al. Bulimia: symptoms and syndromes in an urban population. Aust N Z J Psychiatry. 1989;23:73–80.
24. Fairburn, CG, Beglin, SJ. Studies of the epidemiology of anorexia nervosa. Am J Psychiatry. 1990;147:401–408.
25. Brownley, KA, Berkman, ND, Sedway, JA, et al. Binge eating disorder treatment: a systematic review of randomized controlled trials. Int J Eat Disord. 2007;40:337–348.
26. Strober, M. Family-genetic studies. In: Halmi KA, ed. Psychobiology and Treatment of Anorexia Nervosa and Bulimia Nervosa. Washington, DC: American Psychiatric Press; 1992:61–76.
27. Lilenfeld, LR, Strober, M, Kaye, WH. Genetics and family studies of anorexia nervosa and bulimia nervosa. In: Kaye WH, Jimerson DC, eds. Eating Disorders, Ballière’s Clinical Psychiatry. London: Ballière Tindall; 1997:177–197.
28. The McKnight Investigators. Risk factors for the onset of eating disorders in adolescent girls: results of the McKnight longitudinal risk factor study. Am J Psychiatry. 2003;160:248–254.
29. Striegel-Moore, RH, Bulik, CM. Risk factors for eating disorders. Am. Psychologist. 2007;62:181–198.
30. Procopio, M, Marriott, P. Intrauterine hormonal environment and risk of developing anorexia nervosa. Arch Gen Psychiatry. 2007;64:1402–1408.
31. Watson, TL, Anderson, AE. A critical examination of the amenorrhea and weight criteria for diagnosing anorexia nervosa. Acta Psychiatr Scand. 2003;108:175–182.
32. Wilfley, DE, Bishop, ME, Wilson, GT, et al. Classification of eating disorders: towards DSM-V. Int J Eat Disord. 2007;40:S123–S129.
33. Striegel-Moore, RH, Franko, DL. Should binge eating disorder be included in the DSM-V? A critical review of the state of the evidence. Annu Rev Clin Psychol. 2008;4:305–324.
34. Woodside, DB, Garfinkel, PE, Lin, E, et al. Comparisons of men with full or partial eating disorders, men without eating disorders, and women with eating disorders in the community. Am J Psychiatry. 2001;158:570–574.
35. Becker, AE, Grinspoon, SK, Klibanski, A, et al. Eating disorders. N Engl J Med. 1999;340:1092–1098.
36. Attia, E, Walsh, BT. Anorexia nervosa. Am J Psychiatry. 2007;164:1805–1810.
37. First, MB, Spitzer, RL, Gibbon, M, et al. Structured Clinical Interview for DSM-IV Axis I Disorders. New York: Biometrics Research Department, New York State Psychiatric Institute; 1996.
38. Eddy, KT, Dorer, DJ, Franko, DL, et al. Diagnostic crossover in anorexia nervosa and bulimia nervosa: implications for DSM-V. Am J Psychiatry. 2008;165:245–250.
39. Favaro, A, Santonastaso, P. The value of anorexia nervosa subtypes. Am J Psychiatry. 2008;165:772–773.
40. Bulik, CM, Sullivan, PF, Kendler, KS. An empirical study of the classification of eating disorders. Am J Psychiatry. 2000;157:886–895.
41. Nielsen, S, Palmer, B. Diagnosing eating disorders—AN, BN and the others. Acta Psychiatr Scand. 2003;108:161–162.
42. Herzog, DB, Keller, MB, Sacks, NR, et al. Psychiatric comorbidity in treatment-seeking anorexics and bulimics. J Am Acad Child Adolesc Psychiatry. 1992;31:810–818.
43. Braun, DL, Sunday, SR, Halmi, KA. Psychiatric comorbidity in patients with eating disorders. Psychol Med. 1994;24:859–867.
44. Holderness, CC, Brooks-Gunn, J, Warren, WP. Co-morbidity of eating disorders and substance abuse: review of the literature. Int J Eat Disord. 1994;16:1–34.
45. Brewerton, TD, Lydiard, RB, Herzog, DB, et al. Comorbidity of Axis I psychiatric diagnosis in bulimia nervosa. J Clin Psychiatry. 1995;56:77–80.
46. Godart, NT, Flament, MF, Curt, F, et al. Anxiety disorders in subjects seeking treatment for eating disorders: a DSM-IV controlled study. Psychiatry Res. 2003;117:245–258.
47. Halmi, KA, Sunday, SR, Klump, KL, et al. Obsessions and compulsions in anorexia nervosa subtypes. Intl J Eat Disord. 2003;33:308–319.
48. Berkman, ND, Lohr, KN, Bulik, CM. Outcomes of eating disorders: a systematic review of the literature. Int J Eat Disord. 2007;40:293–309.
49. Matsunaga, H, Kiriike, N, Iwasaki, Y, et al. Clinical characteristics in patients with anorexia nervosa and obsessive-compulsive disorder. Psychol Med. 1999;29:407–414.
50. Srinivasagam, NM, Plotnicov, KH, Greeno, C, et al. Persistent perfectionism, symmetry, and exactness in anorexia nervosa after long-term recovery. Am J Psychiatry. 1995;152:1630–1634.
51. Kaye, WH, Greeno, CG, Moss, H, et al. Alterations in serotonin activity and psychiatric symptomatology after recovery from bulimia nervosa. Arch Gen Psychiatry. 1998;55:927–935.
52. Anderluh, MB, Tchanturia, K, Rabe-Hesketh, S, et al. Childhood obsessive-compulsive personality traits in adult women with eating disorders: defining a broader eating disorder phenotype. Am J Psychiatry. 2003;160:242–247.
53. Fernandez-Arand, F, Pinheiro, AP, Thornton, LM, et al. Impulse control disorders in women with eating disorders. Psychiatry Res. 2008;157:147–157.
54. Leckman, JF, Grice, DE, Boardman, J, et al. Symptoms of obsessive-compulsive disorder. Am J Psychiatry. 1997;154:911–917.
55. Ward, A, Tiller, J, Treasure, J, et al. Eating disorders: psyche or soma? Int J Eat Disord. 2000;27:279–287.
56. Klump, KL, Kaye, WH, Strober, M. The evolving genetic foundations of eating disorders. Psychiatr Clin North Am. 2001;24:215–225.
57. Tozzi, F, Bulik, CM. Candidate genes in eating disorders. Curr Drug Targets—CNS Neurol Disord. 2003;2:31–39.
58. Stein, D, Lilenfeld, LR, Plotnicov, K, et al. Familial aggregation of eating disorders: results from a controlled family study of bulimia nervosa. Intl J Eat Disord. 1999;26:211–215.
59. Strober, M, Freeman, R, Lampert, C, et al. Controlled family study of anorexia nervosa and bulimia nervosa: evidence of shared liability and transmission of partial syndromes. Am J Psychiatry. 2000;157:393–401.
60. Strober, M, Freeman, R, Lampert, C, et al. Males with anorexia nervosa: a controlled study of eating disorders in first-degree relatives. Intl J Eat Disord. 2001;29:263–269.
61. Klump, KL, Gobrogge, KL. A review and primer of molecular genetic studies of anorexia nervosa. Int J Eat Disord. 2005;37:S43–S48.
62. Bulik, CM. Exploring the gene-environment nexus in eating disorders. J Psychiatry Neurosci. 2005;30:335–339.
63. Klump, KL, Burt, SA, McGue, M, et al. Changes in genetic and environmental influences on disordered eating across adolescence. Arch Gen Psychiatry. 2007;64:1409–1415.
64. Grice, DE, Halmi, KA, Fichter, MM, et al. Evidence for a susceptibility gene for anorexia nervosa on chromosome 1. Am J Hum Genet. 2002;70:787–792.
65. Devlin, B, Bacanu, SA, Klump, KL, et al. Linkage analysis of anorexia nervosa incorporating behavioral covariates. Hum Mol Genet. 2002;11:689–696.
66. Bulik, CM, Devlin, B, Bacanu, SA, et al. Significant linkage on chromosome 10p in families with bulimia nervosa. Am J Hum Genet. 2003;72:200–207.
67. Bergen, AW, Yeager, M, Welch, R, et al. Candidate gene analysis of the Price Foundation anorexia nervosa affected relative pair dataset. Curr Drug Targets—CNS Neurol Disord. 2003;2:41–51.
68. Kaye, WH, Frank, GK, Bailer, UF, et al. The neurobiology of anorexia nervosa: clinical implications of alterations of the function of serotonin and other neuronal systems. Int J Eat Disord. 2005;37:S15–S19.
69. Lask, B, Gordon, I, Christie, D, et al. Functional neuroimaging in early onset anorexia nervosa. Int J Eat Disord. 2005;37:S49–S51.
70. Ramacciotti, CE, Coli, E, Paoli, R, et al. Serotonergic activity measured by platelet [3H]paroxetine binding in patients with eating disorders. Psychiatry Res. 2003;118:33–38.
71. de Krom, M, Hendriks, J, Hillebrand, J, et al. A polymorphism in the 3′ untranslated region of the CCK gene is associated with anorexia nervosa in Dutch patients. Psychiat Genet. 2006;16:239.
72. Comerci, GD. Medical complications of anorexia nervosa and bulimia nervosa. Med Clin North Am. 1990;74:1293–1310.
73. Mitchell, JE, Crow, S. Medical complications in adolescents with anorexia nervosa: a review of the literature. Curr Opin Psychiatry. 2006;19:438–443.
74. Casper, RC. Carbohydrate metabolism and its regulatory hormones in anorexia nervosa. Psychiatry Res. 1996;62:85–96.
75. Mitan, LAP. Menstrual dysfunction in anorexia nervosa. J Pediatr Adolesc Gynecol. 2004;17:81–85.
76. Cook, VJ, Coxson, HO, Mason, AG, et al. Bullae, bronchiectasis and nutritional emphysema in severe anorexia nervosa. Canad Resp J. 2001;8:361–365.
77. Fisher, M. Treatment of eating disorders in children, adolescents, and young adults. Pediatr Rev. 2006;27:5–16.
78. Hadley, SJ, Walsh, BT. Gastrointestinal disturbances in anorexia nervosa and bulimia nervosa. Curr Drug Targets—CNS Neurol Disord. 2003;2:1–9.
79. Katzman, DK. Osteoporosis in anorexia nervosa: a brittle future? Curr Drug Targets—CNS Neurol Disord. 2003;2:11–15.
80. Soyka, LA, Misra, M, Frenchman, A, et al. Abnormal bone mineral accrual in adolescent girls with anorexia nervosa. J Clin Endocrinol Metab. 2002;87:4177–4185.
81. Misra, M, Prabhakaran, R, Miller, KK, et al. Weight gain and restoration of menses as predictors of bone mineral density change in adolescent girls with anorexia nervosa-1. J Clin Endocrinol Metab. 2008;93:1231–1237.
82. Misra, M, Prabhakaran, R, Miller, KK, et al. Prognostic indicators of changes in bone density measures in adolescent girls with anorexia nervosa-2. J Clin Endocrinol Metab. 2008;93:1292–1297.
83. Katzman, DK. Medical complications in adolescents with anorexia nervosa: a review of the literature. Int J Eat Disord. 2005;37(Suppl):S52–S59.
84. Kerem, NC, Katzman, DK. Brain structure and function in adolescents with anorexia nervosa. Adol Med. 2003;14:109–118.
85. Barbarich, NC, Kaye, WH, Jimerson, D. Neurotransmitter and imaging studies in anorexia nervosa: new targets for treatment. Curr Drug Targets—CNS Neurol Disord. 2003;2:61–72.
86. Hendren, RL, De Backer, I, Pandina, GJ. Review of neuroimaging studies of child and adolescent psychiatric disorders from the past 10 years. J Am Acad Child Adolesc Psychiatry. 2000;39:815–828.
87. Neumarker, KJ, Bzufka, WM, Dudek, U, et al. Are there specific disabilities of number processing in adolescent patients with anorexia nervosa? Evidence from clinical and neuropsychological data when compared to morphometric measures from magnetic resonance imaging. Eur Child Adolesc Psychiat. 2000;9(Suppl 2):II111–II121.
88. Drevelengas, A, Chourmouzi, D, Pitsavas, G, et al. Reversible brain atrophy and subcortical high signal on MRI in a patient with anorexia nervosa. Neuroradiology. 2001;43:838–840.
89. Mühlau, M, Gaser, C, Ilg, R, et al. Gray matter decrease of the anterior cingulate cortex in anorexia nervosa. Am J Psychiatry. 2007;164:1850–1857.
90. Katzman, DK, Lambe, EK, Mikulis, DJ, et al. Cerebral gray matter and white matter volume deficits in adolescent girls with anorexia nervosa. J Pediatr. 1996;129:794–803.
91. Wagner, A, Greer, P, Bailer, UF, et al. Normal brain tissue volumes after long-term recovery in anorexia and bulimia nervosa. Biol Psychiatry. 2006;59:291–293.
92. Roser, W, Bubl, R, Buergin, D, et al. Metabolic changes in the brain of patients with anorexia and bulimia nervosa as detected by proton magnetic resonance spectroscopy. Int J Eat Disord. 1999;26:119–136.
93. Rzanny, R, Freesmeyer, D, Reichenbach, JR, et al. 31P-MR spectroscopy of the brain in patients with anorexia nervosa: characteristic differences in the spectra between patients and healthy control subjects. ROFO–Fortschr Gebiet Rontgenstr Bildgebend V. 2003;175:75–82.
94. Kuruoglu, AC, Kapucu, O, Atasever, T, et al. Technitium-99m-HMPAO brain SPECT in anorexia nervosa. J Nucl Med. 1998;39:304–306.
95. Kojima, S, Nagai, N, Nakabeppu, Y, et al. Comparison of regional cerebral blood flow in patients with anorexia nervosa before and after weight gain. Psychiatry Res. 2005;30:251–258.
96. Wagner, A, Aizenstein, H, Venkatraman, VK, et al. Altered reward processing in women recovered from anorexia nervosa. Am J Psychiatry. 2007;164:1842–1849.
97. Wagner, A, Aizenstein, H, Mazurkewicz, L, et al. Altered insula response to taste stimuli in individuals recovered from restricting-type anorexia nervosa. Neuropsychopharmacology. 2008;33:513–523.
98. Ellison, Z, Foong, J, Howard, R, et al. Functional anatomy of calorie fear in anorexia nervosa. Lancet. 1998;352:1192.
99. Uher, R, Brammer, MJ, Murphy, T. Recovery and chronicity in anorexia nervosa: brain activity associated with differential outcomes. Biol Psychiatry. 2003;54:934–942.
100. Audenaert, K, Van Laere, K, Dumont, F, et al. Decreased 5-HT2a receptor binding in patients with anorexia nervosa. J Nucl Med. 2003;44:163–169.
101. Kaye, W, Frank, G, Bailer, UF, et al. Serotonin alterations in anorexia and bulimia nervosa: new insights from imaging studies. Physiol Behav. 2005;85:73–81.
102. Krassas, GE. Endocrine abnormalities in anorexia nervosa. Pediatr Endocrinol Rev. 2003;1:46–54.
103. Södersten, P, Bergh, C, Zandian, M. Psychoneuroendocrinology of anorexia nervosa. Psychoneuroendocrinology. 2006;31:1149–1153.
104. Torsello, A, Brambilla, F, Tamiazzo, L, et al. Central dysregulations in the control of energy homeostasis and endocrine alterations in anorexia and bulimia nervosa. J Endocrinol Invest. 2007;30:962–976.
105. Monteleone, P, Castaldo, E, Maj, M. Neuroendocrine dysregulation of food intake in eating disorders. Regul Pept. 2008.
106. Paz-Filho GJ, Licinio J: Anorexia nervosa and bulimia nervosa. In Pfaff D, Arnold AP, Etgen AM, Fahrbach SE, Rubin RT, editors: Hormones, Brain and Behavior, ed 2, Oxford, Elsevier (in press).
107. Pirke, KM, Pahl, J, Schweiger, U. Metabolic and endocrine indices of starvation in bulimia: a comparison with anorexia nervosa. Psychiatry Res. 1985;15:33–39.
108. Judge, BS, Eisenga, BH. Disorders of fuel metabolism: medical complications associated with starvation, eating disorders, dietary fads, and supplements. Emerg Med Clin N Am. 2005;23:789–813.
109. Vande Wiele, RI. Anorexia nervosa and the hypothalamus. Hosp Pract. 1977;12:45–51.
110. Fichter, MM, Pirke, KM. Starvation models and eating disorders. In: Szmukler G, Dare C, Treasure J, eds. Handbook of Eating Disorders: Theory, Treatment and Research. West Sussex, England: Wiley and Sons; 1995:83–107.
111. Licinio, J, Wong, ML, Gold, PW. The hypothalamic-pituitary-adrenal axis in anorexia nervosa. Psychiatry Res. 1996;62:75–83.
112. Kaye, WH, Gendall, K, Kye, C. The role of the central nervous system in the psychoneuroendocrine disturbances of anorexia and bulimia nervosa. Psychiatr Clin North Am. 1998;21:381–396.
113. Tomova, A, Kumanov, P, Kirilov, G. Factors related to sex hormone binding globulin concentrations in women with anorexia nervosa. Horm Metab Res. 1995;27:508–510.
114. Fichter, MM, Pirke, KM, Pollinger, J, et al. Disturbances in the hypothalamo-pituitary-adrenal and other neuroendocrine axes in bulimia. Biol Psychiatry. 1990;27:1021–1037.
115. Kaye, WH. Neuropeptide abnormalities in anorexia nervosa. Psychiatry Res. 1996;62:65–74.
116. Rubin, RT, Poland, RE, Lesser, IM, et al. Neuroendocrine aspects of primary endogenous depression. I. Cortisol secretory dynamics in patients and matched control subjects. Arch Gen Psychiatry. 1987;44:329–336.
117. Rubin, RT, Carroll, BJ. The neuroendocrinology of mood disorders. In Pfaff D, Arnold AP, Etgen AM, Fahrbach SE, Rubin RT, eds.: Hormones, Brain and Behavior, Second Edition, Oxford: Elsevier, 2009.
118. Rubin, RT, Phillips, JJ, McCracken, JT, et al. Adrenal gland volume in major depression: Relationship to basal and stimulated pituitary-adrenal cortical axis function. Biol Psychiatry. 1996;40:89–97.
119. Demitrack, MA, Kalogeras, KT, Altemus, M, et al. Plasma and cerebrospinal fluid measures of arginine vasopressin secretion in patients with bulimia nervosa and in healthy subjects. J Clin Endocrinol Metab. 1992;74:1277–1283.
120. Antoni, FA. Vasopressinergic control of pituitary adrenocorticotropin secretion comes of age. Front Neuroendocrinol. 1993;14:76–122.
121. Scott, LV, Dinan, TG. Vasopressin and the regulation of hypothalamic-pituitary-adrenal axis function: implications for the pathophysiology of depression. Life Sci. 1998;62:1985–1998.
122. Connan, F, Lightman, SL, Landau, S, et al. An investigation of hypothalamic-pituitary-adrenal axis hyperactivity in anorexia nervosa: the role of CRH and AVP. J Psychiatr Res. 2007;41:131–143.
123. Monteleone, P, Luisi, M, Colurcio, B, et al. Plasma levels of neuroactive steroids are increased in untreated women with anorexia nervosa or bulimia nervosa. Psychosom Med. 2001;63:62–68.
124. Natori, Y, Yamaguchi, N, Koike, S, et al. Thyroid function in patients with anorexia nervosa and depression. Rinsho Byori. 1994;42:1268–1272.
125. Altemus, M, Hetherington, M, Kennedy, B, et al. Thyroid function in bulimia nervosa. Psychoneuroendocrinology. 1996;21:249–261.
126. Chopra, IJ. Clinical review 86: euthyroid sick syndrome: is it a misnomer? J Clin Endocrinol Metab. 1997;82:329–334.
127. De Groot, LJ. Dangerous dogmas in medicine: the nonthyroidal illness syndrome. J Clin Endocrinol Metab. 1999;8:151–164.
128. De Groot, LJ. Non-thyroidal illness syndrome is a manifestation of hypothalamic-pituitary dysfunction, and in view of current evidence, should be treated with appropriate replacement therapies. Crit Care Clin. 2006;22:57–86.
129. Adler, SM, Wartofsky, L. The nonthyroidal illness syndrome. Endocrinol Metab Clin N Am. 2007;36:657–672.
130. Lesem, MD, Kaye, WH, Bissette, G, et al. Cerebrospinal fluid TRH immunoreactivity in anorexia nervosa. Biol Psychiatry. 1994;35:48–53.
131. Devlin, MJ, Walsh, BT, Kral, JG, et al. Metabolic abnormalities in bulimia nervosa. Arch Gen Psychiatry. 1990;47:144–148.
132. Altemus, M, Hetherington, MM, Flood, M, et al. Decrease in resting metabolic rate during abstinence from bulimic behavior. Am J Psychiatry. 1991;148:1071–1072.
133. Spalter, AR, Gwirtsman, HE, Demitrack, MA, et al. Thyroid function in bulimia nervosa. Biol Psychiatry. 1993;33:408–414.
134. Gianotti, L, Lanfranco, F, Ramunni, J, et al. GH/IGF-I axis in anorexia nervosa. Eat Weight Disord. 2002;7:94–105.
135. Scacchi, M, Pincelli, AI, Cavagnini, F. Nutritional status in the neuroendocrine control of growth hormone secretion: the model of anorexia nervosa. Front Neuroendocrinol. 2003;24:200–204.
136. Hadigan, CM, Walsh, BT, Buttinger, C, et al. Behavioral and neuroendocrine responses to metaCPP in anorexia nervosa. Biol Psychiatry. 1995;37:504–511.
137. Brewerton, TD, Jimerson, CD. Studies of serotonin function in anorexia nervosa. Psychiatry Res. 1996;62:31–42.
138. Goldbloom, DS, Garfinkel, PE, Katz, R, et al. The hormonal response to intravenous 5-hydroxytryptophan in bulimia nervosa. J Psychosom Res. 1996;40:289–297.
139. Jimerson, DC, Wolfe, BE, Metzger, ED, et al. Decreased serotonin function in bulimia nervosa. Arch Gen Psychiatry. 1997;54:529–534.
140. Levitan, RD, Kaplan, AS, Joffe, RT, et al. Hormonal and subjective responses to intravenous meta-chlorophenylpiperazine in bulimia nervosa. Arch Gen Psychiatry. 1997;54:521–527.
141. Monteleone, P, Brambilla, F, Bortolotti, F, et al. Prolactin response to d-fenfluramine is blunted in people with anorexia nervosa. Br J Psychiatry. 1998;172:439–442.
142. Monteleone, P, Brambilla, F, Bortolotti, F, et al. Plasma prolactin response to d-fenfluramine is blunted in bulimic patients with frequent binge episodes. Psychol Med. 1998;28:975–983.
143. Mortola, JF, Laughlin, GA, Yen, SS. Melatonin rhythms in women with anorexia nervosa and bulimia nervosa. J Clin Endocrinol Metab. 1993;77:1540–1544.
144. Kennedy, SH. Melatonin disturbances in anorexia nervosa and bulimia nervosa. Int J Eat Disord. 1994;16:257–265.
145. Hoffmann, G, Pollow, K, Nowara, D, et al. Circadian blood serotonin and melatonin level in anorexia nervosa patients in comparison with normally menstruating women. Geburtshilfe Frauenheilkd. 1996;56:485–490.
146. Ferrari, E, Magri, F, Pontiggia, B, et al. Circadian neuroendocrine functions in disorders of eating behavior. Eat Weight Disord. 1997;2:196–202.
147. Pirke, KM, Schweiger, U, Lemmel, W, et al. The influence of dieting on the menstrual cycle of healthy young women. J Clin Endocrinol Metab. 1985;60:1174–1179.
148. Fichter, MM, Pirke, KM, Holsboer, F. Weight loss causes neuroendocrine disturbances: experimental studies in healthy starving subjects. Psychiatry Res. 1986;17:61–72.
149. Keys, A, Brozek, J, Henschel, A. The Biology of Human Starvation. Minneapolis: University of Minnesota Press; 1950.
150. Turtzo, LC, Lane, MD. Completing the loop: neuron-adipocyte interactions and the control of energy homeostasis. Horm Metab Res. 2002;34:607–615.
151. Kalra, SP, Dube, MG, Pu, S, et al. Interacting appetite-regulating pathways in the hypothalamic regulation of body weight. Endocr Rev. 1999;20:68–100.
152. Luckman, SM, Lawrence, CB. Anorectic brainstem peptides: more pieces to the puzzle. Trends Endocr Metab. 2003;14:60–65.
153. Taylor, MM, Samson, WK. The other side of the orexins: endocrine and metabolic actions. Am J Physiol Endocrinol Metab. 2003;284:E13–E17.
154. Zigman, JM, Elmquist, JK. Minireview: from anorexia to obesity—the yin and yang of body weight control. Endocrinology. 2003;144:3749–3756.
155. Bailer, UF, Kaye, WH. A review of neuropeptide and neuroendocrine dysregulation in anorexia and bulimia nervosa. Curr Drug Targets—CNS Neurol Disord. 2003;2:53–59.
156. Rajala, MW, Scherer, PE. Minireview: the adipocyte—at the crossroads of energy homeostasis, inflammation, and atherosclerosis. Endocrinology. 2003;144:3765–3773.
157. Jimerson, DC, Wolfe, BE. Neuropeptides in eating disorders. CNS Spectr. 2004;9:516–522.
158. Germain, N, Galusca, B, Le Roux, CW, et al. Constitutional thinness and lean anorexia nervosa display opposite concentrations of peptide YY, glucagon-like peptide 1, ghrelin, and leptin. Am J Clin Nutr. 2007;85:967–971.
159. Kishi, T, Elmquist, JK. Body weight is regulated by the brain: a link between feeding and emotion. Mol Psychiatry. 2005;10:132–146.
160. Batterham, RL, Bloom, SR. The gut hormone peptide YY regulates appetite. Ann NY Acad Sci. 2003;994:162–168.
161. Elmquist, JK, Maratos-Flier, E, Saper, CB, et al. Unraveling the central nervous system pathways underlying leptin. Nature Neuroscience. 1998;1:445–450.
162. Baratta, M, Leptin—from a signal of adiposity to a hormonal mediator in peripheral tissues. Med Sci Monit 2002;8:RA282–RA292. http://www.MedSciMonit.com/pub/vol_8/no_12/2990.pdf.
163. Considine, RV, Sinha, M, Heiman, ML, et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N Engl J Med. 1996;334:292–295.
164. Hamann, A, Matthaei, S. Regulation of energy balance by leptin. Exp Clin Endocrinol Diabetes. 1996;104:293–300.
165. Brichard, SM, Delporte, ML, Lambert, M. Adipocytokines in anorexia nervosa: a review focusing on leptin and adiponectin. Horm Metab Res. 2003;35:337–342.
166. Chan, L, Mantzoros, CS. Role of leptin in energy-deprivation states: normal human physiology and clinical implications for hypothalamic amenorrhoea and anorexia nervosa. Lancet. 2005;366:74–85.
167. Hebebrand, J, Muller, TD, Holtkamp, K, et al. The role of leptin in anorexia nervosa: clinical implications. Mol Psychiatry. 2007;12:23–35.
168. Holtkamp, K, Herpertz-Dehlman, B, Mika, C, et al. Elevated physical activity and low leptin levels co-occur in patients with anorexia nervosa. J Clin Endocrinol Metab. 2003;88:5169–5174.
169. Lob, S, Pickel, J, Bidlingmaier, M, et al. Serum leptin monitoring in anorectic patients during refeeding therapy. Exp Clin Endocrinol Diabetes. 2003;111:278–282.
170. Holtkamp, K, Hebebrand, J, Mika, C, et al. High serum leptin levels subsequent to weight gain predict renewed weight loss in patients with anorexia nervosa. Psychoneuroendocrinology.. 2004;29:791–797.
171. Steiner, J, LaPaglia, N, Kirsteins, L, et al. The response of the hypothalamic-pituitary-gonadal axis to fasting is modulated by leptin. Endocrine Res. 2003;29:107–117.
172. Blüher, S, Mantzoros, CS. The role of leptin in regulating neuroendocrine function in humans. J Nutr. 2004;134:2469S–2474S.
173. Welt, CK, Chan, JL, Bullen, J, et al. Recombinant human leptin in women with hypothalamic amenorrhea. N Engl J Med.. 2004;35:787–797.
174. Baranowska, B, Baranowska-Bik, A, Bik, W, Martynska, L. The role of leptin and orexins in the dysfunction of hypothalamo-pituitary-gonadal regulation and in the mechanism of hyperactivity in patients with anorexia nervosa. Neuro Endocrinol Lett. 2008;29:37–40.
175. Inui, A. Feeding and body-weight regulation by hypothalamic neuropeptides—mediation of the actions of leptin. Trends Neurosci. 1999;22:62–67.
176. Stefan, N, Stumvoll, M. Adiponectin—its role in metabolism and beyond. Horm Metab Res. 2002;34:469–474.
177. Beltowski, J. Adiponectin and resistin—new hormones of white adipose tissue. Med Sci Monit. 2003;9:RA55–RA61.
178. Nedvídková, J, Smitka, K, Kopský, V, et al. Adiponectin, an adipocyte-derived protein. Physiol Res. 2005;54:133–140.
179. Gavrila, A, Chan, JL, Yiannakouris, N, et al. Serum adiponectin levels are inversely associated with overall and central fat distribution but are not directly regulated by acute fasting or leptin administration in humans: cross-sectional and interventional studies. J Clin Endocrinol Metab. 2003;88:4823–4831.
180. Ott, V, Fasshauer, M, Dalski, A, et al. Direct peripheral effects of ghrelin include suppression of adiponectin expression. Horm Metab Res. 2002;34:640–645.
181. Pannacciulli, N, Vettor, R, Milan, G, et al. Anorexia nervosa is characterized by increased adiponectin plasma levels and reduced nonoxidative glucose metabolism. J Clin Endocrinol Metab. 2003;88:1748–1752.
182. Iwahashi, H, Funahashi, T, Kurokawa, N, et al. Plasma adiponectin levels in women with anorexia nervosa. Horm Metab Res. 2003;35:537–540.
183. Tagami, T, Satoh, N, Usui, T, et al. Adiponectin in anorexia nervosa and bulimia nervosa. J Clin Endocrinol Metab. 2004;89:1833–1837.
184. Housova, J, Anderlova, K, Krizová, J, et al. Serum adiponectin and resistin concentrations in patients with restrictive and binge/purge form of anorexia nervosa and bulimia nervosa. J Clin Endocrinol Metab. 2005;90:1366–1370.
185. Monteleone, P, Fabrazzo, M, Martiadis, V, et al. Opposite changes in circulating adiponectin in women with bulimia nervosa or binge eating disorder. J Clin Endocrinol Metab. 2003;88:5387–5391.
186. Dostálová, I, Smitka, K, Papezová, H, et al. Increased insulin sensitivity in patients with anorexia nervosa: the role of adipocytokines. Physiol Res. 2007;56:587–594.
187. Broglio, F, Gottero, C, Arvat, E, et al. Endocrine and non-endocrine actions of ghrelin. Horm Res. 2003;59:109–117.
188. Hosoda, H, Kohima, M, Kangawa, K. Biological, physiological, and pharmacological aspects of ghrelin. J Pharmacol Sci. 2006;100:398–410.
189. Nonogaki, K. Ghrelin and feedback systems. Vitam Horm. 2008;77:149–170.
190. Tolle, V, Kadem, M, Bluet-Pajot, M-T, et al. Balance in ghrelin and leptin plasma levels in anorexia nervosa patients and constitutionally thin women. J Clin Endocrinol Metab. 2003;88:109–116.
191. Tanaka, M, Naruo, T, Yasuhara, D, et al. Fasting plasma ghrelin levels in subtypes of anorexia nervosa. Psychoneuroendocrinology. 2003;28:829–835.
192. Monteleone, P, Fabrazzo, M, Tortorella, A, et al. Circulating ghrelin is decreased in non-obese and obese women with binge eating disorder as well as in obese non-binge eating women, but not in patients with bulimia nervosa. Psychoneuroendocrinology. 2005;30:243–250.
193. Nakai, Y, Hosoda, H, Nin, K, et al. Plasma levels of active form of ghrelin during oral glucose tolerance test in patients with anorexia nervosa. Eur J Endocrinol. 2003;149:R1–R3.
194. Nedvídková, J, Krykorková, I, Barták, V, et al. Loss of meal-induced decrease in plasma ghrelin levels in patients with anorexia nervosa. J Clin Endocrinol Metab. 2003;88:1678–1682.
195. Akamizu, T, Kangawa, K. Emerging results of anticatabolic therapy with ghrelin. Curr Opin Clin Nutr Metab Care. 2007;10:278–283.
196. Moran, TH, Kinzig, KP. Gastrointestinal satiety signals II. Cholecystokinin. Am J Physiol Gastrointest Liver Physiol. 2004;286:G183–G188.
197. Rehfeld, JE. Clinical endocrinology and metabolism. Cholecystokinin. Best Pract Res Clin Endocrinol Metab. 2004;18:569–586.
198. Tomasik, PJ, Sztefko, K, Starzyk, J. Cholecystokinin, glucose dependent insulinotropic peptide and glucagon-like peptide 1 secretion in children with anorexia nervosa and simple obesity. J Pediatr Endocrinol Metab. 2004;17:1623–1631.
199. Jimerson, DC, Wolfe, BE. Neuropeptides in eating disorders. CNS Spectr. 2004;9:516–522.
200. Zipfel, S, Sammet, I, Rapps, N, et al. Gastrointestinal disturbances in eating disorders: clinical and neurobiological aspects. Auton Neurosci. 2006;129:99–106.
201. Keel, PK, Wolfe, BE, Liddle, RA, et al. Clinical features and physiological response to a test meal in purging disorder and bulimia nervosa. Arch Gen Psychiatry. 2007;64:1058–1066.
202. Adan, RAH, Hillebrand, JJG, de Rijke, C, et al. Melanocortin system and eating disorders. Ann NY Acad Sci. 2003;994:267–274.
203. Inui, A. Melanocortin signaling, single nucleotide polymorphism, and eating disorders. Intl J Psychiatry Med. 2004;34:399–403.
204. Foster, AC, Joppa, M, Markison, S, et al. Body weight regulation by selective MC4 receptor agonists and antagonists. Ann NY Acad Sci. 2003;994:103–110.
205. Moriya, J, Takimoto, Y, Yoshiuchi, K, et al. Plasma agouti-related protein levels in women with anorexia nervosa. Psychoneuroendocrinology. 2006;31:1057–1061.
206. de Rijke, CE, Jackson, PJ, Garner, KM, et al. Functional analysis of the Ala67Thr polymorphism in agouti related protein associated with anorexia nervosa and leanness. Biochem Pharmacol. 2005;70:308–316.
207. Lu, X-Y, Barsh, GS, Akil, H, Watson, SJ. Interaction between α-melanocyte-stimulating hormone and corticotropin-releasing hormone in the regulation of feeding and hypothalamo-pituitary-adrenal responses. J Neurosci. 2003;23:7863–7872.
208. Masaki, T, Yoshimichi, G, Chiba, S, et al. Corticotropin-releasing hormone-mediated pathway of leptin to regulate feeding, adiposity, and uncoupling protein expression in mice. Endocrinology. 2003;144:3547–3554.
209. Sawchenko, P. Toward a new neurobiology of energy balance, appetite, and obesity: the anatomists weigh in. J Comp Neurol. 1998;402:435–441.
210. Heisler, LK, Cowley, MA, Kishi, T, et al. Central serotonin and melanocortin pathways regulating energy homeostasis. Ann NY Acad Sci. 2003;994:169–174.
211. Nishita, JK, Ellinwood, EH, Jr., Rockwell, WJ, et al. Abnormalities in the response of plasma arginine vasopressin during hypertonic saline infusion in patients with eating disorders. Biol Psychiatry. 1989;26:73–86.
212. Frank, GK, Kaye, WH, Altemus, M, et al. CSF oxytocin and vasopressin levels after recovery from bulimia nervosa and anorexia nervosa, bulimic subtype. Biol Psychiatry. 2000;48:315–318.
213. Bergen, MBM, van den Bree, M, Yeager, R, et al. Candidate genes for anorexia nervosa in the 1 p33–36 linkage region: serotonin 1D and delta opioid receptor loci exhibit significant association to anorexia nervosa. Mol Psychiatry. 2003;8:397–406.
214. Brown, KM, Bujac, SR, Mann, ET, et al. Further evidence of association of OPRD1 & HTR1D polymorphisms with susceptibility to anorexia nervosa. Biol Psychiatry. 2007;61:367–373.
215. Jonas, JM, Gold, MS. Treatment of antidepressant resistant bulimia with naltrexone. Int J Psychiatry Med. 1987;16:305–309.
216. Jonas, JM, Gold, MS. The use of opiate antagonists in treating bulimia: a study of low dose versus high dose naltrexone. Psychiatry Res. 1988;24:195–199.
217. Mitchell, JE, Christenson, G, Jennings, J, et al. A placebo-controlled, double-blind crossover study of naltrexone hydrochloride in outpatients with normal weight bulimia. J Clin Psychopharmacol. 1989;9:94–97.
218. Agger, SA, Schwalberg, MD, Bigaouette, JM, et al. Effect of a tricyclic antidepressant and opiate antagonist on binge eating behavior in normal weight bulimia and obese, binge-eating subjects. Am J Clin Nutr. 1991;53:865–871.
219. Marrazzi, MA, Bacon, JP, Kinzie, J, et al. Naltrexone use in the treatment of anorexia nervosa and bulimia nervosa. Int Clin Psychopharmacol. 1995;10:163–172.
220. Ratnasuriya, RH, Eisler, I, Szmuckler, GI, et al. Anorexia nervosa: outcome and prognostic factors after 20 years. Br J Psychiatry. 1991;158:495–502.
221. Garner DM, Garfinkel PE, eds. Handbook of Treatment for Eating Disorders, ed 2, New York: Guilford Press, 1997.
222. American Psychiatric Association. Practice guideline for the treatment of patients with eating disorders, ed 3. Am J Psychiatry. 2006;163(Jul Suppl):4–54.
223. Wilson, GT, Grilo, CM, Vitousek, KM. Psychological treatment of eating disorders. Am Psychologist. 2007;62:199–216.
224. Wilson, GT, Fairburn, CG. Cognitive treatments for eating disorders. J Consult Clin Psychol. 1993;61:261–269.
225. Kaye, W, Strober, M, Stein, D, et al. New directions in treatment research of anorexia nervosa and bulimia nervosa. Biol Psychiatry. 1999;45:1285–1292.
226. Shapiro, JR, Berman, ND, Brownley, KA, et al. Bulimia nervosa treatment: a systematic review of randomized controlled trials. Int J Eat Disord. 2007;40:321–336.
227. Bulik, CM, Berkman, ND, Brownley, KA, et al. Anorexia nervosa treatment: a systematic review of randomized controlled trials. Int J Eat Disord. 2007;40:310–320.
228. Lilenfeld, LR, Kaye, WH, Greeno, CG, et al. A controlled family study of anorexia nervosa and bulimia nervosa: psychiatric disorders in first-degree relatives and effects of proband comorbidity. Arch Gen Psychiatry. 1998;55:603–610.
229. Channon, S, de Silva, P, Hemsley, D, et al. A controlled trial of cognitive-behavioural and behavioural treatment of anorexia nervosa. Behav Res Ther. 1989;27:529–535.
230. Crisp, AH, Norton, K, Gowers, S, et al. A controlled study of the effect of therapies aimed at adolescent and family psychopathology in anorexia nervosa. Br J Psychiatry. 1991;159:325–333.
231. Treasure, J, Todd, G, Brolly, M, et al. A pilot study of a randomized trial of cognitive analytical therapy vs. educational behavioral therapy for adult anorexia nervosa. Behav Res Ther. 1995;33:363–367.
232. McIntosh, V, Jordan, J, Carter, F, et al. Three psychotherapies for anorexia nervosa: a randomized controlled trial. Am J Psychiatry. 2005;162:741–747.
233. Pike, KM, Walsh, BT, Vitousek, K, et al. Cognitive behavior therapy in the posthospitalization treatment of anorexia nervosa. Am J Psychiatry. 2003;160:2046–2049.
234. Ramsay, R, Ward, A, Treasure, J, Russell, GF. Compulsory treatment in anorexia nervosa. Short-term benefits and long-term mortality. Br J Psychiatry. 1999;175:147–153.
235. Watson, TL, Bowers, WA, Anderson, AE. Involuntary treatment of eating disorders. Am J Psychiatry. 2000;157:1806–1810.
236. McCallum, KE, Bruton, JR. The continuum of care in the treatment of eating disorders. Primary Psychiatry. 2003;10:48–54.
237. Guarda, AS, Heinberg, LJ. Inpatient and partial hospital approaches to the treatment of eating disorders. In: Thompson JK, ed. Handbook of Eating Disorders and Obesity. Hoboken, NJ: Wiley; 2004:297–322.
238. Kaye, W, Gendall, K, Strober, M, Nutrition, serotonin and behavior in anorexia and bulimia nervosa. Nestlé Nutrition Workshop Series Clinical and Performance Program. Fernstrom, JD, Uany, R, Arroyo, P, eds. Nestlé Nutrition Workshop Series Clinical and Performance Program, vol 5. Basel: Vevey/S. Karger, 2001:153–168.
239. Ferguson, CP, La Via, MC, Crossan, PJ, et al. Are serotonin selective reuptake inhibitors effective in underweight anorexia nervosa? Int J Eat Disord. 1999;25:11–17.
240. Walsh, BT, Kaplan, AS, Attia, E, et al. Fluoxetine after weight restoration in anorexia nervosa: a randomized controlled trial. JAMA. 2006;295:2605–2612.
241. Malina, A, Gaskill, J, McConaha, C. Olanzapine treatment of anorexia nervosa: a retrospective study. Int J Eat Disord. 2003;33:234–237.
242. Mondraty, N, Birmingham, CL, Touyz, S, et al. Randomized controlled trial of olanzapine in the treatment of cognitions in anorexia nervosa. Australas Psychiatry. 2005;13:72–75.
243. Bissada, H, Tasca, GA, Barber, AM, et al. Olanzapine in the treatment of low body weight and obsessive thinking in women with anorexia nervosa: a randomized, double-blind, placebo-controlled trial. Am J Psychiatry. 2008 Jun 16.
244. Ward, A, Campbell, IC, Brown, N, et al. Anorexia nervosa subtypes: differences in recovery. J Nerv Ment Dis. 2003;191:197–201.
245. Gordon, CM, Grace, E, Emans, SJ, et al. Effects of oral dehydroepiandrosterone on bone density in young women with anorexia nervosa: a randomized trial. J Clin Endocrinol Metab. 2002;87:4935–4941.
246. Hsu, LK, Rand, W, Sullivan, S, et al. Cognitive therapy, nutritional therapy and their combination in the treatment of bulimia nervosa. Psychol Med. 2001;31:871–879.
247. Mehler, PS. Bulimia nervosa. N Engl J Med. 2003;349:875–881.
248. Lilly, RZ. Bulimia nervosa. BMJ. 2003;327:380–383.
249. Fairburn, CG, Jones, R, Peveler, RC, et al. Psychotherapy and bulimia nervosa: the longer-term effects of interpersonal psychotherapy, behavior therapy and cognitive-behavior therapy. Arch Gen Psychiatry. 1993;50:419–428.
250. Gendall, KA, Joyce, PR, Carter, FA, et al. Thyroid indices and treatment outcome in bulimia nervosa. Acta Psychiatr Scand. 2003;108:190–195.
251. Carter, WP, Pindyck, LJ. Pharmacologic treatment of binge-eating disorder. Primary Psychiatry. 2003;10:31–36.
252. McElroy, SL, Arnold, LM, Shapira, NA, et al. Topiramate in the treatment of binge eating disorder associated with obesity: a randomized, placebo-controlled trial. Am J Psychiatry. 2003;160:255–261.
253. Appolinario, JC, Bacaltchuk, J, Sichieri, R, et al. A randomized, double-blind placebo-controlled study of sibutramine in the treatment of binge-eating disorder. Arch Gen Psychiatry. 2003;60:1109–1116.
254. Grilo, CM, Masheb, RM, Salant, SL. Cognitive behavioral therapy guided self-help and orlistat for the treatment of binge eating disorder: a randomized, double-blind, placebo-controlled trial. Biol Psychiatry. 2005;57:1193–1201.
255. Steffen, KJ, Roerig, JL, Mitchell, JE, et al. Emerging drugs for eating disorder treatment. Expert Opin Emerging Drugs. 2006;11:315–336.
256. Wilfley, DE, Crow, SJ, Hudson, JI, et al. Efficacy of sibutramine for the treatment of binge eating disorder: a randomized multicenter placebo-controlled double-blind study. Am J Psychiatry. 2008;165:51–58.
[/level-membership-for-endocrinology-diabetes-and-metabolism-category][not-level-membership-for-endocrinology-diabetes-and-metabolism-category]Chapter 4
Anorexia Nervosa, Bulimia Nervosa, and Other Eating Disorders
“A young woman thus afflicted, her clothes scarcely hanging together on her anatomy, her pulse slow and slack, her temperature two degrees below the normal mean, her bowels closed, her hair like that of a corpse—dry and lustreless, her face and limbs ashy and cold, her hollow eyes the only vivid thing about her—this wan creature whose daily food might lie on a crown piece, will be busy with mother’s meetings, with little sister’s frocks, with university extension and with what you please else of unselfish effort, yet on what funds God only knows.”1
Anorexia nervosa (AN) and bulimia nervosa (BN) are eating disorders that have been recognized for hundreds of years, yet their etiologies remain poorly understood.2–9 Both illnesses carry significant physical and psychological morbidity, and in the case of AN, death can occur in severe, untreated cases. Early recognition and aggressive treatment thus are particularly important. Because the physical manifestations of these illnesses are so prominent, most patients have their first encounters with nonpsychiatric physicians. Lengthy diagnostic workups for underlying physical illness may be conducted, and a psychiatric disturbance may be considered only when the results of the workup do not fit a known physical illness. The careful application of psychiatric diagnostic criteria for AN and BN permits these illnesses to be suspected early in the medical workup and facilitates referral to the psychiatric specialist in a manner acceptable to the patient and family.
History and Epidemiology
AN and BN are diseases primarily of adolescent girls; approximately 95% of AN cases are female.10 Although there is documentary evidence of AN occurring in the Middle Ages, the first medical accounts appeared in the 17th century as A Discourse upon Prodigious Abstinence by John Reynolds in 1669 and Phthisiologia; or, a Treatise of Consumptions by Richard Morton in 1689.3,11,12 AN as a modern clinical entity derives from publications in 1873 by Charles Lasègue and William Gull, who referred to the illness as hysterical anorexia,13,14 and Gull’s publication in 1874 entitled Anorexia Nervosa.15 Bliss and Branch3 commented, “It is revealing to note the extraordinary differences in the description of the same condition by the two men. While Gull’s comments were as direct and precise as a pathologic report, Lasègue conveyed a sense of the spirit and feeling of these people, the nuances of their disturbed relationships, and the subtleties of their intrapsychic turmoil” (p. 14). The early 20th-century history of AN primarily involves its distinction from physical illnesses such as Simmonds’ cachexia.16
Although binge eating (bulimia) has long been recognized as part of the symptomatology of AN, BN as a distinct syndrome was first proposed by Russell in 1979.17 He elaborated two criteria for BN: an irresistible urge to overeat followed by self-induced vomiting or purging, and a morbid fear of becoming fat. In common with AN, such patients kept their body weight somewhat below normal but not to the same extent as AN patients. They also tended not to develop amenorrhea and were more active sexually. Russell considered BN to be an “ominous variant” of AN, in that comorbid depressive symptoms were often severe and distressing, leading to a high risk of suicide.
In the 1990s, there were opposing views about whether to treat binge eating disorder (BED) as a separate entity. Some argued that too little was known about it and other recurrent forms of overeating, and inclusion would be a source of diagnostic confusion. Others argued that there was a relatively pure group of individuals with Eating Disorder Not Otherwise Specified (EDNOS) who met criteria for BED and not BN, and that recognizing BED as a new disorder would stimulate research and clinical programs for these patients. Since 1994, BED became a provisional eating-disorder diagnosis and was included in the appendix of the fourth edition text revision of the Diagnostic and Statistical Manual of Mental Disorders (DSM-IV-TR).18
The reported incidence of AN has varied between about 0.5 and 15 cases per 100,000 population per year.19–21 The large variation is related to the diagnostic criteria used, the methods of case ascertainment, and the population studied (e.g., clinic versus community). The prevalence of AN ranges from 0.5% to 1.0% among females, with males being affected about one tenth as frequently, although the prevalence has been considerably higher in some studies.20,21 A meta-analysis of 42 studies revealed the crude mortality rate in AN (due to all causes of death) to be 5.9%, which is substantially greater than that for female inpatients and for the general population.22
The prevalence of BN varies between 2% and 4%.20,21,23,24 Both AN and BN, as well as subsyndromal anorexia and bulimia, are far more common in certain groups of young women, notably athletes and ballet dancers, where occupational demands place a premium on thinness. Primary and secondary amenorrheas in these individuals are extraordinarily common. Overweight and obesity are common comorbidities of BED, which has an estimated prevalence rate between 0.7% and 3%.25
Since the 1980s, large-scale twin and family studies have suggested that eating disorders are familial.26,27 There is cross-transmission between AN and BN, and they appear to share a common vulnerability, but the transmissible elements remain elusive. Twin and family studies suggest that major depressive disorder and substance dependence most likely do not share a common etiology with the eating disorders. The genetic contribution is likely predisposing rather than determining, in that sociocultural circumstances, as well as personal psychological stressors, are risk factors.28,29 The risk of developing AN is higher in female twins than in male twins, but male members of opposite-sex pairs have almost the same risk as in female twins.30 This suggests that an intrauterine factor, such as sex steroid hormones, also influences the development of AN.
Diagnosis
The DSM-IV-TR18 provides the current clinical criteria for AN (Table 4-1) and BN (Table 4-2) and EDNOS, which includes the provisional diagnostic category of BED. Since the DSM became a criterion-based system with DSM-III in 1980, it has undergone two revisions, and several diagnostic categories have undergone considerable change across these revisions. In contrast, the criteria for AN have remained relatively stable, although future revision might be useful, such as the elimination of amenorrhea as a criterion.31 The criteria for bulimia are in greater flux. For example, BED, characterized as recurrent episodes of binge eating and associated distress without inappropriate compensatory purging or other behaviors, has been included in the DSM-IV-TR Appendix as a set of criteria for further study.18 There have been recommendations to include BED as a distinct eating disorder in the upcoming DSM-V.32,33 Although there are far fewer men with eating disorders than women, the clinical features are similar.34
Table 4-1
DSM-IV-TR Diagnostic Criteria for Anorexia Nervosa
A Refusal to maintain body weight at or above a minimally normal weight for age and height (e.g., weight loss leading to maintenance of body weight less than 85% of that expected; or failure to make expected weight gain during period of growth, leading to body weight less than 85% of that expected).
B Intense fear of gaining weight or becoming fat, even though underweight.
C Disturbance in the way in which one’s body weight or shape is experienced, undue influence of body weight or shape on self-evaluation, or denial of the seriousness of the current low body weight.
D In postmenarcheal females, amenorrhea—i.e., the absence of at least three consecutive menstrual cycles. (A woman is considered to have amenorrhea if her periods occur only following hormone [e.g., estrogen] administration.)
Type
Restricting type: During the current episode … the person has not regularly engaged in binge-eating or purging behavior (i.e., self-induced vomiting or the misuse of laxatives, diuretics, or enemas).
Binge-eating/purging type: During the current episode … the person has regularly engaged in binge-eating or purging behavior (i.e., self-induced vomiting or the misuse of laxatives, diuretics, or enemas).
Reprinted with permission from American Psychiatric Association: Diagnostic and Statistical Manual of Mental Disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association, 2000.
Table 4-2
DSM-IV-TR Diagnostic Criteria for Bulimia Nervosa
A Recurrent episodes of binge eating. An episode of binge eating is characterized by both of the following:
(1) Eating, in a discrete period of time (e.g., within any 2-hour period), an amount of food that is definitely larger than most people would eat during a similar period of time and under similar circumstances.
(2) A sense of lack of control over eating during the episode (e.g., a feeling that one cannot stop eating or control what or how much one is eating).
B Recurrent inappropriate compensatory behavior in order to prevent weight gain, such as self-induced vomiting; misuse of laxatives, diuretics, enemas, or other medications; fasting; or excessive exercise.
C The binge eating and inappropriate compensatory behaviors both occur, on average, at least twice a week for 3 months.
D Self-evaluation is unduly influenced by body shape and weight.
E The disturbance does not occur exclusively during episodes of anorexia nervosa.
Type
Purging type: During the current episode … the person has regularly engaged in self-induced vomiting or the misuse of laxatives, diuretics, or enemas.
Nonpurging type: During the current episode … the person has used other inappropriate compensatory behaviors, such as fasting or excessive exercise, but has not regularly engaged in self-induced vomiting or the misuse of laxatives, diuretics, or enemas.
Reprinted with permission from American Psychiatric Association: Diagnostic and Statistical Manual of Mental Disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association, 2000.
The DSM-IV-TR criteria for AN, BN, and BED highlight the many identifiable behavioral and psychological aspects of these illnesses.35 If the patient and her family are queried about these aspects, considerable information pointing toward the diagnosis can be gleaned, psychiatric consultation can be considered early in the diagnostic process, and specialized and expensive laboratory testing often can be avoided.
For AN, a key feature of the weight loss is the patient’s refusal to maintain body weight, which often manifests as an active resistance to increasing caloric intake. As illustrated in Fig. 4-1, the cachexia can be severe, and as mentioned, self-starvation can lead to death. If the onset is in childhood or early adolescence, there may be a failure to make the expected weight gain during the active growth phase. This refusal to gain or maintain body weight is rooted psychologically in the second criterion, an intense fear of gaining weight or of becoming fat, even though the patient is underweight. There is a subjective distortion of body image such that the emaciated individual appears to herself to be either at an acceptable weight or even fat. Indeed, initiation of treatment by insisting on the patient’s eating can lead to purging behavior that did not occur prior to treatment. How inflexible a given patient may be in refusing to gain weight, however, can vary considerably.36
FIGURE 4-1 Extreme cachexia in an anorexia nervosa patient. (From Bliss EL, Branch CHH: Anorexia Nervosa: Its History, Psychology, and Biology. New York: Paul B. Hoeber, 1960.)
A corollary of this is the third criterion, disturbance of the experience of one’s body weight or shape (e.g., undue influence of body weight or shape on self-evaluation/self-esteem and/or denial of the seriousness of the weight loss), even though there may be clear adverse physical sequelae in addition to the current fourth criterion, secondary amenorrhea. For the endocrinologist, questions that should be asked of the patient and her family to address these psychological and behavioral aspects of AN include the following (adapted from the Structured Clinical Interview for DSM-IV37):
Why do you limit yourself to those foods?
Are you concerned that you might become fat if you ate more?
Do you weigh less than other people think you should weigh?
Do you need to be very thin in order to feel good about yourself?
Has anyone told you it can be dangerous to be as thin as you are?
What kinds of things do you do to keep from gaining weight?
Have you ever made yourself vomit or take laxatives, enemas, or water pills? How often?
Before now, were you having periods? Were they regular? When did they stop?
Rather than rigidly adhering to the DSM-IV-TR algorithm for diagnosing AN—that is, the reduction of body weight to less than 85% of expected and complete amenorrhea for at least 3 months—the clinician should consider both the patient’s core symptoms and current medical impairment; such impairment may require treatment even though body weight may be greater than 85% of expected and/or menses still may occur sporadically.31
For BN, the distinguishing features are uncontrollable binge eating of a definitely larger-than-normal amount of food (first criterion) that is clearly repetitive (third criterion). Here too, one’s sense of self is unduly influenced by weight and body shape (fourth criterion). Compensatory behaviors such as self-induced vomiting, purging, fasting between binges, and exercise are invoked to prevent weight gain (second criterion). The frequency and chronicity of the bingeing, and especially the compensatory behaviors, help distinguish BN from overeating in general. And because binge eating and purging can occur as part of AN as a subtype, a fifth criterion for BN is that it does not occur exclusively during episodes of AN. The validity of the DSM-IV-TR eating disorders classification has been questioned, owing to the high rates of diagnostic crossover among the AN subtypes and BN. Recent findings support the longitudinal distinction between AN and BN, but they may not support the AN subtyping schema.38,39 BED also is being distinguished from BN18,40 because obesity rather than inanition often results, and the endocrinologic and other metabolic changes are different from those of AN and BN.
For the endocrinologist, questions that should be asked of the patient and her family to address the criteria for BN include the following (adapted from the Structured Clinical Interview for DSM-IV37):
Do you have times when your eating gets out of control? Tell me about these times.
During these times, do you often eat within a 2-hour period what most people would regard as an unusual amount of food?
Can you give me an example of the kinds and amounts of food that you might eat during one of these times?
How often do these times occur?
Do you do anything to counteract the effects of eating that much—like making yourself vomit; taking laxatives, enemas, or water pills; strict fasting between periods of eating a lot of food; or exercising a lot?
How important is your body shape and size in how you feel about yourself?
These questions and those regarding AN should be phrased in a way that is comfortable to both the health care professional and the patient. Depending on the patient, questions about both bulimic and anorexic behaviors may need to be asked; the patient’s history and presenting symptoms should dictate the emphasis of the interview.41 The sample questions are given to indicate that one can (and should) ask about eating behavior forthrightly. If the topics of the questions are followed, information about all the diagnostic criteria for both AN and BN will be gleaned. The patient should also be asked if this is the first time these behaviors have occurred or if there have been past episodes; in the case of the latter, a careful history taking about each episode, its severity and duration, and treatments and their success should be done. This information may yield important clues as to how to approach the patient therapeutically during the current episode.
Comorbid symptoms of depression and anxiety occur with some frequency in both AN and BN and should be assessed, because their persistence during treatment may portend poorer outcomes.42–48 Symptoms of depression and anxiety are exaggerated by malnutrition and improved with nutrition. AN patients with obsessive-compulsive disorder tend to have an obsessional need for symmetry/exactness and compulsions toward ordering and arranging.49 Mild to moderate negative mood states and obsessive symptoms can persist after recovery in both anorexic and bulimic individuals,50,51 suggesting that these traits may contribute to the pathogenesis of eating disorders.52 As well, comorbid impulse control disorders (ICD), the most common being compulsive buying disorder and kleptomania, occur more frequently in individuals with binge eating subtypes and are associated with greater use of laxatives, diuretics, appetite suppressants, and fasting.53
There also is a high incidence of comorbid obsessive-compulsive disorder (OCD) in anorexic and bulimic women and their families, as well as increased rates of AN and BN in persons with primary OCD. It may be that the core eating disorder symptoms (e.g., fear of fatness, pursuit of thinness) are a specific type of obsession. Symmetry, ordering, and perfectionism are the most common target symptoms in women with AN and BN and often persist after recovery. Leckman and others54 delineated four symptom dimensions of OCD, one of which was symmetry and ordering; these occurred most commonly in men. OCD in men and eating disorders in women may be gender-specific expressions of a common psychobiology related to obsessions with symmetry and ordering compulsions.
There are primary central nervous system (CNS) conditions for which anorexia or bulimia may be an associated symptom; these include both organic pathology55 and “functional” illnesses. As an example of the latter, one of the authors was asked to consult on a hospitalized adolescent girl with electrolyte disturbances suggestive of Bartter’s syndrome. With no evidence of somatic causes of the patient’s cachexia, and with nurses’ observations of bizarre eating habits, a diagnosis of AN was entertained, and a psychiatric interview of the patient during endocrine rounds was requested. After introductions as to who the psychiatrist was and why he was there, questioning began with what the patient ate (lettuce and carrots only) and why she ate only those two items (the computer in the hospital basement was giving her instructions to do so). Elucidation of additional symptoms of psychosis led to a presumptive diagnosis of schizophrenia with secondary anorexia and referral for inpatient psychiatric care.
Genetics
Eating disorders in both women and men are familial, as shown by both family and twin studies.56–61 The heritability of both AN and BN has been estimated between 33% and 84%.62 Recent findings corroborate previous research implicating the transition from early to mid-adolescence as a critical time for the emergence of a genetic diathesis for disordered eating.63 Studies of the genetics of AN and BN have been primarily by linkage analysis and association studies. In linkage analysis, correlations are determined between the occurrence of a disease in families and the inheritance of specific chromosomal regions in those families. In association studies, nucleotide polymorphisms are searched for in specific candidate genes suggested by chromosomal linkage studies and/or the known functions of the gene product.
Linkage studies in AN and BN families have suggested susceptibility genes on several chromosomes, but the strength of the associations has depended on the diagnostic stringency of the cases. For example, in 192 families with at least one affected relative pair with AN, BN, and related eating disorders, modest linkage was found with a marker on chromosome 4.64 However, when the analysis was restricted to 37 families in which at least two relatives had a diagnosis of the restricting subtype of AN (without bingeing and purging), there was a much stronger linkage to a marker on chromosome 1p. With reference to specific behavioral traits, two variables were identified in a sample of 196 families with an AN proband: drive for thinness and obsessionality.65 When these variables were incorporated into a linkage analysis, there again was highly significant linkage to a region on chromosome 1, as well as linkages of lesser significance to regions on chromosomes 2 and 13. In contrast, linkage analysis of 308 families with a BN proband yielded a high linkage score to chromosome 10.66 In a subset of 133 families in which two or more BN members reported self-induced vomiting, an even higher linkage was found to a region on chromosome 10p. Another region on chromosome 14q was suggestive of linkage. These studies can serve to suggest specific chromosomal regions for association studies.
In association studies, researchers have targeted serotonin, dopamine, and norepinephrine-related genes. The most promising results have been with the serotonergic system, because serotonin (5-HT) neurotransmission inhibits feeding behavior.56,67 Several studies have suggested an association between polymorphisms in the promoter region of the 5-HT2A receptor gene and both AN and BN, but the results have been inconsistent, the association has been rather weak, and the functionality of the 5-HT2A polymorphism is unknown.68,69 Studies reporting an association between a polymorphism in the promoter region of the 5-HT transporter gene and eating disorders have been inconsistent as well. In one study, a relationship was suggested between a polymorphism in the 5-HT1B receptor gene and BN; in another study, a relationship was suggested between a polymorphism in the 5-HT2C receptor gene and vulnerability to rapid weight loss following reduced food intake in AN. On the other hand, studies of the 5-HT receptor genes 2C, 7, and 1Dβ, as well as the tryptophan hydroxylase gene (the rate-limiting enzyme in the biosynthesis of 5-HT), have been negative. Phenotypically, reduced serotonergic activity, as measured by platelet 3H-paroxetine binding, has been found in both AN and BN and was unrelated to diagnostic subtype or ancillary dimensions such as impulsive behavior or depression.70 The use of serotonin uptake inhibiting antidepressants in the treatment of AN and BN will be discussed later.
The dopaminergic system also has received attention because dopamine (DA) neurotransmission has been implicated in feeding behavior and in motor activity, amenorrhea, and distortion of body image.56,57 One study each of D3 and D4 receptor polymorphisms and food restriction in AN have been negative. One of three studies of polymorphisms in the catechol-O-methyl transferase gene (an enzyme in DA metabolism) in AN was positive, but the other two were negative.
Other candidate genes controlling hormones and proteins putatively related to food intake that have been studied in eating disorders include the estrogen receptor, the uncoupling proteins UCP-2 and UCP-3, pro-opiomelanocortin, the melanocortin-4 receptor, leptin, agouti-related protein, neuropeptide Y, cholecystokinin (CCK), the β3-adrenergic receptor, and tumor necrosis factor. All but one have been negative.56,71 Thus, although the familial clustering of both AN and BN is clear, there is no solid evidence that single nucleotide polymorphisms in any of the aforementioned genes are other than mildly related to either of these eating disorders.
General Physical and Laboratory Findings
The general physical and laboratory findings in AN and BN are presented in Tables 4-3 and 4-4.72,73 Metabolic changes and medical complications occur secondary to chronic starvation and malnutrition and to bingeing and purging.7,73–75 Malnutrition-associated disturbances as severe as pulmonary bronchiectasis and emphysema have been reported.76 Cardiac complications are also well recognized in patients with eating disorders, including the more common abnormalities of bradycardia, orthostatic hypotension, and (although less common) the risk for more serious abnormalities, including prolongation of the QT interval and silent pericardial effusion.77
Table 4-3
Physical and Laboratory Findings in Anorexia Nervosa
Physical
Cachexia, emaciation, dehydration, shock or impending shock
Covert infectious processes, immunologic problems late in process
Xerosis (dry, scaly skin), desquamation, yellowish palms and soles
Scalp and pubic hair loss, lanugo, increased pigmented body hair
Hypothermia, decreased metabolic rate, bradycardia, hypotension
Acrocyanosis (circulatory changes resulting in cold, blue, and occasionally sweaty hands and feet)
Bradypnea (respiratory compensation for alkalosis)
Edema of lower extremities, heart murmur (infrequent)
Signs of estrogen deficiency (dry skin, osteoporosis, small uterus and cervix, dry vaginal mucosa) and androgen deficiency (no acne or oily skin)
Chemical
Normal laboratory results early in process
Elevated BUN secondary to dehydration
Elevated serum cholesterol early in process; may decrease later
Decreased plasma transferrin, complement, fibrinogen, prealbumin; usually normal protein and albumin:globulin ratio
Elevated serum lactic dehydrogenase and alkaline phosphatase
Depressed serum magnesium, calcium, phosphorus, the last a late and ominous sign
Hematologic
Panleukopenia with relative lymphocytosis
Thrombocytopenia (causing purpura)
Very low erythrocyte sedimentation rate
Mild anemia (folate or iron deficiency); severe anemia (rare and late in process), especially with rehydration
Adapted from Comerci GD: Medical complications of anorexia nervosa and bulimia nervosa. Med Clin North Am 74:1293–1310, 1990; and Mitchell JE, Crow S: Medical complications in adolescents with anorexia nervosa: a review of the literature. Curr Opin Psychiatry 19:438–443, 2006.
Table 4-4
Physical and Laboratory Findings in Bulimia Nervosa
Physical
Usually well groomed with good hygiene
Usually normal weight or mild to moderate obesity
Generalized or localized edema of lower extremities
Swelling of parotid and other salivary glands
Bruises and lacerations of posterior pharynx, scar/callus formation over dorsum of hands (Russell’s sign) secondary to induced vomiting
Dental enamel discoloration and dysplasia secondary to vomiting of gastric acid
Pyorrhea and other gum disorders
Diminished reflexes, muscle weakness, paralysis, infrequently peripheral neuropathy
Muscle cramping (with induced hypoxia or positive Trousseau’s sign)
Signs of hypokalemia (hypotension, weak pulse, arrhythmias, decreased cardiac output, poor-quality heart sounds; shortness of breath; ileus, abdominal distension, acute gastric dilatation; depression, mental clouding)
Additional physical features of anorexia nervosa, if food restriction is part of syndrome
Chemical
No abnormalities reported; possible abnormal glucose metabolism
Metabolic alkalosis (hypochloremia, elevated serum bicarbonate)
Hypokalemia (secondary to metabolic alkalosis)
Hypovolemia with secondary hyperaldosteronism (also contributes to hypokalemia), pseudo-Bartter’s syndrome
Adapted from Comerci GD: Medical complications of anorexia nervosa and bulimia nervosa. Med Clin North Am 74:1293–1310, 1990.
Gastrointestinal (GI) disturbances occur in both AN and BN.78 Delayed gastric emptying and delayed colonic transit time resulting in constipation are common in AN patients and are associated with complaints of early satiety, bloating, and abdominal distension, leading the patient to feel fat and avoid eating. GI disturbances in BN include increased gastric capacity, decreased gastric relaxation, delayed gastric emptying, and abnormal function of the enteric autonomic nervous system. Abnormalities of GI function in AN and BN are reversible to various degrees with improvement in the underlying disorders. Treatments targeted at these abnormalities, such as agents to improve gastric emptying in AN and neurotransmitter modulators to reduce bingeing in BN, have met with varying success and require further study.
Increasing recognition is being given to osteopenia as an early and serious complication of AN.79–82 Adolescence is a time when optimal bone mineralization supporting physical growth is critical, and adolescent girls with AN have relatively poor bone mineral accrual. Several factors contribute to osteopenia in AN, including estrogen, androgen, and insulin-like growth factor deficiency; elevated cortisol, ghrelin, and peptide YY; excessive exercise; and nutritional deficiencies such as calcium and vitamin D. Multifaceted treatment of AN is required to restore bone mineral density to the normal range.
The electrolyte disturbances of AN and BN depend on whether purging is primarily by vomiting or by abuse of laxatives or diuretics (or both) and can be life threatening. The medical management of these cases can be complex and must be individualized. Refeeding syndrome may occur in severely malnourished patients as a result of severe shifts in fluids and electrolytes, in particular phosphate, from extracellular to intracellular spaces. These electrolyte shifts can lead to cardiovascular, neurologic, and hematologic complications and can be associated with significant morbidity and mortality.83 Comerci72 presented detailed case histories of an anorexic and a bulimic patient, including critiques of their treatment, which highlight the intricacies of successful medical management of these disorders.
Neuroimaging
This is a growing area of eating-disorders research supported by major improvements in imaging technology.84,85 In AN, enlargement of cortical sulci and subarachnoid spaces suggestive of cerebral, and on occasion cerebellar, atrophy have been reported, the changes being positively correlated with poor neuropsychological test performance and being reversible to varying degrees with weight gain.86–88 Reduced gray matter volume in the anterior cingulate cortex has been reported in AN compared to healthy controls, the amount of the decrease being correlated with illness severity.89 Studies in recovered AN and BN patients have differed, showing either persistent alterations or normalization of gray- and white-matter volumes.90,91
With regard to CNS metabolites, proton magnetic resonance spectroscopy (1H-MRS) indicates higher ratios of choline-containing compounds to creatine and N-acetylaspartate, as well as reduced myoinositol, in the frontal white matter and occipital gray matter in association with decreased body mass index (BMI), suggesting starvation-associated increased cell membrane turnover.92 Consistent with these findings, 31P-MRS has shown altered phosphodiester and phosphomonoester peaks in malnourished anorexics, suggesting that reduced body mass alters CNS cellular membrane phospholipid metabolism.93 Reduced blood flow in frontal, parietal, and frontotemporal cortex, as shown by single photon emission computed tomography (SPECT), has been reported in AN, which reverted to normal perfusion with clinical remission.94 Another SPECT study suggested reduced blood flow in the anterior cingulate in patients with AN compared to controls, both before and after weight restoration.95
Functional neuroimaging (fMRI) also is delineating activation of limbic and paralimbic areas that may be involved with calorie fear, including exaggerated activation of the caudate and reduced activation of the insula to taste stimuli in women recovered from AN compared to normal women.96,97 These areas also have been implicated as neural substrates of obsessive-compulsive and depressive symptoms.98 Decreased food-stimulated activation of several cortical areas has also been shown by fMRI in chronically ill anorexics, compared to those exhibiting long-term recovery.99 Positron emission tomography (PET) imaging has demonstrated reduced 5-HT1A and 5-HT2A receptor binding in frontal, parietal, and occipital cortex in both AN and BN patients, which may persist after recovery in both conditions.8,100,101 This may tie in with the candidate gene studies of these receptors discussed earlier; correlative studies have yet to be undertaken.
Neuroendocrinology
Abnormal hormone profiles and responses to challenge are closely related to the “starvation” status of AN and BN patients.102–106 Hormone abnormalities also may be present (but to a lesser extent) in normal-weight women with BN. The presence of starvation in AN is evident from the weight loss, but it may not be recognized in normal-weight BN: Although bulimic women often maintain a normal weight, they do so by restricting food intake when not bingeing and purging, and they often have poorly balanced meals. Starvation-induced depletion of hepatic glycogen stores results in free fatty acids and ketone bodies replacing glucose as the primary energy source. This shift from glycogenolysis to lipolysis and ketogenesis is associated with an increase in free fatty acids and their metabolites. β-Hydroxybutyric acid levels are elevated in both AN and BN,107 indicating that bulimic patients are nutritionally depleted despite their normal body weight.108
The relationship of starvation and eating disorders to neuroendocrine function is most clearly seen for the pituitary-gonadal axis. As mentioned, secondary amenorrhea is one of the criteria for AN in postmenarcheal women,74 and oligomenorrhea occurs in about 50% of bulimics. Table 4-5 lists the major endocrine disturbances that occur in AN and BN.103,109–112 The secondary amenorrhea is a direct result of altered gonadotropin secretion. Serum sex hormone–binding globulin may be increased, and both estrogen and testosterone are decreased.113 As indicated in Table 4-3, there are physical signs of severe estrogen deficiency. The luteinizing hormone (LH) response to LH-releasing hormone stimulation is blunted, but the follicle-stimulating hormone response is usually normal.
Fig. 4-2 illustrates circadian serum LH profiles of girls at different pubertal stages and in a 21-year-old woman with AN of 14 months’ duration whose body weight was 60% of ideal weight.109 This patient’s menarche was at age 14, and during the period of weight loss, her LH profile was that of a prepubertal girl. BN patients also may have decreased gonadotropin secretion if they have lost 15% or more of their body weight, but they usually have normal circulating gonadotropins and continue their menses.
FIGURE 4-2 Circadian serum luteinizing hormone (LH) profiles in girls at different pubertal stages and in a 21-year-old woman with anorexia nervosa of 14 months’ duration whose body weight was 60% of ideal weight. Menarche was at age 14 years, and her LH profile is that of a prepubertal girl. (From Vande Wiele RL: Anorexia nervosa and the hypothalamus. Hosp Pract 12:45–51, 1977. © 1977, The McGraw-Hill Companies Inc. Illustration by Albert Miller.)
With reference to the hypothalamic-pituitary–adrenal cortical (HPA) axis (see Table 4-5), the abnormalities in AN and in reduced-weight BN111,114,115 are strikingly similar to those occurring in 30% to 50% of patients with major depression.111,116,117
[/not-level-membership-for-endocrinology-diabetes-and-metabolism-category]
