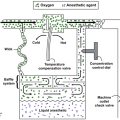
Anesthetic considerations in the patient with down syndrome
Michael J. Murray, MD, PhD and Jennifer A. Rabbitts, MB, ChB
Clinical manifestations
Head and neck abnormalities
Because patients with DS often have abnormalities of the head and neck, the anesthesia provider should assess any patient with DS seen in the preoperative clinic for these abnormalities (Box 207-1). Despite the many airway abnormalities described, patients with DS are typically not difficult to intubate, though they may be difficult to ventilate by mask. Cervical spinal malformations have been reported in association with trisomy 21, with 10% of patients having cervical spinal stenosis and 30% having atlantoaxial instability, although only 1% to 2% of these patients will become symptomatic.