CHAPTER 25 Anesthesia for Plastic Surgery
For the pediatric plastic surgeon, a significant portion of reconstructive surgery involves pathologies of the craniofacial skeleton and soft tissue, and these may be of significant concern for the pediatric anesthesiologist. In this chapter, the pathologies that result in surgery are set in the framework established by Whitaker and associates in 1979. The anesthesiologist who cares for these children will find it easier to organize the varied syndromes and sequences when they are structured in this manner.
Craniofacial surgery
Craniofacial anomalies are characterized by congenital or acquired deformities of the cranial or facial skeleton. Although rare, they comprise a diverse group of defects, and the goal of surgical intervention is to restore both form and function. The classification of craniofacial anomalies is difficult because of their variability, rarity, and degree of severity, and often because of our lack of understanding of their etiology and pathogenesis. The Committee on Nomenclature and Classification of Craniofacial Anomalies of the American Cleft Palate Association has proposed the following categories: synostosis, clefts, hypoplasia, hyperplasia, and unclassified (Whitaker and Bartlett, 1990).
Craniosynostosis
Craniosynostosis is defined as premature closure of one or more of the cranial sutures. It is a relatively common defect, occurring in one in 2000 to 2500 live births (Posnick, 2000). It can result in abnormalities in the size and shape of the calvarium, cranial base, and orbits, and can cause dental occlusion, and thus it constitutes a diverse group of deformities. Craniosynostosis is not only a cosmetic issue, for it can also affect brain growth, intracranial pressure (ICP), and vision, resulting in developmental delay and vision impairment (Slater et al., 2008).
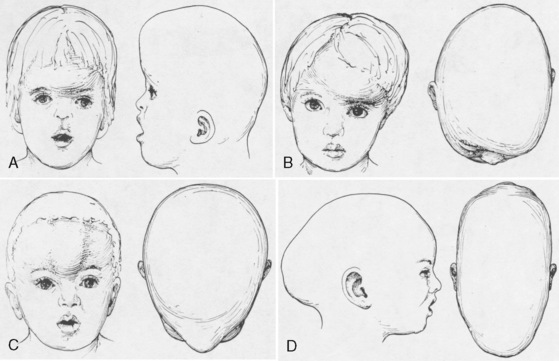
Premature fusion of a cranial suture results in an abnormal head shape. Growth of the skull perpendicular to the suture is impaired. Compensatory growth parallel to the suture creates the characteristic abnormal skull shape. The shape of the skull defines craniosynostosis. Figure 25-1 shows the different types of abnormal skull shapes and the corresponding synostotic sutures.
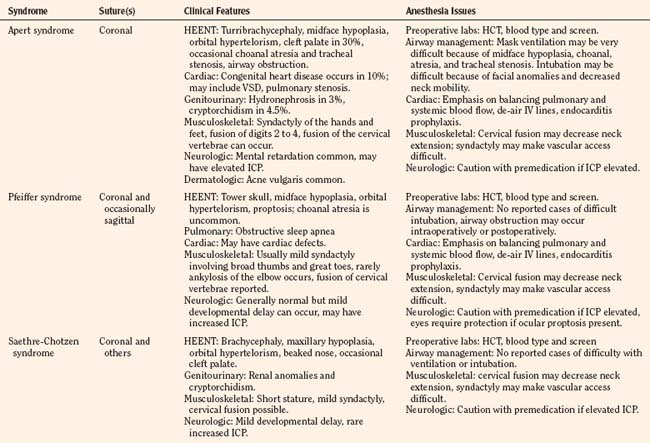
Craniosynostosis can occur by itself (simple) or as a major component of a syndrome (complex, or syndromic). Over 100 craniosynostosis syndromes have been described, but six are more common than the others (Slater et al., 2008). These are Apert, Pfeiffer, Saethre-Chotzen, Carpenter’s, Crouzon, and Muenke’s syndromes. Of these, Apert and Crouzon syndromes are the most common. Table 25-1 lists the various syndromes and their associated anomalies and anesthetic concerns. The timing of surgery usually occurs before 1 year of age. The importance of early intervention is related to improved ability of the infant to reossify, the malleable nature of the calvaria during infancy, and the rapid brain growth that occurs during the first year of life (Panchal and Uttchin, 2003). The motivation for and goal of surgical intervention are to reduce ICP, prevent brain injury, and enhance appearance. Repair of syndromic craniosynostosis may be more complicated and appears to be associated with increased blood loss. The etiology of the increased bleeding is unclear, but it might be related to the length of surgery (Fearon and Weinthal, 2002).
Apert Syndrome
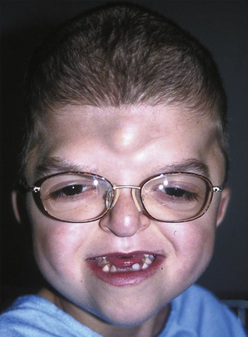
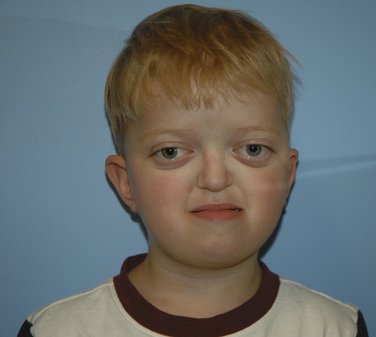
Apert syndrome, also referred to as acrocephalosyndactyly, occurs at a frequency of 1 in 160,000 live births (Cohen et al., 1992). Acrocephalosyndactyly, a defect involving the cranium and the extremities, occurs in Apert, Pfeiffer, Carpenter’s, and Saethre-Chotzen syndromes. The characteristic features of Apert syndrome include turribrachycephaly (high steep flat forehead and occiput), mid-face hypoplasia, and orbital hypertelorism (Fig. 25-2). Cleft palate occurs in approximately 30% of these patients. Choanal atresia and occasionally tracheal stenosis have been reported and can cause airway obstruction. Congenital cardiac disease is one of the more common associated visceral anomalies, occurring in approximately 10% of patients. Genitourinary anomalies (hydronephrosis, cryptorchidism) also occur in 10% of patients with Apert syndrome (Cohen and Kreiborg, 1993). Severe synostosis can result in increased ICP and, if uncorrected, developmental delay. Syndactyly of the hands and feet with the fusion of digits two to four can occur and can make intravenous (IV) access difficult. Kreiborg and coworkers (1992) have reported cervical spine fusion, and they have suggested that cervical spine films prior to anesthesia may help predict difficult intubation. Although infants and children with Apert syndrome are often difficult to intubate, many have been intubated uneventfully. In some cases, suboptimal laryngoscopic views secondary to abnormal anatomy may require flexible fiberoptic intubation. The laryngeal mask airway (LMA) may be a reasonable adjunct in those patients who are difficult to ventilate or intubate. However, to date there are no reported instances of their use in infants and children with Apert syndrome. The clinical features and anesthetic implications of Apert syndrome and the other acrocephalosyndactylies are outlined in Table 25-1. Unlike Apert syndrome, the other acrocephalosyndactylies are not typically associated with difficult airways. However, midface hypoplasia is common in these infants and may cause significant upper airway obstruction intraoperatively and postoperatively (Perkins, 1997).
Pfeiffer Syndrome
Pfeiffer syndrome is another example of an acrocephalosyndactyly. The incidence of Pfeiffer syndrome is approximately 1 in 100,000 live births. This syndrome is characterized by bicoronal synostosis, proptosis, midface hypoplasia, and broad thumbs and great toes (Moore et al., 1995). Patients with Pfeiffer syndrome can also present with hydrocephalus, which may contribute to increased intracranial pressure. There are three types of Pfeiffer syndrome, and typically the clinical features, degree of airway obstruction, and mortality rates increase with types 2 and 3 (Moore et al., 1995).
Saethre-Chotzen and Carpenter’s Syndromes
The clinical features of Saethre-Chotzen include brachycephaly, facial asymmetry, low hairline, proptosis, beaked nose, large halluces (great toes), and pectus excavatum. Some patients may have renal anomalies, cryptorchism, developmental delay, and epilepsy. It can be difficult to differentiate it from the other acrocephalosyndactylies because there can be significant clinical variability (Nascimento et al., 2004). Carpenter’s syndrome is the most rare of the syndromic craniosynostosis, with only 45 reported cases since the mid 1990s. As with all of the syndromic craniosynostosis, patients with Carpenter’s syndrome have synostosis, midface hypoplasia, and musculoskeletal deformities. They may also have hypogonadism, developmental delay, and obesity (Idestrand et al., 2009).
Crouzon Syndrome
Crouzon syndrome, also known as craniofacial dysostosis, is one of the syndromic craniosynostoses. These infants present with craniosynostosis, proptosis, and midface hypoplasia but without visceral or extremity involvement (Fig. 25-3). As in other patients with midface hypoplasia, significant airway obstruction can occur and may require early tracheostomy (Sirotnak et al., 1995). Table 25-1 outlines the main clinical features and anesthetic issues as they relate to patients with Crouzon syndrome. During infancy, patients with Crouzon syndrome may come to the operating room for tracheostomy or cranial vault remodeling.
Surgical Management
Cranial Vault
Surgical correction for craniosynostosis can be performed with an open or an endoscopic procedure. The calvarial vault reconstruction typically involves both plastic surgery (by craniofacial surgeons) and neurosurgery. The surgical approach for an open cranial vault reconstruction is through a bicoronal incision (Fig. 25-4, A). A blocking stitch or clips may be applied to the skin flaps to minimize blood loss. The clips may be more effective in preventing bleeding, but some surgeons have expressed concern about the risk for ischemia of the underlying hair follicles (Ray Harschberger, personal communication, 2009). The scalp flap is dissected off the forehead and mobilized down to expose the superior orbital rim (see Fig. 25-4, B). The calvarium is typically removed by the neurosurgeons in one or several pieces. A bandeau osteotomy is then performed along the lateral temporal bones and the nasion to mobilize the superior orbital rim (see Fig. 25-4, C). Once the osteotomies are complete, the surgical field is protected with moist gauze. The calvarium and the orbital bandeau are sectioned and the pieces are reshaped and replaced in a manner that replicates a normal head shape. The bone is secured with a craniofacial plating system, the scalp flaps are replaced, and the coronal incision is closed.
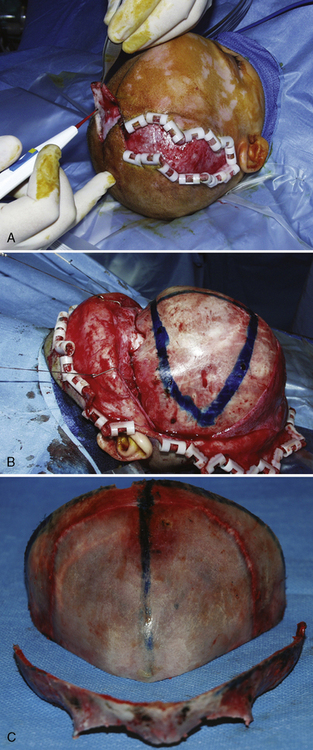
Strip Craniectomy with Helmet
The endoscopic approach is less invasive and has been reported to cause less blood loss (Jimenez et al., 2002). This procedure is more commonly used for sagittal synostosis, although it has been described for the repair of other single suture and even multiple-suture synostosis. The surgical approach is through smaller incisions. As with open approaches, significant blood loss can occur if the sagittal sinus is entered. Jiminez reported a small percentage of patients who required transfusions, and most were discharged on the first postoperative day. Unlike those who had an open calvarial reconstruction, these patients do require helmet therapy after this repair.
Midface Advancement
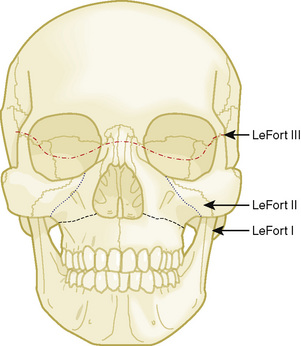
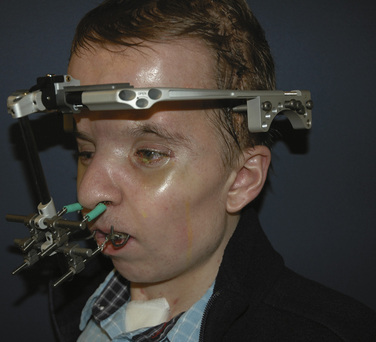
Midface advancements are performed to correct midface hypo-plasia. The different types of advancements include the Le Fort I maxillary advancement, the Le Fort III maxillary and upper face advancement (Fig. 25-5), and the monobloc advancement, which includes the maxilla, upper face, orbits, and forehead. The Le Fort I osteotomy is typically performed for patients with midface hypoplasia secondary to cleft lip and palate. The Le Fort III and monobloc osteotomies are typically performed for patients with midface hypoplasia secondary to syndromic craniosynostosis (Apert, Pfeiffer, Crouzon syndromes). The surgical approach for the Le Fort III and monobloc osteotomies are through bicoronal, intraoral, and often eyelid incisions. Significant blood loss can occur during the surgical dissection and osteotomies. Once the midface is mobilized, the advancement can be performed immediately with rigid fixation or gradually with internal or external distraction osteogenesis. Distraction osteogenesis became a common in craniofacial surgery in 1997, and the advantages include less morbidity, more long-term stability, and improved aesthetic outcome (Fearon, 2001; Shetye et al., 2007). The external distraction device has a frame that is anchored to the skull, with a distraction bar positioned perpendicular to the face. The distraction bar is anchored to the newly mobilized midface with wires (Fig. 25-6). Once in place, the midface may be distracted at a rate of 1 to 2 mm a day.
Anesthesia Management
The anesthetic management of infants with craniosynostosis begins with a complete preoperative evaluation. The history should define the cranial sutures involved, the planned surgical procedure, and previous craniofacial reconstruction, and it should identify any associated syndrome. Infants and children with syndromes may have difficult airways, other organ involvement, and more complicated surgical repair with more bleeding. Associated anomalies that can present a challenge to the anesthesiologist include facial and airway features that make mask ventilation and intubation difficult. Airway pathology can also cause obstruction, and some of these children have obstructive sleep apnea (OSA) (Mixter et al., 1990; Pijpers et al., 2004).
Children with OSA may present with daytime somnolence, enuresis, behavioral changes, and snoring. As many as 40% to 50% of infants and children with syndromic craniosynostosis have clinical features of OSA. This compares with the 0.2% to 0.7% incidence of OSA in the general pediatric population (Lo and Chen, 1999; Pijpers et al., 2004). The most common treatment for OSA is adenotonsillectomy. If this fails to reduce the symptoms, the sequence of recommended steps in this population includes nasal continuous positive airway pressure (CPAP), Le Fort III osteotomy with midface advancement, and as a last resort, tracheostomy. Several studies have demonstrated a reduction in the symptoms of OSA after midface advancement. In one study by Nelson and colleagues (2008), all of the patients reported a decrease in snoring, and five of the six patients with a tracheostomy could be decannulated.
A history of fatigue (or sweating with feedings), cyanosis, and syncope are suggestive of an underlying cardiac anomaly. Cardiac pathology is associated with some of the syndromes (e.g., Apert, Pfeiffer, Carpenter’s). Congenital heart disease is most common in Apert syndrome, and the most common cardiac defect is ventricular septal defect (Hidestrand et al., 2009). Infants and children with clinical signs or symptoms suggestive of heart disease or a heart murmur should be preoperatively evaluated by a pediatric cardiologist.
Some infants and children with craniosynostosis may have increased ICP. The incidence of increased ICP in nonsyndromic craniosynostosis varies from 8% to 47%, depending on the number of sutures involved (Arnaud et al., 1995; Renier et al., 2000; Mathijssen et al., 2006). The incidence appears to be higher in syndromic craniosynostosis, with approximately 50% having funduscopic evidence of papilledema (Bannink et al., 2008). This may manifest as headaches, vomiting, and somnolence. However, infants and children with chronically elevated ICP may be asymptomatic. Elevations in ICP manifest as papilledema on ophthalmologic examination, so all patients with craniosynostosis should have a funduscopic examination by an ophthalmologist.
Airway
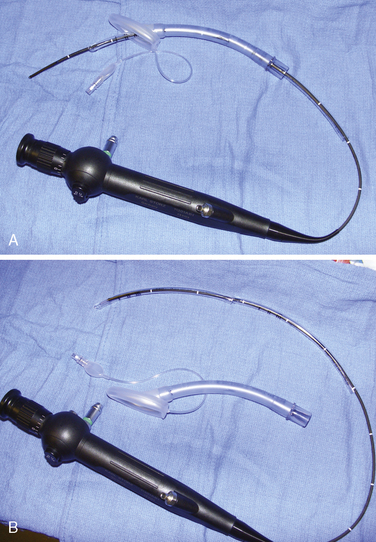
Airway management in these patients can be very challenging during attempts at ventilation, intubation, or both. Fortunately, difficult airways are not common. However, the incidence is higher in those patients with syndromes and in those patients who have had previous reconstruction. Successful techniques described for infants and children include using the Bullard laryngoscope, laryngeal mask airway, flexible fiberoptic scope, and retrograde intubation (Cooper and Murray-Wilson, 1987; Blanco et al., 2001; Brown et al., 1993, 2004) (see Chapter 10, Equipment, and Chapter 12, Airway Management). A combination of techniques may be required to secure the airway. For example, the LMA has been used to facilitate the passage of the fiberoptic scope and endotracheal tube (Inada et al., 1995) (Fig. 25-7). Although those authors report this technique in a patient with Treacher Collins syndrome, it could be used for any craniofacial patient with a difficult airway. An endotracheal tube and LMA size chart helps in choosing the appropriate equipment (Table 25-2, and see also Table 10-3 in Chapter 10, Equipment). Cuffed endotracheal tubes may be more challenging, because the cuff on the endotracheal tube may not fit through the smaller LMAs. However, the air-Q (Trudell Medical Marketing, London, Ontario, Canada) has its short stalk and detachable adapter that make it easy to pass a cuffed endotracheal tube with the pilot balloon. If an uncuffed endotracheal tube is used, it can be secured while removing the LMA by telescoping a smaller endotracheal tube through the top of the larger tube (see Fig. 25-7, B). When passing an endotracheal tube through an LMA that is a size 3 or smaller, an uncuffed endotracheal tube is easier to use, but it runs the risk for creating a large air leak.
| Laryngeal Mask Airway (size) | Endotracheal Tube (mm) |
| 1.0 | 3.0 uncuffed |
| 1.5 | 4.0 uncuffed |
| 2 | 4.5-5.0 uncuffed |
| 2.5 | 5.5 uncuffed |
| 3 | 6.0 uncuffed |
| 4 | 6.0 cuffed |
Patients having Le Fort osteotomies require a nasal or an oral intubation depending on the type of Le Fort. Those having a Le Fort I osteotomy (for clefts) will need a nasal endotracheal tube so the maxillae and the mandible can be aligned properly. Nasal endotracheal tubes have been damaged during Le Fort osteotomies, resulting in difficulty with ventilation (Bidgoli et al., 1999), so care must be taken by the surgeons when mobilizing the maxillae. Patients having a Le Fort III osteotomy for midface distraction can be intubated orally with an oral RAE tube.
Some infants with craniofacial anomalies require tracheostomy because of significant upper airway obstruction (Perkins, 1997; Sculerati et al., 1998). Adequate preparation entails having all the necessary equipment available and having personnel who are trained and experienced in the use these airway instruments. It may also mean having a pediatric otorhinolaryngologist immediately available.
Blood Conservation and Transfusion Medicine
Craniofacial procedures are often long, exposing infants to the risks of hypovolemia, hypothermia, blood loss, and venous air emboli. The craniofacial procedures typically performed during the first year of life include cranial vault remodeling (including fronto-orbital, posterior, and total vault advancement) and strip craniectomy. One of the most pressing concerns related to anesthesia care in craniofacial surgery is the management of intraoperative bleeding. The cranial procedures can involve significant blood loss because of the duration of the procedure, the many exposed skin and bone surfaces, and the rare complication of entering large vessels such as the sagittal sinus. Blood loss during these procedures has remained an issue since Whitaker and colleagues’ description of perioperative blood loss in 1979. Blood loss is often as high as half to one blood volume, and nearly 90% to 100% of the infants undergoing these procedures may require a blood transfusion (Faberowski et al., 1999; Tuncbilek et al., 2005; Stricker et al., 2010). Even the strip craniectomy, which is typically performed to correct nonsyndromic isolated sagittal synostosis and results in less blood loss, can produce significant hemorrhage. In a recent study by White and others (2009), predictors of blood loss during craniofacial surgery included surgery time greater than 5 hours, age less than 18 months, the presence of multiple-suture craniosynostosis, and syndromic craniosynostosis. Infants undergoing strip craniectomy usually experience less blood loss, but this population may be at increased risk because they come to the operating room at the nadir of their physiologic anemia (i.e., at 2 to 3 months of age). Preparation for these procedures requires a baseline hematocrit, and blood typing and cross-match. Some centers also obtain coagulation studies. Adequate IV access needs to be obtained for resuscitation. In an infant, at least two large-bore (22- to 20-gauge) peripheral IV catheters should be placed to provide adequate access. Arterial pressure monitoring is recommended for beat-to-beat analysis of blood pressure and intravascular volume status, as well as for blood gas monitoring. A central venous catheter may be placed for central venous pressure monitoring, or in patients with difficult IV access.
Preoperative blood donation has been described in children as small as 8 kg (Mayer et al., 1996), but this technique has significant limitations in the infant. Their young age, small size, and lower hematocrit may make blood collection more challenging and less feasible. Acute normovolemic hemodilution has been described in older children and adolescents and may be effective. Hemodilution is relatively contraindicated in the infant less than 6 months old. The normal physiologic advantage with hemodilution (increased preload, increased stroke volume, and decreased systemic vascular resistance, increased tissue oxygenation) may be lost in the infant because of fetal hemoglobin, which more avidly binds oxygen, and a naturally less compliant myocardium.
Because most infants presenting for cranial vault reconstruction have a normal physiologic anemia, erythropoietin was proposed as a therapy to address this concern. Preoperatively, recombinant erythropoietin decreases the transfusion requirements in infants having craniosynostosis repair. Fearon and Weinthal (2002) reported the dosage of erythropoietin as 600 units/kg given subcutaneously once per week along with oral iron supplementation. Erythropoietin was started 3 to 4 weeks before surgery, and the incidence of blood transfusions in infants having craniosynostosis repair decreased from 93% to 57%.
In March of 2007, the U.S. Food and Drug Administration placed a black-box warning on synthetic erythropoietin because of the concern of increased death, deep venous thrombosis, and cancer spread in adult patients receiving synthetic Epogen (Singh et al., 2006; see also www.fda.gov/for consumers consumers). These concerns have occurred only in the adult population, but some centers have stopped using synthetic erythropoietin for craniofacial surgery. A multicenter retrospective review of 396 pediatric craniofacial patients receiving erythropoietin did not reveal an increase in perioperative complications, specifically death or deep venous thrombosis (Naran et al., 2010).
Antifibrinolytic therapy has been described in pediatric surgery, but the data for craniofacial surgery are limited. Aprotinin, a serine protease inhibitor, decreases perioperative blood loss and transfusion requirements during craniofacial procedures. In a prospective, randomized, and blinded placebo-controlled study evaluating the effect of aprotinin in infants and children having cranial vault remodeling and frontal orbital advancements, D’Errico and colleagues (2003) noted a reduction in the amount of packed red blood cells being transfused intraoperatively and postoperatively in those receiving aprotinin. However, aprotinin did not reduce the number of patients requiring transfusion. Although no adverse events were reported, the option of aprotinin has been curtailed with its removal from the market. Tranexamic acid and aminocaproic acid are two antifibrinolytics that remain available for potential pediatric use. The data to guide the use of tranexamic acid and aminocaproic acid in this population are limited, but a small study by Durán de la Fuente and colleagues (2003) demonstrated a reduction in estimated blood loss and transfusion requirements. The tranexamic acid was not infused continuously but instead was administered every 8 hours at a dosage of 15 mg/kg. Table 25-3 shows standard dosage regimens for tranexamic acid and aminocaproic acid.
| Drug | Dosage |
| Aminocaproic acid (Amicar) | Load 100 mg/kg |
| Infuse 10 mg/kg per hr | |
| Tranexamic acid | Load 10-100 mg/kg |
| Infuse 1-10 mg/kg per hr |
In the past, the use of a cell saver was reported as being impractical for small pediatric patients because of the size of the collection reservoir (De Ville, 1997). Recently, the cell-saver reservoirs are available in sizes as small as 25 mL. This technology may reduce the rate of autologous blood transfusion in infants having craniofacial surgery. In fact, the most significant benefit may occur when cell-saver technology is combined with erythropoietin pretreatment. In a prospective analysis evaluating the use of cell saver with a 55-mL pediatric bowl for patients pretreated with erythropoietin, only 30% of those infants having cranial vault remodeling required allogeneic blood (Fearon, 2005). Krajewski and coworkers (2008) in a prospective randomized trial describe a 5% transfusion rate in infants having elective craniosynostosis surgery when both cell saver (25-mL collection reservoir) and erythropoietin were used, compared with a 100% transfusion rate for those not receiving cell-saver techniques or erythropoietin.
Temperature and Positioning
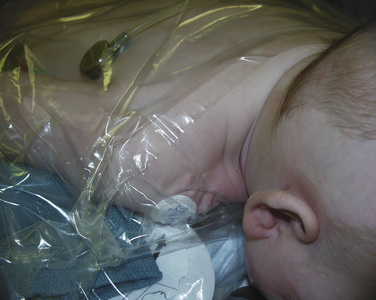
Craniofacial procedures can be very long—sometimes several hours. Complications resulting from long surgical procedures include skin breakdown, neuropathic injury, and hypothermia. Attention must be paid to the initial setup to ensure adequate positioning and padding to minimize these intraoperative injuries. Infants having cranial vault remodeling may be positioned prone, and attention to protecting the face and eyes is important. Patients with midface hypoplasia and proptosis may present a challenge when placed prone, because adequately protecting the face and eyes may be more difficult. The infant is placed on a full-access forced-hot-air blanket to minimize hypothermia, and the surgical site (head) is then isolated from the body using plastic drapes (Fig. 25-8). This not only minimizes convective and radiant heat losses but also prevents conductive heat loss to a bed wet from irrigation and blood. Blood products should be warmed through a fluid warmer before administration (except for platelets). Intravenous fluid warmers may be used, particularly if large volumes of cold allogeneic blood are being administered.
Venous Air Embolism
Venous air embolism (VAE) is a potential complication of craniofacial and neurosurgical procedures (see Chapter 22, Anesthesia for Neurosurgery). VAE can present with hemodynamic instability and can result in death. VAE occurs commonly in pediatric patients having cranial procedures. A prospective study using precordial Doppler in infants and children having craniosynostosis repair detected VAE in 82% of the patients; 31% percent developed hypotension secondary to VAE, but none developed cardiovascular collapse (Faberowski et al., 2000). This is higher than the previously reported incidence of 66% (Harris et al., 1987). Infants may be at increased risk for VAE, because significant hemorrhage during cranial vault remodeling can result in low central venous pressures. In addition, the relatively large size of the infant head may raise the surgical site above the level of the heart, thereby increasing the pressure gradient for air entrainment. Some advocate the placement of central venous catheters to monitor the trend of central venous pressures and minimize the risk for air embolism. However, no data suggest that central venous pressure monitoring decreases the risk for VAE.
Management of VAE begins with preventing hypovolemic states by providing adequate volume resuscitation and using a precordial Doppler for early detection of VAE. The precordial Doppler should be placed over the left or right parasternal border between the third and sixth intercostal spaces (Schubert et al., 2006). Lowering the head of the bed, left lateral positioning, flooding the surgical field with saline, applying bone wax, discontinuing nitrous oxide, and providing inotropic support are all measures that have been used to acutely manage VAE. Pediatric advanced life support with chest compressions and epinephrine is required for patients who develop symptomatic low blood pressure, bradycardia (heart rate < 60 bpm), or pulseless electrical activity.
Postoperative Care
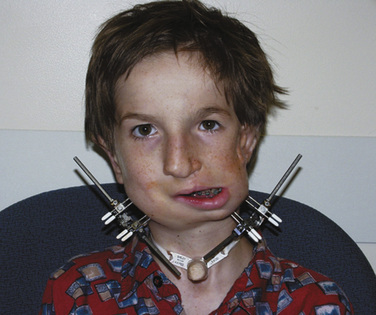
The postoperative treatment of infants having craniofacial surgery depends on coexisting morbidities and the procedure performed. Most infants and children presenting for cranial vault reconstruction or midface advancement can be extubated in the operating room at the completion of surgery. Factors that may preclude extubation include intraoperative complications resulting in hemodynamic instability, known difficult airway, significant tongue swelling, and intranasal cerebrospinal fluid drainage after Le Fort III osteotomy. Infants in whom distractors were placed may have a more difficult airway after extubation (Fig. 25-9). Mask ventilation can be very difficult with mandibular distractors and impossible with midface distractors. Airway equipment, including oral and nasal airways and appropriately sized LMAs, should be available after extubation. Equipment and personnel to remove part of the distractor device are also important in the operating room (Wong et al., 2004).
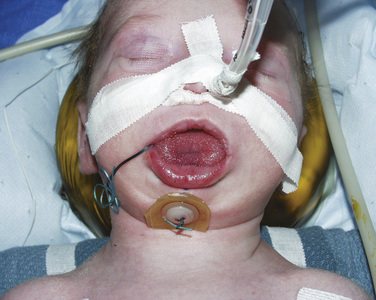
Infants having cranial vault remodeling and frontal orbital advancements can experience significant blood loss intraoperatively. If these patients are adequately resuscitated and are hemodynamically stable, they can often be extubated in the operating room. Infants with difficult airways or significant airway obstruction, or those who have experienced intraoperative complications, may benefit from delayed extubation in the intensive care unit (or in the operating room) after their condition has stabilized. Ongoing blood loss is common after major craniofacial surgery, and infants may require repeat transfusions in the immediate postoperative setting. Other complications include cerebral edema (Levine et al., 2001), visual changes (Lo et al., 2002), cerebrospinal fluid leak (Fearon, 2003), infection (Fialkov et al., 2001), metabolic acidosis, and transfusion reactions.
Hyponatremia has been reported after major intracranial procedures in pediatric patients. This was originally attributed to syndrome of inappropriate diuretic hormone (SIADH). However, this diagnosis has come into question. Although it is not entirely clear, cerebral salt wasting syndrome (CSW) may be the more likely cause. Both SIADH and CSW manifest with hyponatremia. CSW is differentiated from SIADH by hypovolemia, high urine output, and low or normal antidiuretic hormone levels (Levine et al., 2001) (Table 25-4). The proper diagnosis is important because the two are managed differently. SIADH is treated with free water restriction, whereas CSW is managed with isotonic fluid replacement (normal saline) to cover maintenance requirements and urine losses. Frequent sodium evaluations are important to guide therapy.
TABLE 25-4 Comparison of Syndrome of Inappropriate Antidiuretic Hormone (SIADH) and Cerebral Salt Wasting (CSW)
| Monitoring | SIADH | CSW |
| Central venous pressure | High (>5) | Low (<5) |
| Urine output | Decreased | Increased |
| Urine sodium level | High (>20 mmol/L) | High (>20 mmol/L) |
| Urine osmolality | High | Low or normal |
| Serum osmolality | Low | Low or normal |
| Antidiuretic hormone | High | Normal |
Hypoplasia
Pierre Robin Sequence
Neonates and infants with PRS will have some degree of airway obstruction. They often also present with significant reflux and feeding difficulties. Mild airway obstruction may require only lateral or prone positioning to relieve the airway obstruction; however, as many as one quarter of infants with PRS have more severe obstruction, necessitating surgical intervention (Bijnen et al., 2009). The optimal surgical intervention is not clear, and there is significant regional variation. Preoperative evaluation by a pediatric plastic surgeon and pediatric otolaryngologist is essential to completely evaluate the airway and identify coexisting airway pathology. The management options include tongue-lip adhesion, mandibular distraction, and tracheostomy. A tongue-lip adhesion involves suturing the inferior portion of the tongue to the lower lip to prevent the tongue from falling to the back of the pharynx and causing obstruction (Fig. 25-10). Once the mucosal flaps from the tongue and the lower lip are approximated, a temporary retention suture with a button is placed through the floor of the mouth and around the mandible to secure the repair. The infant is intubated postoperatively for several days to ensure the success of the adhesion. The goal of the tongue-lip adhesion is to relieve the airway obstruction and improve feeding until the mandible has had a chance to grow. Some evidence suggests that this repair is effective at decreasing airway obstruction and improving feeding (Kirschner et al., 2003). Bijnen and coworkers (2009) found that 73% of their patients had improved airway patency, based on sleep studies, and improved feeding. The tongue-lip adhesion is left intact until the patient is approximately 1 year old. At that time, the mandible has had time to grow and the adhesion can be taken down.
An alternative therapeutic approach involves mandibular distraction. In infants with severe obstruction, mandibular distraction can be performed to rapidly distract the jaw. It can be performed in infants less than 6 months old. Genecov and colleagues (2009) recently described their 10-year experience with mandibular distraction for PRS. Of the 67 patients that had preoperative polysomnography, 65 experienced an improvement. Also, several of the patients who required tracheostomy for PRS could be decannulated after their mandibles were distracted.
Tracheostomy is a final option for those patients who fail conservative treatment, tongue-lip adhesion, or distraction. Some have advocated for early tracheostomy for PRS, but there is evidence that it involves increased morbidity and mortality. Also, once infants receive a tracheostomy, it takes on average 3 years for decannulation to occur (Tomaski et al., 1995).
Anesthesia Management
Airway management in the infant with Pierre Robin sequence can be very challenging because of difficulty with mask ventilation and intubation. The laryngeal mask has been successfully used to ventilate and to assist in the intubation of these patients (Markakis et al., 1992). The LMA has even been placed in the awake patient with PRS after regional nerve blockade (Stricker et al., 2008). Nasal intubation with the flexible fiberoptic scope has also been described (Blanco et al., 2001). Other airway devices used for patients with PRS and difficult intubation include the Storz video laryngoscope (KARL STORZ Endoscopy-America, Inc.) and the Airtraq optical laryngoscope (PRODOL MEDITEC S.A.). In addition, when infants have significant difficulty with ventilation or intubation, aside from oral pharyngeal and nasal airways, a suture (0-silk) can be placed at the base of the tongue to displace the tongue anteriorly to assist with ventilation or intubation.
Hemifacial Microsomia
Anesthesia Management
As with PRS, the most significant anesthetic challenge with hemifacial microsomia is airway management (see Pierre Robin Sequence, Anesthesia Management, earlier). Mask ventilation may be difficult because of facial asymmetry. Intubation may be more challenging because of micrognathia and asymmetric mandibular hypoplasia, and also potentially because of decreased cervical range of motion. This difficulty may decrease with age, but it may increase after surgical reconstruction. Successful ventilation and intubation of the infant have been reported with an LMA and fiberoptic laryngoscopy (Johnson, 1994).
Parry-Romberg Syndrome
Parry-Romberg syndrome is a rare disorder characterized by progressive hemifacial atrophy. Many believe this is a form of linear scleroderma. The process often starts in infancy or later childhood as a pigmented line (coup de sabre) on the forehead, and then progressive atrophy (which ultimately burns itself out) affects the subcutaneous tissues and skeleton of the hemiface. Reconstruction, which occurs after the atrophy is complete, is often complicated and may require multiple surgeries. Microvascular flaps and fat injections are surgical options (Hunt and Hobar, 2003).
Clefting
Historically, repair of the cleft lip and palate was performed with no anesthesia (Jones, 1971). It consisted of reapproximating pared tissue edges. These simple and quick procedures were performed on older children or adults who could tolerate the pain and inconvenience of the procedure. At most, patients were allowed to gargle with ice water to produce local numbing. In 1847, Snow described using ether for a cleft lip repair. Later in 1850, chloroform was reported for the repair of a bilateral lip and palate in a 7-year-old (Jones, 1971). The airway was unprotected, and blood could drain into the posterior pharynx. In 1921, Magill provided the first endotracheal insufflation technique on an infant and later applied this technique for the repair of a cleft palate in 1924 (Jones, 1971).
Today, general anesthesia is routinely and safely used for infants, children, and young adults having cleft lip or palate repairs. Typically, surgical repair of the cleft lip is performed at 3 to 6 months and the repair of the cleft palate at 9 to 18 months. Pharyngoplasty, performed for velopalatal insufficiency, is usually performed later, at 5 to 15 years of age. The safe age for cleft lip repair was established to be 6 to 12 weeks in 1964 by an audit of American plastic surgeons (Lewin, 1964) and later supported in 1966 by a large retrospective review (Wilhelmsen and Musgrave, 1966) that showed an increased rate of complications in children with a weight of less than 10 lb, hemoglobin level less than 10 g/dL, and white cell counts higher than 10,000. More recent studies have highlighted the safety of anesthesia for full-term and preterm neonates (Rudolph, 1974; Van Boven et al., 1993). The authors of these studies stress the importance of having a team of surgeons, anesthesiologists, and nursing staff that are experienced and comfortable with the intraoperative and postoperative care of the neonate. The preoperative preparation of the infant for cleft lip and palate surgery begins with a sound understanding of their anatomy, physiology, and associated anomalies.
Lip and Palate
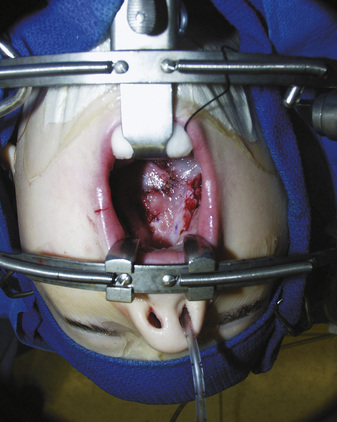
Anesthesia Management
For the patient with an isolated cleft lip and palate, difficulty with laryngoscopy is common. Some studies have described the incidence of difficult laryngoscopy (laryngoscopic view of Cormack and Lehane laryngoscope view grades 3 and 4) to be as high as 4% to 7% (Xue et al., 2006). Factors predicting a more difficult laryngoscopic view include bilateral clefts and retrognathia. Despite this high percentage of poor laryngoscopic view, only 1% of the patients were difficult to intubate, and only one patient had a failed intubation (Gunawardana, 1996).
Cleft patients with associated craniofacial anomalies or retrognathia may be difficult to ventilate or intubate. In these patients, preparations for alternative airway management need to be made. If general anesthesia is induced, the patient should remain spontaneously ventilating until the trachea is secured with an endotracheal tube. Successful intubation through a laryngeal mask with the aid of a fiberoptic scope has been described for pediatric patients with difficult airways (Inada et al., 1995). Other intubating devices include the Shikani Optical Stylet (Shukry et al., 2005; Clarus Medical). Surgical repair of the cleft palate using an LMA has even been described (Beveridge, 1989). However, disruption of a previous cleft palate repair during the placement of an LMA has also been described, suggesting that care should be taken when placing an LMA in a patient with a history of cleft palate repair (Somerville et al., 2004).
The oral RAE preformed tracheal tube is routinely used for the intubation because the preformed bend in the tube facilitates the use of the mouth retractor. Care should be paid to securing the tube and protecting it from unintentional extubation. Surgical preparation of the field can result in removal of the tape from the tube if it is not secured well (Fig. 25-12). Because of the fixed length of the preformed oral RAE tube, there is a risk of main stem intubation in a smaller infant. Cotton rolls can be placed in the Dingman-Dott retractor to minimize this risk. There is also an increased risk of extubation if an unexpectedly small endotracheal tube is required. An armor-reinforced endotracheal tube (see Fig. 10-11 in Chapter 10, Equipment) can be used to secure the airway in this situation. These reinforced endotracheal tubes do not kink with flexion, making them ideal for infants or children who require a smaller than expected endotracheal tube. Breath sounds should be reevaluated after the mouth gag is in place because of possible compression of the tube, and because advancement may cause single-lung ventilation.
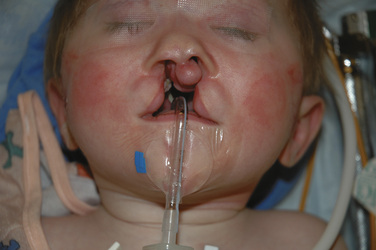
There is no standard technique for the maintenance of anesthesia. Both inhalation and total IV anesthetics have been described for patients having cleft lip and palate surgery. A balanced anesthetic of an inhalation agent (with or without nitrous oxide), an opioid, and a muscle relaxant (optional) is effective and safe. For patients having palate surgery, keeping the mean arterial pressure at 50 to 60 mm Hg may prevent excessive bleeding from the surgical site. This can be achieved by deepening the anesthetic with inhalation agents, opioids (remifentanil), or α2-agonists (clonidine or dexmedetomidine). Some studies (Rolla et al., 2003; Steinmetz et al., 2007) have attempted to determine whether the IV or the inhalation technique is better. However, there does not appear to be a clear benefit of either one.
The surgeon infiltrates the palate with epinephrine to prevent excessive bleeding and to facilitate mucosal dissection. It has been demonstrated that inhaled anesthetics sensitize the myocardium and decrease the arrhythmia threshold of epinephrine. Katz and others (1962), almost 50 years ago, described ventricular dysrhythmias when epinephrine was infiltrated during halothane anesthesia. The safe dosage of epinephrine is different for adults and for children. Dosages as low as 1 mcg/kg were found to be arrhythmogenic in adults under halothane anesthesia (Katz et al., 1962). In contrast, a 1983 study found that 10 mcg/kg was safe in children under halothane anesthesia (Karl, 1983). The children in this study ranged from 3 months to 17 years old and were ventilated to maintain an end-tidal CO2 of 40 mm Hg. The other inhaled anesthetics appear to be as safe or safer than halothane. In adults, the arrhythmogenic dosage of epinephrine was greater than 5 mcg/kg and did not differ between isoflurane, sevoflurane, or desflurane (Moore et al., 1993; Navarro et al., 1994). In children, 10 mcg/kg appears to be a safe maximal dosage of epinephrine for infiltration (see Box 7-3 in Chapter 7, Pharmacology of Pediatric Anesthesia).
Precautions with the airway management should be taken before extubation, especially for patients with difficult airways. Airway obstruction has been described after palatoplasties (Anthony and Sloan, 2002). Patients undergoing a palatoplasty who have a history of a difficult airway or have an associated congenital anomaly should have a nasopharyngeal airway placed by the surgeon before emergence. This may help maintain a patent airway and decrease the work of breathing after extubation (Fig. 25-13). Throat packs can result in airway obstruction if unintentionally left in place, and confirmation of their removal must take place before extubation.
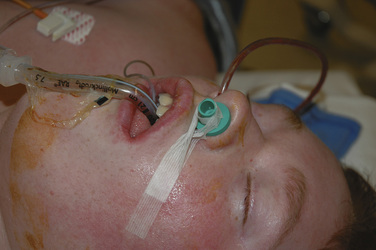
Tongue edema is a particular concern for the anesthesiologist taking care of an extubated patient after cleft palate surgery. Profound tongue, palate, and pharyngeal edema can occur from venous engorgement after use of the Dingman-Dott mouth retractor. It is prudent for surgeons to release the retractor repeatedly throughout the procedure to minimize this risk (Anthony and Sloan, 2002; Oste et al., 2004). Patients experiencing respiratory distress need to be reintubated and mechanically ventilated (Fig. 25-14). It may take several for days for the edema to resolve (Anthony and Sloan, 2002).
Postoperative pain management for cleft lip and palate repair should provide analgesia without respiratory depression, nausea, or vomiting. The best solution seems to be a combination of different medications, usually acetaminophen with the occasional addition of an opioid. Acetaminophen, when administered rectally, is given at an initial dosage of 20 to 40 mg/kg, followed by 20 mg/kg every 6 hours. The oral dosage is 10 to 15 mg/kg every 4 to 6 hours (Birmingham et al., 1997, 2001). Acetaminophen may have an opioid-sparing effect, and some infants after cleft lip repair require none, or very small amounts, in the postoperative period (Stephens et al., 1997).
Nonsteroidal antiinflammatory agents have been described for analgesia after cleft palate surgery (Sylaidis and O’Neill, 1998). Although some evidence suggests there may be no increased risk for bleeding, many surgeons and anesthesiologists choose not to use them routinely. Dosages of opioids, if administered, should be appropriate for weight, to minimize the risk for respiratory compromise in infants. A safe starting dosage of morphine for infants is 0.02 mg/kg every 3 to 4 hours, as needed. Infants undergoing cleft surgery and being given postoperative parenteral narcotics should be appropriately monitored (e.g., for apnea and bradycardia, electrocardiography, oxygen saturation).
The bilateral infraorbital nerve block works very well as an adjunct or as the sole analgesic technique for cleft lip repair (Eipe et al., 2006). The block, which can be performed by anesthesiologists or surgeons, provides better analgesia than periincisional infiltration or opioids alone (Pradeep et al., 1999; Rajamani et al., 2007). The infraorbital nerve block has been described for all age groups including neonates (Bosenberg and Kible, 1995).
The maxillary division of the trigeminal nerve exits from the infraorbital foramen. The nerve provides the sensory supply to the upper lip, the choana, the maxillary sinus, and part of the nasal septum. This block can be used to provide analgesia for cleft lip surgery, nasal septal repair, and endoscopic sinus surgery. There are two approaches to the maxillary division of the trigeminal nerve: extraoral (percutaneous) and intraoral. For the extraoral approach, the needle is directed toward the infraorbital foramen externally through the skin. The location of the foramen varies with the age. In children and adults, the foramen is located approximately 5 to 10 mm below the infraorbital rim in a vertical line from the pupil of the eye (Fig. 25-15). The thumb or a finger of the hand not holding the syringe can be placed on the inferior orbital rim as a guide and to protect the eye during the injection. The needle is placed just inferior to finger. Once it makes contact with the bone, it is withdrawn slightly and the local anesthetic is injected.
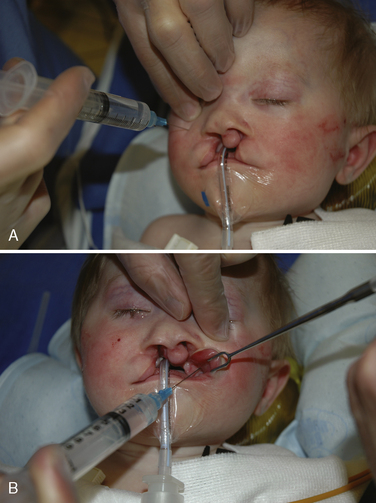
The duration of postoperative analgesia depends on the local anesthetic and its concentration. For children and adults, 0.125% bupivacaine provided analgesia for approximately 4 to 8 hours (Pradeep et al., 1999). Bupivacaine (0.5%) provides analgesia for 6 to 24 hours (Nicodemus et al., 1991; Eipe et al., 2006). In neonates, the block may be shorter (2 to 3 hours) (Bosenberg and Kible, 1995).
Anesthesia in Developing Countries
Medical missions are performed in developing countries for a variety of procedures including cardiac, orthopedic, and genitourinary. Cleft lip and palate missions are some of the most common and are carried out by multiple organizations around the world. The barriers to providing safe effective surgical and anesthesia care in developing countries include lack of hospital resources (electricity, lighting), medical resources (anesthesia machines, ventilators, oxygen, surgical equipment, medications), personnel resources (lack of training, temperament), standards of care, language skills, and cultural and social differences (Ward and James, 1990; Hodges and Hodges, 2000; Fisher et al., 2001). Despite these barriers, many health care providers volunteer their time and skills to help children with cleft deformities.
Pathologies that occur in developing countries and complicate anesthesia care include malnutrition, anemia, and parasitic infections, to name a few. Unfortunately, many of these pathologies are not identified, and it is not clear what impact this has on outcomes. Also, families may withhold information for fear of having the surgery cancelled, which may result in increased morbidity and even mortality (Fisher et al., 2001).
The induction of, maintenance of, and emergence from anesthesia need to be tailored to the available resources. Patients who present for isolated lip repair, and who are old enough, can have the procedure under local anesthesia (Hodges and Hodges, 2000). Patients who require general anesthesia are induced with either an inhalational or an IV technique. Halothane is a commonly used inhalational agent because of its cost. The airway is secured with an endotracheal tube in most circumstances; however, endotracheal tubes may have to be reused. Muscle relaxants in some practices (Fisher et al., 2001) are rarely used because of the lack of mechanical ventilation. In this population, spontaneous ventilation may be safer for the patient, and the anesthetist’s hands are free. Some groups preferred muscle relaxants because it facilitated rapid recovery in a setting that had no recovery area (Ward and James, 1990).
Outcome data for patients having cleft repairs in developing countries are limited. In a recent survey, (Fisher et al., 2001) identified complications associated with surgical patients from Operation Smile. Most of the surgeries were for cleft lip and palate repair, but other common plastic and orthopedic procedures were included. The data, collected from voluntary submission of perioperative data sheets, were collected on 6037 of the 9422 patients. Complications in the operating room, which included laryngospasm, bronchospasm, and unintentional extubation, occurred in 5% of these patients. In the recovery area, complications such as upper airway obstruction, postextubation croup, bronchospasm, and delayed discharge (pain, agitation) occurred in 3.3%. Cardiovascular complications including ventricular ectopy and supraventricular tachycardia occurred in 1.5%. The complications from arrhythmias are most likely underreported because not all patients had electrocardiographic monitoring. Significant negative events that occurred in 1.8% of the patients included return to the operating room for bleeding, intraoperative reintubation, cancellation of surgery after induction, blood transfusion, cardiac arrest, and death. The age of the patient predicted respiratory complications. A significantly larger percentage of children less than 1 year old experienced intraoperative respiratory complications. Death was an uncommon event; during the 18-month collection period, there were four deaths among all of the Operation Smile patients. This death rate of 0.4 per 1000 (Fisher et al., 2001) is higher than some other reports of pediatric perioperative deaths (<1 per 10,000) (Tiret et al., 1988; Morray et al., 2000), but it is difficult to compare the different patient populations. Despite this information, countless lives have benefited from medical missions to developing countries, and most of these procedures are accomplished safely. However, it must be recognized that performing surgery and anesthesia in these environments poses unique cultural, patient care, and system-based challenges for the entire surgical team.
Facial Clefting
Craniofacial clefts involve a defect of the underlying cranial or facial skeleton (or both), as well as the soft tissue envelope, and they can involve the entire face. Tessier classified these defects by the location of the facial clefts. In his classification, the orbit is the center of the defect, and the clefts radiate out like the spokes of a wheel (Whitaker and Bartless, 1990) (Fig. 25-16).
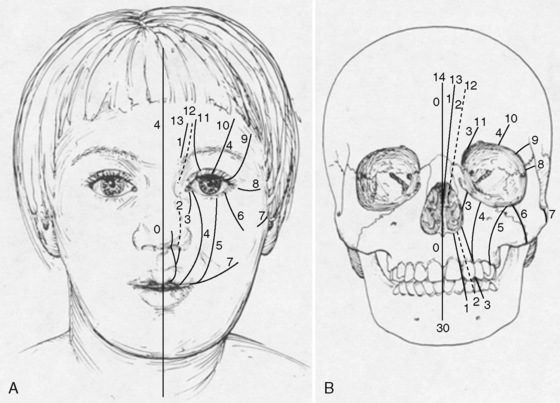
FIGURE 25-16 Topographic map of craniofacial clefting. Treacher Collins syndrome is a facial cleft located at 6, 7, and 8.
(From Whitaker LA, Bartlett SP: Craniofacial anomalies. In Jurkiewicz J, Krizek T, Mathes S, et al, editors: Plastic surgery: principles and practice, St Louis, 1990, Mosby, p 99.)
Treacher Collins Syndrome
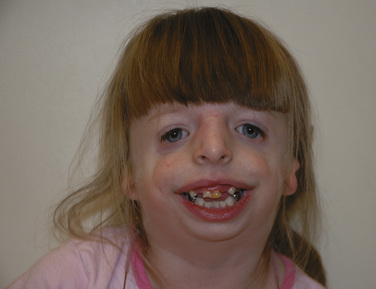
Treacher Collins syndrome (TCS), also know as mandibulofacial dysostosis, is an example of a bilateral 6, 7, and 8 craniofacial cleft (Fig. 25-17). It was first described in 1846 by Thompson and was further elaborated on by Treacher Collins. Inheritance is as an autosomal dominant pattern with variable penetration. The syndrome is characterized by poorly developed supraorbital ridges, aplastic/hypoplastic zygomas, ear deformities, hearing loss, cleft palate (in one third), and mandibular and midface hypoplasia. From birth, the adequacy of the airway is of primary concern. The degree of airway obstruction is related to the degree of maxillary and mandibular hypoplasia, choanal atresia, and glossoptosis. Tracheostomy may be required during infancy for those at highest risk for OSA and sudden infant death syndrome. The morbidity associated with tracheostomy is significant and some have advocated distraction osteogenesis or tongue-lip adhesion for these patients (Genecov et al., 2009; Thompson et al., 2009). Approximately 50% of patients with TCS have hearing loss, which occurs secondary to anomalies of the inner ear. They also suffer from external ear anomalies (microtia) (Marres, 2002). Congenital heart disease has been described in patients with TCS but it is uncommon (Gollin, 1976). Developmental delay is also uncommon in this population. These patients are followed by a multidisciplinary craniofacial team and can come to the operating room for a variety of procedures. Aside from cleft lip and palate repair, the timing of major reconstruction typically occurs during childhood or adolescence when the cranioorbital zygomatic bony development is nearly complete.
Surgical Management
The focus of care of the patient with TCS during the first 1 to 2 years of life is on the airway and feeding. These patients may have significant airway obstruction and may require positioning, tongue-lip adhesion, mandibular distraction, or tracheostomy. Patients with an associated cleft palate ideally have it repaired before 1 year of age. After 2 years of age, the surgical care of these patients focuses on restoration of hearing, and on reconstruction of the upper face. Ear surgery can involve the external ear (microtia repair) for cosmesis and the middle ear for hearing. Several surgical procedures have been described to reconstruct the upper face. Autologous and vascularized bone flaps are used to reconstruct the zygoma and orbits, and local flaps have been described to repair the eyelid defects. Unfortunately, good cosmetic results are very difficult to achieve and do not last long. Repeat surgery is common. Once dental development is complete, orthodontic and orthognathic surgery is scheduled. These procedures usually involve a sagittal split osteotomy, with or without a Le Fort I (Thompson et al., 2009).
Anesthesia Management
Anesthetic concerns specific to this syndrome primarily involve the airway. Infants and children with Treacher Collins syndrome may be very difficult or impossible to mask ventilate or intubate, and this airway difficulty may increase with age (Nargozian, 2004). Several techniques have been successfully used to manage the airway safely in these patients. Direct laryngoscopy regardless of the blade used may be difficult. However, the Bullard laryngoscope has been used to successfully intubate a child with TCS (Brown et al., 1993). The LMA has also been used successfully to assist in the intubation of these children (Ebata et al., 1991; Inada et al., 1995). In one series, the LMA was placed in neonates with PRS and TCS awake, and it was then used to guide intubation (Asai et al., 2008). The LMA has even been used successfully to ventilate a newborn with TCS for an extended period of time (Bucx et al., 2003). Other techniques used to intubate infants and children with TCS include the GlideScope (Verathon) (Bishop et al., 2009), a video-assisted laryngoscope (Sugawara et al., 2009), and the Airtraq optical stylet (PRODOL MEDITEL S.A.)(Hirabayashi et al., 2009). Given the potential for difficult mask ventilation and intubation, this population may be best managed with a sedated fiberoptic intubation. Successful sedation in a 6-year-old with TCS using dexmedetomidine and ketamine has been described (Iravani and Wald, 2008).
Another concern for the anesthesiologist is protecting the patient’s eyes. Because of the maxillary and zygomatic hypoplasia, prone positioning may increase the risk for orbital compression and perioperative blindness. Infants and children with TCS can have congenital cardiac defects and require prophylaxis for subacute endocarditis when indicated. Those most affected by upper airway obstruction and OSA may have reduced opioid intraoperative and postoperative requirements (Brown et al., 2004).
Hypertrophy and Vascular Anomalies
Vascular anomalies—disorders of the arteries, veins, capillaries, and lymphatics—were classified by Glowacki and Mulliken (1982). They are divided into two groups: vascular tumors and vascular malformations. The most common vascular tumor is the hemangioma. Vascular malformations are further divided into arterial, venous, capillary, and lymphatic malformations.
Hemangiomas
Hemangiomas, the most common vascular tumor, are characterized by endothelial proliferation and angiogenesis and then postnatal regression. Examples include “strawberry” and capillary hemangiomas. Hemangiomas develop after birth and can be common, with 4% to 12% of infants having one by the first year of life. There is a higher incidence in female children. By 9 years of age, most have involuted. A rule of thumb is that 50% involute by 5 years of age, 70% by 7 years of age, and 90% by 9 years of age (Bruckner and Frieden, 2003). Most hemangiomas appear on the head and neck (60%) or the trunk (24%) (Finn et al., 1983). Airway hemangiomas, although rare, can be seen in infants. The typical clinical findings are cough and stridor. Many airway hemangiomas (60%) have associated cutaneous lesions on the preauricular region, chin, lower lip, and neck (Orlow et al., 1997).
Some hemangiomas have associated anomalies. For example, facial hemangiomas can be associated with central nervous system vessel anomalies, coarctation of the aorta, right-sided aortic arch, and eye abnormalities. Some hemangioma-associated syndromes are of importance to the anesthesiologist. PHACES syndrome occurs with facial hemangiomas and is composed of posterior fossa abnormalities (Dandy-Walker), hemangiomas, arterial anomalies (coarctation), congenital heart disease (patent ductus arteriosus, ventricular septal defect), eye abnormalities (cataracts), and sternal defects (Poetke et al., 2002).
Vascular Malformations
Arterial
Arterial malformations are often asymptomatic. However, when they do become symptomatic they may have systemic sequelae. These malformations may cause limb hypertrophy or if extensive may result in a consumptive coagulopathy or congestive heart failure. Treatment is more effective with staged excision and embolization (Upton et al., 1985).
Capillary
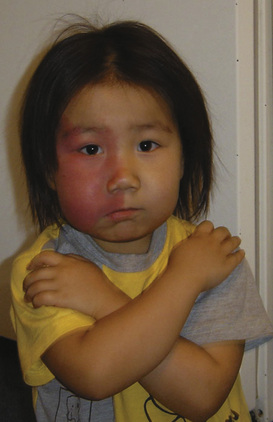
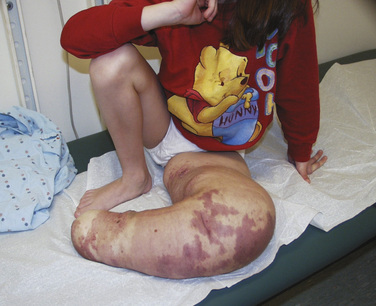
Capillary malformations occur in 0.3% to 0.5 % of infants. They often appear on the face and neck. Port-wine stains and nevus flammeus are examples. Facial port-wine stains typically occur in the trigeminal nerve distributions V1 (ophthalmic) and V2 (maxillary). Glaucoma and retinal arterial venous malformations are associated findings, and these patients need to be evaluated by an ophthalmologist. Head and brain MRI may also be required to identify underlying leptomeningeal malformations and choroidal angioma. Associated syndromes of capillary malformations include Sturge-Weber and Klippel-Trénaunay. Sturge-Weber syndrome consists of port-wine stain in the V1 trigeminal nerve distribution, glaucoma, overgrowth of the facial skeleton, leptomeningeal malformations, choroidal angiomas, and seizures (Fig. 25-18). Klippel-Trénaunay syndrome consists of capillary, venous, and lymphatic malformations. These lesions may involve the trunk and the extremities (Fig. 25-19). Treatment for capillary vascular malformations consists of multiple treatments with flashlamp pulsed dye lasers (Garzon et al., 2007).
Venous
Venous malformations are low-flow lesions that typically appear on the extremities and are often asymptomatic. However, they may occur in the gastrointestinal tract and can cause bleeding. Because most venous malformations are asymptomatic, treatment is conservative and consists of compressive garments. Treatment for symptomatic lesions includes embolization, sclerotherapy, and surgical excision (Hein et al., 2002).
Lymphatic
Lymphatic malformations are low-flow lesions that can involve the skin and underlying muscle. They typically appear on the head, neck, axillae, and chest. Lesions in the neck can be massive and can cause airway compromise, especially if they become infected and enlarged (Greinwald et al., 2008). These lesions do not regress spontaneously. They can be associated with syndromes including Klippel-Trénaunay, Turner’s, Noonan’s, and trisomies 13, 18, and 21. Intralesional sclerotherapy, laser treatment (CO2, Nd:YAG), and surgical excision have been described for their treatment (Padwa et al., 1995).
Anesthesia Management
Patients with hemangiomas or capillary malformations such as the port-wine stain may present for repeat laser treatments. For those patients with few or no comorbidities, these treatments can be performed in the outpatient setting. Typically, an inhalational anesthetic via a mask with or without IV access is all that is required. Lesions on the face or back may require an LMA or an endotracheal tube to secure the airway (Isago et al., 2006). Nitrous oxide, although safe for induction, may pose a fire hazard when laser treatments are near the face. Postoperative analgesia can be achieved with acetaminophen.
Vascular malformations involving the airway can lead to respiratory obstruction and OSA (Arneja and Gosain, 2008). In the anesthetized patient, there may be problems with ventilation and intubation (Nargozian, 2004; McLoughlin and McBrien, 2009). Some of the vascular malformations may not initially be obstructing lesions, but after sclerotherapy treatment or dependent positioning may swell dramatically leading to an obstructed airway (Sun et al., 2009). These patients may require alternative techniques to secure the endotracheal tube, such as a fiberoptic scope, the GlideScope, esophageal intubating devices, or intubating through an LMA. A hallmark of the management of the difficult pediatric airway is to secure the airway under general anesthesia while preserving spontaneous ventilation. When preservation of spontaneous ventilation under general anesthesia is not feasible, an awake or sedated intubation or tracheostomy is required. Patients with a consumptive process (Kasabach-Merritt syndrome) may be predisposed to intraoperative bleeding secondary to thrombocytopenia, and they may not be candidates for a regional anesthetic technique. However, regional anesthesia (intrathecal block) has been described in patients with Kasabach-Merritt syndrome when their platelet count is stable (Holak and Pagel, 2010).
Congenital Melanocytic Nevi
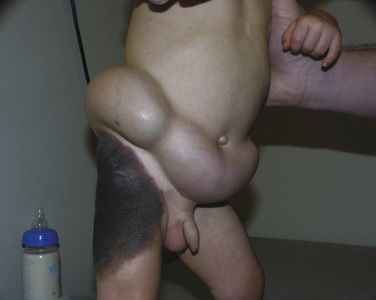
Congenital melanocytic nevi are pigmented, plaquelike skin lesions that are typically present at birth and persist and become more pigmented with age. They are classified by their size, which ranges from small to giant. Small nevi are 1.5 cm or less in diameter. Medium-sized nevi measure 1.5 to 19.9 cm in diameter, and large nevi have a diameter of greater than 20 cm. The giant nevi are significantly larger, with a diameter of greater than 50 cm. Small and medium nevi are more common and occur in approximately 1 in 1000 live births. Large nevi are less common and occur in 1 in 20,000 live births. Giant nevi are far less common and occur in only 1 in 50,000 births (Zaal et al., 2005). Besides the cosmetic concerns about congenital melanocytic nevi, there is the risk for malignant transformation, and this is an important consideration for removing these lesions. (Zaal et al., 2005) demonstrated in a large Dutch study that patients with congenital melanocytic nevi have a higher risk for developing malignant melanoma (15 cases in 19,253 person-years versus 1.2 expected cases), and the risk appears to be even higher in those with giant congenital melanocytic nevi. Some have reported melanoma rates as high as 10% in large nevi, with the majority of the malignant transformations in the largest lesions occurring by puberty (Tromberg et al., 2005).
Prophylactic removal of large nevi is typically performed early in life. The complexity of the surgical management of congenital nevi depends on the size and the location of the lesion. Large nevi may require a staged excision or the use of tissue expanders (Fig. 25-20). Placement of tissue expanders may occur at as early as 6 months of age. The expanders can be placed in the scalp, forehead, trunk, back, and lower extremities. Expansion is performed gradually over weeks to months and may allow complete closure after the nevi is surgically removed. Other treatment modalities include dermabrasion, lasers (CO2), and curettage. All of these techniques typically require general anesthesia. Patients with large congenital nevi may also require anesthesia for MRI evaluation for neurocutaneous melanosis. Patients with large nevi in axial locations (head, neck, back) are at increased risk for having melanocyte proliferation in the leptomeninges (pia and arachnoid mater). These melanocytes may malignantly transform and require radiologic surveillance (Lovett et al., 2009).
Trauma
Facial Fractures
Trauma to the facial skeleton may result in one or more fractures and may require surgical fixation. Typical facial fractures include zygoma fractures, Le Fort II, Le Fort III, mandible, orbital, nasal, and panfacial fractures. Depending on the mechanism of injury, there may be associated injuries, the most concerning of which is traumatic brain injury. In one study, 25% of patients diagnosed with facial fractures had associated injuries, including limb, brain, chest, spine, and abdominal injuries. Up to 10% of these patients had a brain injury (Thoren et al., 2010). Although only 2.7% of the patients had spinal injuries, almost all of these involved the cervical spine. Another study demonstrated that up to 69% of patients with mandible fractures had an associated traumatic brain injury. Although most of these patients had mild traumatic brain injury, up to 40% of them had a moderate to severe injury (intraparenchymal hemorrhage, epidural or subdural hemorrhage). Of concern, 20% of the patients with mild brain injuries had minimal signs or symptoms (Glasgow Coma Scale [GCS] 15, or no loss of consciousness), but they had an injury documented by computed tomography scan (Czerwinski et al., 2008). The hazard is that these brain injuries may be missed.
Anesthesia Management
Facial fractures involving the midface commonly result in airway compromise because of the displacement of the midface posteriorly into the oropharynx. Blood and secretions may also contribute to airway obstruction. One third of adult patients in one study with midface fractures required intubation (Ng et al., 1998). Patients with Le Fort III fractures developed more severe airway obstruction than patients with Le Fort I or II fractures. In fact, up to 40% of Le Fort III fractures required tracheostomy in one study of adults, compared with 9% of Le Fort I and II fractures (Bagheri et al., 2005).
This technique, however, is not an ideal option for the injured pediatric patient. The retromolar approach has been described for patients requiring MMF. This technique requires placing the endotracheal in the retromolar space, and it is meant for short-term intubation. The endotracheal tube can be sutured or wired in place by the surgeon. A smaller tube is recommended, and the anesthesiologist needs to be aware of the peak airway pressures once the tube is manipulated into this space (Lee et al., 2009).
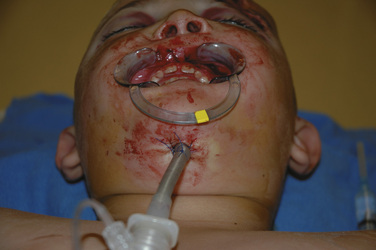
Traditionally, a short-term tracheostomy was recommended for patients with panfacial fractures because of the prolonged intubation in a patient with a potentially difficult airway or MMF. However, tracheostomy has an associated morbidity, and some have advocated a submental or submandibular approach to the airway (Amin et al., 2002; Anwer et al., 2007). The technique is performed after the patient is anesthetized and orally intubated. An armor-reinforced tube is recommended to prevent kinking. A small submandibular incision is made by the surgeon and bluntly dissected until it communicates with the floor of the mouth. The endotracheal tube cuff and then the end of the endotracheal tube (without the adaptor for the circuit) can be passed through this opening with the assistance of curved forceps or hemostats. The tube is sutured in place (Fig. 25-21). The submandibular airway can be removed at the end of surgery or can be left in place for several days (Amin et al., 2002; Anwer, 2009). This technique has been described for young adolescents (12 years old) (Eipe et al., 2005) and children as young as 6 years (Amin et al., 2002). Patients that are placed in MMF should have wire cutters at the bedside to release the fixation in case of an airway emergency. Even in skilled hands, this may not be quick. In one study, surgeons were able to remove the fixation in 30 seconds, but nonsurgical hospital staff took more than 2 minutes (Goss et al., 1979).
Extremity surgery
Hand Surgery
Congenital hand anomalies are numerous and varied. A classification of hand anomalies and descriptions of specific defects follow. Approximately 1 in 600 infants is born with some form of congenital hand anomaly. The terminology of hand anomalies (Table 25-5) reflects the type, classification, and degree of the defect (Netscher and Baumholtz, 2007). Surgical correction is needed for many of them. The American Society of Surgery of the Hand has created a classification of these anomalies (Box 25-1).
| Term | Definition |
| Brachy- | Short |
| Clino- | Bent |
| Campto- | Flexed |
| Phoco- | Shortened or “seal-like” |
| -Melia- | Extremity |
| Syn- | Side-to-side fusion |
| Sym- | Longitudinal fusion |
Box 25- Types of Congenital Hand Anomalies
From Netscher DT, Baumholtz MA: Treatment of congenital upper extremity problems, Plast Reconstr Surg 119:101e–129e, 2007.
From Netscher DT, Baumholtz MA: Treatment of congenital upper extremity problems, Plast Reconstr Surg 119:101e–129e, 2007.
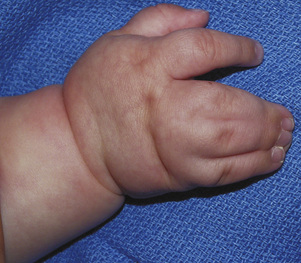
Anomalies that require surgical correction after the neonatal period but before 1 year of age are the syndactylies that occur between fingers of unequal length (Fig. 25-22). Hands exhibit a growth spurt by 2 years of age. Digits of unequal length that are fused together may need to be separated before significant hand growth occurs, because the anomalous growth of one of the digits may alter the growth of the other digit if they remain tethered together. Also, surgeries that affect critical hand function, such as opposition, need to be performed early so the development of grasping skills is not delayed. Thus, pollicization, movement of the index finger into the thumb position for a baby without a thumb, may be done on the early side, so that the child can grasp things with the thumb and perform the critical function known as opposition.
Brachial Plexus Palsy
Obstetric brachial plexus palsy is defined as an injury or a disruption to one or more of the trunks (C5 to T1) of the brachial plexus. The injury occurs at an incidence of 0.4 to 4 per 100 vaginal births and decreases significantly for cesarean sections but does not decrease to zero. Usually the injury occurs during passage through the birth canal. Perinatal risk factors for a brachial injury are thought to include large size for gestational age, shoulder dystocia, breech presentation, prolonged labor, and history of previous brachial plexus injuries (Waters, 2005). Patients present with varying degrees of motor and sensory loss of the upper extremity, depending on the type of injury.
The injuries are categorized into four groups, and the groupings predict outcome and guide recommendations for surgical intervention. Group one, involving an injury to the C5 to C6 trunks only, is the most common injury and accounts for approximately 50% of brachial plexus injuries. This upper trunk lesion is commonly called an Erb’s palsy. This group has the best prognosis. Group two involves an injury or disruption to the C5 to C7 trunks. Nearly 30% of patients with brachial plexopathy have this injury, and its prognosis is worse than for group one. Group three involves the entire brachial plexus (C5 to T1), with complete loss of function of the entire upper extremity. This injury pattern is seen in 20% of patients. Group four is the most severe, with injury to the entire brachial plexus, including the sympathetic chain. This injury most likely involves an avulsion at or near the spinal cord. These patients have complete loss of motor function of the upper extremity and Horner’s syndrome (ptosis, miosis, and anhydrosis). They may also have other motor injuries involving the phrenic, long thoracic, dorsoscapular, and suprascapular nerves (Hale, 2010).
Most brachial plexus injuries are transient and resolve with time alone. Physical therapy often helps to keep the shoulder joint supple while the nerves are recovering. Surgical repair is recommended for patients with group four injuries who are less than 3 months old. The timing and role of brachial plexus surgery become more controversial in those patients in groups one to three who do not demonstrate clinical recovery by 6 to 9 months of age. Surgical repair often involves exploration and resection of the neuroma and placement of a nerve graft. Nerve grafts can be harvested from several locations but the sural nerve is most often used (Waters, 2005; Hale et al., 2010). Neurolysis may be performed if the nerve appears completely normal but is encased by scar (Clarke, 2007).
Anesthesia Management
The anesthesia management of the pediatric patient presenting for upper extremity surgery begins with a thorough preoperative evaluation. Infants and children with hand anomalies may have underlying systemic disorders. Some of the syndromic craniofacial anomalies (Apert and Pfeiffer syndromes) may have associated upper extremity anomalies that need hand surgery. The anesthesia issues concerning these syndromes are outlined in Table 25-1. Patients with epidermolysis bullosa present for hand surgery because of the pseudosyndactyly from dermal loss. Anesthesia for these patients is described in Chapter 36, Systemic Disorders.
Other syndromes involving the hand may also involve the heart. Holt-Oram syndrome, otherwise known as hand-heart syndrome, exhibits varying degrees of congenital cardiac disease and arrhythmias (Shono et al., 1998). The degree of hand pathology does not predict the degree of cardiac involvement. A history consistent with cardiac pathology (poor feeding, sweating with feeding, cyanosis, syncope) or a heart murmur on examination warrants a cardiac evaluation before surgery. Patients with radial anomalies (aplasia) may have an associated VATER (vertebral defects, imperforate anus, tracheoesophageal fistula, radial and renal dysplasia) association. These patients require a preoperative evaluation to identify cardiac, renal, and vertebral anomalies. Radial anomalies also occur with the TAR (thrombocytopenia–absent radius) syndrome. The Poland sequence, also associated with upper extremity anomalies, starts with an initial chest wall deformity and then subsequent ipsilateral upper extremity anomalies. These patients may have significant thoracic insufficiency (Sethuraman et al., 1998).
When patients having hand surgery are otherwise healthy, general anesthesia can be used, with or without peripheral nerve blockade. Older patients can have hand procedures with a regional technique as the primary anesthetic (see Chapter 16, Regional Anesthesia). Anesthesia can be achieved with an inhalation or IV induction. Airway management depends on the size of the patient, coexisting disease, and duration of surgery. Infants having procedures with surgical field avoidance most likely require endotracheal intubation. Otherwise, larger healthy children can have their airway managed with an LMA. Patients with congenital heart disease do not require subacute endocarditis prophylaxis for hand surgery.
Patients presenting for surgical repair of the brachial plexus require a general anesthetic. The airway is managed with an endotracheal tube, as the surgery is long and the surgical field is close to the airway. It is important to secure the endotracheal tube well, because the head and shoulder may be moved during the operation. Use of a transparent drape for the uppermost sterile drape can help the anesthesiologist monitor the endotracheal tube position (Clarke, 2007). Neuromuscular blockade may be used for intubation, but a short- or intermediate-acting agent should be used and should not be repeated if the surgeons are using neuromuscular monitoring. Remifentanil with an inhalation or IV anesthetic will help produce a profound level of anesthesia and akinesis in those patients where neuromuscular blockade is avoided. The surgical repair of the brachial plexus may be lengthy and may require exposure of large areas of the infant. Sometimes both lower extremities are exposed and prepared so as to harvest the sural nerve. Proper padding is essential to minimize risk for pressure injuries, and multiple warming devices (forced hot air devices and warming lamps) may be required to maintain normothermia.
Summary
For questions and answers on topics in this chapter, go to “Chapter Questions” at www.expertconsult.com.
Amin M., Dill-Russell P., Manisali M., et al. Facial fractures and submental tracheal intubation. Anaesthesia. 2002;57:1195-1212.
Anthony A.K., Sloan G.M. Airway obstruction following palatoplasty: analysis of 247 consecutive operations. Cleft Palate Craniofac J. 2002;39:145-148.
Anwer H., Zeitoun I., Shehata E. Submandibular approach for tracheal intubation in patients with panfacial fractures. Br J Anaesth. 2007;98(6):835-840.
Arnaud E., Renier D., Marchac D. Prognosis for mental function in scaphocephaly. J Neurosurg. 1995;83:476-479.
Arneja J.S., Gosain A.K. Vascular malformations. Plast Reconstr Surg. 2008;121:195e-206e.
Asai T., Nagata A., Shingu K. Awake tracheal intubation through the laryngeal mask in neonates with upper airway obstruction. Paediatr Anaesth. 2008;18:77-80.
Bagheri S.C., Holmgren E., Kademani D., et al. Comparison of the severity of bilateral Le Fort injuries in isolated midface trauma. J Oral Maxillofac Surg. 2005;63:1123-1129.
Bannink N., Joosten K., van Veelen M., et al. Papilledema in patients with apert, crouzon, and pfeiffer syndrome: prevalence, efficacy of treatment, and risk factors. J Craniofac Surg. 2008;19(1):121-127.
Beveridge M.E. Laryngeal mask anesthesia for repair of cleft palate. Anaesthesia. 1989;44:656-657.
Bidgoli S.J.H., Dumont L., Mattys M., et al. A serious anaesthetic complication of a Le Fort I osteotomy. Eur J Anaesthesiol. 1999;16(3):201-203.
Bijnen C.L., Don Griot J.W., Mulder W.J., et al. Tongue lip adhesion in the treatment of pierre robin sequence. J Craniofac Surg. 2009;20:315-320.
Birmingham P.K., Tobin M.J., Fisher D.M., et al. Initial and subsequent dosing of rectal acetaminophen in children: a 24-hour pharmacokinetic study of new dose recommendations. Anesthesiology. 2001;94(3):385-389.
Birmingham P.K., Tobin M.J., Henthom T.K., et al. Twenty four hour pharmacokinetics of rectal acetaminophen in children: an old drug with new recommendations. Anesthesiology. 1997;87(2):244-252.
Bishop S., Clement P., Kale K., et al. Use of GlideScope Ranger in the management of a child with Treacher Collins syndrome in a developing world setting. Pediatric Anesthesia. 2009;9:695-715.
Blanco G., Melman E., Cuairan V., et al. Fibreoptic nasal intubation in children with anticipated and unanticipated difficult intubation. Paediatr Anaesth. 2001;11:49-53.
Bosenberg A.T., Kible F.W. Infraorbital nerve block in neonates for cleft lip repair: anatomical study and clinical application. Br J Anaesth. 1995;74:506-508.
Brown K.A., Laferriere A., Moss I.R., et al. Recurrent hypoxemia in young children with obstructive sleep apnea is associated with reduced opioid requirements for analgesia. Anesthesiology. 2004;100(4):806.
Brown R.E., Vollers I.M., Rader G.R., et al. Nasotracheal intubation in a child with Treacher Collins syndrome using the Bullard intubating laryngoscope. J Clin Anesth. 1993;5:492-493.
Bruckner A.L., Frieden I.J. Hemangiomas of infancy. J Am Acad Dermatol. 2003;48:477-493.
Bucx M.J., Grohman W., Kruisinga F.H., et al. The prolonged use of the laryngeal mask airway in a neonate with airway obstruction and Treacher Collins syndrome. Paediatr Anaesth. 2003;13:530-533.
Clarke H.M. Management of obstetrical brachial plexus. In: Bentz M.L., Bauer B.S., Zuker R.M., editors. Principles and practice of pediatric plastic surgery. St Louis: Quality Medical, 2007.
Cohen M.M., Kreiborg S. Visceral anomalies in the Apert syndrome. Am J Genetics. 1993;45(6):758-760.
Cohen M.M., Kreiborg S., Lammer E.J., et al. Birth prevalence study of the Apert syndrome. Am J Med Genet. 1992;42(5):655-659.
Cooper C.M., Murray-Wilson A. Retrograde intubation: management of a 4.8 kg, 5 month old. Anesthesia. 1987;42:1197-1200.
Czerwinski M., Parker W.L., Williams H.B. Algorithm for head computed tomography imaging in patients with mandible fractures. J Oral Maxillofac Surg. 2008;66:2093-2097.
D’Errico C.C., Munro H.M., Buchman S.R., et al. Efficacy of aprotinin in children undergoing craniofacial surgery. J Neurosurg. 2003;99:287-290.
De Ville A. Editorial: blood saving in pediatric patients. Paediatr Anaesth. 1997;7:181-182.
Durán de la Fuente P., García-Fernández J., Pérez-López C., et al. Usefulness of tranexamic acid in cranial remodeling surgery. Rev Esp Anestesiol Reanim. 2003;50(8):388-394. Oct
Ebata T., Nishiki S., Masuda A., et al. Anaesthesia for Treacher Collins syndrome using a laryngeal mask airway. Can J Anaesth. 1991;38(8):1043-1045.
Eipe N., Choudhrie A., Pillai A.D., et al. Regional anesthesia for cleft lip repair: a preliminary study. Cleft Palate Craniofac J. 2006;43(2):138-141.
Eipe N., Neuhoefer E., La Rosee G., et al. Submental intubation for cancrum oris: a case report. Pediatr Anesthesia. 2005;15:1009-1012.
Faberowski L.W., Black S., Mickle J.P. Blood loss and transfusion practice in the perioperative management of craniosynostosis repair. J Neurosurg Anesthesiol. 1999;11:167.
Faberowski L.W., Black S., Mickle J.P. Incidence of venous air embolism during craniectomy for craniosynostosis repair. Anesthesiology. 2000;92:20-23.
Fearon J.A., Weinthal J. The use of recombinant erythropoietin in the reduction of blood transfusion rates in craniosynostosis repair in infants and children. Plast Reconstr Surg. 2002;109(7):2190-2196.
Fearon J.A. Rigid fixation of the calvaria in craniosynostosis without using “rigid” fixation. Plast Reconstr Surg. 2003;111(1):27.
Fearon J.A. The Le Fort III osteotomy: to distract or not to distract? Plast Reconstr Surg. 2001;107:1091.
Fearon J.A. Reducing allogenic blood transfusions during cranial vault surgical procedures: a prospective analysis of blood recycling. Plast Reconstr Surg. 2005;113(4):1126-1130.
Fialkov J.A., Holy C., Forrest C.R., et al. Postoperative infections in craniofacial reconstructive procedures. J Craniofac Surg. 2001;12(4):362-368.
Finn M.C., Glowacki J., Mulliken J.B. Congenital vascular lesions: clinical application of a new classification. J Pediatr Surg. 1983;18:894-900.
Fisher Q.A., Nichols D., Stewart F., et al. Assessing pediatric anesthesia practices for volunteer medical services abroad. Anesthesiology. 2001;95:1315-1322.
Garzon M.C., Huang J.T., Enjolras O., et al. Vascular malformations: Part 2: Associated syndromes. J Am Acad Dermatol. 2007;56(4):541-564.
Genecov D.G., Barcelo C.R., Steinberg D., et al. Clinical experience with the application of distraction osteogenesis for airway obstruction. J Craniofac Surg. 2009;20:1817-1821.
Glowacki J., Mulliken J.B. Classification of pediatric vascular anomalies. Plast Reconstr Surg. 1982;70(1):120-121.
Gollin R.J. Syndromes of the head and neck, ed 2. New York: McGraw Hill, 1976.
Goss A.N., Chau K.K., Mayne L.H. Intermaxillary fixation: how practicable is emergency jaw release? Anaesth Intensive Care. 1979;7:253-257.
Greinwald J.R., Cohen A.P., Hemanackah S., et al. Massive lymphatic malformations of the head, neck, and chest. J Otolaryngol Head Neck Surg. 2008;37(2):169-173.
Gunawardana R.H. Difficult laryngoscopy in cleft lip and palate surgery. Br J Anaesthesia. 1996;76:757-759.
Hale H.B., Bae D.S., Waters P.M. Current concepts in the management of brachial plexus birth palsy. J Hand Surg. 2010;35A:322-331.
Harris M.M., Davidson A., Strafford M.A., et al. Venous air embolism during craniectomy in supine infants. Anesthesiology. 1987;67:816-819.
Hein K.D., Mulliken J.B., Kozakewich H.P., et al. Venous malformations of skeletal muscle. Plast Reconstr Surg. 2002;110:1625-1635.
Hidestrand P., Vasconez H., Cottrill C. Carpenter syndrome. J Craniofac Surg. 2009;20(1):254-256.
Hirabayashi Y., Shimada N., Nagashima S. Tracheal intubation using pediatric Airtraq optical laryngoscope in a patient with Treacher Collins syndrome. Pediatric Anesthesia. 2009;19:908-928.
Hodges S.C., Hodges A.M. A protocol for safe anesthesia for cleft lip and palate surgery in developing countries. Anaesthesia. 2000;55:436-441.
Holak E.J., Pagel P.S. Successful use of spinal anesthesia in a patient with severe Klippel-Trénaunay syndrome associated with upper airway abnormalities and chronic Kasabach-Merritt coagulopathy. J Anesth. 2010;24:134-138.
Hunt J.A., Hobar P.C. Common craniofacial anomalies: conditions of craniofacial atrophy/hypoplasia and neoplasia. Plast Reconstr Surg. 2003;111(4):1497-1508.
Idestrand P., Vasconez H., Cottrill C. Carpenter syndrome. J Craniofac Surg. 2009;20(1):254-256.
Inada T., Fujise K., Tachibana K., et al. Orotracheal intubation through the laryngeal mask airway in paediatric patients with Treacher Collins syndrome. Paediatr Anesth. 1995;5:129-132.
Iravani M., Wald S. Dexmedetomidine and ketamine for fiberoptic intubation in a child with severe mandibular hypoplasia. J Clin Anesth. 2008;20:455-457.
Isago T., Kono T., Nozaki M., et al. Ambulatory anesthesia for children undergoing laser treatment. Surg Today. 2006;36:765-768.
Jimenez D.F., Barone C.M., Cartwright C.C., et al. Early management of craniosynostosis using endoscopic-assisted strip craniectomies and cranial orthotic molding therapy. Pediatrics. 2002;110(1):97-104.
Johnson C.M., Sims C.P. Awake fiberoptic intubation via a laryngeal mask airway in an infant with Goldenhar syndrome. Anaesth Intensive Care. 1994;22:194-197.
Jones R.G. A short history of anaesthesia for hare-lip and cleft palate repair. Br J Anaesth. 1971;43:796.
Karl H., Swedlow D.B., Lee K.W., et al. Epinephrine-halothane interactions in children. Anesthesiology. 1983;58:142-145.
Katz R.L., Matteo R.S., Papper E.M. The injection of epinephrine during general anesthesia with halogenated hydrocarbons and cyclopropane in man. Anesthesiology. 1962;23:579-600.
Kirschner R.E., Low D.W., Randall P., et al. Surgical airway management in Pierre Robin sequence: is there a role for tongue lip-adhesion? Cleft Palate Craniofac J. 2003;40(1):13-17.
Krajewski K., Ashley R.K., Pung N., et al. Successful blood conservation during craniosynostotic correction with dual therapy using Procrit and cell saver. J Craniofac Surg. 2008;19(1):101-105.
Kreiborg S., Barr M., Cohen M.M. Cervical spine in the Apert syndrome. Am J Med Genet. 1992;43:704-708.
Lee S., Huang S., Wu S., et al. A review of intraoperative airway management for midface facial bone fracture patients. Ann Plast Surg. 2009;63:162-166.
Levine J.P., Stelnicki E., Weiner H.L., et al. Hyponatremia in the postoperative craniofacial pediatric patient population: a connection to cerebral salt wasting syndrome and management of the disorder. Plast Reconstr Surg. 2001;108:1501-1508.
Lewin M.L. Management of cleft lip and palate in the United States and Canada. Plastic Reconstr Surg. 1964;33:383-394.
Lo L.J., Chen Y.R. Airway obstruction in severe syndromic craniosynostosis. Ann Plast Surg. 1999;43:258-264.
Lo L.J., Hung K.F., Chen Y.R. Blindness as a complication of Le Fort I osteotomy for maxillary distraction. Plast Reconstr Surg. 2002;109(2):688.
Lovett A., Maari C., Decarie J., et al. Large congenital melanocytic nevi and neurocutaneous melanocytosis: one pediatric center’s experience. J Am Acad Dermatol. 2009;61(5):766-767.
Markakis D.A., Sayson S.C., Schreiner M.S. Insertion of the laryngeal mask airway in awake infants with the Robin sequence. Anesth Analg. 1992;75(5):822-824.
Marres H.A. Hearing loss in the Treacher-Collins syndrome. Adv Otorhinolaryngol. 2002;61:209-215.
Mathijssen I., Arnaud E., Lajeunie E., et al. Postoperative cognitive outcome for synostotic frontal plagiocephaly. J Neurosurg. 2006;105:16-20.
Mayer M.N., de Montalembert M., Audat F., et al. Autologous blood donation for elective surgery in children weighing 8-25 kg. Vox Sang. 1996;70:224-228.
McLoughlin J.M., McBrien M.E. Acute upper airway obstruction secondary to late presentation of a massive oropharyngeal arteriovenous malformation. J Clin anesth. 2009;21:278-281.
Mixter R.C., David D.J., Perloff W.H., et al. Obstructive sleep apnea in Apert’s and Pfeiffer’s syndrome: more than a craniofacial abnormality. Plast Reconstr Surg. 1990;86(3):457-463.
Moore M.H., Cantrell S.B., Trott J.A., et al. Pfeiffer syndrome: a clinical review. Cleft Palate Craniofac J. 1995;32(1):62-70.
Moore M.A., Weiskopf R.B., Eger E., et al. Arrhythmogenic doses of epinephrine are similar during desflurane or isoflurane anesthesia in humans. Anesthesiology. 1993;79:943-947.
Morray J.P., Geiduschek J.M., Ramamoorthy C., et al. Anesthesia related cardiac arrest in children: the initial findings of the Pediatric Perioperative Cardiac Arrest (POCA) Registry. Anesthesiology. 2000;93:6-14.
Naran S., Cladis F., Fearon J., et al. Safety of preoperative epoetin alpha in surgical calvarial remodeling: an 8 year retrospective analysis. Presented at the 67th Annual meeting of the American Cleft Palate Association, Fort Worth, Tex. March 2010.
Nargozian C. The airway in patients with craniofacial abnormalities. Paediatr Anaesth. 2004;14:53-59.
Nascimento S.R., De Mello M.P., Batista J.C., et al. Clinical findings in four Brazilian families affected by Saethre-Chotzen syndrome without TWIST mutations. Cleft Palate Craniofac J. 2004;41:250-255.
Navarro R., Weiskopf R.B., Moore M.A., et al. Humans anesthetized with sevoflurane or isoflurane have similar arrhythmic response to epinephrine. Anesthesiology. 1994;80:545-549.
Nelson T.E., Mulliken J.B., Padwa B.L. Effect of midfacial distraction on the obstructed airway in patients with syndromic bilateral coronal synostosis. J Oral Maxillofac Surg. 2008;66(11):2318-2321. Nov
Netscher D.T., Baumholtz M.A. Treatment of congenital upper extremity problems. Plast Reconstr Surg. 2007;119:101e-129e.
Ng M., Saadat D., Sinha U.K. Managing the emergency airway in Le Fort fractures. J Craniomaxillofac Trauma. 1998;4:38.
Nicodemus H.F., Ferrer M.J., Cristobal V.C., et al. Bilateral infraorbital block with 0.5% bupivacaine as postoperative analgesia following cheiloplasty in children. Scand J Reconstr Surg Hand. 1991;25:253-257.
Orlow S.J., Isakoff M.S., Blei F. Increased risk of symptomatic hemangiomas of the airway in association with cutaneous hemangiomas in the “beard” distribution. J Pediatr. 1997;131:643-646.
Oste C.D., Savron F., Pelizzo G., et al. Acute airway obstruction in an infant with Pierre Robin syndrome after palatoplasty. Acta Anaesthesiol Scand. 2004;48:787-789.
Padwa B.L., Hayward P.G., Ferraro N.F., et al. Cervicofacial lymphatic malformation: clinical course, surgical intervention, and pathogenesis of skeletal hypertrophy. Plast Reconstr Surg. 1995;95:951-960.
Padwa B.L., Hayward P.G., Ferraro N.F., et al. Cervicofacial lymphatic malformation: clinical course, surgical intervention, and pathogenesis of skeletal hypertrophy. Plast Reconstr Surg. 2003;133:238-242.
Panchal J., Uttchin V. Management of craniosynostosis. Plast Reconstr Surg. 2003;111:2032.
Perkins J.A. Airway management in children with craniofacial anomalies. Cleft-craniofacial Journal. 1997;34(2):135-140.
Pijpers M., Poels P.J., Vaandrager M., et al. Undiagnosed obstructive sleep apnea syndrome in children with syndromal craniofacial synostosis. J Craniofac Surg. 2004;15(4):670-674.
Poetke M., Frommeld T., Berlien H.T. PHACE syndrome: new views on diagnostic criteria. Eur J Pediatr Surg. 2002;12:366-374.
Posnick J.C. Craniofacial syndromes and anomalies in children and young adults. vol 1. Philadelphia: Saunders; 2000.
Pradeep K., Prabhu K., Wig J., et al. Bilateral infraorbital nerve block is superior to peri-incisional infiltration for analgesia after repair of cleft palate. Scand J Plast Reconstr Hand Surg. 1999;33:83-87.
Rajamani A., Kamat V., Rajavel V.P., et al. A comparison of bilateral infraorbital nerve block with intravenous fentanyl for analgesia following cleft lip repair in children. Pediatr Anesthesia. 2007;17(2):133-139.
Renier D., Lajeunie E., Arnaud E., et al. Management of craniosynostoses. Childs Nerv Syst. 2000;16:645-658.
Rolla P., Gall O., Dejesus L., et al. Remifentanil for cleft palate surgery in young infants. Paediatr Anaesth. 2003;13:701-707.
Rudolph A.M. Congenital diseases of the heart. Chicago: Year Book Medical, 1974.
Schubert A., Deogaonkar A., Drummond J. Precordial Doppler probe placement for optimal detection of venous air embolism during craniotomy. Anesth Analg. 2006;102:1543-1547.
Sculerati N., Gottlieb M.D., Zimbler M.S. Airway management in children with major craniofacial anomalies. Laryngoscope. 1998;108(12):1806-1812.
Sethuraman R., Kannan S., Bala I., et al. Anaesthesia and Poland syndrome. Can J Anaesth. 1998;45:277-279.
Shetye P.R., Boutros S., Grayson B.H., et al. Midterm follow-up of midface distraction for syndromic craniosynostosis: a clinical and cephalometric study. Plast Reconstr Surg. 2007;120:1621-1632.
Shono S., Higa K., Kumano K., et al. Holt-Oram syndrome. BJA. 1998;80:856-857.
Shukry M., Hanson R.D., Koveleskie J., et al. Management of the difficult pediatric airway with Shikani optical stylet. Pediatr Anesth. 2005;15:342-345.
Singh A.K., Szczech L., Tang K.L., et al. for the CHOIR Investigators: Correction of anemia with epoetin alfa in chronic kidney disease. N Engl J Med. 2006;355:2085-2098.
Sirotnak J., Brodsky L., Pizzuto M. Airway obstruction in the Crouzon syndrome: case report and review of the literature. Int J Ped Otolaryng. 1995;31(2–3):235-246.
Slater B., Lenton K.A., Kwan M.D., et al. Cranial sutures: a brief review. Plast Reconstr Surg. 2008;121:170e.
Somerville N.S., Fenlon S., Boorman J., et al. Disruption of cleft repair following the use of the laryngeal mask airway. Anaesthesia. 2004;59:401-403.
Steinmetz J., Holm-Knusen R., Sorensen M.K., et al. Hemodynamic differences between propofol-remifentanil and sevoflurane anesthesia for repair of cleft lip and palate in infants. Pediatr Anesthesia. 2007;17:32-37.
Stephens P., Saunders P., Bingham R. Neonatal cleft lip repair: a retrospective review of anaesthetic complications. Pediatr Anaesth. 1997;7:33-36.
Stricker P., Shaw T., Desouza D.G., et al. Blood loss, replacement, and associated morbidity in infants and children undergoing craniofacial surgery. Pediatr Anesth. 2010;20:150-159.
Stricker P.A., Budac S., Fiadjoe J.E., et al. Awake laryngeal mask insertion followed by induction of anesthesia in infants with the Pierre Robin sequence. Acta Anaesthesiol Scand. 2008;52(9):1307-1308.
Sugawara Y., Inagawa G., Satoh K., et al. Successful intubation using a simple fiberoptic assisted laryngoscope for Treacher Collins syndrome. Pediatric Anesthesia. 2009;19:1025-1033.
Sun Y., Jiang H., Zhu Y. Acute upper airway obstruction secondary to late presentation of a massive oropharyngeal arteriovenous malformation. Pediatr Anesth. 2009;19:623-625.
Sylaidis P., O’Neill T. Diclofenac analgesia following cleft palate surgery. Cleft Palate Craniofac J. 1998;35(6):544-545.
Thompson J., Anderson P., David D.J. Treacher Collins syndrome: protocol management from birth to maturity. J Craniofac Surg. 2009;20:2028-2035.
Thorén H., Snall J., Salo J., et al. Occurrence and types of associated injuries in patients with fractures of the facial bones. J Oral Maxillofac Surg. 2010;68(4):805.
Tiret L., Nivoche Y., Hatton F., et al. Complications related to anesthesia in infants and Children. Br J Anaesth. 1988;61:263-269.
Tomaski S., Zalzal G., Saal H. Airway obstruction in the Pierre Robin sequence. Laryngoscope. 1995;105:111-114.
Tromberg J., Bauer B., Bienvenuto-Andrade C., et al. Congenital melanocytic nevi needing treatment. Dermatol Ther. 2005;18:136-150.
Tunçbilek G., Vargel I., Erdem A., et al. Blood loss and transfusion rates during repair of craniofacial deformities. J Craniofac Surg. 2005;16(1):59-62. Jan
Upton J., Mulliken J.B., Murray J.E. Classification and rationale for the management of vascular anomalies in the upper extremities. J Hand Surg. 1985;10(6):970-975.
Van Boven M.J., Penderville P.E., Veyckemans F., et al. Neonatal cleft lip repair: the anesthetist’s point of view. Cleft Palate Craniofac J. 1993;30:574-577.
Ward C.M., James I. Surgery of 346 patients with unoperated cleft lip and palate in Sri Lanka. Cleft Palate J. 1990;27(1):11-17.
Waters P.M. Update on management of pediatric brachial plexus palsy. J Pediatr Orthop. 2005;25(1):116-126.
Whitaker L.A., Bartlett S.P. Craniofacial anomalies. In: Jurkiewicz M.J., Krizek T.J., Mathes S.J., et al, editors. Plastic surgery: principles and practice. St Louis: Mosby; 1990:99.
Whitaker L.A., Munro I.R., Salyer K.E., et al. Combined report of problems and complications in 793 craniofacial operations. Plast Reconstr Surg. 1979;64(2):198-203.
White N., Marcus R., Dover S., et al. Predictors of blood loss in fronto-orbital advancement and remodeling. J Craniofac Surg. 2009;20(2):378-381.
Wilhelmsen H.R., Musgrave R.H. Complications of cleft lip surgery. Cleft Palate J. 1966;3:223-231.
Wong G.B., Nargozian C., Padwa B.L. Anesthetic concerns of external maxillary distraction osteogenesis. J Craniofac surg. 2004;15(1):78-82.
Xue F.S., Zhang G.H., Li P., et al. The clinical observation of difficult laryngoscopy and difficult intubation in infants with cleft lip and palate. Pediatr Anesthesia. 2006;16:283-289.
Zaal L.H., Mooi W.J., Klip H., et al. Risk of malignant transformation of congenital melanocytic nevi: a retrospective nationwide study from The Netherlands. Plast Reconstr Surg. 2005;116:1902-1909.