39
Amyloidosis
Amyloidosis encompasses a wide range of disorders, several of which have cutaneous manifestations. The common thread is the formation of extracellular deposits that are composed of a β-pleated sheet by x-ray crystallography. In general, amyloid deposits exhibit a red color with Congo red stain and demonstrate birefringence (apple green color) under polarized light. Precursor proteins of amyloid vary from keratin to immunoglobulin (Ig) light chains to gelsolin. If necessary, tandem mass spectrometry can be performed to characterize the specific protein.
In dermatology, the initial distinction is between amyloidosis that is systemic versus skin-limited (Fig. 39.1). The former is less common than the latter.
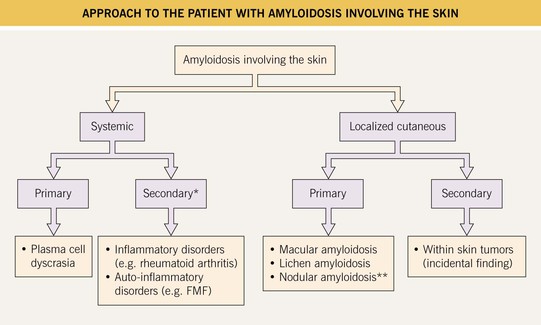
Fig. 39.1 Approach to the patient with amyloidosis involving the skin. *Cutaneous lesions are rare; **A minority of patients may develop systemic amyloidosis. FMF, familial Mediterranean fever.
Systemic Amyloidosis
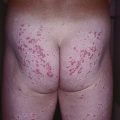
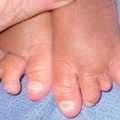
• Firm skin-colored to pink to yellow-brown waxy papules or plaques occur most commonly on the face (Fig. 39.2); infiltration of the tongue can lead to macroglossia (Fig. 39.3), and occasionally there is diffuse waxy induration of the skin (sclerodermoid presentation); purpura due to trauma or pinching of the skin can also be seen and is due to the fragility of blood vessels surrounded by amyloid deposits (Fig. 39.4).
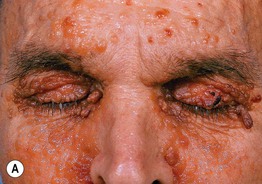
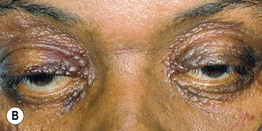
Fig. 39.2 Primary systemic amyloidosis. A Numerous waxy translucent facial papules. Some have a yellow to yellow-brown color. B Some of the waxy papules have become purpuric, but this may be difficult to appreciate because of the pigmentation of the skin; the periorbital region is a common site of involvement. A, Courtesy, Jean Bolognia, MD; B, Courtesy, Judit Stenn, MD.
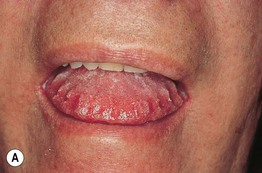
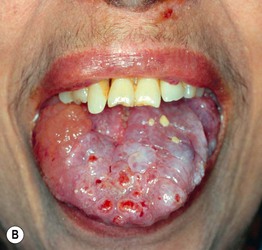
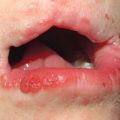
Fig. 39.3 Primary systemic amyloidosis. A Macroglossia with dental impressions on the tongue. B Papulonodues of the tongue and external nares; some are purpuric while others are translucent with a yellowish hue. The combination of macroglossia plus carpal tunnel syndrome is a classic clinical presentation. B, Courtesy, Dennis Cooper, MD.
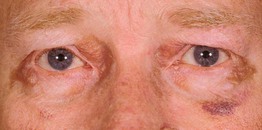
Fig. 39.4 Primary systemic amyloidosis. Purpura and yellow-brown plaques in a periorbital distribution. Courtesy, Joyce Rico, MD.
• Secondary systemic amyloidosis develops in the setting of chronic inflammation, e.g. rheumatoid arthritis, tuberculosis, and in several auto-inflammatory disorders (see Chapter 3); the precursor protein is (apo) serum AA, which is produced by the liver; cutaneous lesions rarely occur.
• Amyloid deposits can also lead to a restrictive cardiomyopathy and congestive heart failure as well as renal dysfunction, especially nephrotic syndrome (Fig. 39.5).
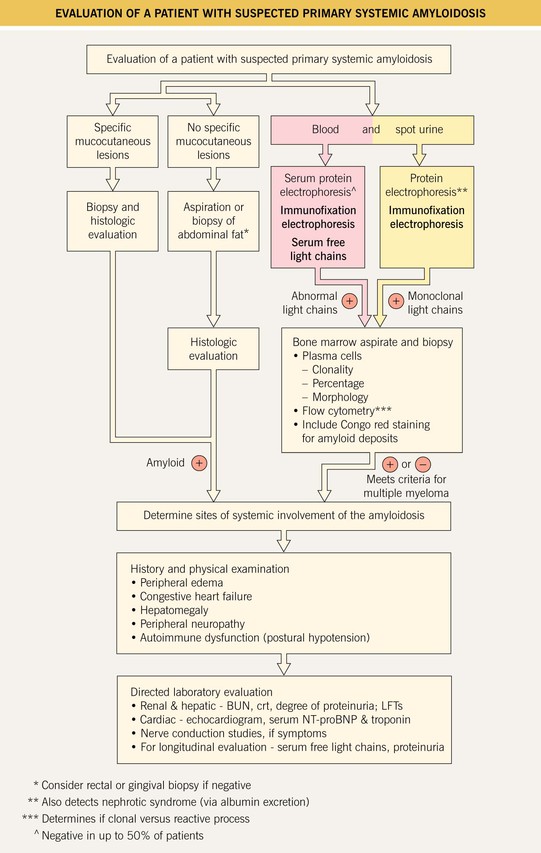
Fig. 39.5 Evaluation of a patient with suspected primary systemic amyloidosis. BUN, blood urea nitrogen; crt, creatinine; LFTs, liver function tests; NT-proBNP, N-terminal pro-brain natriuretic peptide.
• DDx: for facial papules, lipoid proteinosis, mucinoses, colloid milium, multiple adnexal tumors (see Fig. 91.15).
• Rx: for primary systemic amyloidosis, it is similar to myeloma, e.g. dexamethasone, melphalan, bortezomib, lenalidomide, and hematopoietic stem cell transplant (HSCT) (for younger patients).
Localized Cutaneous Amyloidosis
• The three major forms of primary localized cutaneous amyloidosis are macular, lichen, and nodular amyloidosis (see Fig. 39.1); an overlap of the first two forms is referred to as biphasic because these two entities exist on a spectrum.
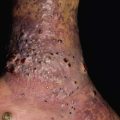
• Both macular and lichen amyloidosis are due to rubbing, scratching, or friction and have a rippled appearance as well as hyperpigmentation; the macular form favors the upper back of adults while the lichenoid form favors the extensor surfaces of the extremities, primarily the anterolateral shins, and has a palpable component (Figs. 39.6 and 39.7).
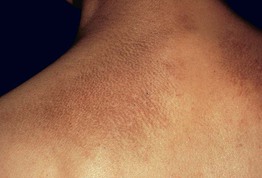
Fig. 39.6 Primary localized cutaneous amyloidosis. Characteristic rippled hyperpigmentation of macular amyloidosis (superiorly) as well as papules of lichenoid amyloidosis (inferiorly). This combination is referred to as biphasic amyloidosis. Courtesy, Richard W. Groves, MD, and Martin M. Black, MD.
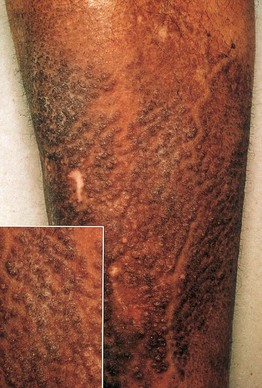
Fig. 39.7 Lichen amyloidosis. Keratotic hyperpigmented papules in a rippled pattern (see insert). The shin is the most common location for this form of primary localized cutaneous amyloidosis. Courtesy, St. John’s Institute of Dermatology.
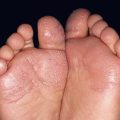
• Nodular amyloidosis presents as one or more waxy skin-colored to pink-orange plaques or nodules, most commonly on the trunk or extremities (Fig. 39.8); the precursor protein is Ig light chains ± β2-microglobulin, thought to be produced by cutaneous infiltrates of plasma cells.
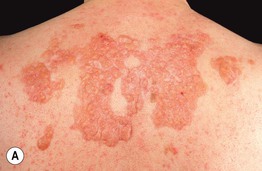
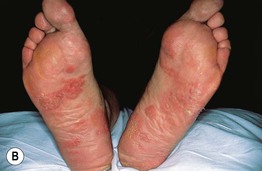
Fig. 39.8 Nodular amyloidosis. A, B Firm pink to pink-orange plaques on the back and plantar surface of the foot. A, Courtesy, Richard W. Groves, MD, and Martin M. Black, MD.
• DDx: back – notalgia paresthetica, lichen simplex chronicus (LSC) with post-inflammatory hyperpigmentation; lower extremity – LSC, hypertrophic lichen planus, pretibial myxedema; nodular – cutaneous lesions of systemic amyloidosis, pretibial myxedema, cutaneous lymphoma.
For further information see Ch. 47. From Dermatology, Third Edition.