[level-membership-for-basic-science-category]
Chapter 41 Androgens and Antiandrogens
| Abbreviations | |
|---|---|
| AR | Androgen receptor |
| cGMP | Cyclic guanosine monophosphate |
| DHEA | Dehydroepiandrosterone |
| DHT | Dihydrotestosterone |
| FSH | Follicle-stimulating hormone |
| GnRH | Gonadotropin-releasing hormone |
| hCG | Human chorionic gonadotropin |
| IM | Intramuscular |
| LH | Luteinizing hormone |
| SHBG | Sex hormone-binding globulin |
| StAR | Steroidogenic acute regulatory protein |
Therapeutic Overview
Androgens are produced by the testis, ovary, and adrenal glands. Testosterone is the most potent androgen. It stimulates virilization and spermatogenesis. Within the ovary, testosterone and androstenedione are precursor steroids for estradiol production (see Chapter 40). In both sexes androgens stimulate body hair growth, positive nitrogen balance, bone growth, muscle development, and erythropoiesis. The mechanism of action of testosterone at its target organs is similar to that of other steroid hormones (see Chapter 1). The primary clinical use of androgens is replacement therapy in men with diagnosed testosterone deficiency. Testosterone synthesis inhibitors, referred to as antiandrogens, and competitive androgen receptor (AR) antagonists, are used to reduce the effects of androgens in patients with androgen-dependent disorders such as prostatic cancer, benign prostate hyperplasia hirsutism, and precocious puberty. Another male-specific phenomenon is treatment of erectile dysfunction, which employs peripherally acting vasodilators such as cyclic guanosine monophosphate (cGMP) phosphodiesterase type 5 inhibitors or synthetic prostaglandin E1 analogs.
| Therapeutic Overview |
|---|
| Androgens |
| Primary testicular insufficiency |
| Hypogonadotropic hypogonadism |
| Constitutional delay of growth and adolescence |
| Osteoporosis, anemia |
| Male contraception |
| Antiandrogens and Androgen Receptor Antagonists |
| Virilization in women |
| Precocious puberty in boys |
| Prostate cancer, hyperplasia |
responsible for stimulating the development of male external genitalia during the first trimester of fetal life. Therefore, when the fetal synthesis of androgen is insufficient (e.g., due to an inborn enzymatic error) or the action of androgen is ineffective at its target tissues (e.g., androgen resistance), the genital phenotype may be female or ambiguous.
Mechanisms of Action
Androgens are synthesized from cholesterol in testicular Leydig cells, the adrenal cortex, and ovarian thecal cells (see Fig. 38-1). In the adult gonads the principal regulator of testosterone synthesis and secretion is luteinizing hormone (LH), which is produced by the anterior pituitary gland (Chapter 38). The precursor cholesterol is synthesized in the Leydig cells from acetate and stored as cholesterol esters in lipid droplets. A cholesterol ester hydrolase mobilizes free cholesterol from the lipid droplets, which in turn is transferred to the inner mitochondrial membrane. Stimulation of this transfer represents a major action of LH and is mediated by the steroidogenic acute regulatory (StAR) protein. Leydig cells can convert a small fraction of testosterone to estradiol (see Chapter 40 and Fig. 38-1). LH increases the level of enzymes in this synthetic pathway.
Androgen Production by the Adrenal Glands
Although glucocorticoids and mineralocorticoids are the principal products of the adult adrenal gland, dihydroepiandrosterone (DHEA), androstenedione, and testosterone, as well as some DHEA sulfate and estrone, can be also secreted (see Fig. 38-1). The concentrations of DHEA, DHEA sulfate, and androstenedione in the circulation increase between 7 to 10 years of age. This process has been termed adrenarche, to distinguish it from gonadarche, which is the onset of adult gonadal function at puberty. Adrenal androgen secretion declines in the elderly and during severe illness.
Regulation of Testosterone Synthesis and Secretion
The major site of testosterone synthesis and secretion is the Leydig cell, which has cell-surface LH receptors that associate with the Gs subunit of adenylyl cyclase. Steroidogenesis mediated by LH requires mobilization of intracellular Ca++ and the Ca++-binding protein calmodulin, and activation of phospholipase C (see Chapter 1). The effects of LH involve rapid stimulation of testosterone production (within minutes), which is mediated by the StAR. Other hormones that influence testosterone synthesis include prolactin, cortisol, insulin, insulin-like growth factors, estradiol, activin, and inhibin. There is a growing appreciation of the multiple factors involved in testosterone synthesis that are produced within the seminiferous tubules by germ cells and Sertoli cells or peritubular myoid cells. These factors maintain the serum concentration of testosterone in adult men at 0.3 to 1.0 mg/dL (10 to 30 nM). During illness, LH production declines, and cytokines suppress testosterone production.
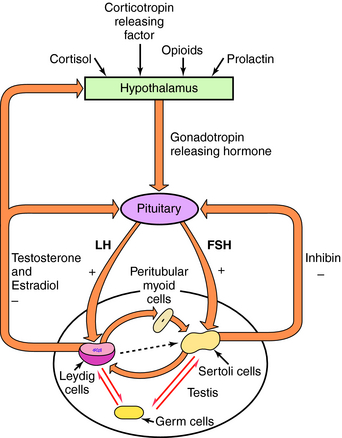
The hormones of the hypothalamus, pituitary, and testes form an internally regulated unit (Fig. 41-1). Not only are the testes stimulated by pituitary gonadotropins, but the testes also regulate LH and FSH secretion through negative-feedback mechanisms. Testosterone suppresses gonadotropin secretion by slowing the pulsatile release of GnRH. Estradiol, which is synthesized from testosterone in the ovary, testes, adipose tissue, liver, and brain, inhibits gonadotropin release through effects on both the hypothalamus and pituitary. Inhibin-B selectively reduces FSH synthesis and secretion.
Normally, women produce approximately 0.25 mg/day of testosterone compared with the 5 to 7 mg/day for adult men. Most testosterone circulating in women is derived from the peripheral conversion of androstenedione secreted by the ovaries and adrenals (see Chapter 40). Benign and malignant tumors of the adrenal and ovary, congenital steroidogenic enzyme defects, and disturbances of gonadotropin secretion can be associated with increased androgen production in women.
Depending on the tissue, intracellular testosterone or a more active metabolite DHT can interact directly with ARs (Fig. 41-2). When testosterone enters the prostate gland or any tissue with significant 5α-reductase activity, nearly 90% of it is metabolized to DHT. There are two isoenzymes of 5α-reductase, encoded by two different genes. Type I 5α-reductase is found in liver, skin, sebaceous glands, most hair follicles, and prostate, whereas 5α-reductase type II predominates in genital skin, beard and scalp hair follicles, and prostate. The presence of ambiguous genitalia in patients with inactivating mutations of the 5α-reductase type II gene is indicative of the importance of this enzyme in normal development of male external genitalia. The distribution of the isozymes has been exploited to develop tissue-specific inhibitors of 5α-reductase activity.
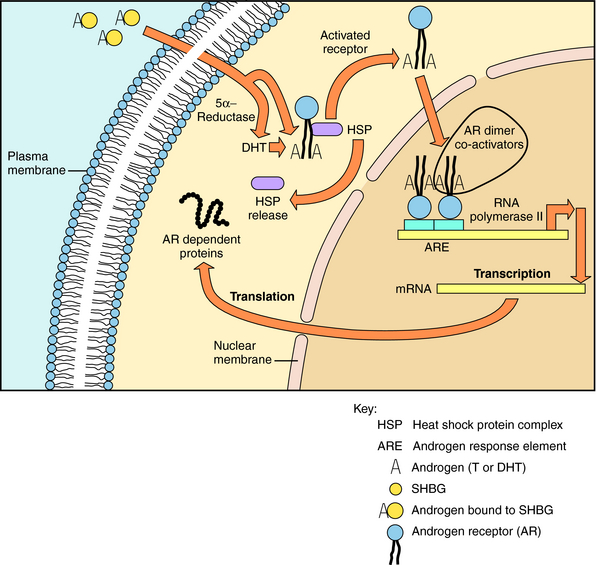
Androgen binding to ARs and the events that follow are similar to those of other steroid hormones (see Chapter 1). The AR is encoded by a gene on the X chromosome and is expressed in most tissues. When a ligand binds to the AR, the conformation of the receptor is altered, it binds to DNA response elements, multiple coactivator proteins are recruited, and the transcription of messenger RNAs for tissue-specific proteins ensues. Although most actions of androgens are mediated by transcriptional activity of the receptor, others are mediated through second messengers, such as the mitogen-activated protein kinase pathway.
Antiandrogens and Androgen Receptor Antagonists
There are two basic mechanisms to block the activity of androgen. These include inhibition of androgen formation or antagonism of androgen-AR interactions. Strategies for suppression of androgen formation include blockade of gonadotropin formation using GnRH analogs and the use of spironolactone and ketoconazole to competitively inhibit the activities of steroid 17α-hydroxylase (CYP17) and cholesterol side-chain cleavage enzyme (CYP11A), respectively (see Fig. 38-1). Another agent, finasteride, is a competitive inhibitor of the 5α-reductase isozymes and antagonizes the formation DHT, which has potent androgenic action. All these drugs are substrate analogs and compete with the natural substrates for active sites on the enzymes.
Pharmacokinetics
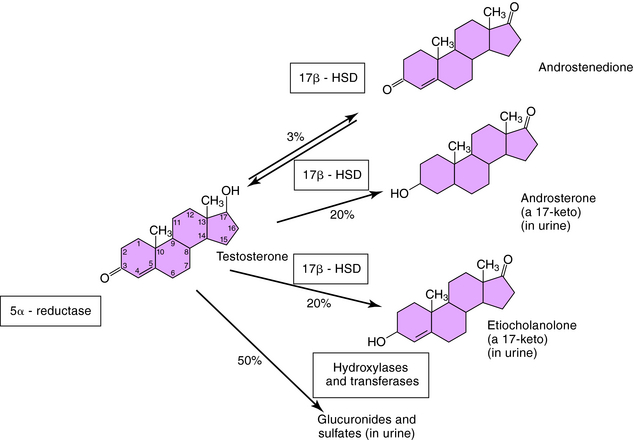
The nonhepatic metabolism of testosterone is summarized in Figure 38-1; the hepatic metabolism is shown in Figure 41-3. Nearly half of hepatic testosterone is metabolized to the 17-ketosteroids, 5α-androsterone and 5β-etiocholanolone, both of which are excreted in the urine. These compounds, however, constitute only a small fraction of the 17-ketosteroids in urine. Most urinary 17-ketosteroids are metabolites of androstenedione and DHEA produced by adrenals. In addition, testosterone is also conjugated and excreted as glucuronides and sulfates.
The pharmacokinetic properties for the androgens available for clinical use are listed in Table 41-1. Testosterone administered orally is rapidly cleared by the liver by first-pass metabolism and is ineffective for clinical use.
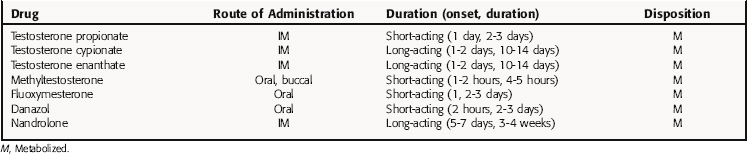
The synthetic alkylated androgens, methyltestosterone and fluoxymesterone, are less extensively metabolized by the liver than testosterone and are available for sublingual or oral use. Methyltestosterone and fluoxymesterone have relatively short durations of action. These drugs are used only after sexual development, because they lack the potency to facilitate sexual development.
Relationship of Mechanisms of Action to Clinical Response
A decline in testicular function begins in middle age. As men age, Leydig cell volume decreases, and less testosterone is produced. Although the primary defect is in Leydig cells, GnRH secretion is also modified. Because many signs of aging are similar to those of hypogonadism, healthy middle-aged and older men are sometimes treated with testosterone. The risks and benefits of androgen replacement for healthy older men are difficult to assess, although a family or personal history of prostate cancer or benign prostatic hyperplasia complicates replacement therapy.
Androgens have been shown to stimulate erythropoiesis by increasing renal erythropoietin production (see Chapter 26). Androgens also have a direct effect on erythrocyte maturation. Because 5β-androgens (which bind weakly to ARs) are more effective than 5α-androgens, this may constitute a novel mechanism explaining the direct effect of androgens on bone marrow cells. Androgens may be used to treat patients with aplastic anemia, although responses vary. Danazol is an androgen derivative that has been used for the treatment of endometriosis, fibrocystic disease of the breast, and premenstrual tension syndrome. Danazol is used in women, rather than testosterone, because it is weakly androgenic. Danazol is also used to prevent attacks of hereditary angioneurotic edema, a disorder characterized by recurrent edema of the skin and mucosa. These patients lack the function of the inhibitor of the activated first component of complement, and androgens increase serum concentrations of this protein.
Antiandrogens and Androgen Receptor Antagonists
Finasteride is a competitive inhibitor of 5α-reductase type II that blocks conversion of testosterone to 5α-DHT in tissues containing this enzyme but has little activity against 5α-reductase type I. Finasteride reduces prostate DHT content by 80%, decreasing prostate size, and is used to treat benign prostatic hyperplasia. Another therapy that can be used to improve urinary flow is monotherapy with α1-adrenergic receptor antagonists (see Chapter 11) or combination therapy with finasteride, as symptoms progress. Finasteride has little effect in treating established prostate cancer. In a large multicenter primary prevention trial, finasteride prevented or delayed the appearance of prostate cancer (6.3% vs. 8.7% of men followed developed cancer), but unexpectedly, the risk of high-grade prostate cancer increased. Consequently, it is not recommended for prevention of prostate cancer. Finasteride at a reduced dose of 1 mg/day is also approved to treat male-pattern baldness. After 12 months of therapy, visible improvement occurs in approximately 50% of treated men.
Spironolactone is a synthetic steroid that is used primarily as an aldosterone antagonist in the treatment of primary and secondary hyperaldosteronism. It can be used to treat hypertension and as a treatment for heart failure (see Chapters 20 and 23). In addition to effects at aldosterone receptors, spironolactone interacts with ARs. Further, spironolactone inhibits testosterone synthesis. Progesterone concentration increases, because its further metabolism is inhibited. However, a decrease in serum androgen concentration in men produces an increase in gonadotropin secretion, which may return serum testosterone concentrations to normal. Because of its ability to block testosterone synthesis and impede androgen action, spironolactone is used in treatment of hirsute women.
Ketoconazole is a broad-spectrum antimycotic agent used in treatment of systemic fungal infections (see Chapter 50). It inhibits the synthesis of ergosterol in fungi, resulting in altered membrane permeability. It also inhibits the synthesis of cholesterol and interferes with the action of cytochrome P450 enzymes in several mammalian cell types, including Leydig cells. The result is a dose-dependent decline in circulating testosterone concentrations in adult men and a rise in serum 17α-hydroxyprogesterone concentrations. Serum LH and FSH concentrations rise because of the decline in testosterone negative feedback. This action of ketoconazole has prompted its investigational use in treatment of prostate cancer and gonadotropin-independent precocious puberty in boys. However, the extent to which it suppresses testosterone synthesis in men is highly variable. Ketoconazole also inhibits cortisol biosynthesis and is used as an adjunct therapy in patients with Cushing’s syndrome. Gynecomastia may develop in ketoconazole-treated men.
The histamine receptor antagonist cimetidine, used to decrease gastric acid secretion in treatment of peptic ulcer disease and esophagitis (see Chapter 14), also acts as an antiandrogen. Thus it has been reported to produce gynecomastia when given in large doses, such as those used in the treatment of patients with Zollinger-Ellison syndrome. Gynecomastia occurs in less than 1% of patients treated with the doses used in peptic ulcer disease. Cimetidine interacts with ARs approximately 0.01% as effectively as testosterone and has been used with limited effectiveness to treat hirsutism in women.
Pharmacovigilance: Clinical Problems, Side Effects, and Toxicity
Professional and amateur athletes often use multiple androgens in doses that far exceed physiological concentrations. These androgens, like testosterone, suppress gonadotropin secretion and reduce testicular function, including spermatogenesis. Recovery of normal function may take several years. These drugs also cause increased concentrations of low-density lipoprotein cholesterol and decreased high-density lipoprotein synthesis and
concentrations. This may increase the risk of atherosclerosis in these men. Long-term, high-dose androgen treatment may also increase risk of benign prostatic hyperplasia and cause prostate cancer when these men age.
The testosterone precursor, androstenedione, is available as a nutritional supplement in the United States (see Chapter 7) and is used by amateur and professional athletes as a performance-enhancer. The ingestion by young men of 100 mg of androstenedione three times daily did not increase total serum testosterone levels but did increase androstenedione, free testosterone, estradiol, and DHT. Most of the orally administered androstenedione is metabolized to testosterone glucuronide and other metabolites.
Anonymous. Dehydroepiandrosterone (DHEA). Med Lett. 2005;47:37-38.
Anonymous. Testim and striant: Two new testosterone products. Med Lett. 2003;45:70-72.
Anonymous. Performance enhancing drugs. Med Lett. 2004;46:57-59.
Anonymous. Tadalafil (Cialis) for erectile dysfunction. Med Lett. 2003;45:101-102.
Anonymous. Alfuzosin (Uroxatral): Another alpha1-blocker for benign prostatic hyperplasia. Med Lett. 2004;46:1-2.
Bagatell CJ, Bremner WJ. Androgens in health and disease. Towata: NJ, 2003.
Lue TF, Broderick GA. Evaluation and nonsurgical management of erectile dysfunction and premature ejaculation. In Wein A, editor: Campbell-Walsh urology, 9, Philadelphia: Saunders, 2007.
Singh et al. 2000 Singh SM, Gauthier S, Labrie F. Androgen receptor antagonists (antiandrogens): Structure-activity relationships. Curr Med Chem. 2000;7:211-247.
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
Chapter 41 Androgens and Antiandrogens
| Abbreviations | |
|---|---|
| AR | Androgen receptor |
| cGMP | Cyclic guanosine monophosphate |
| DHEA | Dehydroepiandrosterone |
| DHT | Dihydrotestosterone |
| FSH | Follicle-stimulating hormone |
| GnRH | Gonadotropin-releasing hormone |
| hCG | Human chorionic gonadotropin |
| IM | Intramuscular |
| LH | Luteinizing hormone |
| SHBG | Sex hormone-binding globulin |
| StAR | Steroidogenic acute regulatory protein |
Therapeutic Overview
Androgens are produced by the testis, ovary, and adrenal glands. Testosterone is the most potent androgen. It stimulates virilization and spermatogenesis. Within the ovary, testosterone and androstenedione are precursor steroids for estradiol production (see Chapter 40). In both sexes androgens stimulate body hair growth, positive nitrogen balance, bone growth, muscle development, and erythropoiesis. The mechanism of action of testosterone at its target organs is similar to that of other steroid hormones (see Chapter 1). The primary clinical use of androgens is replacement therapy in men with diagnosed testosterone deficiency. Testosterone synthesis inhibitors, referred to as antiandrogens, and competitive androgen receptor (AR) antagonists, are used to reduce the effects of androgens in patients with androgen-dependent disorders such as prostatic cancer, benign prostate hyperplasia hirsutism, and precocious puberty. Another male-specific phenomenon is treatment of erectile dysfunction, which employs peripherally acting vasodilators such as cyclic guanosine monophosphate (cGMP) phosphodiesterase type 5 inhibitors or synthetic prostaglandin E1 analogs.
| Therapeutic Overview |
|---|
| Androgens |
| Primary testicular insufficiency |
| Hypogonadotropic hypogonadism |
| Constitutional delay of growth and adolescence |
| Osteoporosis, anemia |
| Male contraception |
| Antiandrogens and Androgen Receptor Antagonists |
| Virilization in women |
| Precocious puberty in boys |
| Prostate cancer, hyperplasia |
responsible for stimulating the development of male external genitalia during the first trimester of fetal life. Therefore, when the fetal synthesis of androgen is insufficient (e.g., due to an inborn enzymatic error) or the action of androgen is ineffective at its target tissues (e.g., androgen resistance), the genital phenotype may be female or ambiguous.
Mechanisms of Action
Androgens are synthesized from cholesterol in testicular Leydig cells, the adrenal cortex, and ovarian thecal cells (see Fig. 38-1). In the adult gonads the principal regulator of testosterone synthesis and secretion is luteinizing hormone (LH), which is produced by the anterior pituitary gland (Chapter 38). The precursor cholesterol is synthesized in the Leydig cells from acetate and stored as cholesterol esters in lipid droplets. A cholesterol ester hydrolase mobilizes free cholesterol from the lipid droplets, which in turn is transferred to the inner mitochondrial membrane. Stimulation of this transfer represents a major action of LH and is mediated by the steroidogenic acute regulatory (StAR) protein. Leydig cells can convert a small fraction of testosterone to estradiol (see Chapter 40 and Fig. 38-1). LH increases the level of enzymes in this synthetic pathway.
Androgen Production by the Adrenal Glands
Although glucocorticoids and mineralocorticoids are the principal products of the adult adrenal gland, dihydroepiandrosterone (DHEA), androstenedione, and testosterone, as well as some DHEA sulfate and estrone, can be also secreted (see Fig. 38-1). The concentrations of DHEA, DHEA sulfate, and androstenedione in the circulation increase between 7 to 10 years of age. This process has been termed adrenarche, to distinguish it from gonadarche, which is the onset of adult gonadal function at puberty. Adrenal androgen secretion declines in the elderly and during severe illness.
Regulation of Testosterone Synthesis and Secretion
The major site of testosterone synthesis and secretion is the Leydig cell, which has cell-surface LH receptors that associate with the Gs subunit of adenylyl cyclase. Steroidogenesis mediated by LH requires mobilization of intracellular Ca++ and the Ca++-binding protein calmodulin, and activation of phospholipase C (see Chapter 1). The effects of LH involve rapid stimulation of testosterone production (within minutes), which is mediated by the StAR. Other hormones that influence testosterone synthesis include prolactin, cortisol, insulin, insulin-like growth factors, estradiol, activin, and inhibin. There is a growing appreciation of the multiple factors involved in testosterone synthesis that are produced within the seminiferous tubules by germ cells and Sertoli cells or peritubular myoid cells. These factors maintain the serum concentration of testosterone in adult men at 0.3 to 1.0 mg/dL (10 to 30 nM). During illness, LH production declines, and cytokines suppress testosterone production.
The hormones of the hypothalamus, pituitary, and testes form an internally regulated unit (Fig. 41-1). Not only are the testes stimulated by pituitary gonadotropins, but the testes also regulate LH and FSH secretion through negative-feedback mechanisms. Testosterone suppresses gonadotropin secretion by slowing the pulsatile release of GnRH. Estradiol, which is synthesized from testosterone in the ovary, testes, adipose tissue, liver, and brain, inhibits gonadotropin release through effects on both the hypothalamus and pituitary. Inhibin-B selectively reduces FSH synthesis and secretion.
Normally, women produce approximately 0.25 mg/day of testosterone compared with the 5 to 7 mg/day for adult men. Most testosterone circulating in women is derived from the peripheral conversion of androstenedione secreted by the ovaries and adrenals (see Chapter 40). Benign and malignant tumors of the adrenal and ovary, congenital steroidogenic enzyme defects, and disturbances of gonadotropin secretion can be associated with increased androgen production in women.
Depending on the tissue, intracellular testosterone or a more active metabolite DHT can interact directly with ARs (Fig. 41-2). When testosterone enters the prostate gland or any tissue with significant 5α-reductase activity, nearly 90% of it is metabolized to DHT. There are two isoenzymes of 5α-reductase, encoded by two different genes. Type I 5α-reductase is found in liver, skin, sebaceous glands, most hair follicles, and prostate, whereas 5α-reductase type II predominates in genital skin, beard and scalp hair follicles, and prostate. The presence of ambiguous genitalia in patients with inactivating mutations of the 5α-reductase type II gene is indicative of the importance of this enzyme in normal development of male external genitalia. The distribution of the isozymes has been exploited to develop tissue-specific inhibitors of 5α-reductase activity.
Androgen binding to ARs and the events that follow are similar to those of other steroid hormones (see Chapter 1). The AR is encoded by a gene on the X chromosome and is expressed in most tissues. When a ligand binds to the AR, the conformation of the receptor is altered, it binds to DNA response elements, multiple coactivator proteins are recruited, and the transcription of messenger RNAs for tissue-specific proteins ensues. Although most actions of androgens are mediated by transcriptional activity of the receptor, others are mediated through second messengers, such as the mitogen-activated protein kinase pathway.
Antiandrogens and Androgen Receptor Antagonists
There are two basic mechanisms to block the activity of androgen. These include inhibition of androgen formation or antagonism of androgen-AR interactions. Strategies for suppression of androgen formation include blockade of gonadotropin formation using GnRH analogs and the use of spironolactone and ketoconazole to competitively inhibit the activities of steroid 17α-hydroxylase (CYP17) and cholesterol side-chain cleavage enzyme (CYP11A), respectively (see Fig. 38-1). Another agent, finasteride, is a competitive inhibitor of the 5α-reductase isozymes and antagonizes the formation DHT, which has potent androgenic action. All these drugs are substrate analogs and compete with the natural substrates for active sites on the enzymes.
[/not-level-membership-for-basic-science-category]
