Chapter 7 Anatomy and Physiology of Congenital Spinal Lesions
Preoperative Considerations
In general, congenital spinal anomalies are sporadic, isolated cases.1,2 A wide variety of associated anomalies often accompany the spinal deformity and often need to be addressed prior to treatment of spinal lesions.3,4 In fact, the incidence of associated renal abnormalities is roughly 25% and that for cardiac abnormalities is 10%.5 Intraspinal anomalies, such as stenosis, diastematomyelia, and tethering of the spinal cord, may also occur (5–35%) in association with congenital spinal deformities.6–9 Therefore, before making treatment decisions, the entire bony vertebral column and spinal cord must be thoroughly analyzed using radiographs, CT, and MRI. Pulmonary function testing and/or arterial blood gases should be obtained when a thoracotomy is planned or when severe thoracic lordosis is present.
Craniovertebral Junction Abnormalities
Basilar Invagination
Basilar invagination results from a defect in the chondrocranium and is often associated with both skeletal and neural axis abnormalities.10–12 It results from deformation in all three parts of the occipital bone (basiocciput, exocciput, and squamous occipital bone).11 Two types of basilar invagination have been identified: ventral and paramedian.10–12 In ventral basilar invagination, the basiocciput is shortened and the associated platybasia raises the plane of the foramen magnum. In paramedian basilar invagination, the exoccipital bone is hypoplastic and the medial portion of the occipital bone is elevated. Clinically, the distinction is not important, and there is admixture between types. One should evaluate patients for an associated elevation of the floor of the posterior fossa that is most pronounced in the region of the foramen magnum. This anomaly may compromise the space available within the foramen magnum.13 Associated skeletal developmental anomalies found with basilar invagination include occipitalization of the atlas and Klippel-Feil syndrome in addition to neural axis abnormalities such as Chiari malformation, syringobulbia, syringomyelia, and hydrocephalus.11,12 Although the term basilar impression is often used synonymously with basilar invagination, this condition refers to an acquired form of basilar invagination caused by softening of the occipital bone, which occurs in conditions such as rheumatoid arthritis, Paget disease, hyperparathyroidism, achondroplasia, and osteogenesis imperfecta.14
Patients who are symptomatic from pure basilar invagination most commonly present with weakness and paresthesias in the limbs, whereas those patients with symptomatic Chiari malformations typically have cerebellar and vestibular complaints. Both groups may have evidence of lower cranial nerve dysfunction. Many patients do not develop symptoms until the second or third decade of life.15 This may be related to increasing instability from ligamentous laxity caused by aging, similar to that from delayed myelopathies reported after atlantoaxial dislocations.16 If chronic instability is present, granulation tissue may develop and act as a space-occupying mass in the ventral portion of the foramen magnum. Fibrous bands and dural adhesions are common in the dorsal cervicomedullary junction and around the cerebellar tonsils in basilar invagination.11 A high incidence of vertebral artery anomalies in basilar invagination has been reported (important for surgical planning), and symptoms of vertebral artery insufficiency can occur.12,17 Diagnosis of basilar impression (or invagination) is based on radiographic evaluation that demonstrates the altered relationship between the occipital bone and the upper cervical spine. Classically, this diagnosis is made by radiographic evaluation of the craniovertebral junction. A series of reference lines have been described to assist with this evaluation.12 MRI assists in delineating the relationship between the neural structures and the bony abnormalities; however, bony pathology should be evaluated with a thin-section CT.
Assimilation of the Atlas
Assimilation of the atlas represents a failure of segmentation between the atlas and the base of the skull. Embryologically, this entity represents a failure of segmentation between the fourth occipital and first spinal sclerotomes. This condition occurs in 0.25% of the population.10 The assimilation may be partial or complete and can involve the ventral arch of the atlas, the lateral masses, or the entire atlas. In many instances, assimilation of the atlas occurs with other spinal abnormalities, such as basilar invagination; Klippel-Feil syndrome; or Chiari malformation; as well as systemic congenital abnormalities such as cleft palate or urinary tract abnormalities.18 Patients with atlanto-occipital fusion often present with symptoms much like those for patients with classic Klippel-Feil syndrome, that is, restricted motion, short neck, low hairline, and torticollis.10 An increased incidence of atlantoaxial instability occurs with assimilation of the atlas, especially if there is failure of segmentation between the second and third vertebrae.11
The onset of symptoms in assimilation of the atlas generally occurs in the third and fourth decades of life. Dull headache and scalp tenderness in the distribution of the greater occipital nerve occurs frequently. Ventral compression of the brainstem from the odontoid processes is also a common finding. Weakness, spasticity, gait disturbances, or cranial nerve dysfunction can be associated problems.10 Neurologic symptoms have been related to the position of the odontoid process as an indication of the degree of actual or relative basilar impression. Vertical and horizontal nystagmus is related to cerebellum and tonsillar abnormalities. Decreased posterior column function from dorsal compression by the foramen magnum or a dural band is a less common finding. Symptoms may develop precipitously, but in the majority of cases the onset is gradual.
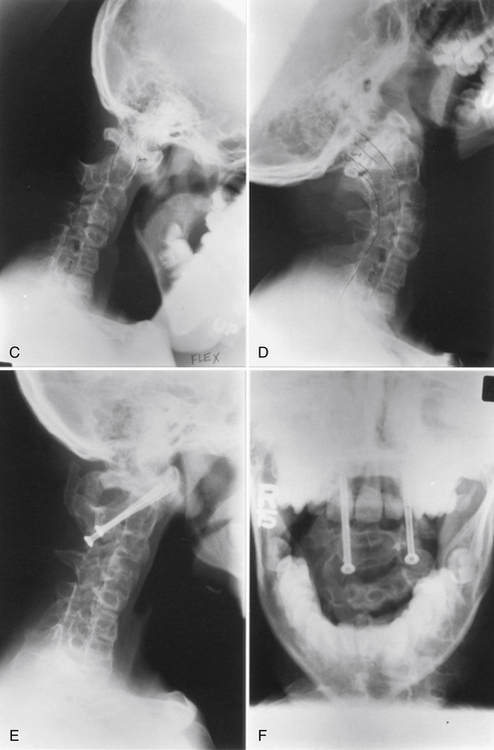
Atlantoaxial Instability
Atlantoaxial instability may result from aplasia or hypoplasia of the odontoid process, from laxity of the transverse ligament, or with assimilation of the atlas. Atlantoaxial instability is associated with Down syndrome, Klippel-Feil syndrome, numerous skeletal dysplasias, osteogenesis imperfecta, neurofibromatosis, and congenital scoliosis.19 The clinical significance of this condition is the potential for neurologic compromise, which can range from pain and dysesthesias in the distribution of the greater occipital nerve to tetraplegia or death.10,11 The articulation between the first and second cervical vertebrae is the most mobile segment in the vertebral column and is the least inherently stable. The odontoid process acts as a bony buttress that prevents hyperextension. However, the remainder of the normal range of motion is maintained and depends on the integrity of the ligamentous and capsular structures. Neurologic compromise can occur despite a normal odontoid process. With an attenuated or ruptured transverse atlantal ligament, a relative ventral shift of the atlas over the axis can result in spinal cord injury by impingement against the intact odontoid process such as occurs with atlantal assimilation.11,20 The risk is less if the odontoid process is absent, fractured, or moves with the axis during flexion, as occurs with most cases of os odontoideum.21 The stability of the atlantoaxial complex can often be determined using lateral radiographs. The atlantodental interval (ADI) is the distance between the dorsal edge of the ventral ring of the atlas and the ventral edge of the odontoid process. The normal ADI is less than 3 mm in adults and less than 4 mm in children.22,23 It has been suggested that an ADI greater than 3 mm in adults indicates a disruption of the transverse ligament. An ADI of 5 to 10 mm represents additional ligamentous damage, with total ligamentous disruption occurring in patients with an ADI greater than 10 mm.22 In congenital anomalies, such as hypoplasia of the odontoid process or os odontoideum, the space available for the cord (SAC) is often a better predictor of potential for neurologic compromise. The SAC is the distance from the ventral edge of the dorsal ring of the atlas or foramen magnum to the dorsal aspect of the odontoid process or the dorsal aspect of the axis. Greenberg suggested that in the adult, spinal cord compression always occurred when the SAC was 14 mm or less and never occurred if the SAC was 18 mm or more.24 In cases of os odontoideum, a SAC of 13 mm or less is associated with neurologic sequelae.25 In cases with persistent concerns about atlantoaxial stability, flexion-extension lateral radiographs can be performed. An awake patient should voluntarily perform the flexion-extension movements. MRI should be considered for any patient with a neurologic deficit before obtaining flexion-extension radiographs. MRI with flexion and extension views provides an excellent method of determining the potential for neural impingement with movement.
Anomalies of the Odontoid Process
Aplasia-Hypoplasia of the Dens
Aplasia-hypoplasia of the dens is a rare condition with a spectrum of presentations ranging from a hypoplastic rudimentary dens to complete absence of the dens. Usually, the rudimentary dens does not reach the upper edge of the ventral arch of the atlas, and an associated incompetence of the cruciate ligaments and alar ligaments results in atlantoaxial instability. Distinguishing aplasia or hypoplasia is of limited clinical importance because both conditions can lead to atlantoaxial instability and the treatment is identical. Vascular compromise from stretching and torsion of the vertebral arteries has been reported. Chronic atlantoaxial dislocation may provoke the formation of granulation tissue that can cause neurologic deficit because of constriction of the cervicomedullary junction.11 The presentation in children with atlantoaxial dislocation and congenital anomalies varies and includes syncope, torticollis, dysesthesia, and tetraplegia.
Os Odontoideum
Os odontoideum is an independent ossicle located rostral to the axis bone in the position of the odontoid process that is separated from a hypoplastic dens by a variable distance.26 The space between the os odontoideum and the remnant of the odontoid process is above the level of the superior facet of the axis. This leads to potential incompetence of the transverse ligament, which can lead to atlantoaxial instability.2 In children younger than 5 years, the normal epiphyseal line may be confused with the presence of an os odontoideum or a fracture. In os odontoideum, the free ossicle is rounded or oval, with a smooth cortical border. In the case of an odontoid fracture, the gap is usually narrow and irregular and often extends into the body of the axis at the level of the superior facet of the axis vertebra.2 The incidence of os odontoideum is increased in Down syndrome, spondyloepiphyseal dysplasia, Morquio syndrome, and after upper respiratory tract infections.26,27
There are two types of os odontoideum: orthotopic and dystopic. In the orthotopic variety, the ossicle lies in the location of the normal dens and moves with the axis body and the ventral arch of the atlas. This type is often associated with an intact cruciate ligament. In the dystopic variety, the os is located near the basion and is often fused to the clivus. The ventral arch of the atlas is hypertrophied, and the dorsal arch is hypoplastic. Dystopic os odontoideum has a greater likelihood of causing neurologic compromise than the orthotopic variety. This may occur because of dorsal compromise of the spinal cord by the ventrally located dorsal arch of the atlas during flexion and ventral compromise by the odontoid ossicle.26 Evaluation of atlantoaxial instability should be performed with flexion-extension films. In cases of chronic subluxation, dense granulation tissue may form, leading to an irreducible state. Os odontoideum has been ascribed to congenital, vascular, and traumatic causes.26 Trauma or infection during childhood is the most likely cause for the vast majority of cases of os odontoideum. Several cases have been reported in children with a normal odontoid process before trauma who subsequently developed os odontoideum. Many patients have a significant episode of trauma before the diagnosis of os odontoideum. After fracture or vascular compromise, a separation of the bone fragments occurs, probably because of contracture of the apical ligaments. The ossicle continues to receive a blood supply via the apical arcade, but the blood supply in the region of the fracture is disrupted, resulting in poor healing. It is probable that congenital forms of os odontoideum also exist. The congenital form results from failure of fusion of the portions of the dens derived from the proatlas and first cervical sclerotome. Dystopic os odontoideum is thought to be congenital in origin.
Disorders of the Subaxial Cervical Spine
Klippel-Feil Syndrome
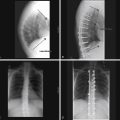
Klippel-Feil syndrome was first described as a case report with the clinical triad of a short neck, low dorsal hairline, and marked limitation of cervical range of motion resulting from a single unsegmented vertebral mass extending from the craniocervical junction through the fourth thoracic vertebra.28 Fewer than half of patients have all three signs. The most consistent finding is limitation of cervical motion.10 Currently, the term Klippel-Feil syndrome is used to describe any congenital fusion of the cervical spine with or without the clinical features of the original description (Fig. 7-1).
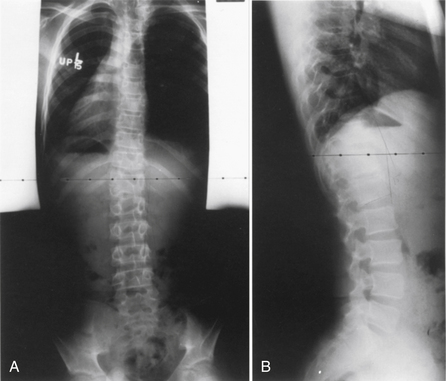
Often patients with Klippel-Feil syndrome have associated congenital abnormalities, which are often the conditions that prompt evaluation. The Klippel-Feil syndrome occurs in 25% of patients with congenital scoliosis.29 Therefore, all patients with congenital scoliosis should have radiographs of the entire spine to exclude the coexistence of the Klippel-Feil syndrome, and, conversely, all patients with a diagnosis of Klippel-Feil syndrome should have radiographs of the thoracic and lumbar spine. Approximately 50% of patients with congenital cervical or cervicothoracic scoliosis have associated Klippel-Feil anomalies. Renal anomalies occur in more than one third of patients. It has been suggested that routine screening ultrasonography be performed in all patients with Klippel-Feil syndrome.30
Sprengel deformity (an abnormal elevation of the scapula) occurs in 25% to 35% of cases of Klippel-Feil syndrome. It may be unilateral or bilateral.10 Other less commonly associated anomalies include deafness, synkinesis (involuntary paired movements of the hands and occasionally the arms), congenital heart disease, cervical ribs, ptosis, Duane syndrome (an abducens nerve palsy in which the adducted eye becomes retracted), lateral rectus palsy, facial nerve palsy, syndactyly, hypoplastic thumb, and upper extremity hypoplasia.
Symptoms related to the Klippel-Feil syndrome can be classified as mechanical or secondary to neural compression. Cervical instability and stenosis are potential problems in Klippel-Feil patients. The spinal cord area adjacent to the vertebral fusion may be compressed because of cervical instability, particularly at the occiput-C1 and C1-2 levels. All of these patients should have flexion-extension cervical spine radiographs. Neurologic deficits can range from radiculopathy to sudden death from minor trauma. Overall, 20% of patients who develop neurologic symptoms do so during the first 5 years of life and 65% by the age of 30 years.10 Neurologic symptoms detected during infancy are usually related to craniovertebral junction abnormalities. Children often have pain with atlantoaxial fusions. Subaxial cervical fusions often do not become problematic until the third decade or later, when degenerative changes begin to develop. Patients with short-segment fusions are less likely to develop symptoms, because of compensatory movement at uninvolved segments.
Iniencephaly
Iniencephaly is a disorder of the cervical spine consisting of congenital cervical synostoses, fixed retroflexion of the head, severe cervical lordosis, and varying degrees of deficits of the dorsal occiput and cervical vertebrae. This condition probably belongs to the spectrum of neural tube defects. The majority of fetuses with this condition are not viable.10 Parents of a child with iniencephaly have a 5% risk of having another child with a neural tube defect. Ultrasonography and serum or amniotic α-fetoprotein levels can be used to detect this condition in utero.31 Surviving patients are often handicapped by the cervical lordosis and hyperextension of the head. This posture makes it impossible to see straight without flexing the low back and hips.
Disorders of the Thoracolumbar Spine
Congenital Scoliosis
Congenital scoliosis is an abnormal curvature of the spine in the coronal plane that develops when anomalous vertebrae are present at birth. Congenital scoliosis is distinct from infantile idiopathic scoliosis, although both present with deformity during childhood. Infantile idiopathic scoliosis has no structural vertebral abnormality. Although vertebral abnormalities are present at birth in congenital scoliosis, the spinal deformity is rarely noticeable during infancy and usually presents during childhood or adolescence. Patients with mild or compensated deformities often receive diagnosis as adults when vertebral anomalies are discovered incidentally during routine radiographs. Congenital scoliosis can be associated with a variety of cardiac, genitourinary, and skeletal abnormalities.1,32 The spectrum of clinical presentations ranges widely based on number, location, and type of vertebral abnormalities. Certain vertebral anomalies result in rapidly progressive scoliosis during early childhood, resulting in severe morbidity, whereas other anomalies cause little or no deformity at any time.33 In general, 25% of congenital scolioses do not progress, 50% progress slowly, and 25% progress rapidly.33 Major advancements in the treatment of congenital scoliosis are improved imaging of the spine by CT and MRI, classification by type of vertebral anomaly, improved understanding of the natural history, and clarification of the indications and timing of surgery.
Advances in imaging have aided the diagnosis of associated neural axis abnormalities, such as occult spinal dysraphism and tethering of the spinal cord. Between 10% to 20% of all congenital scoliosis patients have some anomaly of the neural axis.8 Dorsal midline skin lesions (e.g., hairy patches or deep dimples), asymmetrical foot deformities (cavus or flat feet), muscle weakness, or spasticity all suggest underlying nervous system abnormalities. A thorough imaging evaluation is therefore indicated.
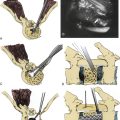
Usually, the vertebral abnormalities can be classified by the anomaly in the mesenchymal precursor that results in either a failure of formation or a failure of segmentation. Failure of formation results from a defect in the developmental process that produces an absence of part or all of the vertebrae. The defects range from mild wedging to total absence of the vertebra. A hemivertebra occurs with the complete absence of half of a vertebra and is one of the most common causes of congenital scoliosis. The hemivertebra consists of a wedged vertebral body with a single pedicle and hemilamina (Fig. 7-2).
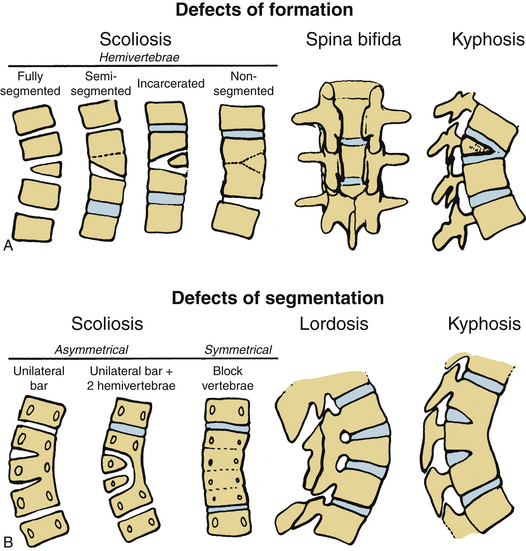
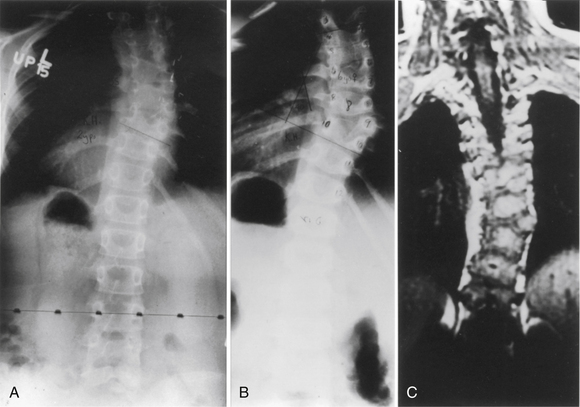
Segmentation failure causes unilateral or bilateral bony fusion between vertebrae. The defect can involve ventral elements, dorsal elements, or both. The most common segmentation failure is the unilateral unsegmented bar, which results in a bony block that involves the disc spaces and facet joints (Fig. 7-3). A combination of defects of formation and defects of segmentation can coexist in the same patient. An unsegmented bar with contralateral hemivertebrae can cause severe progressive scoliosis.1
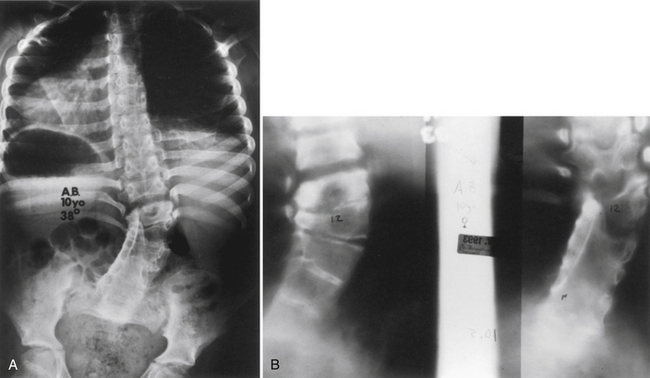
Three major types of hemivertebrae are classified by the positioning of the hemivertebra and whether the disc spaces above and below the hemivertebra are morphologically normal (Fig. 7-4). A fully segmented hemivertebra has a normal disc space above and below the vertebral body that allows near-normal longitudinal growth. A portion of the vertebral body and growth plates are absent on the side of the unformed vertebra, resulting in limited growth potential. Because of full growth potential on one side of the spine and none on the other at the level of the hemivertebra, the potential for a hemivertebra located at the apex of the scoliosis is significant in these cases. The rate of progression and the need for treatment of the scoliosis caused by a fully segmented hemivertebra depends on its location in the spine, with the thoracolumbar and the lumbosacral junction being the most problematic. In general, these scoliotic curves progress at 1 to 2 degrees per year.34
The incarcerated hemivertebra is a variant of the fully segmented hemivertebra. This type of hemivertebra is set into defects in the vertebrae above and below it. The incarcerated hemivertebra is small, oval, and has poorly formed disc spaces. The defects in the adjacent vertebrae tend to compensate for the hemivertebra, and the poor potential growth of the malformed growth plates results in less scoliotic deformity than with the standard fully segmented vertebrae.34
A semisegmented hemivertebra is connected to either the vertebra above or below it and causes the absence of one disc space on the side of the hemivertebra with obliteration of two growth plates. Theoretically, this would result in similar growth on both sides of the spine because two active growth plates coexist on each side. However, the wedge shape of the hemivertebra and differences in growth (between sides) can result in some scoliosis.
Another common cause of congenital scoliosis is a unilateral unsegmented bar.1 This condition results from a failure of segmentation of two or more vertebrae. The unsegmented bar contains no growth plates, but the unaffected side of the spine continues to grow. The imbalance in growth results in the scoliosis with the unsegmented bar in the concavity. On average, these curves deteriorate at a rate of 5 degrees or greater per year and often result in a significant deformity.33
Congenital Kyphosis
Congenital kyphosis is an uncommon sagittal plane deformity, which, if left untreated, is often associated with a neurologic deficit.35 As with congenital scoliosis, congenital kyphosis is caused by segmentation failure. Winter et al.36 classified congenital kyphosis into three types: type I is the failure of formation of the vertebral body; type II is the failure of segmentation of the vertebral body, resulting in a ventral unsegmented bar; and type III is the mixed failure of formation and segmentation. The type I kyphosis is the most common and the most likely to lead to both severe deformity and neurologic compromise.36 The severity of type I kyphosis is directly proportional to the amount of vertebral body or bodies that fail to form. The type II kyphosis is less common, produces less severe deformity, and is much less frequently associated with neurologic compromise than type I. The amount of kyphosis produced is proportional to the discrepancy between the ventral vertebral growth and the growth of the dorsal elements. Type III kyphosis is very rare and probably behaves like type I kyphosis.
Congenital Lordosis
Congenital lordosis is rarer than either congenital scoliosis or congenital kyphosis. This condition results from dorsal defects in segmentation, with normal ventral growth.37 Often, it has some component of coronal plane deformity, leading to lordoscoliosis because of a dorsolateral location of the unsegmented bar. The most severe consequence of congenital lordosis is an impairment of pulmonary function.37
Lumbar Spine Abnormalities
Congenital Spinal Stenosis
Congenital spinal stenosis occurs in a very small number of patients who present with spinal stenosis.1 It results from a malformation present at birth that predisposes the patient to the development of stenosis, which often manifests itself later in life.
Congenital spinal stenosis can occur as a part of spinal dysraphism.1 The signs and symptoms are usually not the consequence of stenosis alone, but also of myelodysplasia. Serious radicular pain or dysfunction occurs frequently in this condition.
Congenital stenosis can also result from an area of failure of vertebral segmentation (block vertebrae). Stenosis in the area of the block vertebrae results from a reduction of the midsagittal diameters of the vertebral canal. The signs and symptoms do not differ from those observed with idiopathic developmental stenosis.
Developmental Spinal Stenosis
Developmental spinal stenosis usually occurs as the result of an inborn chromosomal error or mutation that alters the fetal and postnatal spinal canal formation. Developmental spinal stenosis commonly occurs in conditions such as achondroplasia, hypochondroplasia, diastrophic dwarfism, Morquio syndrome, and hereditary multiple exostoses. This condition may involve only the lumbar spine or can be associated with developmental stenosis of the cervical spine.1
Segmented Spinal Dysgenesis
Segmental spinal dysgenesis is a localized congenital defect in which severe stenosis occurs with malalignment and focal agenesis or dysgenesis in the thoracolumbar or lumbar spine.1 It was initially described as a form of congenital spinal stenosis with focal narrowing of the spinal canal in the area of the thoracolumbar junction. Neurologic deficits are often present at birth and may range from mild paresis to complete paraplegia. Patients may have congenital absence of nerve root or spinal cord segments. The spinal canal above and below the involved segment is usually normal, and the sacrum is well formed, differentiating this condition from sacral agenesis.38
Spondylolisthesis
Spondylolisthesis is the slippage of all or part of one vertebra in relationship with another. The most widely accepted classification of spondylolisthesis is by Wiltse et al.39 They divided spondylolisthesis into five types: I—dysplastic, II—isthmic, III—degenerative, IV—traumatic, and V—pathologic.
Dysplastic spondylolisthesis accounts for 14% to 21% of cases, with a 2:1 female to male ratio.40 This type is characterized by structural anomalies of the lumbosacral junction, including dysplasia of the lamina and facet joints. The lack of the normal facet buttress provided by normal facet joints predisposes toward a slippage of the rostral vertebra on its caudal counterpart. The dysplastic articular processes may be oriented in the axial or sagittal planes. In axial dysplasia, the articular processes have a horizontal orientation. This condition is often associated with spina bifida. In sagittal dysplasia the facet joints are often asymmetrical, but the neural arch is usually intact. Therefore, high-grade slippage seldom occurs. Both types can present with hamstring spasm, back or leg pain, or neurologic deficit, including paresthesia, weakness, or, rarely, incontinence of the bowel or bladder. Neurologic deficits are usually associated with high-grade slips.
Axially oriented facet joints associated with spina bifida have an increased risk for high-grade spondylolisthesis. The pars interarticularis is often poorly developed and may elongate, develop a defect, or remain intact. If the pars interarticularis remains intact, neurologic symptoms usually occur only when the spondylolisthesis exceeds 35%. Progression of spondylolisthesis is more likely in younger or skeletally immature patients and in patients with wide spina bifida. Initial treatment should be nonoperative unless progression is documented in younger patients or slippage greater than 50% is observed at the time of the initial evaluation. Fusion in situ is a frequently performed surgical procedure, although some surgeons use reduction and fixation, especially with high-grade abnormalities.1
Disorders of the Sacral Spine
Sacral Agenesis
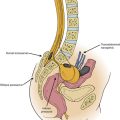
Sacral agenesis is a group of disorders characterized by an absence of variable portions of the caudal spine. Williams and Nixon41 coined the term sacral agenesis in 1957. Sacral agenesis belongs within the spectrum of aplastic vertebral malformations that are loosely grouped under the entity of caudal regression syndrome. It can range from agenesis of the coccyx to absence of sacral, lumbar, and lower thoracic vertebrae.
The clinical severity parallels the number of spinal segments involved with the aplasia or dysplasia. Associated anomalies of the genitourinary, GI, and urinary systems often occur.42 Patients with sacral agenesis usually lack motor function below the level of the last normal vertebra. It is interesting that in sacral agenesis, compared with other dysplastic syndromes of the lower spine (e.g., myelomeningocele), sensation is relatively spared below the level of the lesion. In the development of the human embryo, the notochord induces the formation of the ventral spinal elements and cells derived from neural crest independently from the dorsal root ganglia. Thus, an insult specific to the notochord/ventral spine could lead to the observed clinical picture in sacral agenesis.43
The exact incidence of sacral agenesis is difficult to determine because mild caudal agenesis is often not clinically apparent, and severe cases can result in stillbirth or neonatal death. Sacral agenesis is a relatively rare lesion. An incidence of 1 in 60,000 live births has been reported.44 Sacral agenesis is considered to have a sporadic, nonfamilial inheritance pattern, although cases of siblings with the disorder have been reported. Maternal diabetes appears to increase the risk of sacral agenesis.45 Embryonal trauma producing longitudinal kinking of the long embryonic axis, dietary deficiencies, and teratogenic chemicals have caused caudal agenesis in experimental models.45 Caudal agenesis, as well as other associated congenital anomalies such as imperforate anus and cloacal exstrophy, result from alterations in the normal formation and development of the caudal eminence. The caudal eminence is a mass of undifferentiated cells at the caudal end of the embryo that gives rise to the distal spinal cord, nerve roots, and the vertebral column of the sacral and coccygeal regions.
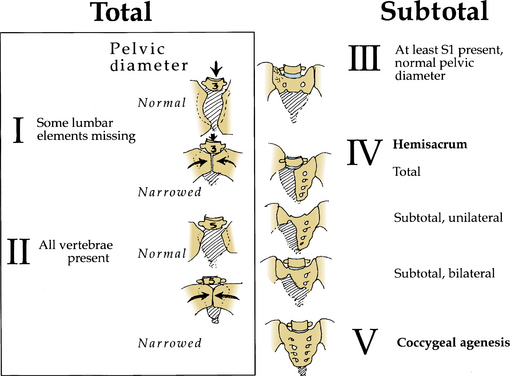
Pang42 devised a new classification scheme that combined salient features from other classification schemes (Fig. 7-5). By this method, lumbosacral agenesis is divided into five types, with some of these divided into subtypes. Type I is total sacral agenesis with some lumbar vertebrae also missing. Type II is total sacral agenesis with the lumbar vertebrae not involved. Type III is subtotal sacral agenesis with at least S1 present and the ilia articulate with the side of the rudimentary sacrum. Type IV is a hemisacrum, and type V is coccygeal agenesis.
The clinical features of sacral agenesis can be quite severe. Because of the lack of motor innervation of the lower limbs, intrauterine contractures develop. In severe forms of sacral agenesis, the malformation in the spine-pelvis articulation causes a severe kyphosis to develop. Affected children sit in the “Buddha” position with legs flexed and crossed and lean forward because of the kyphosis. Other spinal deformities develop in children with sacral agenesis. Congenital and developmental scoliosis is common. Klippel-Feil syndrome has also been reported.1
Multiple musculoskeletal deformities can present in patients with sacral agenesis. Hip dysplasia, clubfoot, and knee flexion contractures are common.44 The etiologic factor responsible for sacral agenesis, such as an insult to the caudal eminence, seems to occur during the time of organogenesis. Therefore, children with sacral agenesis can present with multiple abnormalities of the GI, cardiac, and renal systems. Abnormalities of the terminal spinal cord can be associated with sacral agenesis. These include elongated conus medullaris with hydromyelia, tethering of the spinal cord by a thickened filum terminale, lipomas, split-cord malformations, and terminal myelocystoceles. Neurogenic bladder almost always results in cases of sacral agenesis above S2.
Teratomas
Teratomas in the spine almost exclusively occur in the sacrococcygeal region due to their origin from pluripotent tissue derived from the area around Hensen node.46 This tissue migrates rostrally to lie in the coccyx. These usually benign tumors can undergo malignant transformation if diagnosis and treatment are delayed.
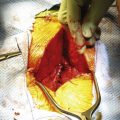
Sacrococcygeal teratoma (SCT) is a common neoplasm in the newborn, with a reported incidence of 1 in 35,000 live births, with a 3:1 female preponderance.1 The majority of tumors are large, external, and cystic. The tumor mass usually protrudes from between the anus and the coccyx, although some tumors are located predominantly in the presacral space of the pelvis. Although the diagnosis is often possible prenatally by ultrasound, small presacral tumors can be missed in the newborn. The tumors range in size but can average approximately 8.5 cm. The cystic component is usually cerebrospinal fluid (CSF), but is not generally connected with circulating spinal fluid within the thecal sac, instead arising from the choroid plexus contained within the tumor mass. SCTs have been classified into four types: I—totally external, II —almost totally external, III—almost completely internal, and IV—completely internal.47 Symptoms are largely related to the degree of displacement or obstruction of the bladder, urethra, or rectum.
Surgical therapy by midline or chevron incision is the mainstay for benign SCT. After removal of the tumor with coccygectomy, survival is high.1 Presacral tumors may require an abdominal approach combined with the usual sacral approach. Multiagent adjuvant chemotherapy is added to surgical therapy for malignant tumors.
Spinal Dysraphism
Dysraphism is an abnormal development of the spinal cord and column, resulting from malformations that arise from the failure of normal embryologic structures to fuse in the midline. These malformations are broadly divided into two groups: spina bifida aperta and spina bifida occulta. The first group involves midline lesions that are (or potentially can) open at birth. These lesions include spina bifida cystica (myelomeningocele, meningocele) and myelodysplasia. The second group encompasses malformations that are hidden by complete layers of dermis and epidermis. These lesions include lipomyelomeningocele, neurenteric cyst, and diastematomyelia.1 In the United States, the incidence of neural tube defects is roughly 1 per 1000. Folic acid supplementation has been credited with decreased rates worldwide.48
Diastematomyelia
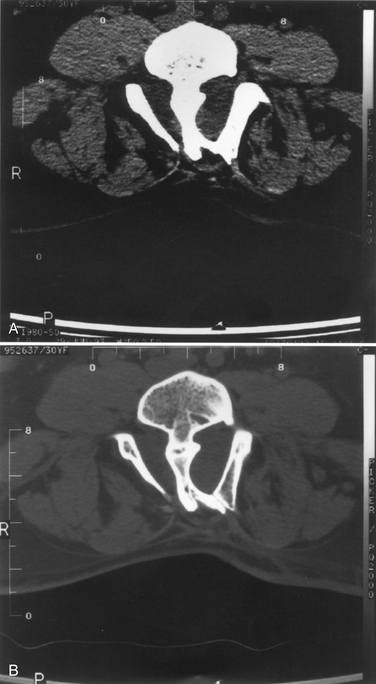
Diastematomyelia is a disorder in which the spinal cord develops into two hemicords, separated by a cartilaginous or bony septum (Fig. 7-6).49 Diastematomyelia usually occurs from the third thoracic to the fourth lumbar vertebrae. Diastematomyelia is among a spectrum of split-cord malformations. These can occur with two hemicords (each within its own dural tube and split with a rigid osseocartilaginous septum), or they can occur with both hemicords in the same dural covering, with only a fibrous band for separation.
Neurenteric Cysts
If endoderm is retained in the tract between hemicords, a neurenteric cyst can result.50 These rare lesions are retained cystic structures, ventrally located in the spinal canal, derived from embryonic foregut. These cysts occur most commonly in the thoracic and cervical spine. The epithelium of these cysts varies from ciliated columnar lining that suggests a respiratory origin to linings that can resemble gut mucosa. Because of embryonic gut rotation, neurenteric cysts tend to lie to the right of the vertebral column. Neurenteric cysts can cause spinal cord compression usually appearing in childhood.
Lipomas
Lipomas of the spine are a commonly encountered developmental spinal abnormality, often seen with occult spinal dysraphism. They occur in the lumbosacral area 90% of the time. In contrast, intraspinal lipomas not associated with spina bifida occulta account for about 5% of intraspinal tumors in children and show a predilection for the thoracic spine. These lesions most likely result from inclusion of adipose cells from the overlying mesodermal tissue into the developing spinal canal or the folding neural tube. A tethered spinal cord occurs when these lesions traverse both the bony and neural elements of the spine.51
Lipomas associated with spinal dysraphism take three principal forms: dorsal, terminal, or transitional. In the dorsal form, the lipoma extends from the subcutaneous space through incomplete neural arches and attaches to the dorsal spinal cord. It is rare for nerve roots to be contained within the substance of a dorsal lipoma. Terminal lipomas insert into the distal conus and may be entirely intraspinal, many times containing nerve roots. Features of both dorsal and terminal lipomas appear in transitional lipomas. The embryology of caudal lipomas most likely arises during secondary neurulation. During secondary neurulation the caudal end of the neural tube blends with a large collection of undifferentiated cells, the caudal cell mass. The last phase of secondary neurulation involves regression of the previously formed tail structures, leaving the filum terminale, coccygeal ligament, and terminal ventricle of the conus as its only remnants. Cell rests with the potential for differentiation may be left in these elements and account for the development of lipomas, hamartomas, teratomas, and the rare malignancy.51
Dermoids and Dermal Sinus Tracts
Dermal sinus tracts are lined by squamous epithelium and may penetrate the spinal cord at any level in the midline from the lumbosacral spine to the occiput. Dermoid and epidermoid nodules can frequently accompany dermal sinus tracts. Dermoid and epidermoid tumors may arise within the tract in approximately half of all dermal sinuses.52 These tumors are also encountered within the subarachnoid space, arising from isolated congenital rests of cells derived from the multipotential caudal cell mass. The embryology of dermal sinus tracts and dermoids of the spine is probably a result of incomplete dysjunction of ectoderm from endoderm during the fourth week of embryologic development. The dermal tract becomes elongated during ascent of the spinal cord within the spinal canal and may traverse several layers of dermis and epidermal space before entering the subarachnoid space. Dermal sinus tracts may frequently be missed on initial examination of the infant and only become apparent when the child has symptoms of recurrent meningitis.
Tethered Cord
The degree of spinal cord traction, rather than the type or distribution of the tethering lesions, most likely determines the age of symptom onset. Severe traction on the spinal cord results in presentation in childhood, whereas less severe traction is asymptomatic in childhood but appears later in life (because of repeated tugging of the conus during head and neck flexion), or when abnormal tension is aggravated by trauma or spondylotic spinal canal stenosis.53
Syringomyelia
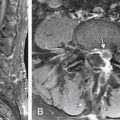
Syringomyelia is a fluid-filled cavity of the spinal cord (Fig. 7-7). Syringomyelia occurs frequently in the cervical and thoracic spine. Although not directly a congenital malformation, it may be considered a developmental abnormality because of its frequent association with Chiari malformations. There is up to a 50% to 75% incidence of cavitation (syrinx) of the spinal cord in the setting of Chiari malformation. The posterior fossa is small because of flattening of the squamous occipital bone. The foramen magnum is enlarged to accommodate the descended cerebellar tonsils.
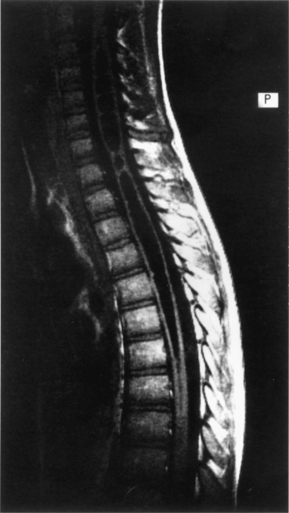
Syringomyelia may present with pain in the spine, limb, and trunk. Radiographic features may be widening of the spinal canal and erosion of the vertebrae.54 In support of the theories of misguided CSF flow around the herniated cerebellar tonsils, posterior fossa decompression without cyst drainage can improve symptoms.
Basu P.S., Elsebaie H., Noordeen M.H. Congenital spinal deformity: a comprehensive assessment at presentation. Spine (Phila Pa 1976). 2002;27:2255-2259.
Belmont P.J.Jr., Kuklo T.R., Taylor K.F., et al. Intraspinal anomalies associated with isolated congenital hemivertebra: the role of routine magnetic resonance imaging. J Bone Joint Surg [Am]. 2004;86:1704-1710.
Cavalier R., Herman M.J., Cheung E.V., Pizzutillo P.D. Spondylolysis and spondylolisthesis in children and adolescents: I. Diagnosis, natural history, and nonsurgical management. J Am Acad Orthop Surg. 2006;14:417-424.
Lonstein J.E. Congenital spine deformities: scoliosis, kyphosis, and lordosis. Orthop Clin North Am. 1999;30:387-405, viii.
McMaster M.J., Singh H. Natural history of congenital kyphosis and kyphoscoliosis. A study of one hundred and twelve patients. J Bone Joint Surg [Am]. 1999;81:1367-1383.
Pang D. Sacral agenesis and caudal spinal cord malformations. Neurosurgery. 1993;32:755-778. discussion 778–779
Smith J.S., Shaffrey C.I., Abel M.F., et al. Basilar invagination. Neurosurgery. 2010;66:39-47.
1. Oskouian R.J.Jr., Sansur C.A., Shaffrey C.I. Congenital abnormalities of the thoracic and lumbar spine. Neurosurg Clin North Am. 2007;18:479-498.
2. Winter R. Congenital deformities of the spine. New York: Thieme-Stratton; 1983.
3. Basu P.S., Elsebaie H., Noordeen M.H. Congenital spinal deformity: a comprehensive assessment at presentation. Spine (Phila Pa 1976). 2002;27:2255-2259.
4. Reckles L.N., Peterson H.A., Weidman W.H., Bianco A.J.Jr. The association of scoliosis and congenital heart defects. J Bone Joint Surg [Am]. 1975;57:449-455.
5. Chan G., Dormans J.P. Update on congenital spinal deformities: preoperative evaluation. Spine (Phila Pa 1976). 2009;34:1766-1774.
6. Belmont P.J.Jr., Kuklo T.R., Taylor K.F., et al. Intraspinal anomalies associated with isolated congenital hemivertebra: the role of routine magnetic resonance imaging. J Bone Joint Surg [Am]. 2004;86:1704-1710.
7. Gillespie R., Faithfull D.K., Roth A., Hall J.E. Intraspinal anomalies in congenital scoliosis. Clin Orthop Relat Res. 1973:93:103-109.
8. McMaster M.J. Occult intraspinal anomalies and congenital scoliosis. J Bone Joint Surg [Am]. 1984;66:588-601.
9. Prahinski J.R., Polly D.W.Jr., McHale K.A., Ellenbogen R.G. Occult intraspinal anomalies in congenital scoliosis. J Pediatr Orthop. 2000;20:59-63.
10. Klimo P.Jr., Rao G., Brockmeyer D. Congenital anomalies of the cervical spine. Neurosurg Clin North Am. 2007;18:463-478.
11. Menezes A.H. Craniocervical developmental anatomy and its implications. Childs Nerv Syst. 2008;24:1109-1122.
12. Smith J.S., Shaffrey C.I., Abel M.F., Menezes A.H. Basilar invagination. Neurosurgery. 2010;66:39-47.
13. Wackenheim A. [Radiologic diagnosis of congenital forms, intermittent forms and progressive forms of stenosis of the spinal canal at the level of the atlas]. Acta Radiol Diagn (Stockh). 1969;9:759-768.
14. Krauss W.E., Bledsoe J.M., Clarke M.J., et al. Rheumatoid arthritis of the craniovertebral junction. Neurosurgery. 2010;66:83-95.
15. Caetano de Barros M., Farias W., Ataide L. Lins S: Basilar impression and Arnold-Chiari malformation. A study of 66 cases. J Neurol Neurosurg Psychiatr. 1968;31:596-605.
16. Fromm G.H., Pitner S.E. Late progressive quadriparesis due to odontoid agenesis. Arch Neurol. 1963;9:291-296.
17. Okahara M., Kiyosue H., Mori H., et al. Anatomic variations of the cerebral arteries and their embryology: a pictorial review. Eur Radiol. 2002;12:2548-2561.
18. Spillane J.D., Pallis C., Jones A.M. Developmental abnormalities in the region of the foramen magnum. Brain. 1957;80:11-48.
19. Burke S.W., French H.G., Roberts J.M., et al. Chronic atlanto-axial instability in Down syndrome. J Bone Joint Surg [Am]. 1985;67:1356-1360.
20. Ahmed R., Traynelis V.C., Menezes A.H. Fusions at the craniovertebral junction. Childs Nerv Syst. 2008;24:1209-1224.
21. Fielding J.W., Hensinger R.N., Hawkins R.J. Os odontoideum. J Bone Joint Surg [Am]. 1980;62:376-383.
22. Fielding J.W., Hawkins R.J., Ratzan S.A. Spine fusion for atlanto-axial instability. J Bone Joint Surg [Am]. 1976;58:400-407.
23. Locke G.R., Gardner J.I., Van Epps E.F. Atlas-dens interval (ADI) in children: a survey based on 200 normal cervical spines. Am J Roentgenol Radium Ther Nucl Med. 1966;97:135-140.
24. Greenberg A.D. Atlanto-axial dislocations. Brain. 1968;91:655-684.
25. Spierings E.L., Braakman R. The management of os odontoideum. Analysis of 37 cases. J Bone Joint Surg [Br]. 1982;64:422-428.
26. Arvin B., Fournier-Gosselin M.P., Fehlings M.G. Os odontoideum: etiology and surgical management. Neurosurgery. 2010;66:22-31.
27. Dyck P. Os odontoideum in children: neurological manifestations and surgical management. Neurosurgery. 1978;2:93-99.
28. Klippel M., Feil A. The classic: a case of absence of cervical vertebrae with the thoracic cage rising to the base of the cranium (cervical thoracic cage). Clin Orthop Relat Res. 1975;109:3-8.
29. Winter R.B., Moe J.H., Lonstein J.E. The incidence of Klippel-Feil syndrome in patients with congenital scoliosis and kyphosis. Spine (Phila Pa 1976). 1984;9:363-366.
30. Moore W.B., Matthews T.J., Rabinowitz R. Genitourinary anomalies associated with Klippel-Feil syndrome. J Bone Joint Surg [Am]. 1975;57:355-357.
31. Bulas D. Fetal evaluation of spine dysraphism. Pediatr Radiol. 2010;40:1029-1037.
32. Hensinger R.N. Congenital scoliosis: etiology and associations. Spine (Phila Pa 1976). 2009;34:1745-1750.
33. McMaster M.J., Singh H. Natural history of congenital kyphosis and kyphoscoliosis. A study of one hundred and twelve patients. J Bone Joint Surg [Am]. 1999;81:1367-1383.
34. McMaster M.J., David C.V. Hemivertebra as a cause of scoliosis. A study of 104 patients. J Bone Joint Surg [Br]. 1986;68:588-595.
35. Marks D.S., Qaimkhani S.A. The natural history of congenital scoliosis and kyphosis. Spine (Phila Pa 1976). 2009;34:1751-1755.
36. Winter R.B., Moe J.H., Wang J.F. Congenital kyphosis. Its natural history and treatment as observed in a study of one hundred and thirty patients. J Bone Joint Surg [Am]. 1973;55:223-256.
37. Lonstein J.E. Congenital spine deformities: scoliosis, kyphosis, and lordosis. Orthop Clin North Am. 1999;30:387-405, viii.
38. Faciszewski T., Winter R.B., Lonstein J.E., et al. Segmental spinal dysgenesis. A disorder different from spinal agenesis. J Bone Joint Surg [Am]. 1995;77:530-537.
39. Wiltse L.L., Newman P.H., Macnab I. Classification of spondylolisis and spondylolisthesis. Clin Orthop Relat Res. 1976;117:23-29.
40. Cavalier R., Herman M.J., Cheung E.V., et al. Spondylolysis and spondylolisthesis in children and adolescents: I. Diagnosis, natural history, and nonsurgical management. J Am Acad Orthop Surg. 2006;14:417-424.
41. Williams D.N., Nixon H.H. Agenesis of the sacrum. Surg Gynecol Obstet. 1957;105:84-88.
42. Pang D. Sacral agenesis and caudal spinal cord malformations. Neurosurgery. 1993;32:755-778. discussion 778–779
43. Ignelzi R.J., Lehman R.A. Lumbosacral agenesis: management and embryological implications. J Neurol Neurosurg Psychiatr. 1974;37:1273-1276.
44. Singh S.K., Singh R.D., Sharma A. Caudal regression syndrome—case report and review of literature. Pediatr Surg Int. 2005;21:578-581.
45. Adra A., Cordero D., Mejides A., et al. Caudal regression syndrome: etiopathogenesis, prenatal diagnosis, and perinatal management. Obstet Gynecol Surv. 1994;49:508-516.
46. Altman R.P., Randolph J.G., Lilly J.R. Sacrococcygeal teratoma: American Academy of Pediatrics Surgical Section Survey-1973. J Pediatr Surg. 1974;9:389-398.
47. Ein S.H., Adeyemi S.D., Mancer K. Benign sacrococcygeal teratomas in infants and children: a 25 year review. Ann Surg. 1980;191:382-384.
48. Botto L.D., Moore C.A., Khoury M.J., Erickson J.D. Neural-tube defects. N Engl J Med. 1999;341:1509-1519.
49. Miller A., Guille J.T., Bowen J.R. Evaluation and treatment of diastematomyelia. J Bone Joint Surg [Am]. 1993;75:1308-1317.
50. Ein S.H., Mancer K., Adeyemi S.D. Malignant sacrococcygeal teratoma—endodermal sinus, yolk sac tumor—in infants and children: a 32-year review. J Pediatr Surg. 1985;20:473-477.
51. Finn M.A., Walker M.L. Spinal lipomas: clinical spectrum, embryology, and treatment. Neurosurg Focus. 2007;23:1-12.
52. Kanev P.M., Park T.S. Dermoids and dermal sinus tracts of the spine. Neurosurg Clin North Am. 1995;6:359-366.
53. Cardoso M., Keating R.F. Neurosurgical management of spinal dysraphism and neurogenic scoliosis. Spine (Phila Pa 1976). 2009;34:1775-1782.
54. Williams B. Orthopaedic features in the presentation of syringomyelia. J Bone Joint Surg [Br]. 1979;61:314-323.