[level-membership-for-neurosurgery-category]
Chapter 8 Anatomy and Pathophysiology of Acquired Spinal Disorders
Degenerative Disc Disease and Spondylosis
Degenerative disc disease (with its characteristic clinical syndromes of disc herniation, spondylosis, and radiculopathy) is associated with vascular, biochemical, and anatomic changes in the disc. There is a consistent anatomic pattern of disc degeneration in the spine, with most changes occurring in the midcervical, thoracolumbar, and lower lumbar regions. This pattern is thought to reflect the distribution of the mechanical stresses caused by spine movement and loading, as well as those due to erect posture.1
The intervertebral disc consists of three components: (1) the nucleus pulposus; (2) the annulus fibrosus, which surrounds the nucleus pulposus; and (3) the cartilaginous end plates, which attach these structures to the rostral and caudal vertebrae of the motion segment. The annulus is formed by a series of lamellae that have high collagen content and thereby provide significant resistance to tensile forces. The ventral annulus is usually wider and more organized than the dorsal annulus, with which discontinuous lamellae may be present.2 The nucleus pulposus, derived embyologically from the primitive notochord,3 has a much higher proteoglycan and water content than the annulus fibrosus. The hyaline cartilage end plates are similar in collagen type to the inner annulus fibrosus and the nucleus pulposus.4
The disc acts as a deformable, fluid-like material, whose tendency to bulge is resisted by the tensile stress in the annular lamellae and the end plates. Therefore, a substantial intradiscal surface strength is required to resist a high circumferential annular stress and thus prevent excessive disc deformation (bulging). When disruption of the nucleus pulposus and annulus fibrosus reduces intradiscal pressure, bulging occurs.5
The disc receives its nutrients through small vessels in the cartilage end plates and from the periphery of the annulus.6 With aging, however, the end plates calcify, and vessel loss occurs, until nearly the entire disc becomes avascular.7 The loss of vasculature promotes increased anaerobic metabolism, increasing lactic acid production and cellular necrosis. The water content of the annulus fibrosus decreases from 78% at birth to 70% by the fourth decade, and the nucleus pulposus water content decreases from 90% to less than 70% with maturation.8,9 With this change in vascularity, and with water loss in the region of the inner annulus and nucleus pulposus, there is a relative increase in fibrocytes and chondrocytes, which are more tolerant of a low-pH environment.3
Before the age of 2 years, the nucleus pulposus is translucent and anatomically different from the annulus fibrosus.10 By the second decade, the inner annulus and nucleus grow increasingly fibrous and lose both height and proteoglycans.9 In the third decade, nuclear fragmentation and fibrosis appear. Progressive myxomatous degeneration, swelling, and fissure formation occur in the annulus by the fourth decade.11,12 Eventually the nucleus pulposus may become disorganized, dehydrated, and fragmented with circumferential and radial tears.10,11,13 Grading systems for these patterns of disc degeneration, using plain radiographs or MRI studies, have been published.10,14
On plain radiographs, the degenerative disc changes range from grade I to grade IV. Grade I represents a normal disc. Grade II demonstrates sclerosis along with disc space narrowing or osteophyte formation. Grade III shows moderate sclerosis, and grade IV is associated with severe sclerosis with disc space narrowing or osteophyte formation.14 Yu et al.10 classified changes in the disc, with reference to the age of the subject and to the stage of degeneration, by comparing the anatomic characteristics with the appropriate MRI findings in cadaveric dissections (Fig. 8-1 and Table 8-1). The primitive notochord is present up to age 10. In the second decade of life, a distinct fibrous band forms in the nucleus and disc height diminishes. In the third decade fragmentation and fibrosis of the nucleus occurs. By the fourth decade there is swelling, separation, and myxomatous degeneration of the annular lamellae with fissure formation.12
| Type of Disc | Anatomic Characteristics | MRI Features |
|---|---|---|
| Immature | Nucleus pulposus and annulus fibrosus differentiated, primitive notochord may be present | High-signal intensity from nucleus and annulus |
| Transitional | Fibrous tissue in equator of annulus | High-signal intensity from nucleus and annulus, low-signal intensity in ventral and/or dorsal region of nucleus pulposus, corresponding to dense fibrous tissue |
| Adult | Annulus and nucleus not differentiated, annulus intact or marked by small concentric or transverse tears | Moderately high-signal intensity from nucleus and annulus, low-signal intensity from Sharpey fibers and fibrous tissue in midportion of disc |
| Early degenerated | Radial tear of annulus, diminishing amount and discoloration of fibrocartilage in nucleus | Diminishing signal intensity from nucleus pulposus, low signal from Sharpey fibers disrupted by region of higher-signal intensity at location of annular tear, slightly diminished disc height |
| Severely degenerated | Replacement of nuclear and annular fibrocartilage with amorphous fiber and cysts | Severely reduced disc height, low (fibrous tissue) or high (fluid) signal intensity from intervertebral disc |
From Yu S, Haughton VM, Sether LA, et al: Criteria for classifying normal and degenerated lumbar intervertebral disks. Radiology 170:523–526, 1989, with permission.
One of the most common disc-related clinical syndromes is a herniated disc with sciatica. With degeneration, fissure formation occurs in a radial distribution. It is likely that the biomechanical cause of disc herniation is related to a combination of complex movements involving compression, lateral flexion, and/or rotation.15–18 With flexion, the nucleus pulposus moves dorsally. The dorsal annulus has fewer and more disorganized lamellae and may be inherently weaker than the thicker ventral annulus. Degeneration of the annulus results in the development of peripheral, circumferential, and, subsequently, radial tears. With complex stresses applied to the dorsally migrating nucleus, herniation may occur along a radial tear.
Disc herniation can cause associated nerve-root impingement. The typical dorsolateral herniated disc affects the nerve root passing to the next lower foramen, but a more laterally herniated disc can affect the nerve root above. Masaryk et al.19 used MRI findings to classify the stages of disc herniation. A bulging disc has an MRI signal similar to the rest of the disc, but the bulge is beyond the adjacent vertebral margins. A prolapsed or protruding disc has nearly breached the outer annular fibers and is barely contained. The disc remains contiguous with the rest of the nucleus pulposus by a pedicle that has a high signal on T2-weighted MRI. The disc is extruded when it completely breaches the outer annular fibers and the posterior longitudinal ligament, but remains in continuity with the disc proper. If the fragment is no longer in continuity with the main part of the disc, it is termed a sequestered, or “free,” disc fragment. The International Society for the Study of the Lumbar Spine20 classified the disc as either contained or noncontained, with the latter group including extruded and sequestered discs. Free fragments may migrate in a rostral or caudal direction. It appears that far-lateral herniated discs are more likely to migrate in a rostral direction, thus affecting the nerve root above the disc space.21
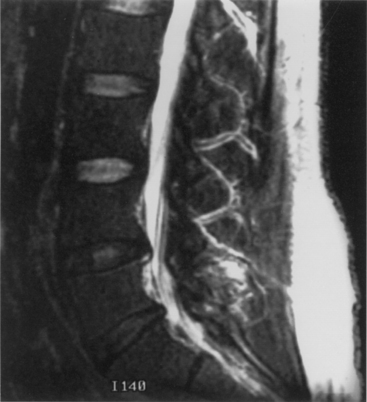
Disc degeneration without herniation may also lead to changes affecting the biomechanical function and stability of both the intervertebral and articular facet joints. Although opinions differ regarding whether facet or disc degeneration is the initial event that causes spondylosis, the three-joint intervertebral-motion–segment concept emphasizes that disease in each component affects the others. This is to say that unilateral or bilateral facet disease or disc degeneration may lead to progressive changes in the other segmental units. Adjacent bone changes are associated with cartilaginous degeneration in these three joints. Spurs and osteophytes form at the site of peripheral annular attachment to the end plates. These osteophytes are thought to be formed in regions of excessive motion. Kirkaldy-Willis22 incorporated this concept into a theory regarding the natural history of spinal degeneration. He believed that facet and disc disease occurred with progressive reciprocal dysfunction. This resulted in ligamentous laxity around the facet joint and increased stresses that lead to internal disc disruption. This condition causes subluxation, disc resorption, and, finally, paradiscal osteophyte formation. Enlargement of the facets also occurs as a result of osteophyte formation. These changes may contribute to lumbar stenosis (Fig. 8-2) or a lateral recess syndrome.23–25
Patients with significant lumbar spinal canal narrowing resulting in stenosis complain primarily of pain, weakness, and leg numbness while walking. This pain can be relieved when the patient flexes the spine by sitting, by leaning forward while walking (shopping cart sign), or by leaning against counters. The symptomatic improvement associated with these maneuvers is related to an increase in lateral recess and spinal canal dimensions. Flexion results in stretching of the protruding ligamentum flavum and posterior longitudinal ligament, as well as reduction of overriding laminae and facets.26 This small amount of change in the circumferential spinal canal, lateral recess, and foraminal region alleviates the pressure on the nerve roots and subsequently relieves the symptoms. Returning to the erect posture leads to repeated compression and a further exacerbation of symptoms. During ambulation, some patients experience the onset of symptoms because of an increased metabolic demand in nerve roots that have become ischemic as a result of stenotic compression. Such “neurogenic claudication” is relieved when the subject sits. Often, bicycling (which is associated with flexion of the lumbar spine) is well tolerated.
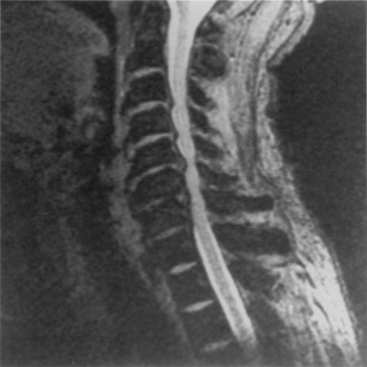
Aging discs in the cervical spine cause characteristic spine alterations that may lead to cervical myelopathy or radicular pain and deficit. In young subjects, the cervical spine assumes a lordotic posture. This results in a greater ventral height of the annulus, compared with the dorsal annular height. With aging, however, intradiscal water loss and disc narrowing occur, thus leading to progressive spine straightening. In young patients, the range of intervertebral motion is greatest at C5-6 and C6-7. Narrowing and degeneration with osteophyte formation is most marked at these levels (Fig. 8-3). With these changes there is progressively less movement. In patients older than age 60, motion at C3-4 and C4-5 increases. Increased degenerative instability in older patients, therefore, is associated with translational subluxation, especially retrolisthesis at C3-4 and C4-5.27 In this scenario the spinal cord of the patient with cervical spondylotic myelopathy may not only be compressed by osteophytes, but may also suffer repeated injuries secondary to intervertebral hypermobility or instability. Dynamic flexion-extension radiographs are necessary to diagnose degenerative spondylolisthesis since static films in neutral position do not demonstrate subluxation, if present. A treatment protocol that does not take these factors into account may be associated with less than optimal success.
Rheumatoid Arthritis of the Spine
Rheumatoid arthritis (RA) affects both the spine and the peripheral joints. It has a prevalence of approximately 1%, with the greatest incidence in the fourth through sixth decades.28 RA is a disease of the synovial joints. The earliest change in the joints is synovitis, followed by an acute inflammatory response as a result of antibody-antigen complex formation. These processes activate the complement cascade and generate biologically active substances, ultimately resulting in complete destruction of the joint. This acute process is followed by a chronic granulomatous process, or pannus formation, which produces collagenase and other enzymes that destroy surrounding cartilage and bone.29 This may lead to instability because of ligamentous incompetence.28,30,31
Considerable controversy regarding the pathogenesis of cervical spine rheumatoid joint disease revolves around whether the initial site of involvement is (1) the apophyseal joint, with resultant facet destruction and progressive secondary instability of the intervertebral disc, or (2) inflammation in the uncovertebral joint, which leads to primary disc destruction with secondary degenerative involvement of the apophyseal joints. Martel32 examined 20 RA patients and found instability associated with apophyseal joint involvement. This leads to vertebral end plate destruction, disc space narrowing, and erosion. At autopsy, the discs showed evidence of necrosis and degeneration, with minimal inflammation. Martel proposed that apophyseal changes caused the instability with secondary disc destruction and end plate microfractures. The relative infrequency of cervical spine disease in juvenile-onset RA was explained by the early bony ankylosis of the apophyseal joints observed in these subjects.
Ball33 reviewed the pathology of 14 RA patients with no radiologic evidence of cervical disease and found that the earliest histologic lesions were in the uncovertebral joints. He suggested that the disc and adjacent bone are then secondarily involved with resultant inflammatory destruction and progressive instability. The fact that uncovertebral joints are not completely developed in the first two decades of life34 might also explain the infrequency with which cervical rheumatoid disease is seen in juvenile-onset RA.35
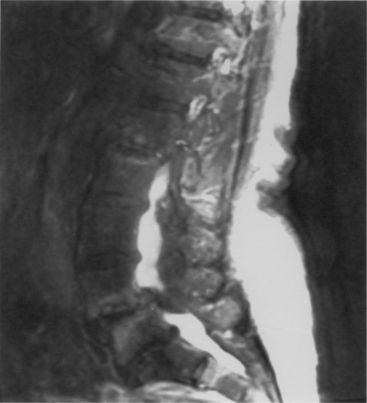
Cervical spine disease is observed in as many as 88% of patients with RA.36 The manifestations include C1-2 instability, occipitocervical (OC) instability (with or without vertical displacement of the dens), and subaxial cervical RA. C1-2 instability is the most common form of cervical rheumatoid involvement and may occur in up to 74% of the patients.37 The dens is surrounded by two synovial joints, one ventrally, between the atlas and dens, and another between the transverse ligament and the dens. With involvement of the synovial joints there is progressive inflammation, destruction, and subsequent transverse ligament laxity, with destruction of the osseous attachments of the ligamentous complex. This loss of ligamentous integrity allows C1 to move ventrally on C2. If there is further significant disruption and osteomalacia of the dens itself, then dorsal C1-2 subluxation can also occur.38 If the synovial apophyseal joints between C1-2 are involved as well, lateral rotation may also be evident in addition to subluxation at C1-2. OC instability results from involvement of the atlanto-occipital articulations. With significant articular facet destruction, there is progressive collapse of the occiput at C1 and vertical displacement of the residual dens (Figs. 8-4 and 8-5). This has also been termed atlantoaxial impaction, vertical subluxation, cranial settling, and basilar invagination.38 Vertical displacement of the dens occurs in 5% to 32% of RA patients.29,39,40 It is believed that vertical displacement of the dens represents a more advanced stage of systemic disease burden; indeed, one 10-year retrospective review of patients with RA cervical instability treated with OC fusion noted significantly worse long-term outcomes in the subset of patients with vertical displacement of the dens.41
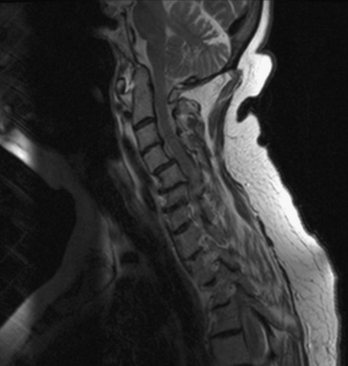
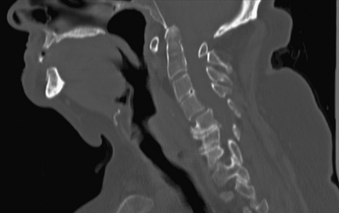
In the subaxial region, the levels most commonly involved with rheumatoid synovitis are C2-3 and C3-4. Subluxation (Fig. 8-6) may occur in approximately 7% to 29% of the patients with RA.38 Subaxial region subluxation rates as high as 31% have been noted after rostral surgical fusion; however, there was no increased incidence of myelopathy or pain with fusion-adjacent subluxations.41 These “staircase” subluxations are thought to be caused by significant ligamentous laxity and facet degeneration.36,42 At any of the various sites of rheumatoid involvement, osseous erosion of adjacent bone, caused by osteoclastic resorption, occurs frequently.43
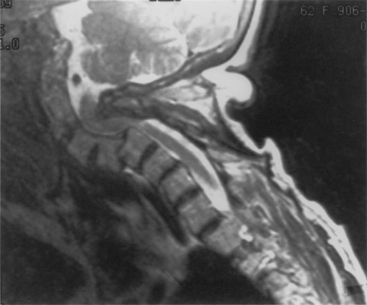
With the significant bony destruction, ligamentous laxity, and the potential for neural compression observed in the rheumatoid cervical spine, the primary emphasis of treatment is reduction of subluxation and fusion/fixation to prevent spinal cord injury. The optimal time to proceed with operative intervention is yet to be determined. Omura et al. stratified their RA population and found that the subset of patients with seropositive disease and systemic evidence of mutilating-type joint involvement are at the highest risk of deterioration of their known cervical lesion.44 Furthermore, retrospective review of RA patients with cervical disease found that best-medical management faired significantly worse when compared with surgical fusion with respect to both morbidity and mortality.44 When substratifying the patient population undergoing surgical fusion, patients operated on earlier in their course and with a better functional preoperative score had a more pronounced overall improvement than those undergoing late surgical management. There is strong evidence for early operative intervention for the stabilization of RA-associated cervical disease.44,45
Surgical fusion yields multiple benefits, including reduction of both pain and neurologic sequelae; retrospective analysis of long-construct dorsal fusion demonstrates significant recovery of these two characteristics, compared with best-medical management, with improvement of an average of one to two grades on the Ranawat scales for pain and neurologic symptoms (Boxes 8-1 and 8-2).41,44,45 These improvements were persistent, even in the setting of failed permanent postoperative reduction of deformity and imbalance.41 The chronic granulomatous pannus decreases in size with the elimination of abnormal movement after successful arthrodesis.46,47 There is no consensus regarding the optimal type of intervention, but one must keep in mind the inherent poor quality of RA bone, the laxity of ligaments, the insidious inflammatory nature of RA itself, and the destructive effects of the myriad pharmacologic interventions, especially with respect to treatment with corticosteroids.45
Scheurmann Disease (Juvenile Kyphosis)
Scheuermann48 first described the progressive dorsal kyphosis of adolescent children in 1920. The deformity is usually evident as a fixed thoracic kyphosis that does not correct with hyperextension, thereby differentiating it from a postural kyphosis. Compensatory hyperlordosis of the lumbar and cervical spine may also be present. A mild scoliosis is noted in 20% to 30% of patients.49 Sorenson50 described the characteristic feature of ventral wedging of 5 degrees or more in at least three adjacent vertebrae. Other characteristics include kyphosis of greater than 40 degrees, vertebral end plate irregularity, and disc space narrowing.51 The prevalence of the disease ranges from 0.4% to 8%.50 It occurs predominantly in males (91% in one series).52 Hereditary patterns of transmission have been identified, though genetic loci have yet to be determined.53
Basic biomechanical factors and forces may play a role in this disorder. The thoracic spine has a natural kyphosis determined primarily by the shape of the vertebrae; in the adolescent thoracic spine 20 to 40 degrees of kyphosis is normal. The dorsal elements, including the ligamentum flavum and the laminae, resist forward flexion of the spine in tension, whereas the ventral bony elements (vertebral bodies) and disc resist compression.54 However, the facet joint capsules in the thoracic region are mechanically “weaker” than those in the lumbar region, so that any factor that increases the torque of the spine can result in greater deformity. The more marked the initial angulation of the spine, the larger the load (subject’s weight), and the longer the duration of load application, the greater the likelihood of the progression of the deformity.
Scheuermann disease must be differentiated from juvenile postural kyphosis, which, as the name attests, is a kyphosis seen during flexion that will correct with improved posture and extension. The apex of the curve is smooth. The condition will improve with therapy that targets improved posture and core strengthening.53
The pathogenesis of the disease remains unclear. Scheuermann believed that aseptic necrosis of the ring apophyses caused interruption of growth, which resulted in ventral vertebral body wedging.55 Subsequent work has refuted this theory by demonstrating that the apophyses do not contribute to longitudinal growth. Such growth is now known to result from endochondral ossification of the end plates.55 Schmorl56 felt that damage to the end plate by herniated disc material was of importance. Schmorl nodes are, however, not limited to the kyphotic region of the spine and are common in otherwise normal patients. It has been postulated that osteoporosis is involved,57,58 but recent investigations have found no differences in the trabecular bone density between patients with Scheuermann disease and controls matched for age, gender, and race.52,59 Other factors such as inflammation,60 hormonal influences,61 genetic factors,62 altered calcium metabolism,63 hypovitaminosis,64 neuromuscular disorders,65 extradural cyst formation,66,67 defective collagen formation of the end plate,68 and a decrease in the collagen-proteoglycan ratio of the end plate61 have been implicated, but their roles in the development of the disease have not been substantiated.
There is a high association (>90%) between ventral osseous extensions from the anterior margin of the vertebral body and the diseased vertebrae, a feature that is absent in normal specimens.52 Histologic examination reveals disorganized endochondral ossification, which may be a result of abnormal stress. Traumatic features of vascular and fibrocartilage proliferation are evident in the ventral end plates in Scheuermann disease.52,68,69 The dorsal vertebral height in cases of Scheuermann disease is not significantly different from that of controls, implying that either the ventral and dorsal stresses are different or that the kyphotic changes occur after dorsal growth is completed (the normal pattern of ring apophysis closure starts dorsolaterally, then works ventrally).52 Possibly, the natural thoracic kyphosis, being exacerbated by a rounded back, results in the development of the abnormal kyphosis.
Back pain is uncommon in the growing child with Scheuermann disease. Low back pain has been reported to be common (up to 50%)51 in adults with progressive, untreated dorsal kyphotic deformities. In other studies pain was not a significant problem.51 Progression of deformity is documented in 80% of patients older than 25 years of age, but the extent of deformity and pain is generally not severe.70 The kyphosis most commonly progresses before skeletal maturity, but can occur in adulthood.71 Disc degeneration is also associated with the deformity. Development of neurologic complications is rare, but is due to thoracic disc hernation, dural tenting, extradural cysts, and vascular compromise.72
Examination of the patient with Scheuermann disease can reveal a hyperpigmented lesion at the apex of the thoracic curve—a result of friction injury from the abnormally protruded spinous process. Patients often have a forward-protruding head, flexion contractures of the shoulders and hips, as well as tight hamstrings.53
Treatment is often indicated to correct the deformity, prevent its progression, and alleviate pain. The extent of the kyphosis and the age of the patient are important criteria for intervention. The nonoperative forms of treatment, such as bracing (Milwaukee brace) or casting, are the first line of treatment for most cases in which the kyphotic deformity is less than 65 degrees. These cases have a high success rate in correcting the deformity, especially if treatment begins before closure of the iliac apophyses (i.e., skeletal maturation).73 Operative treatment with fusion is reserved for cases of progressive deformity, pain not responsive to an adequate trial of casting or bracing, degenerative changes in adults associated with the kyphosis, cardiopulmonary compromise, and for a deformity greater than 65 degrees.71
Dorsal long-construct instrumentation that extends rostrally and caudally well above the thoracic apex is often adequate for stabilization and correction of the deformity. In the event of extreme kyphotic deformity, both a ventral and dorsal surgical approach is necessary for a more definitive correction,53 as well as the maintenance of correction until fusion in the setting of greater tension forces opposing the correction. A large retrospective review comparing 78 patients treated with either dorsal instrumentation alone or combined anterior-posterior instrumentation showed a comparable degree of deformity correction. The rates of proximal junctional kyphosis and surgical complications were clinically and statistically significantly increased in the combined procedure. A decreased rate of postoperative loss of correction was observed with the combined procedure. A higher rate of proximal junctional kyphosis was correlated with a greater degree of postoperative kyphosis, greater pelvic incidence, and less imbalance correction. The authors conclude that dorsal arthrodesis and fixation alone should be the favored procedure whenever possible due to the lower complication rate.74 On the other hand, anterior ligamentous and disc release with video-assisted thoracoscopic surgery (VATS) combined with dorsal spinal fusion may yield lower complication rates and increased sagittal deformity correction, due to the anterior tension band release.75
Paget Disease
Paget disease is a metabolic bone disorder thought to be of possible viral origin. Prevalence of the disease has marked geographic variation. In the United States, Paget disease is found radiographically in 3% to 4% of patients older than age 40.76 Histologically, the disease is characterized by areas of bone resorption and new bone deposition resulting from focal increases in the population of osteoclasts. The individual cells are larger than normal and contain inclusion bodies similar to paramyxovirus capsids. This suggests viral induction of the osteoclastic activity and results in a greater surface bone resorption. There is no disturbance of reactive bone formation; therefore, increased osteoblastic activity compensates for the bone resorption and, in fact, produces a net-positive balance of bone. The bone is usually lamellar, and it is normally mineralized.76 However, woven bone and occasionally osteoid bone are also present and result in reduced bone quality with disruption of the lamellar structure of both cortical and trabecular bone.
The pelvic bones are the most commonly affected, followed by the spine. Approximately 70% of patients have lumbar spine involvement, 45% have thoracic spine lesions, and the cervical spine is involved in 15% of cases.77 The frequent involvement of the lumbar spine is thought to be caused by increased loading.78 The lesions are primarily in cancellous bone. Approximately two thirds of the radiographically evident lesions are asymptomatic.77 Back pain in Paget disease is related to the combination of the bone deformity and subchondral bone enlargement that alters the contours of the joint surfaces and leads to joint degeneration. The subchondral changes include increased bone deposition and subchondral infarcts from abnormal pressure on expanded bone, each of which causes the bone to lose its normal flexibility and usual biomechanical properties.76 The involved vertebral body can interfere with nutrition of the intervertebral disc, thus leading to early degenerative sclerotic changes.
Radiographically, the majority of patients with Paget disease have involvement of both the vertebral body and dorsal osseus elements—involvement of only ventral or dorsal structures is rare. Consistent with histologic analysis supporting periosteal bone formation and endosteal absorption, early radiographs show increased density in the osseous periphery contrasted with a central lucency.79,80 Commonly, sclerotic areas are present as well as localized osteolytic lesions, which may coalesce with time. As a result of the disorganized pattern of bone deposition, biomechanical efficiency is reduced and the risk of fracturing is increased. Healing of fractures is usually efficient. The histologic features of Paget disease are observed in the fracture line.76 The incidence of neurologic sequelae with thoracic and cervical spine involvement is increased, perhaps caused by the narrower diameter of the spinal canal due to stenosis in these regions.81,82 Some advocate that a component of epidural fat ossification is a factor, though this may be simply a component of advancing periosteal bone formation that projects into the canal.79
Back pain is the most frequent presenting symptom, resulting from multiple possible etiologies, including periosteal stretching, deranged vascularity with resulting zones of ischemia, stenosis, nerve root compression, facet arthropathy, and osseous microfracture.79 Neurologic sequelae have been reported in 25% to 30% of cases of Paget disease.81,83 The neurologic deficits are most often caused by bony compression of the spinal canal or the foramina, with the neural arch and the facet joints most commonly affected by the proliferative bone deposition.81 Fractures and subluxations can also compromise the spinal canal, and progressive platybasia can result in compression of the medulla. Vascular “steal,” resulting from the increased vascularity of the pathologic bone, has also been implicated in the development of neurologic deficits.84
Treatment centers on reducing the burden of hypertrophied and abnormal bone. Despite the prevalence of stenosis with resultant neural element compression, the first intervention is medical treatment with bisphosphonates and calcitonin, among other agents. Surgical decompression is rarely indicated, owing to the success of medical intervention.79 If surgery is to be considered, an aggressive preoperative course of treatment should be considered to reduce the volume of abnormal and highly vascularized tissue, which can lead to voluminous blood loss.80 Pagetic lesions rarely degenerate to benign and malignant neoplasms that require more aggressive surgical management, with osteosarcomas predominating in the latter category.79
Ankylosing Spondylitis
The cause of the disease remains unknown, but it appears to be multifactorial with both genetic and acquired factors playing a role. There is a male predominance, varying from 3:1 to 8:1.85 Peak age of onset is between 15 and 29 years, with less than 5% beginning after age 50.86 Prevalence in the United States population is about 0.1%.87
The earliest signs of ankylosing spondylitis occur in the region of ligamentous attachment to bone (the enthesis).88,89 In ankylosing spondylitis the enthesis shows multiple, focal, microscopic inflammatory lesions that eventually destroy the ligament and erode the adjacent cortical bone. This process leads to an osteitis, primarily at the ventral and ventrolateral aspects of the attachment of the annulus fibrosus to the vertebral bone. This is the “anterior spondylitis,” or Romanus lesion, that is observed radiographically.90,91 As the reparative process occurs, woven bone replaces the cortical erosion (ossification in fibrous tissue without preceding cartilage formation). Ultimately, this is replaced by lamellar bone.33 Syndesmophytes are formed, most conspicuously on the ventrolateral aspects of the vertebrae adjacent to each disc. This results in new enthesis formation above the original level of cortical bone. Further thickening and growth of the syndesmophyte may be caused by inflammatory lesions in this new bone33 or chondroid metaplasia with ossification.92
In the apophyseal joint, osteitis and enthesiopathy occur at the junction of capsule and bone and result in reactive bone formation and ossification of the capsule,93,94 usually in the presence of well-preserved articular cartilage, implying that the capsule-ligamentous attachment is of primary importance in the apophyseal joint pathology.33,93 Ultimately the joint may become ankylosed by endochondral ossification. This may be the result of capsular ossification or the general immobility of the spine as a result of discovertebral syndesmophyte formation as described previously.33,95 However, the observation that apophyseal joint ankylosis may occur in the absence of vertebral ankylosis at the same level makes the former more likely.94
Concomitant ossification of the supraspinous and interspinous ligaments also occurs where there is a nonspecific inflammatory process at the attachment of the ligaments.91 The anterior longitudinal ligament, however, does not usually become ossified, except at its deep fibers adjacent to the annulus fibrosus.92
Bone resorption (resulting in squaring of the vertebrae), syndesmophyte formation, bony ankylosis of the intervertebral discs, and apophyseal joint and ligament ossification complete the classic radiographic “bamboo-spine” appearance. Although bone formation at the attachments of the ligaments and at the apophyseal joints is increased, the vertebrae in ankylosing spondylitis are generally osteoporotic. This may be a result of the systemic effects of the disease, immobilization of the vertebrae, the inflammatory process, or drug treatment.96
As the bony ankylosis in the discovertebral region and the apophyseal joints progresses, the normal flexibility of the spine is lost. The spine is much stiffer than normal and is unable to absorb and dissipate loading energy in an efficient manner. Indeed, the ankylosing process itself may introduce a “lever-arm” quality to regions of affected neuroaxis, increasing the magnitude of injury that may be focused at specific spinal levels.97 Because of these factors and osteoporosis, the bone is much more prone to fracture and subluxation after trivial trauma (Fig. 8-7).98 Due to the long lever-arm effect of inflexible segments adjacent to the fracture, the spinal cord is significantly vulnerable when dislocation occurs in these fractures. The cervical spine appears to be particularly susceptible; approximately 75% of the spinal fractures occur in this region, primarily in the lower cervical spine.99 These fractures tend to pass through the ventrodorsal width of the vertebra and may involve the calcified ligaments in the spinous processes. This process may occur either at the level of the disc space or through the vertebral body.98 A cervical kyphosis is often present, and the neck is especially vulnerable to hyperextension injuries.99–101 Some authors have attempted to match the mechanism of injury to the fracture location, considering extension to cause transdiscal fractures and flexion to cause transvertebral fractures.101 Others have not found this relationship.100
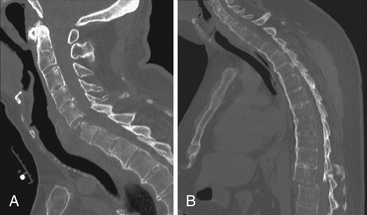
Injuries to the thoracolumbar spine, though less frequent than cervical traumatic injury, are themselves significantly more frequent in the ankylosing spondylitis patient, occurring at a rate four times that in the general trauma population.102 The majority of these fractures represent three-column injuries, again an indication of the imbalance, poor osseous quality, and associated disease in adjacent soft tissue structures that is seen with ankylosing spondylitis.
In spondylitic patients with cervical spine fractures, the mortality rate is 35%, as compared with 20% for patients with a normal spine. Also, the risk of severe neurologic sequelae in ankylosing spondylitis is 57% compared with 18% in the normal spine.98 One review documented an American Spinal Injury Association (ASIA) A posttraumatic grade in 41% of ankylosing spondylitis patients; the mechanism for the majority of these patients was a fall from a standing position.103 Without ligamentous support, and with multiple ankylosed vertebrae, any spinal movement is concentrated at the fracture site. Therefore, fractures are usually very unstable. The increased risk of bleeding with fracture in ankylosing spondylitis is thought to be related to the enlarged diploic spaces of the pathologic cancellous bone, the extensive nature of the fracture, and damage to adjacent epidural veins.100,104 Epidural hematomas have been reported to occur in 20% of cases.30 For these reasons, there is greater potential for neurologic deficit. This is especially problematic because fractures often occur after minor trauma, and often in the lower cervical region. These may be difficult to visualize radiographically, especially in osteoporotic bone. Further complicating the radiographic evaluation of neuraxis trauma is the diffuse and active inflammation that forms the basis of the disease, which is seen as increased signal on the short-tau inversion-recovery (STIR) MRI sequence. Acute injury may be masked during radiographic examination due to these chronic MRI changes.
Operative interventions in the acute setting should focus on restoring preoperative sagittal balance, rather than on attempting to improve the kyphotic deformity that is typically present before injury. Further strain to neural elements by excessive traction, or during patient transfers unprotected by external immobilization, may introduce a devastating additional injury. Correction may proceed at a later time when in a more controlled setting. Additionally, halo vest or other cervicothoracic vest fixation should be used judiciously in cases of ankylosing spondylitis, as these patients often have multiple medical morbidities, including decreased vital capacity and pulmonary insufficiency due to ankylosis of the costovertebral joints, which would be further strained by such intervention.97,103
When fractures occur, a normal callus forms at the site, and although inadequate immobilization may lead to pseudarthrosis, healing is typically rapid.99 Pseudarthrosis of transdiscal fractures in undiagnosed ankylosing spondylitis is often confused with disc space infection or tuberculous spondylitis.
Although atlantoaxial instability is far less common in ankylosing spondylitis than in RA, it may occur.105,106 Inflammation of the entheses, the apophyseal joints, and the synovial joint between the dens and the transverse ligament results in both bony and ligamentous damage, with subsequent instability similar to that observed in RA. The atlantoaxial joint may be the only remaining mobile segment and the fulcrum of all craniocervical mobility of the cervical spine in patients with advanced ankylosing spondylitis.
Ossification of the Posterior Longitudinal Ligament
Although OPLL was first reported in 1838 in England,107 it has received increased attention because of the high incidence in Japanese and other Asian populations.61,108 OPLL appears in approximately 2% of the cervical spine radiographs in the Japanese population, and autopsy studies show an incidence of 20% in subjects older than age 60.109 More recently it has been recognized in the non-Asian population, but the prevalence is lower in other countries: 0.1% in West Germany,110 0.12% to 0.7% in the United States,110–112 and 1.7% in Italy.113 The incidence appears to be higher in males, and it increases with age.109 The pathogenesis of OPLL remains unclear, though recent investigation has narrowed the genetic loci of interest to a site near the human leukocyte antigen (HLA) on chromosome 6p.114 Routine tests to determine levels of C-reactive protein, rheumatoid factor, and HLA-B27, as well as the erythrocyte sedimentation rate are all normal.108,109 HLA-BW40 and SA5 alterations are more common in OPLL patients, but there is no clear evidence of an inheritance pattern.109,115 Metabolic abnormalities such as hypoparathyroidism, acromegaly, vitamin D–resistant rickets, and spondyloepiphyseal dysplasia may occur concurrently with OPLL,109,114 implying a disturbance of calcium metabolism. However, the significance of these abnormalities in the pathogenesis of OPLL is unclear. The number of growth hormone receptors are often elevated, and bone morphogenic protein (BMP) levels are elevated even in nonossified tissue compared with controls.114 In one series 28.4% of the OPLL patients were diabetic, and 17.7% had an impaired glucose tolerance test. Patients with diabetes mellitus have an increased incidence of OPLL.109 Myotonic muscular dystrophy has also been reported in association with OPLL.
Other hyperostotic conditions associated with OPLL are diffuse idiopathic skeletal hyperostosis (with a concomitance rate of 50%),116 ankylosing spondylitis (with a 2% concomitance rate), and ossification of the yellow ligament (with a concomitance rate of 6.8%).108,109
Radiographically, this acquired spine abnormality is characterized by abnormal ossification involving the posterior longitudinal ligament along the dorsal border of the vertebral body. Greater than 70% of disease is located within the cervical spine, and thoracic or lumbar involvement without concomitant cervical involvement is unusual.114 OPLL is grouped according to its localization along the vertebrae. It has been classified into segmental, mixed, continuous, and localized forms.109 The segmental type is characterized by calcification or ossification behind each body, with each osteophytic segment separated by the uninvolved disc (Fig. 8-8). The continuous type extends over the bodies and discs of several vertebrae. The mixed type is a combination of these two types. The localized type demonstrates ossification limited to the ligament over the disc space. Early OPLL first presents with small ossification patterns posterior to the disc space, making delineation from more ubiquitous degenerative disc disease difficult. Contrast-enhanced MRI may help with the diagnosis, as the posterior longitudinal ligament (PLL) uniformly enhances in the setting of OPLL, but disc pathology does not.114 The vertebrae at C4, C5, and C6 are most affected, and the average number of vertebrae involved is 3.1.109 Ligamentous ossification substantially reduces the size of the spinal canal, particularly in the mixed and continuous types, especially when underlying developmental stenosis is present.
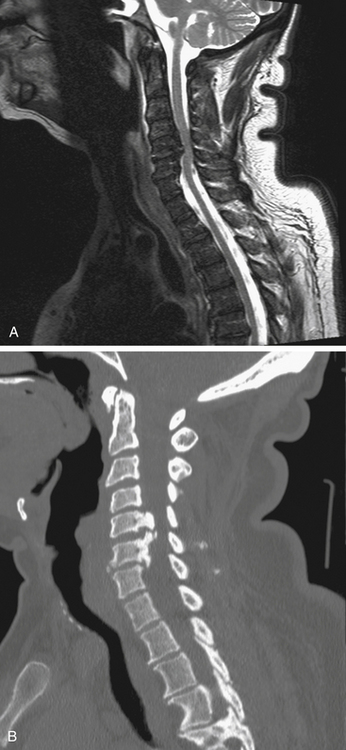
Histologically, the normal PLL contains both type I and type II collagen. In OPLL only type I collagen is identified, suggesting that the process of ossification involves replacement of the original collagen matrix.117 The heterotopic bone formation observed with OPLL occurs in the superficial layer of the PLL, leaving an unossified gap between the dorsal aspect of the vertebral body and the ligament. The ossified ligament has a typical lamellar bone structure with haversian canals and a few bone marrow canals.117 Calcification or ossification may also involve the dura mater.
The average radiographic narrowing of the anteroposterior diameter of the cervical spinal cord has been noted to be more than 40% for the mixed and continuous types.109 Progression of the disease in a single, small series has been documented as a mean annual increase of 4.07 mm rostrocaudally and 0.67 mm in the ventrodorsal direction.118 Myelopathy is the most common neurologic abnormality. It is likely that a large proportion of cases are asymptomatic when developmental stenosis does not aggravate cord compression.119 The relative paucity of symptoms has been attributed to the slow rate of progression observed in most cases, as well as the lack of underlying developmental stenosis. However, a critical spinal canal diameter can be reached, where even minimal trauma can result in severe neurologic deficit.
Management of OPLL must first include a determination of whether a neurologic deficit due to severe stenosis is present or impending, as well as a characterization of other medical morbidity due to OPLL’s association with a myriad of metabolic derangements. Conservative treatment of pain and minor neurologic deficits can include NSAIDs, steroids, and external brace immobilization. Studies have shown that early surgical intervention before the onset of neurologic deficit correlates with significantly improved outcomes. Much like patients with other degenerative/inflammatory pathology of the cervical spine, even very minor trauma can lead to devastating neurologic injury due to the derangement of normal cervical dynamics.114
The surgical treatment of OPLL has been aimed at enlarging the spinal canal by removing the vertebral bodies and ossified ligament by ventral corpectomy and fusion. Internal fixation may obviate the need for postoperative halo or Minerva immobilization. It is important to note that the dorsal approach for laminoplasty or multilevel laminectomy and posterior spinal fusion with instrumentation does not remove the primary pathologic lesion. With rapid disease progression, ventral surgery may still be required as a secondary procedure. In advanced cases with severe developmental and acquired stenosis over multiple levels, combined or staged dorsal and ventral decompression and fusion may be required. Nevertheless, the anterior approach can be fraught with approach-related complications such as dysphagia and dysphonia. Most notably and unique to this pathology is the complication of iatrogenic durotomy and formation of cerebrospinal fluid (CSF) fistula. Investigation into the predictive value of preoperative radiographic findings has helped to stratify patient risk for CSF leak. Hida et al. first described these CT findings: the single-layer sign, defined as a large focal mass of dense OPLL, and the double-layer sign, with ventral and dorsal hyperdense rims of OPLL surrounding a central hypodense (nonossified) ligament.75 Evaluation of these two groups allows for further risks stratification; Min et al. described an incidence of dural penetration in 52.6% of patients with the double-layer sign, 13.6% of patients with the single-layer sign, and 1.5% of patients without either sign.120 Patients with extension of continuous OPLL and cord compression up to the C2 level or caudally to the upper thoracic segments may be best treated with posterior decompression and fusion alone. Whether progression of the OPLL mass effect on the spinal cord in the rostrocaudal or anteroposterior directions is arrested by laminoplasty or laminectomy with fusion remains controversial.
Spondylothesis and Spondylolysis
The pars interarticularis, or isthmus, is the bone between the lamina, pedicle, articular facet, and transverse process. This region is able to resist significant forces in excess of 1251 N.121 It has a cross-sectional area of about 0.75 cm2, with two layers of cortical bone and intervening trabecular bone.122 Developmental or traumatic incompetence or disruption of the pars is associated with anterolisthesis due to instability of the motion segment.
Flexion, extension, and rotation all have effects on the disc and, subsequently, on the facet joints and pars interarticularis. With normal lumbar lordosis, with the discs inclined in a ventrocaudal direction, the load is transmitted by the discs.123 Axial loading therefore places both the disc and the caudal facets under ventral shear stress.124,125 This stress is parallel to the intervertebral disc and is resisted by the caudal facets of the apophyseal joints, the disc, and the muscles attached to the neural arch.122,124,125 In the intact specimen under shear stress, approximately 60% of the stiffness is provided by the disc and 15% by the facet joints.123 The lower lumbar level apophyseal joints lie directly across the plane of the disc and therefore may contribute more to resisting shear than the apophyseal joints in the upper lumbar region, which are at the level of the pedicles.124 In addition, the upper lumbar disc spaces are more dorsocaudally inclined in the upright position, thus making the apophyseal joints less susceptible.
Exactly which movements cause the mechanical deformation and, ultimately, the failure of the pars interarticularis remains unclear. The contribution of flexion, extension, and rotatory movements has been reviewed.121,124,125
It can be demonstrated that as flexion occurs, compression and ventral shear stresses in the lower lumbar region increase.124 Muscular, and then ligamentous, tension resists the shear stress. The simultaneous application of the shear stress and the resisting forces causes stress concentration at the caudal margin of the pedicle, which progresses across the pars.124 The pars, which is not as strong as the pedicle, fails as the stress increases with greater flexion. Debate remains about whether a single episode of overload124 or fatigue126 causes microfractures that lead to a gross fracture with continuing overload. It is likely that a combination of both processes occurs.125 The same mechanism that causes the fracture prevents complete healing, and fibrous nonunion results. This may allow progressive listhesis with elongation of the pars.127
Research and clinical information also implicate extension movement in generating stresses across the pars interarticularis that may lead to fracture.125 It has been suggested that the frequency of spondylolisthesis in gymnasts is a result of hyperextension injuries occurring on landing in the upright position with accentuated lumbar lordosis. If the extended spine is accelerating and then is subjected to sudden deceleration, increased shear stress is generated along the disc space, which in the lower lumbar spine is at an angle to the line of deceleration. This results in further extension, increasing shear, and greater stress across the pars.125 Also, the disc is less stiff in extension, making ventral translation even more probable.121 Microfractures develop, and once the bone is defective, the forces acting on it result in further microfractures and progression of the lytic lesion. Further support for the importance of lordosis in causing the pars defects is observed in patients with Scheuermann disease, in which a compensatory lumbar lordosis occurs. Asymptomatic lumbar spondylolysis often without spondylolisthesis has been reported in as many as 50% of these patients.128
Torque may also play a role in the development of spondylolisthesis, especially in the degenerative type. With degeneration the disc loses its ability to resist shear and torsional stresses.18,124 Torsional stress, conveyed to the caudal facet, distorts the lamina-pedicle angle and results in the facet being less able to resist shear. The contralateral facet then has to resist more shear stress and may also become damaged.124 Stress concentration with injury to the pars may occur when torsional forces are applied to the neural arch, and ultimately, ventral subluxation may occur.124
The most widely used classification of spondylolisthesis is that of Wiltse et al.127 Wiltse et al. divide the listhesis types into dysplastic, isthmic, degenerative, traumatic, and pathologic. Degenerative listhesis has a prevalence of 4% to 10%129,130; isthmic, 4%; and dysplastic, 1%.131 Traumatic and pathologic listhesis implies a history of localized trauma or generalized bony disease, which allows forward subluxation to occur.
Dysplastic Spondylolisthesis
Dysplastic spondylolisthesis, which is caused by a congenital defect of the upper sacrum, or the vertebral arch of L5, presents in young children and adolescents.132 It has two subtypes: type A, with the dysplastic articular facets oriented axially, and type B, with dysplastic articular facets oriented sagittally. When the facets are dysplastic, the ability to resist the ventral shear stress is reduced and can result in listhesis. The pars may be initially intact or even remain intact, but in other cases the ventral shear stress results in microfractures of the pars, with subsequent pars elongation. Thus the pars is not the initiator of the listhesis.133 In dysplastic cases with a subluxation of greater than 35%, neurologic and muscular symptoms are likely,132 usually manifested as symptoms of cauda equina or nerve root compression. Paralysis and bowel dysfunction are uncommon. Hamstring tightness and abnormal gait, however, are common.132
Isthmic Spondylolisthesis
In isthmic spondylolisthesis a defect occurs in the pars interarticularis (spondylolysis). Facet orientation is normal. The three subtypes depend on the integrity of the pars and the nature of the injury. In subtype A there is distinct separation of the pars interarticularis, as a result of fatigue fracture, a single traumatic episode, or a combination of both (Fig. 8-9). In subtype B the pars is elongated, actually appearing intact, which is thought to be a result of the healing of stress fractures of the pars. Fibrous nonunion is observed in these defects. This can appear similar to a dysplastic lesion with pars elongation. Subtype C is characterized by an acute fracture of the pars, in addition to fractures elsewhere in the vertebra, which are usually a result of severe trauma.
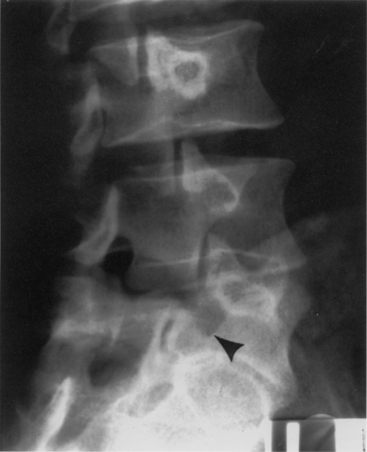
The severity of spondylolisthesis is described by the Meyerding classification of superior vertebral body subluxation over the adjacent inferior vertebral body. The five grades of subluxation are grade 1 (0–25%), grade 2 (25–50%), grade 3 (50–75%), grade 4 (75–100%), and grade 5 (spondyloptysis, >100%).134,135
Isthmic spondylolisthesis occurs at L5-S1 in approximately 82%, L4-5 in 11%, L3-4 in 0.5%, L2-3 in 0.3%, and in other levels, in approximately 6% of the cases.136 The lesion does not appear in other primates, indicating that upright posture is important. Also, true lumbar lordosis, seen only in the human primate, may be a factor.127
Infant cadaveric dissections have demonstrated that lytic pars defects are not present at birth.137 However, bilateral pars interarticularis defects have been documented in a 4-month-old.138 The most common age for development and diagnosis of isthmic spondylolysis is between the ages of 5 and 7 years.127 In a study of 500 children,139 4.4% of the 6-year-olds and 5.2% of the 12-year-olds had unilateral or bilateral pars defects, whereas the incidence is 6% in adults. It is postulated that with assumption of the upright sitting posture and lordosis of the lumbar spine, subluxation is most likely to develop.127,140 In adolescent cases participation in contact sports may be a significant factor.127
There is also evidence that genetics may play a role in isthmic spondylolisthesis.126,141 White males have an incidence of 6.4%, compared with black women, who have an incidence of 1.1%.136 There is an association between the dysplastic and isthmic lesions and spina bifida occulta and hypoplasia of the sacrum.142 The prevalence of spina bifida occulta of L5 or S1 and lumbosacral defects in one series was found to be 94% for the dysplastic type and 32% for the isthmic type.131 The incidence of the two types of spondylolisthesis has been reported to be increased in first-degree relatives. Thirty-three percent and 15%, respectively, of first-degree relatives of patients with dysplastic and isthmic spondylolisthesis have radiographic evidence of subluxation.131
Although the initial degree of slip in isthmic spondylolisthesis can be marked, progression in adulthood is unusual. Slip is more prone to progress at L4-5 than L5-S1, and may be up to 28% in the teenage years.139 Whether subluxation will progress, however, is difficult to predict. Due to the high prevalence of spondylolysis, spondylolithesis, and low back pain in the general population, it is difficult to attribute low back pain in the individual patient to these anatomic lesions. A subgroup analysis from the Framingham Heart Study found not only a higher prevalence of spondylolysis than previously reported (11.5% vs. 6%), but also found no association of spondylolysis or spondylolithesis with low back pain, which suggests that these lesions are not a major cause of back pain in the general population.143
Degenerative Spondylolisthesis
Degenerative spondylolisthesis is more common in women than men,23 with a ratio of 5:1. It is associated with spondylotic changes of the apophyseal joints and disc narrowing. Degeneration of the disc reduces its stiffness and places greater stress on the facets. When subjected to shear forces, subluxation may result without fracture of the pars. Subluxation does not usually exceed 30%.144 Because of the greater inherent stability of L5 and the prevalence of L5 sacralization,136 the L4-5 or L3-4 levels are more frequently affected.145 Stabilization as a result of osteophyte formation usually occurs, and significant progression is rare without destabilizing surgical procedures. Degenerative spondylolisthesis is commonly associated with spinal stenosis and neurogenic claudication caused by lumbosacral radiculopathy. Decompression often relieves symptoms. Fusion with internal fixation may be required in cases with radiographic evidence of instability or severe back pain.
Bouchard-Chabot A., Liote F. Cervical spine involvement in rheumatoid arthritis: a review. Joint Bone Spine. 2002;69:141-154.
Dell’Atti C. The spine in Paget’s disease. Skeletal Radiol. 2007;36:609-626.
Farfan H.F., Osteria V., Lamy C. The mechanical etiology of spondylolysis and spondylolisthesis. Clin Orthop Relat Res. 1976;8:40-55.
Katz J.N., Liang M.H. Differential diagnosis and conservative treatment of rheumatoid disorders. In: Frymoyer J.W., editor. The adult spine: principles and practice. Philadelphia: Lippincott-Raven; 1991:699-718.
Omura K., Hukuda S., Katsuura A., et al. Evaluation of posterior long fusion versus conservative treatment for the progressive rheumatoid cervical spine. Spine (Phila Pa 1976). 2002;27(12):1336-1345.
Scheuermann H. Kyphosis dorsalis juvenilis. Ugeskr Laeger. 1920;82:385-393.
Tsuyama N. Ossification of the posterior longitudinal ligament of the spine. Clin Orthop Relat Res. 1984;184:71-84.
Wiltse L.L., Widell E.H.Jr., Jackson D.W. Fatigue fracture: the basic lesion is isthmic spondylolisthesis. J Bone Joint Surg [Am]. 1975;57:17-22.
1. Frymoyer J.W., Moskowitz R.W. Spinal degeneration: pathogenesis and medical management. In: Frymoyer J.W., editor. The adult spine: principles and practice. Philadelphia: Lippincott-Raven; 1991:611-634.
2. Tsuji H., Hirano N., Ohshima H., et al. Structural variation of the anterior and the posterior anulus fibrosus in the development of the human lumbar disc. Spine (Phila Pa 1976). 1993;18:204-210.
3. Taylor J.R., Twomey L.T. The development of the human intervertebral disc. In: Ghosh P., editor. The biology of the intervertebral disc, vol 1. Boca Raton, FL: CRC Press; 1988:39-82.
4. Hukins D.W.L. Disc structure and function. In: Ghosh P., editor. The biology of the intervertebral disc, vol 1. Boca Raton, FL: CRC Press; 1988:1-37.
5. Brinckmann P., Grootenboer H. Change of disc height, radial disc bulge, and intradiscal pressure from discectomy: an in vitro investigation on human lumbar discs. Spine (Phila Pa 1976). 1991;16:641-646.
6. Maroudas A. Nutrition and metabolism of the intervertebral disc. In: Ghosh P., editor. The biology of the intervertebral disc, vol 2. Boca Raton, FL: CRC Press; 1988:1-37.
7. Taylor J.R., Twomey L.T. Human intervertebral disc acid glycosaminoglycans. J Anat. 1992;180:137-141.
8. Gower W.E., Pedrini V. Age-related variations in protein polysaccharides from human nucleus pulposus, annulus fibrosus, and costal cartilage. J Bone Joint Surg (Am). 1969;51:1154-1162.
9. McDevitt C.A. Proteoglycans of the intervertebral disc. In: Ghosh P., editor. The biology of the intervertebral disc, vol 1. Boca Raton, FL: CRC Press; 1988:151-170.
10. Yu S., Haughton V.M., Sether L.A., et al. Criteria for classifying normal and degenerated lumbar intervertebral disks. Radiology. 1989;170:523-526.
11. Osti O.L., Vernon-Roberts B., Fraser R.D. Annulus tears and intervertebral disc degeneration: an experimental study using an animal model. Spine (Phila Pa 1976). 1990;15:762-767.
12. Yasuma T., Koh S., Okamura T., Yamauchi Y. Histological changes in aging lumbar intervertebral discs: their role in protrusions and prolapses. J Bone Joint Surg [Am]. 1990;72:220-229.
13. Yu S., Haughton V.M., Sether L.A., et al. Anulus fibrosus in bulging intervertebral discs. Radiology. 1988;169:761-763.
14. Gordon S.J., Yang K.H., Mayer P.J., et al. Mechanism of disc rupture: a preliminary report. Spine (Phila Pa 1976). 1991;16:450-456.
15. Adams M.A., Hutton W.C. The relevance of torsion to the mechanical derangement of the lumbar spine. Spine (Phila Pa 1976). 1981;6:241-248.
16. Adams M.A., Hutton W.C. Prolapsed intervertebral disc. A hyperflexion injury. 1981 Volvo Award in Basic Science. Spine (Phila Pa 1976). 1982;7:184-191.
17. Adams M.A., Hutton W.C. Gradual disc prolapse. Spine (Phila Pa 1976). 1985;10:524-531.
18. Farfan H.F., Cossette J.W., Robertson G.H., et al. The effects of torsion on the lumbar intervertebral joints: the role of torsion in the production of disc degeneration. J Bone Joint Surg [Am]. 1970;52:468-497.
19. Masaryk T.J., Ross J.S., Modic M.T., et al. High-resolution MR imaging of sequestered lumbar intervertebral disks. Am J Roentgenol. 1988;150:1155-1162.
20. Weinstein J.N., Wiesel S.W. The lumbar spine: the international society for the study of the lumbar spine. Philadelphia: WB Saunders; 1990. pp 394–395
21. Hood R.S. Far lateral lumbar disc herniations. Neurosurg Clin North Am. 1993;4:117-124.
22. Kirkaldy-Willis W.H. Managing low back pain. New York: Churchill Livingstone; 1983.
23. Naylor A. Factors in the development of the spinal stenosis syndrome. J Bone Joint Surg (Br). 1979;61:306-309.
24. Pennal G.F., Schatzker J. Stenosis of the lumbar spinal canal. Clin Neurosurg. 1971;18:86-105.
25. Yamada H., Oya M., Okada T., et al. Intermittent cauda equina compression due to narrow spinal canal. J Neurosurg. 1972;37:83-88.
26. Penning L., Wilmink J.T. Posture-dependent bilateral compression of L4 or L5 nerve roots in facet hypertrophy. A dynamic CT-myelographic study. Spine (Phila Pa 1976). 1987;12:488-500.
27. Hayashi H., Okada K., Hamada M., et al. Etiologic factors of myelopathy. A radiographic evaluation of the aging changes in the cervical spine. Clin Orthop Relat Res. 1987;214:200-209.
28. Zvaifler N.J. Etiology and pathogenesis of rheumatoid arthritis. In: Hoopman W.J., McCarthy D.J., editors. Arthritis and allied conditions, vol 1. Philadelphia: Lea & Febiger; 1993:723-726.
29. Dirheimer Y. The craniovertebral region in chronic inflammatory rheumatic diseases. Berlin: Springer-Verlag; 1977.
30. Resnick D., Niwayama G. Diagnosis of bone and joint disorders. Philadelphia: WB Saunders; 1988. pp 1103–1170
31. Zvaifler N.J. Rheumatoid arthritis: epidemiology, etiology, rheumatoid factor, pathology, pathogenesis. In: Schumacher H.R., editor. Primer on pheumatic disease. Atlanta: Arthritis Foundation, 1988.
32. Martel W. Pathogenesis of cervical discovertebral destruction in rheumatoid arthritis. Arthritis Rheum. 1977;20:1217-1225.
33. Ball J. Enthesopathy of rheumatoid and ankylosing spondylitis. Ann Rheum Dis. 1971;30:213-223.
34. Ecklin U. Die Altersveranderungen der Halswirbelsaule. Berlin: Springer; 1960. pp 17, 28
35. Ansell B.M. The cervical spine in juvenile rheumatoid arthritis (International Congress Series, 61). In: Carter M.E., editor. Radiological Aspects of Rheumatoid Arthritis. Amsterdam: Excerpta Medica, 1964.
36. Halla J.T., Hardin J.G., Vitek J., Alarcon G.S. Involvement of the cervical spine in rheumatoid arthritis, review. Arthritis Rheum. 1989;32:652-659.
37. Zygmunt S., Saveland H., Brattstrom H., et al. Reduction of rheumatoid periodontoid pannus following posterior occipito-cervical fusion visualised by magnetic resonance imaging. Br J Neurosurg. 1988;2:315-320.
38. Katz J.N., Liang M.H. Differential diagnosis and conservative treatment of rheumatoid disorders. In: Frymoyer J.W., editor. The adult spine: principles and practice. Philadelphia: Lippincott-Raven; 1991:699-718.
39. Morizono Y., Sakou T., Kawaida H. Upper cervical involvement in rheumatoid arthritis. Spine (Phila Pa 1976). 1987;12:721-725.
40. Pellicci P.M., Ranawat C.S., Tsairis P., Bryan W.J. A prospective study of the progression of rheumatoid arthritis of the cervical spine. J Bone Joint Surg (Am). 1981;63:342-350.
41. Matsunaga S. Results of a longer than 10-year follow-up of patients with rheumatoid arthritis treated by occipitocervical fusion. Spine (Phila Pa 1976). 2000;25:1749-1753.
42. Komusi T., Munro T., Harth M. Radiologic review: the rheumatoid cervical spine, review. Semin Arthritis Rheum. 1985;14:187-195.
43. Bywaters E.G. Rheumatoid and other diseases of the cervical interspinous bursae, and changes in the spinous processes. Ann Rheum Dis. 1982;41:360-370.
44. Omura K., Hukuda S., Katsuura A., et al. Evaluation of posterior long fusion versus conservative treatment for the progressive rheumatoid cervical spine. Spine (Phila Pa 1976). 2002;27(12):1336-1345.
45. Bouchard-Chabot A., Liote F. Cervical spine involvement in rheumatoid arthritis: A review. Joint Bone Spine. 2002;69:141-154.
46. Shen F.H. Rheumatoid arthritis: evaluation and surgical management of the cervical spine. Spine J. 2004;4:689-700.
47. Wollin D.G., Botterell E.H. Symmetrical forward luxation of the atlas. Am J Roentgenol Radium Ther Nucl Med. 1958;79:575-583.
48. Scheuermann H. Kyphosis dorsalis juvenilis. Ugeskr Laeger. 1920;82:385-393.
49. Bradford D.S., Moe J.H., Montalvo F.J., et al. Scheuermann’s kyphosis and roundback deformity: results of Milwaukee brace treatment. J Bone Joint Surg (Am). 1974;56:749.
50. Sorenson K.H. Scheuermann’s Juvenile kyphosis. Clinical appearances, radiography, aetiology, and prognosis. Copenhagen: Munksgaard; 1964.
51. Bradford D.S. Juvenile kyphosis. Clin Orthop Relat Res. 1977;128:45-55.
52. Scoles P.V., Latimer B.M., DiGiovanni B.F., et al. Vertebral alterations in Scheuermann’s kyphosis. Spine (Phila Pa 1976). 1991;16:509-515.
53. Papagelopoulos P.J., Mavrogenis A.F., Swidou O.D. Current concepts in Scheuermann’s kyphosis. Orthopedics. 2008;31(1):55-58.
54. White A.A., Punjabi M.M., Thomas C.L. The clinical biomechanics of kyphotic deformities. Clin Orthop Relat Res. 1977;128:8-17.
55. Bick E.M., Copel J.W. The ring apophysis of the human vertebra. Contribution to human osteogeny II. J Bone Joint Surg (Am). 1951;33:783-787.
56. Schmorl G. Die Pathogenese der juvenilen Kyphose. Fortschr Geb Rontgen. 1930;41:359-383.
57. Bradford D.S., Brown D.M., Moe J.H., et al. Scheuermann’s kyphosis: a form of osteoporosis? Clin Orthop Relat Res. 1976;118:10-15.
58. Lopez R.A., Burke S.W., Levine D.B., et al. Osteoporosis in Scheuermann’s disease. Spine (Phila Pa 1976). 1988;13:1099-1103.
59. Gilsanz V., Gibbens D.T., Carlson M., et al. Vertebral bone density in Scheuermann disease. J Bone Joint Surg (Am). 1989;71:894-897.
60. Kemp F.H., Wilson D.C. Some factors in the aetiology of osteochondritis of the spine. A report on two families. Br J Radiol. 1947;20:410-417.
61. Ascani E., Borelli P., LaRosa G., et al. Malattia di Scheuermann. I: studio ormonale, progressi in patologia vertebrale. In: Le Cifosi vol 5. Bologna, Italy: Gaggi; 1982.
62. Halal F., Gledhill R.B., Fraser C. Dominant inheritance of Scheuermann’s juvenile kyphosis. Am J Dis Child. 1978;132:1105-1107.
63. Singh M., Riggs B.L., Beabout J.W., et al. Femoral trabecular pattern index for evaluation of spinal osteoporosis. A detailed methodologic description. Mayo Clin Proc. 1973;48:184-189.
64. Simon R.S. The diagnosis and treatment of kyphosis dorsalis juvenilis (Scheuermann’s kyphosis) in the early stage. J Bone Joint Surg. 1942;24:681-683.
65. Kewalramani L.S., Riggins R.S., Fowler W.M.Jr. Scheuermann’s kyphoscoliosis associated with Charcot-Marie-Tooth syndrome. Arch Phys Med Rehab. 1976;57:391-397.
66. Cloward R.B., Bucy P.C. Spinal extradural cyst and kyphosis dorsalis juvenilis. 1937. Surg Neurol. 1993;39(6):469-473.
67. Wise B.L., Fostey J.J. Congenital spinal extradural cyst. J Neurosurg. 1955;12:421-427.
68. Aufdermaur M. Juvenile kyphosis (Scheuermann’s disease): radiography, histology, and pathogenesis. Clin Orthop Rel Res. 1981;154:166-174.
69. Bradford D.S., Moe J.H. Scheuermann’s juvenile kyphosis. A histologic study. Clin Orthop Relat Res. 1975;110:45-53.
70. Travaglini F., Conte M. Progressi in patologia vertebrale. In: Le Cifosi, vol 5. Bologna, Italy: Gaggi; 1982.
71. Sturm P.F., Crawford-Dobson J., Armstrong G.W.D. The surgical treatment of Scheuermann’s disease. Spine (Phila Pa 1976). 1993;18:685-691.
72. Yablon J.S., Kasdon D.L., Levine H. Thoracic cord compression in Scheuermann’s disease. Spine (Phila Pa 1976). 1988;13:896-898.
73. Montgomery S.P., Erwin W.E. Scheuermann’s kyphosis: long-term results of Milwaukee brace treatment. Spine (Phila Pa 1976). 1981;6:5-8.
74. Lonner B.S. Operative management of Scheuermann’s kyphosis in 78 patients. Spine (Phila Pa 1976). 2006;32(24):2644-2652.
75. Hida K., Iwasaki Y., Koyanagi I., Abe H. Bone window computed tomography for detection of dural defect associated with cervical ossified posterior longitudinal ligament. Neurol Med Chir (Tokyo). 1997;37:173-176.
76. Singer F.R., Mills B.G. Primary bone cell dysfunction: I. Paget’s disease of the bone. In: Tam C.S., Heersche J.N.M., editors. Metabolic bone disease: cellular and tissue mechanisms. Boca Raton, FL: CRC Press; 1989:33-47.
77. Meunier P.J. Bone histomorphometry and skeletal distribution of Paget’s disease of bone. Semin Arthritis Rheum. 1994;23:219-221.
78. Douglas D.L., Duckworth T., Kanis J.A., et al. Spinal cord dysfunction in Paget’s disease of bone. Has medical treatment a vascular basis? J Bone Joint Surg (Br). 1981;63:495-503.
79. Dell’Atti C. The spine in Paget’s disease. Skeletal Radiol. 2007;36:609-626.
80. Hadjipavlou A.G. Paget’s Disease of the spine and its management. Eur Spine J. 2001;10(5):370-384.
81. Hadjipavlou A., Lander P. Paget disease of the spine. J Bone Joint Surg. 1991;73A:1376-1381.
82. Mawhinney R., Jones R., Worthington B.S. Spinal cord compression secondary to Paget’s disease of the axis. Br J Radiol. 1985;58:1203-1206.
83. Hartman J.T., Dohn D.F. Paget’s disease of the spine with cord or nerve-root compression. J Bone Joint Surg (Am). 1966;48:1079-1084.
84. Chen J.R., Rhee R.S., Wallach S., et al. Neurologic disturbances in Paget disease of bone: response to calcitonin. Neurology. 1979;29:448-457.
85. Gran J.T. The epidemiology of rheumatoid arthritis. Monogr Allergy. 1987;21:162-196.
86. Blumberg B., Ragan C. The natural history of rheumatoid spondylitis. Medicine. 1956;35:1-31.
87. Cardenosa G., Deluca S.A. Ankylosing spondylosis. Am Fam Physician. 1990;42:147-150.
88. Cooper R.R., Misol S. Tendon and ligament insertion. A light and electron microscopic study, review. J Bone Joint Surg (Am). 1970;52:1-20.
89. Davies D.V., Young L. The distribution of radioactive sulphur (35S) in the fibrous tissues, cartilages, and bones of the rat following its administration in the form of inorganic sulphate. J Anat. 1954;88(Pt 2):174-183.
90. Romanus R. Pelvo-spondylitis ossificans in the male. Stockholm, Aktiebolaget Godvil. 1953.
91. Romanus R., Yden S. Destructive and ossifying spondylitic changes in rheumatoid ankylosing spondylitis. Acta Orthop Scand. 1952;22:88-99.
92. Cruickshank B. Pathology of ankylosing spondylitis. Clin Orthop Relat Res. 1971;74:43-58.
93. Aufdermaur M. The morbid anatomy of ankylosing spondylitis. Doc Rheumatol Geigy No. 2. 1957.
94. Ball J. Articular pathology of ankylosing spondylitis. Clin Orthop Relat Res. 1979;143:30-37.
95. Baker W., Thomas T.G., Kirkaldy-Willis W.H. Changes in the cartilage of the posterior intervertebral joints after anterior fusion. J Bone Joint Surg (Br). 1969;51:736-746.
96. Hanson C.A., Shagrin J.W., Duncan H. Vertebral osteoporosis in ankylosing spondylitis. Clin Orthop Relat Res. 1971;74:59-64.
97. Westerveld L.A., Verlaan J.J. Spinal fractures in patients with ankylosing spinal disorders: a systematic review of the literature on treatment, neurological status and complications. Oner Eur Spine J. 2009;18:145-156.
98. Murray G.C., Persellin R.H. Cervical fracture complicating ankylosing spondylitis: a report of eight cases and review of the literature, review. Am J Med. 1981;70:1033-1041.
99. Hunter T., Dubo H. Spinal fractures complicating ankylosing spondylitis. Ann Intern Med. 1978;88:546-549.
100. Harding J.R., McCall I.W., Park W.M., et al. Fracture of the cervical spine in ankylosing spondylitis. Br J Radiol. 1985;58:3-7.
101. Kewalramani L.S., Taylor R.G., Albrand O.W. Cervical spine injury in patients with ankylosing spondylitis. J Trauma. 1975;15:931-934.
102. Hitchon P.W., From A.M., Brenton M.D., et al. Fractures of the thoracolumbar spine complicating ankylosing spondylitis. J Neurosurg (Spine 2). 2002;97:218-222.
103. Whang P.G., Goldberg G., Lawrence J.P., et al. The management of spinal injuries in patients with ankylosing spondylitis or diffuse idiopathic skeletal hyperostosis: A comparison of treatment methods and clinical outcomes. J Spinal Disord Tech. 2009;22:77-85.
104. Hunter T. The spinal complications of ankylosing spondylitis, review. Semin Arthritis Rheum. 1989;19:172-182.
105. Martel W. The occipito-atlanto-axial joints in rheumatoid arthritis and ankylosing spondylitis. Am J Roentgenol. 1961;86:223-240.
106. Weinstein P.R., Karpman R.R., Gall E.P., et al. Spinal cord injury, spinal fracture, and spinal stenosis in ankylosing spondylitis. J Neurosurg. 1982;57:609-616.
107. Key C.A. On paraplegia depending on disease of the ligaments of the spine. Guys Hosp Rep (Series 1). 1838;3:17-34.
108. Trojan D.A., Pouchot J., Pokrupa R., et al. Diagnosis and treatment of ossification of the posterior longitudinal ligament of the spine: report of eight cases and literature review. Am J Med. 1992;92:296-306.
109. Tsuyama N. Ossification of the posterior longitudinal ligament of the spine. Clin Orthop Relat Res. 1984;184:71-84.
110. Isawa K. Comparative roentgenographical study on the incidence of ossification of the posterior longitudinal ligament and other degenerative changes of the cervical spine among Japanese, Koreans, Americans, and Germans. Nippon Seikeigeka Gakkai Zasshi (Japan). 1980;54:461-474.
111. Firooznia H., Benjamin V.M., Pinto R.S., et al. Calcification and ossification of posterior longitudinal ligament of spine: its role in secondary narrowing of spinal canal and cord compression. NY State J Med. 1982;82:1193-1198.
112. McAfee P.C., Regan J.J., Bohlman H.H. Cervical cord compression from ossification of the posterior longitudinal ligament in non-orientals. J Bone Joint Surg (Br). 1987;69:569-575.
113. Albisinni U., Merlini L., Terayama K., et al. X-ray epidemiology of ligaments, ossifications, and disc degeneration of the cervical spine [Italian]. Chir Degli Organi Movimento. 1985;70:15-22.
114. Epstein N. Diagnosis and surgical management of cervical ossification of the posterior longitudinal ligament. Spine J. 2002;2:436-449.
115. Tanikawa E., Furuya K., Nakajima H. Genetic study on ossification of posterior longitudinal ligament. Bull Tokyo Med Dent Univ. 1986;33:117-128.
116. Resnick D., Guerra J., Robinson C.A., et al. Association of diffuse idiopathic skeletal hyperostosis (DISH) and calcification and ossification of the posterior longitudinal ligament. Am J Roentgenol. 1978;1319:1049-1053.
117. Yasui N., Ono K., Yamaura I., et al. Immunohistochemical localization of types I, II, and III collagens in the ossified posterior longitudinal ligament of the human cervical spine. Calcif Tissue Int. 1983;35:159-163.
118. Sato M., Turn M., Yada K. The antero-posterior diameter of the cervical spinal canal in the ossification of the posterior longitudinal ligament. No Shinkei Geka. 1977;5:511-517.
119. Harsh G.R., Sypert G.W., Weinstein P.R., et al. Cervical spine stenosis secondary to ossification of the posterior longitudinal ligament. J Neurosurg. 1987;67:349-357.
120. Min J.H., Jang J.S., Lee S.H. Significance of the double-layer and single-layer signs in the ossification of the posterior longitudinal ligament of the cervical spine. J Neurosurg Spine. 2007;6:309-312.
121. Troup J.D.G. The etiology of spondylolysis. Orthop Clin North Am. 1977;117:59-67.
122. Hutton W.C., Cyron B.M. Spondylolysis. The role of the posterior elements in resisting the intervertebral compressive force. Acta Orthop Scand. 1978;49:604-609.
123. Adams M.A., Hutton W.C. The effect of posture on the role of the apophyseal joints in resisting intervertebral compressive forces. J Bone Joint Surg (Br). 1980;62:358-362.
124. Farfan H.F., Osteria V., Lamy C. The mechanical etiology of spondylolysis and spondylolisthesis. Clin Orthop Relat Res. 1976;8:40-55.
125. Troup J.D. Mechanical factors in spondylolisthesis and spondylolysis. Clin Orthop Relat Res. 1976;117:59-67.
126. Wiltse L.L., Widell E.H.Jr., Jackson D.W. Fatigue fracture: the basic lesion is isthmic spondylolisthesis. J Bone Joint Surg (Am). 1975;57:17-22.
127. Wiltse L.L., Newman P.H., Macnab I. Classification of spondylolysis and spondylolisthesis. Clin Orthop Relat Res. 1976;117:23-29.
128. Ogilvie J.W., Sherman J. Spondylolysis in Scheuermann’s disease. Spine (Phila Pa 1976). 1987;12:251-253.
129. Farfan H.F. The pathological anatomy of degenerative spondylolisthesis. A cadaver study. Spine (Phila Pa 1976). 1980;5:412-418.
130. Valkenburg H.A., Haanen H.C.M. The epidemiology of low back pain. White A.A., Gordon S.L., editors. The proceedings of the American association of orthopedic surgery symposium on low back pain. 1982:9-22.
131. Wynne-Davies R., Scott J.H.S. Inheritance and spondylolisthesis: a radiographic family survey. J Bone Joint Surg (Br). 1979;61:301-305.
132. Stillerman C.B., Schneider J.H., Gruen J.P. Evaluation and management of spondylolysis and spondylolisthesis. In: Wilkins R.H., Rengachery S.S., editors. Clinical neurosurgery. Baltimore: Williams & Wilkins; 1992:384-415.
133. Macnab I. Backache. Baltimore: Williams & Wilkins; 1990. pp 84–103
134. Ganju A. Isthmic spondylolisthesis. Neurosurg Focus. 13(1), 2002. E1
135. Meyerding H.W. Spondylolisthesis. Surg Gynecol Obstet. 1932;54:371-377.
136. Grobler L.J., Wiltse L.L. Classification, non-operative, and operative treatment of spondylolisthesis. In: Frymoyer J.W., editor. The adult spine: principles and practice. Philadelphia: Lippincott-Raven; 1991:1655-1704.
137. Rowe G.G., Roche M.B. The etiology of separate neural arch. J Bone Joint Surg (Am). 1953;35:102-110.
138. Borkow S.E., Kleiger B. Spondylolisthesis in the newborn. A case report. Clin Orthop Relat Res. 1971;81:73-76.
139. Fredrickson B.E., Baker D., McHolick W.J., et al. The natural history of spondylolysis and spondylolisthesis. J Bone Joint Surg (Am). 1984;66:699-707.
140. Tailard W.F. Etiology of spondylolisthesis. Clin Orthop Relat Res. 1976;117:30-39.
141. Friberg S. Studies on spondylolisthesis. Acta Chir Orthop (suppl 60). 1939.
142. Rothman S.L.G., Glenn W.V. Spondylolysis and spondylolysthesis. In: Post J.D., editor. CT of the lumbar spine. Baltimore: Williams & Wilkins; 1984:591-615.
143. Kalchman L., Kim D.H., Li L., et al. Spondylolytsis and spondylolisthesis: prevalence and association with low back pain in the adult community-based population. Spine (Phila Pa 1976). 2009;34(2):199-205.
144. Newman P.H., Stone K.H. The etiology of spondylolisthesis with a special investigation. J Bone Joint Surg [Br]. 1963;45:39-59.
145. Rosenberg N.J. Degenerative spondylolisthesis. Predisposing factors. J Bone Joint Surg [Am]. 1975;57:467-474.
[/level-membership-for-neurosurgery-category][not-level-membership-for-neurosurgery-category]
Chapter 8 Anatomy and Pathophysiology of Acquired Spinal Disorders
Degenerative Disc Disease and Spondylosis
Degenerative disc disease (with its characteristic clinical syndromes of disc herniation, spondylosis, and radiculopathy) is associated with vascular, biochemical, and anatomic changes in the disc. There is a consistent anatomic pattern of disc degeneration in the spine, with most changes occurring in the midcervical, thoracolumbar, and lower lumbar regions. This pattern is thought to reflect the distribution of the mechanical stresses caused by spine movement and loading, as well as those due to erect posture.1
The intervertebral disc consists of three components: (1) the nucleus pulposus; (2) the annulus fibrosus, which surrounds the nucleus pulposus; and (3) the cartilaginous end plates, which attach these structures to the rostral and caudal vertebrae of the motion segment. The annulus is formed by a series of lamellae that have high collagen content and thereby provide significant resistance to tensile forces. The ventral annulus is usually wider and more organized than the dorsal annulus, with which discontinuous lamellae may be present.2 The nucleus pulposus, derived embyologically from the primitive notochord,3 has a much higher proteoglycan and water content than the annulus fibrosus. The hyaline cartilage end plates are similar in collagen type to the inner annulus fibrosus and the nucleus pulposus.4
The disc acts as a deformable, fluid-like material, whose tendency to bulge is resisted by the tensile stress in the annular lamellae and the end plates. Therefore, a substantial intradiscal surface strength is required to resist a high circumferential annular stress and thus prevent excessive disc deformation (bulging). When disruption of the nucleus pulposus and annulus fibrosus reduces intradiscal pressure, bulging occurs.5
The disc receives its nutrients through small vessels in the cartilage end plates and from the periphery of the annulus.6 With aging, however, the end plates calcify, and vessel loss occurs, until nearly the entire disc becomes avascular.7 The loss of vasculature promotes increased anaerobic metabolism, increasing lactic acid production and cellular necrosis. The water content of the annulus fibrosus decreases from 78% at birth to 70% by the fourth decade, and the nucleus pulposus water content decreases from 90% to less than 70% with maturation.8,9 With this change in vascularity, and with water loss in the region of the inner annulus and nucleus pulposus, there is a relative increase in fibrocytes and chondrocytes, which are more tolerant of a low-pH environment.3
Before the age of 2 years, the nucleus pulposus is translucent and anatomically different from the annulus fibrosus.10 By the second decade, the inner annulus and nucleus grow increasingly fibrous and lose both height and proteoglycans.9 In the third decade, nuclear fragmentation and fibrosis appear. Progressive myxomatous degeneration, swelling, and fissure formation occur in the annulus by the fourth decade.11,12 Eventually the nucleus pulposus may become disorganized, dehydrated, and fragmented with circumferential and radial tears.10,11,13 Grading systems for these patterns of disc degeneration, using plain radiographs or MRI studies, have been published.10,14
On plain radiographs, the degenerative disc changes range from grade I to grade IV. Grade I represents a normal disc. Grade II demonstrates sclerosis along with disc space narrowing or osteophyte formation. Grade III shows moderate sclerosis, and grade IV is associated with severe sclerosis with disc space narrowing or osteophyte formation.14 Yu et al.10 classified changes in the disc, with reference to the age of the subject and to the stage of degeneration, by comparing the anatomic characteristics with the appropriate MRI findings in cadaveric dissections (Fig. 8-1 and Table 8-1). The primitive notochord is present up to age 10. In the second decade of life, a distinct fibrous band forms in the nucleus and disc height diminishes. In the third decade fragmentation and fibrosis of the nucleus occurs. By the fourth decade there is swelling, separation, and myxomatous degeneration of the annular lamellae with fissure formation.12
| Type of Disc | Anatomic Characteristics | MRI Features |
|---|---|---|
| Immature | Nucleus pulposus and annulus fibrosus differentiated, primitive notochord may be present | High-signal intensity from nucleus and annulus |
| Transitional | Fibrous tissue in equator of annulus | High-signal intensity from nucleus and annulus, low-signal intensity in ventral and/or dorsal region of nucleus pulposus, corresponding to dense fibrous tissue |
| Adult | Annulus and nucleus not differentiated, annulus intact or marked by small concentric or transverse tears | Moderately high-signal intensity from nucleus and annulus, low-signal intensity from Sharpey fibers and fibrous tissue in midportion of disc |
| Early degenerated | Radial tear of annulus, diminishing amount and discoloration of fibrocartilage in nucleus | Diminishing signal intensity from nucleus pulposus, low signal from Sharpey fibers disrupted by region of higher-signal intensity at location of annular tear, slightly diminished disc height |
| Severely degenerated | Replacement of nuclear and annular fibrocartilage with amorphous fiber and cysts | Severely reduced disc height, low (fibrous tissue) or high (fluid) signal intensity from intervertebral disc |
From Yu S, Haughton VM, Sether LA, et al: Criteria for classifying normal and degenerated lumbar intervertebral disks. Radiology 170:523–526, 1989, with permission.
One of the most common disc-related clinical syndromes is a herniated disc with sciatica. With degeneration, fissure formation occurs in a radial distribution. It is likely that the biomechanical cause of disc herniation is related to a combination of complex movements involving compression, lateral flexion, and/or rotation.15–18 With flexion, the nucleus pulposus moves dorsally. The dorsal annulus has fewer and more disorganized lamellae and may be inherently weaker than the thicker ventral annulus. Degeneration of the annulus results in the development of peripheral, circumferential, and, subsequently, radial tears. With complex stresses applied to the dorsally migrating nucleus, herniation may occur along a radial tear.
Disc herniation can cause associated nerve-root impingement. The typical dorsolateral herniated disc affects the nerve root passing to the next lower foramen, but a more laterally herniated disc can affect the nerve root above. Masaryk et al.19 used MRI findings to classify the stages of disc herniation. A bulging disc has an MRI signal similar to the rest of the disc, but the bulge is beyond the adjacent vertebral margins. A prolapsed or protruding disc has nearly breached the outer annular fibers and is barely contained. The disc remains contiguous with the rest of the nucleus pulposus by a pedicle that has a high signal on T2-weighted MRI. The disc is extruded when it completely breaches the outer annular fibers and the posterior longitudinal ligament, but remains in continuity with the disc proper. If the fragment is no longer in continuity with the main part of the disc, it is termed a sequestered, or “free,” disc fragment. The International Society for the Study of the Lumbar Spine20 classified the disc as either contained or noncontained, with the latter group including extruded and sequestered discs. Free fragments may migrate in a rostral or caudal direction. It appears that far-lateral herniated discs are more likely to migrate in a rostral direction, thus affecting the nerve root above the disc space.21
Disc degeneration without herniation may also lead to changes affecting the biomechanical function and stability of both the intervertebral and articular facet joints. Although opinions differ regarding whether facet or disc degeneration is the initial event that causes spondylosis, the three-joint intervertebral-motion–segment concept emphasizes that disease in each component affects the others. This is to say that unilateral or bilateral facet disease or disc degeneration may lead to progressive changes in the other segmental units. Adjacent bone changes are associated with cartilaginous degeneration in these three joints. Spurs and osteophytes form at the site of peripheral annular attachment to the end plates. These osteophytes are thought to be formed in regions of excessive motion. Kirkaldy-Willis22 incorporated this concept into a theory regarding the natural history of spinal degeneration. He believed that facet and disc disease occurred with progressive reciprocal dysfunction. This resulted in ligamentous laxity around the facet joint and increased stresses that lead to internal disc disruption. This condition causes subluxation, disc resorption, and, finally, paradiscal osteophyte formation. Enlargement of the facets also occurs as a result of osteophyte formation. These changes may contribute to lumbar stenosis (Fig. 8-2) or a lateral recess syndrome.23–25
Patients with significant lumbar spinal canal narrowing resulting in stenosis complain primarily of pain, weakness, and leg numbness while walking. This pain can be relieved when the patient flexes the spine by sitting, by leaning forward while walking (shopping cart sign), or by leaning against counters. The symptomatic improvement associated with these maneuvers is related to an increase in lateral recess and spinal canal dimensions. Flexion results in stretching of the protruding ligamentum flavum and posterior longitudinal ligament, as well as reduction of overriding laminae and facets.26 This small amount of change in the circumferential spinal canal, lateral recess, and foraminal region alleviates the pressure on the nerve roots and subsequently relieves the symptoms. Returning to the erect posture leads to repeated compression and a further exacerbation of symptoms. During ambulation, some patients experience the onset of symptoms because of an increased metabolic demand in nerve roots that have become ischemic as a result of stenotic compression. Such “neurogenic claudication” is relieved when the subject sits. Often, bicycling (which is associated with flexion of the lumbar spine) is well tolerated.
Aging discs in the cervical spine cause characteristic spine alterations that may lead to cervical myelopathy or radicular pain and deficit. In young subjects, the cervical spine assumes a lordotic posture. This results in a greater ventral height of the annulus, compared with the dorsal annular height. With aging, however, intradiscal water loss and disc narrowing occur, thus leading to progressive spine straightening. In young patients, the range of intervertebral motion is greatest at C5-6 and C6-7. Narrowing and degeneration with osteophyte formation is most marked at these levels (Fig. 8-3). With these changes there is progressively less movement. In patients older than age 60, motion at C3-4 and C4-5 increases. Increased degenerative instability in older patients, therefore, is associated with translational subluxation, especially retrolisthesis at C3-4 and C4-5.27 In this scenario the spinal cord of the patient with cervical spondylotic myelopathy may not only be compressed by osteophytes, but may also suffer repeated injuries secondary to intervertebral hypermobility or instability. Dynamic flexion-extension radiographs are necessary to diagnose degenerative spondylolisthesis since static films in neutral position do not demonstrate subluxation, if present. A treatment protocol that does not take these factors into account may be associated with less than optimal success.
Rheumatoid Arthritis of the Spine
Rheumatoid arthritis (RA) affects both the spine and the peripheral joints. It has a prevalence of approximately 1%, with the greatest incidence in the fourth through sixth decades.28 RA is a disease of the synovial joints. The earliest change in the joints is synovitis, followed by an acute inflammatory response as a result of antibody-antigen complex formation. These processes activate the complement cascade and generate biologically active substances, ultimately resulting in complete destruction of the joint. This acute process is followed by a chronic granulomatous process, or pannus formation, which produces collagenase and other enzymes that destroy surrounding cartilage and bone.29 This may lead to instability because of ligamentous incompetence.28,30,31
Considerable controversy regarding the pathogenesis of cervical spine rheumatoid joint disease revolves around whether the initial site of involvement is (1) the apophyseal joint, with resultant facet destruction and progressive secondary instability of the intervertebral disc, or (2) inflammation in the uncovertebral joint, which leads to primary disc destruction with secondary degenerative involvement of the apophyseal joints. Martel32 examined 20 RA patients and found instability associated with apophyseal joint involvement. This leads to vertebral end plate destruction, disc space narrowing, and erosion. At autopsy, the discs showed evidence of necrosis and degeneration, with minimal inflammation. Martel proposed that apophyseal changes caused the instability with secondary disc destruction and end plate microfractures. The relative infrequency of cervical spine disease in juvenile-onset RA was explained by the early bony ankylosis of the apophyseal joints observed in these subjects.
Ball33 reviewed the pathology of 14 RA patients with no radiologic evidence of cervical disease and found that the earliest histologic lesions were in the uncovertebral joints. He suggested that the disc and adjacent bone are then secondarily involved with resultant inflammatory destruction and progressive instability. The fact that uncovertebral joints are not completely developed in the first two decades of life34 might also explain the infrequency with which cervical rheumatoid disease is seen in juvenile-onset RA.35
Cervical spine disease is observed in as many as 88% of patients with RA.36 The manifestations include C1-2 instability, occipitocervical (OC) instability (with or without vertical displacement of the dens), and subaxial cervical RA. C1-2 instability is the most common form of cervical rheumatoid involvement and may occur in up to 74% of the patients.37 The dens is surrounded by two synovial joints, one ventrally, between the atlas and dens, and another between the transverse ligament and the dens. With involvement of the synovial joints there is progressive inflammation, destruction, and subsequent transverse ligament laxity, with destruction of the osseous attachments of the ligamentous complex. This loss of ligamentous integrity allows C1 to move ventrally on C2. If there is further significant disruption and osteomalacia of the dens itself, then dorsal C1-2 subluxation can also occur.38 If the synovial apophyseal joints between C1-2 are involved as well, lateral rotation may also be evident in addition to subluxation at C1-2. OC instability results from involvement of the atlanto-occipital articulations. With significant articular facet destruction, there is progressive collapse of the occiput at C1 and vertical displacement of the residual dens (Figs. 8-4 and 8-5). This has also been termed atlantoaxial impaction, vertical subluxation, cranial settling, and basilar invagination.38 Vertical displacement of the dens occurs in 5% to 32% of RA patients.29,39,40 It is believed that vertical displacement of the dens represents a more advanced stage of systemic disease burden; indeed, one 10-year retrospective review of patients with RA cervical instability treated with OC fusion noted significantly worse long-term outcomes in the subset of patients with vertical displacement of the dens.41
In the subaxial region, the levels most commonly involved with rheumatoid synovitis are C2-3 and C3-4. Subluxation (Fig. 8-6) may occur in approximately 7% to 29% of the patients with RA.38 Subaxial region subluxation rates as high as 31% have been noted after rostral surgical fusion; however, there was no increased incidence of myelopathy or pain with fusion-adjacent subluxations.41 These “staircase” subluxations are thought to be caused by significant ligamentous laxity and facet degeneration.36,42 At any of the various sites of rheumatoid involvement, osseous erosion of adjacent bone, caused by osteoclastic resorption, occurs frequently.43
With the significant bony destruction, ligamentous laxity, and the potential for neural compression observed in the rheumatoid cervical spine, the primary emphasis of treatment is reduction of subluxation and fusion/fixation to prevent spinal cord injury. The optimal time to proceed with operative intervention is yet to be determined. Omura et al. stratified their RA population and found that the subset of patients with seropositive disease and systemic evidence of mutilating-type joint involvement are at the highest risk of deterioration of their known cervical lesion.44 Furthermore, retrospective review of RA patients with cervical disease found that best-medical management faired significantly worse when compared with surgical fusion with respect to both morbidity and mortality.44 When substratifying the patient population undergoing surgical fusion, patients operated on earlier in their course and with a better functional preoperative score had a more pronounced overall improvement than those undergoing late surgical management. There is strong evidence for early operative intervention for the stabilization of RA-associated cervical disease.44,45
Surgical fusion yields multiple benefits, including reduction of both pain and neurologic sequelae; retrospective analysis of long-construct dorsal fusion demonstrates significant recovery of these two characteristics, compared with best-medical management, with improvement of an average of one to two grades on the Ranawat scales for pain and neurologic symptoms (Boxes 8-1 and 8-2).41,44,45 These improvements were persistent, even in the setting of failed permanent postoperative reduction of deformity and imbalance.41 The chronic granulomatous pannus decreases in size with the elimination of abnormal movement after successful arthrodesis.46,47 There is no consensus regarding the optimal type of intervention, but one must keep in mind the inherent poor quality of RA bone, the laxity of ligaments, the insidious inflammatory nature of RA itself, and the destructive effects of the myriad pharmacologic interventions, especially with respect to treatment with corticosteroids.45
Scheurmann Disease (Juvenile Kyphosis)
Scheuermann48 first described the progressive dorsal kyphosis of adolescent children in 1920. The deformity is usually evident as a fixed thoracic kyphosis that does not correct with hyperextension, thereby differentiating it from a postural kyphosis. Compensatory hyperlordosis of the lumbar and cervical spine may also be present. A mild scoliosis is noted in 20% to 30% of patients.49 Sorenson50 described the characteristic feature of ventral wedging of 5 degrees or more in at least three adjacent vertebrae. Other characteristics include kyphosis of greater than 40 degrees, vertebral end plate irregularity, and disc space narrowing.51 The prevalence of the disease ranges from 0.4% to 8%.50 It occurs predominantly in males (91% in one series).52 Hereditary patterns of transmission have been identified, though genetic loci have yet to be determined.53
Basic biomechanical factors and forces may play a role in this disorder. The thoracic spine has a natural kyphosis determined primarily by the shape of the vertebrae; in the adolescent thoracic spine 20 to 40 degrees of kyphosis is normal. The dorsal elements, including the ligamentum flavum and the laminae, resist forward flexion of the spine in tension, whereas the ventral bony elements (vertebral bodies) and disc resist compression.54 However, the facet joint capsules in the thoracic region are mechanically “weaker” than those in the lumbar region, so that any factor that increases the torque of the spine can result in greater deformity. The more marked the initial angulation of the spine, the larger the load (subject’s weight), and the longer the duration of load application, the greater the likelihood of the progression of the deformity.
Scheuermann disease must be differentiated from juvenile postural kyphosis, which, as the name attests, is a kyphosis seen during flexion that will correct with improved posture and extension. The apex of the curve is smooth. The condition will improve with therapy that targets improved posture and core strengthening.53
The pathogenesis of the disease remains unclear. Scheuermann believed that aseptic necrosis of the ring apophyses caused interruption of growth, which resulted in ventral vertebral body wedging.55 Subsequent work has refuted this theory by demonstrating that the apophyses do not contribute to longitudinal growth. Such growth is now known to result from endochondral ossification of the end plates.55 Schmorl56 felt that damage to the end plate by herniated disc material was of importance. Schmorl nodes are, however, not limited to the kyphotic region of the spine and are common in otherwise normal patients. It has been postulated that osteoporosis is involved,57,58 but recent investigations have found no differences in the trabecular bone density between patients with Scheuermann disease and controls matched for age, gender, and race.52,59 Other factors such as inflammation,60 hormonal influences,61 genetic factors,62 altered calcium metabolism,63 hypovitaminosis,64 neuromuscular disorders,65 extradural cyst formation,66,67 defective collagen formation of the end plate,68 and a decrease in the collagen-proteoglycan ratio of the end plate61 have been implicated, but their roles in the development of the disease have not been substantiated.
There is a high association (>90%) between ventral osseous extensions from the anterior margin of the vertebral body and the diseased vertebrae, a feature that is absent in normal specimens.52 Histologic examination reveals disorganized endochondral ossification, which may be a result of abnormal stress. Traumatic features of vascular and fibrocartilage proliferation are evident in the ventral end plates in Scheuermann disease.52,68,69 The dorsal vertebral height in cases of Scheuermann disease is not significantly different from that of controls, implying that either the ventral and dorsal stresses are different or that the kyphotic changes occur after dorsal growth is completed (the normal pattern of ring apophysis closure starts dorsolaterally, then works ventrally).52 Possibly, the natural thoracic kyphosis, being exacerbated by a rounded back, results in the development of the abnormal kyphosis.
Back pain is uncommon in the growing child with Scheuermann disease. Low back pain has been reported to be common (up to 50%)51 in adults with progressive, untreated dorsal kyphotic deformities. In other studies pain was not a significant problem.51 Progression of deformity is documented in 80% of patients older than 25 years of age, but the extent of deformity and pain is generally not severe.70 The kyphosis most commonly progresses before skeletal maturity, but can occur in adulthood.71 Disc degeneration is also associated with the deformity. Development of neurologic complications is rare, but is due to thoracic disc hernation, dural tenting, extradural cysts, and vascular compromise.72
Examination of the patient with Scheuermann disease can reveal a hyperpigmented lesion at the apex of the thoracic curve—a result of friction injury from the abnormally protruded spinous process. Patients often have a forward-protruding head, flexion contractures of the shoulders and hips, as well as tight hamstrings.53
Treatment is often indicated to correct the deformity, prevent its progression, and alleviate pain. The extent of the kyphosis and the age of the patient are important criteria for intervention. The nonoperative forms of treatment, such as bracing (Milwaukee brace) or casting, are the first line of treatment for most cases in which the kyphotic deformity is less than 65 degrees. These cases have a high success rate in correcting the deformity, especially if treatment begins before closure of the iliac apophyses (i.e., skeletal maturation).73 Operative treatment with fusion is reserved for cases of progressive deformity, pain not responsive to an adequate trial of casting or bracing, degenerative changes in adults associated with the kyphosis, cardiopulmonary compromise, and for a deformity greater than 65 degrees.71
Dorsal long-construct instrumentation that extends rostrally and caudally well above the thoracic apex is often adequate for stabilization and correction of the deformity. In the event of extreme kyphotic deformity, both a ventral and dorsal surgical approach is necessary for a more definitive correction,53
[/not-level-membership-for-neurosurgery-category]
