[level-membership-for-neurology-category]
Chapter 32 Aminoacidemias and Organic Acidemias
Laboratory Approaches to Diagnosis
Laboratory testing for the symptomatic patient can proceed at many levels, including metabolic screening tests, quantitative metabolite profiles, specific enzyme or other functional assays, and DNA mutation analysis. The specific laboratory approach is often dictated by the clinical and family history and by results of routine laboratory investigations. An extensive description of diagnostic algorithms for the laboratory evaluation of patients suspected of having a metabolic disease has been published [Saudubray and Charpentier, 2001].
For the acutely ill patient, a comprehensive evaluation should include quantitative assessment of plasma amino acids, urine organic acid analyses, plasma carnitine (free and total) levels, and identification and quantitation of acylcarnitines in plasma or serum. These tests should be ordered in conjunction with other basic tests, including hematologic cell counts, electrolytes, blood glucose, blood gases, uric acid, liver transaminases, ammonia, and lactic and pyruvic acid levels. This approach can identify many cases of amino acid disorders and organic acidemias. The interpretation of metabolic tests is greatly enhanced when the laboratory is made aware of the clinical, medication, and dietary history of the patient because these factors can significantly influence results. Depending on the clinical evaluation and results of basic chemistry studies, additional testing may be warranted; this may include levels of urine orotic acid (e.g., elevated in certain urea cycle defects) (see Chapter 33), cerebrospinal fluid glycine (together with the plasma glycine level, which is elevated in glycine encephalopathy), cerebrospinal fluid neurotransmitters (see Chapter 39), and urine S-sulfocysteine (elevated in sulfite oxidase deficiency or molybdenum co-factor deficiency).
Inheritance and Genetic Counseling
Treatment of heritable metabolic disorders involves considerations beyond the acute phase of the illness and even beyond the prognosis of the proband. Because of the importance of genetic counseling to the family, the physician has an obligation to try to arrive at a diagnosis, however poor the prognosis for the proband. Identifying a specific entity enables the family to be counseled about recurrence risks. Most inborn errors of metabolism are inherited as autosomal-recessive traits. There are a few disorders, such as the urea cycle defect of ornithine transcarbamylase deficiency, that are inherited as X-linked disorders. In the case of an autosomal-recessive condition, the affected relative is a sibling of either gender. In X-linked disorders, the affected relative may be a maternal uncle, a brother, or a mildly affected mother or other female relative. Some disorders are caused by mitochondrial DNA mutations (see Chapter 37), and maternal transmission to all children in a sibship is observed. In all circumstances, a detailed family history may reveal an affected relative who has a similar illness, and this can be of diagnostic importance. Special attention should be given to a family history of stillbirths, unexplained deaths, and neurologic diseases or delayed development of any degree or severity.
Aminoacidemias
Phenylketonuria
Phenylketonuria, described by Asbørn Følling in 1934 [Følling, 1994], is caused by deficient activity of phenylalanine hydroxylase (PAH), a hepatic enzyme that converts phenylalanine to tyrosine (Figure 32-1). The biochemical block results in the accumulation of phenylalanine, which is then converted to phenylpyruvic acid and phenyllactic acid, phenylketones that are excreted in the urine. Tetrahydrobiopterin is a necessary co-factor in the PAH reaction, and elevated phenylalanine levels rarely may be caused by inherited disorders of tetrahydrobiopterin synthesis, including guanosine triphosphate (GTP) cyclohydrolase I, 6-pyruvoyltetrahydrobiopterin synthase, pterin-4α-carbinolamine dehydratase, and dihydropteridine reductase deficiencies (see Figure 32-1). Phenylalanine is neurotoxic, and untreated patients with classic phenylketonuria are typically mentally retarded. In the 1950s, a diet in which phenylalanine intake was restricted was shown to normalize plasma phenylalanine levels and stop urinary excretion of phenylpyruvic acid [Bickel et al., 1953]. Selective restriction of phenylalanine intake by using phenylalanine-free medical formulas and foods (and tyrosine supplementation), which provides enough additional protein and nutrients to support normal growth, remains the mainstay of phenylketonuria therapy.
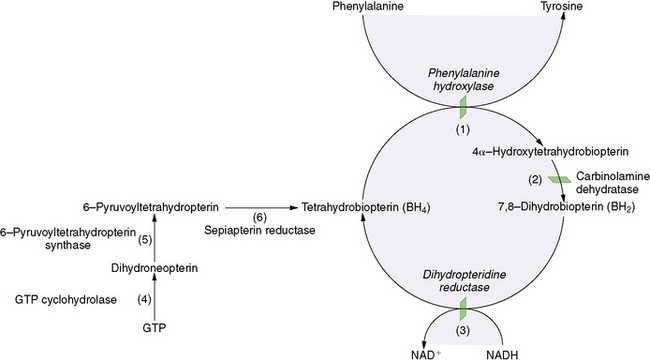
Fig. 32-1 Regulation of phenylalanine hydroxylase activity.
(From Wilcox WR, Cederbaum SD. Amino acid metabolism. In: Rimoin D, Connor J, Pyeritz R, Korf B, eds. Principles and practice of medical genetics, 4th edn. Philadelphia: Churchill Livingstone, 2002:2406.)
Mandatory population newborn screening for phenylketonuria, in combination with postnatal presymptomatic therapy, was begun in the 1960s using the Guthrie bacterial inhibition assay [Guthrie and Susi, 1963; Koch, 1997]. Modern newborn screening programs have switched to techniques that directly assay phenylalanine and tyrosine levels; the most recent innovation is tandem mass spectrometry. The presymptomatic institution of and continued adherence to specific dietary therapy prevents mental retardation. Phenylketonuria is a paradigmatic and landmark success story in biochemical genetics, and it is reviewed in some detail.
Classification
A blood phenylalanine level above the normal range (30–110 μM) is referred to as hyperphenylalaninemia. Patients have been classified as having nonphenylketonuria hyperphenylalaninemia if their blood phenylalanine levels without dietary therapy are 360–600 μM. Classic phenylketonuria is characterized by untreated phenylalanine levels of more than 1000 μM [Scriver and Kaufman, 2001]. A range of reduced PAH-specific activity correlates broadly with the severity of the phenotype. When it has been measured directly (i.e., liver biopsy) or indirectly (i.e., l-[1-13C]phenylalanine breath test), residual liver PAH-specific activity is relatively high in milder hyperphenylalaninemic patients, whereas enzyme activity is zero to low in the more severe cases of classic phenylketonuria [Bartholome et al., 1975]. Measured PAH activity also correlates to some degree with tolerance for dietary protein [Güttler et al., 1996]. Patients with classic phenylketonuria can tolerate very little phenylalanine in the diet (<500 mg/day). The classification of phenylketonuria into subtypes based on blood phenylalanine levels is arbitrary, and some patients (i.e., mild, variant, or atypical phenylketonuria) fall between the two extreme biochemical phenotypes. Because environmental (phenylalanine intake) and genetic (modifier genes) factors alter the biochemical and neurodevelopmental phenotypes in this single-gene disorder, phenylketonuria in many ways behaves more like a complex trait than a monogenic disorder [Kayaalp et al., 1997].
Clinical Manifestations
Profound mental retardation is the most significant clinical finding in untreated or poorly treated phenylketonuria. Acute metabolic encephalopathy, a common feature of many inborn errors of metabolism, does not occur in phenylketonuria. Children with phenylketonuria appear normal at birth and have normal early development, even if untreated. Neurologic manifestations appear insidiously and include reduced rate of growth of head circumference, developmental delay, abnormalities in muscle tone, and hyperactive deep tendon reflexes. Affected children may have lighter pigmentation than other family members (i.e., reduced melanin synthesis) and a musty odor attributed to phenylacetic acid. Eczema and decreased bone mineral density may occur [Zeman et al., 1999]. Patients exposed to chronically elevated phenylalanine levels ultimately develop microcephaly, seizures (e.g., tonic-clonic, myoclonic, infantile spasms), tremors, athetosis, and spasticity, and they may be misdiagnosed as having cerebral palsy. Psychiatric and behavior problems, including autistic behavior and attention-deficit hyperactivity disorder, are common [Pietz et al., 1997; Smith and Knowles, 2000].
Brain magnetic resonance imaging (MRI) may detect dysmyelination, especially T2 enhancement in the periventricular white matter, a finding that is potentially reversible with the initiation of dietary therapy [Cleary et al., 1995]. Abnormal areas of white matter demonstrate restricted diffusion of water, possibly indicating increased myelin turnover [Phillips et al., 2001].
In the past, most untreated phenylketonuria patients were institutionalized, and many born before universal newborn screening remain so. An eloquent and inspiring description of the life of a child with untreated phenylketonuria is given in a short monograph by Pearl S. Buck [Buck, 1950].
As a rule, well-treated classic phenylketonuria patients have normal IQs. However, recent studies have found that children and adults with phenylketonuria may experience cognitive symptoms, such as problems in executive functioning, as well as disturbance in emotional and behavioral functioning despite early and continuous treatment [Enns et al., 2010]. Dietary control of the blood phenylalanine level appears to be the best predictor of ultimate IQ [Waisbren et al., 1987], but careful psychometric testing of well-treated individuals has detected instances and degrees of impairment in visual-motor skills, abstract reasoning, problem solving, specific aspects of executive control, attention, verbal memory, expressive naming, and verbal fluency [Fishler et al., 1987]. Such neuropsychologic impairments may be a consequence of mid-dorsolateral prefrontal cortex dysfunction caused by abnormal catecholamine levels [Huijbregts et al., 2002]. Abnormal EEG patterns, including general slowing and generalized paroxysmal activity with or without spikes, may be demonstrated for children with phenylketonuria, even if they are well treated [Pietz et al., 1988]. Emotional disorders (e.g., depression, anxiety, phobias) and hyperactive behavior are more frequently encountered in persons with classic phenylketonuria than in the general population [Smith and Knowles, 2000]. However, untreated mild hyperphenylalaninemic patients are not at risk for developing neuropsychologic impairment [Weglage et al., 1996].
Maternal Phenylketonuria Syndrome
Elevated maternal blood phenylalanine levels can cross the placenta and cause fetal birth defects, including microcephaly, dysmorphic features, and congenital heart defects. Children with the maternal phenylketonuria syndrome are typically heterozygous for the mutant PAH allele, and they are not affected with phenylketonuria. More than 90 percent of children born to women with untreated classic phenylketonuria have mental retardation; 70 percent have microcephaly, 40 percent have intrauterine growth retardation, and 12 percent have congenital heart disease [Lenke and Levy, 1980]. The risk to the fetus is greatest with increasing phenylalanine levels in maternal blood. Dietary control should ideally be achieved before 3 months prior to conception, and mothers with phenylketonuria should be monitored carefully by an experienced center throughout pregnancy [ACOG, 2009]. Optimal birth outcomes occur when blood phenylalanine levels between 120 and 360 μM are achieved by 8–10 weeks’ gestation [Widaman and Azen, 2003]. Mothers with phenylketonuria can safely breastfeed their children.
Diagnosis
The Guthrie bacterial inhibition assay was a technical breakthrough, allowing newborn screening of large populations. The growth inhibition of Bacillus subtilis by β-2-thienylalanine is prevented by phenylalanine, phenylpyruvic acid, and phenyllactic acid, and this forms the basis of the Guthrie test. Fluorometric assays or tandem mass spectrometry is used in screening and monitoring [Chace et al., 1993]. False-positive results may be seen in neonates with low birth weight or liver disease, or in infants on parenteral alimentation. False-negative results may occur if the newborn screen is performed too early (especially less than 12 hours after birth) [Hanley et al., 1997]. Confirmation of the diagnosis is made by analysis of blood phenylalanine and tyrosine concentrations by means of high-performance liquid chromatography, fluorescent methods, or tandem mass spectrometry. Without the introduction of a phenylalanine-restricted diet, maximal elevation in plasma phenylalanine is typically reached within or soon after the first week of life in patients with classic phenylketonuria. Urine phenylpyruvic acid causes the appearance of a deep green color when ferric chloride is added. This ferric chloride test is sometimes performed as part of a metabolic screening panel for the evaluation of patients suspected of having an inborn error of metabolism, but it should not be used to confirm a diagnosis of phenylketonuria because of a lack of sensitivity and specificity. All patients with confirmed hyperphenylalaninemia must have urine pterins analyzed for defects in tetrahydrobiopterin metabolism. Phenylketonuria may be suspected in a child or adult and should reasonably be included in the differential diagnosis of a given patient of any age presenting with neurodevelopmental delay of unknown origin. In such settings, diagnostic testing for phenylketonuria (i.e., serum phenylalanine levels) must be done, even if there is a history or a record of a normal newborn screen.
Genetics
Phenylketonuria is an autosomal-recessive disorder with an incidence of 1 case per 10,000 people in the general white population of northern European ancestry [Eisensmith and Woo, 1994]. Phenylketonuria is more common in Turkey, Scotland, and Czechoslovakia, and among Arabic populations and Yemenite Jews (1 case per 2500–5000 persons). It is relatively uncommon in Japan and Finland, and among African Americans (1 case per 100,000–200,000 persons) [Scriver and Kaufman, 2001]. Nonphenylketonuria hyperphenylalaninemia has an overall incidence of 1 case per 50,000 persons.
The PAH gene on chromosome 12q24.1 spans 90 kb and contains 13 exons [Kwok et al., 1985]. Almost 500 mutations have been reported throughout all exons and flanking sequences. A detailed account of PAH mutant alleles and other DNA variations is maintained at the PAH Mutation Analysis Consortium website (http://www.pahdb.mcgill.ca/) [Waters and Scriver, 1998]. Most DNA alterations are missense mutations, although splice, nonsense, and frameshift mutations and large deletions and insertions have been identified. Most patients are compound heterozygotes, carrying a different mutant allele on each chromosome. Prevalences of specific mutant alleles differ from population to population [Eisensmith and Woo, 1994]. For example, R408W, IVS12nt1, and IVS10nt11 are severe mutations that account for about 50 percent of mutant alleles in Europeans, but they are rare in Asians. Conversely, mutant alleles R243Q, R413P, and Y204C are common in Asians, but they rare in Europeans [Eisensmith and Woo, 1994].
Pathogenesis
Although PAH is a hepatic enzyme, the major effect of its deficiency is brain dysfunction. Elevated phenylalanine appears to be the cause of neurotoxicity. However, the precise cause of the mental retardation observed in untreated phenylketonuria is not understood. Defective brain myelination may be related to decreased biosynthesis of myelin proteins, because brain protein synthesis is inhibited by excessive phenylalanine [Huether et al., 1982]. CNS effects may be ascribable to more global amino acid imbalances; elevated phenylalanine may affect the CNS concentrations of neutral amino acids by competitive inhibition of a shared amino acid transporter, with relative brain deprivation of tyrosine, tryptophan, and branched-chain amino acids [Huttenlocher, 2000]. Decreased brain tyrosine and tryptophan may lead to decreased neurotransmitter synthesis. The cause of abnormal brain myelination is also unclear. In a phenylketonuria mouse model (i.e., enu2 mouse), there is evidence that oligodendrocytes overexpress glial fibrillary acid protein and become nonmyelinating [Dyer et al., 1996]. Increased myelin turnover has also been observed in the enu2 mouse [Hommes and Moss, 1992]. In phenylketonuria patients studied by positron emission tomography (PET), brain protein synthesis appears to be impaired, which could also affect the myelination process [Paans et al., 1996]. Brain pathology in untreated classic phenylketonuria includes abnormalities in width of the cortical plate, cell density and organization, dendritic arborization and number of synaptic spines, and abnormal myelination [Bauman and Kemper, 1982].
Genotype-Phenotype Correlations
Mutations in the PAH gene cause phenylketonuria and nonphenylketonuria hyperphenylalaninemia. However, the final biochemical phenotype (i.e., blood phenylalanine level and dietary phenylalanine tolerance) and clinical phenotype (i.e., IQ) depend on the severity of the mutations, and are influenced by patient adherence to a strict diet, the effects of modifying genetic factors, and other environmental factors. Potential modifier genes may encode proteins mediating interindividual rates of protein synthesis (and phenylalanine use) or basal metabolism; synthesis and degradation of the PAH enzyme protein; gastrointestinal absorption of phenylalanine; hepatic uptake of circulating phenylalanine; metabolism of the tetrahydrobiopterin co-factor; and rate of phenylalanine transport across the blood–brain barrier [Dipple and McCabe, 2000; Treacy et al., 1997]. Because patients – including sibling patients – with identical mutations can have divergent neurodevelopmental progress, mutation identification may not predict the severity of the disease with certainty in a given patient [DiSilvestre et al., 1991; Enns et al., 1999b].
Genes encoding proteins responsible for transport of amino acids across the blood–brain barrier are especially attractive candidates for modifying factors in phenylketonuria. In a study of two siblings with identical genotypes but widely different IQ, in vivo nuclear MR spectroscopy documented lower peak brain phenylalanine levels and more rapid decreases in brain phenylalanine concentration in the less severely affected sibling after a phenylalanine load [Weglage et al., 1998]. Subsequent studies have confirmed the wide interindividual variation of brain to blood phenylalanine concentrations in classic phenylketonuria patients with divergent cognitive phenotypes [Moller et al., 2003].
Despite these considerations, trends can be identified in whole populations. In general, individuals with classic phenylketonuria and poor dietary control have mental retardation, although exceptions exist. If patients with severe mutations are started on strict dietary therapy in the neonatal period and maintained on such treatment throughout life, cognition will be normal. Using in vitro expression analysis in cultured cells transfected with mutant cDNAs, specific mutant alleles (i.e., genotype) can be categorized as severe or mild; such categorization correlates with biochemical or clinical severity (i.e., phenotype) in most patients in relatively homogenous populations. In a study of German and Dutch subjects, the predicted level of PAH activity correlated strongly with neonatal pretreatment levels of blood phenylalanine and dietary phenylalanine tolerance in both populations [Okano et al., 1991]. In relatively homogeneous German, Swedish, and southeastern U.S. populations, similar genotype-phenotype correlations were observed [Eisensmith et al., 1996; Kayaalp et al., 1997; Trefz et al., 1993]. However, when populations with high ethnic diversity are studied in this way, a clear genotype-phenotype correlation may not be apparent [Enns et al., 1999b; Treacy et al., 1997], and the genotype-phenotype correlation is not straightforward in some patients. Although phenylketonuria is a single-gene mendelian disorder, the observed clinical spectrum is more in keeping with a complex multifactorial trait [Scriver and Waters, 1999].
Genetic Counseling
Patients who are diagnosed in the neonatal period and who adhere to the phenylalanine-restricted diet have normal intelligence. However, learning problems can occur in well-treated patients and include problems in basic spelling, reading, and mathematical calculation skills. Patients may also be more prone to depression, anxiety, phobic tendencies, and isolation from their peers [Smith and Knowles, 2000; Welsh et al., 1990]. Such potential adverse and unpredictable manifestations should be brought to the attention of parents and carefully explained, with care and support, during the on-going genetic counseling process.
Treatment
Significant restriction of dietary phenylalanine is required for treatment, but the exact level of daily phenylalanine intake varies from patient to patient, and varies with age in an individual patient. Because phenylalanine is an essential amino acid, detrimental effects on growth and development may occur if restriction of phenylalanine intake is too severe and the blood level drops to below normal. Although there is no worldwide consensus about optimal plasma phenylalanine levels, most clinics in the United States strive to maintain levels between 120 and 360 μM in children younger than 12 years and between 120 and 600 μM in individuals older than 12 years [Phenylketonuria, 2000]. Phenylalanine levels in unaffected individuals are usually below 120 μM. In general, phenylketonuria patients who harbor severe mutations require a greater limitation of phenylalanine intake to maintain acceptable blood phenylalanine levels. However, individual variations of phenylalanine tolerance may occur, even in patients with identical genotypes. Blood phenylalanine levels therefore are monitored frequently, especially in the first year of life, and the diet is adjusted with care for each individual patient. The regimen must be initiated and overseen by experts in phenylketonuria at a specialized center, and referral of the patient to such a specialized center is mandatory. An expert, coordinated team approach is clearly the most effective way of managing phenylketonuria; stricter management improves developmental outcome [Camfield et al., 2004].
In earlier therapeutic protocols, phenylalanine restriction was continued only through the first few years of life, theoretically corresponding to the age at which brain myelination is complete. As developmental data accumulated, it became evident that treatment throughout childhood and adolescence was the best course to preserve IQ [Smith et al., 1991]. In later studies, it has been found that characteristic periventricular T2 white matter signal abnormalities on conventional MRI, restricted white matter diffusion in diffusion-weighted imaging, and electrophysiologic testing abnormalities referable to the CNS are observed in adults who are on unrestricted phenylalanine intake or poorly compliant with dietary therapy [Phillips et al., 2001]. There is evidence that MRI changes in cases of phenylketonuria are at least partially reversible if patients return to a low-phenylalanine diet [Cleary et al., 1995]. Accordingly, it is reasonable to continue therapy into adulthood, and most centers recommend lifelong treatment. Reassessment of adult phenylalanine tolerance may be necessary as body mass changes with age [MacLeod et al., 2009].
A variety of medical food products is available as special formulas for the treatment of phenylketonuria. These special formulas are low in phenylalanine or do not contain any phenylalanine, and they typically contain supplemental tyrosine and a balanced mixture of the additional amino acids, carbohydrates, essential fatty acids, vitamins, and minerals, including zinc, selenium, and molybdenum. The metabolic medical foods provide a variable amount of calories (up to 70 percent of daily requirement) in the form of starch (e.g., dextrose, cornstarch, dextromaltose, Polycose) and fat (i.e., corn or other oils), and they constitute a major source of nutrition for the lifetime of the patient. The special formula or medical food, containing no phenylalanine, is continued even after solid food is introduced. The special formula or medical food is ingested together with regular food during the same meal, providing the phenylalanine in food plus the amino acids, vitamins, and nutrients in the special formula in a complementary, beneficial manner. The medical and regular foods therefore should be given in a calculated proportion together in intervals throughout the day. Overall, the targeted total amino acid intake for children younger than 2 years is approximately 3 g/kg/day, and it is about 2 g/kg/day for older children [Cockburn and Clark, 1996]. If the medical food is ingested in a single sitting, the supply of amino acids may induce hyperinsulinism and hypoglycemia.
Medical food products continue to be modified to increase palatability and optimize the treatment of phenylketonuria. Amino acid powders and gels with added carbohydrate and with or without fats, vitamins, and minerals are examples of commonly used protein substitutes. Newer protein substitutes include amino acid tablets and capsules, which do not contain carbohydrate, vitamins, or minerals. Special amino acid bars and a protein that is almost phenylalanine-free (glycomacropeptide) are also available [van Spronsen and Enns, 2010]. Phenylketonuria dietary research has focused on making the medical food products more palatable. Although low-phenylalanine flour, pastas, cookies, and nutrition bars are available, the phenylketonuria diet remains very bland, and poor dietary compliance can be a major problem, especially after childhood. Early efforts to make a more palatable amino acid mixture have met with preliminary success, with some patients preferring the new products to traditional medical foods.
It is important to monitor complete blood cell counts and serum vitamin B12 levels periodically, because clinical and subclinical B12 deficiency has been reported in adolescents and adults with classic phenylketonuria, even in those poorly compliant with the restricted diet [Hanley et al., 1996].
In contrast to the strict dietary control required in the treatment of classic phenylketonuria, patients with nonphenylketonuria hyperphenylalaninemia (i.e., untreated blood phenylalanine levels of 360–600 μM) are not necessarily placed on the special diet as long as their phenylalanine levels are in treatment range; many of these patients are able to maintain acceptable blood phenylalanine levels with protein restriction alone. These patients have normal intelligence, and they do not have the psychologic findings or head MRI changes that have been documented in classic phenylketonuria [Weglage et al., 1996]. Dietary therapy may be recommended in some instances for pregnant women with nonphenylketonuria hyperphenylalaninemia to minimize the risk of maternal phenylketonuria syndrome.
Additional and Novel Therapies
A complementary therapeutic approach has received U.S. Food and Drug Administration approval: administration of dietary supplementation of large, neutral amino acids. Large, neutral amino acids compete with phenylalanine for transport across the blood–brain barrier by the L-type amino acid carrier and consequently decrease the level of phenylalanine in the CNS [Matalon et al., 2003; van Spronsen and Enns, 2010]. In a study of six adult subjects with classic phenylketonuria, large, neutral amino acid supplementation resulted in increased blood concentrations of tyrosine and tryptophan (the respective precursors for dopamine and serotonin) and decreased brain phenylalanine concentration, as measured by 1H-MR spectroscopy, toward the carrier range. All patients reported improvements in well-being and energy levels [Koch et al., 2003].
Deficiencies of carnitine and long-chain polyunsaturated fatty acids (i.e., arachidonic and docosahexaenoic acids) may contribute to CNS toxicity in uncontrolled phenylketonuria. Dietary supplementation of these fatty acids and of carnitine may benefit phenylketonuria patients who have low plasma levels of these essential metabolites [Infante and Huszagh, 2001]. Supplementation with omega-3, long-chain, polyunsaturated fatty acids resulted in improvement in visual-evoked potential latencies in 36 children with early-treated phenylketonuria [Beblo et al., 2001].
A novel therapeutic approach uses the nonmammalian enzyme phenylalanine lyase [van Spronsen and Enns, 2010]. This enzyme converts phenylalanine to trans-cinnamic acid, a harmless compound, and it has been found to reduce hyperphenylalaninemia in phenylketonuria rat and mouse models [Bourget and Chang, 1986; Sarkissian et al., 1999]. Enteral phenylalanine lyase therapy has the theoretic potential to increase dietary phenylalanine tolerance substantially, but significant practical hurdles need to be overcome; phenylalanine lyase is destroyed by gastric acidic pH and intestinal proteolysis. Alternative approaches being considered include the use of polyethylene glycol derivatization to produce protected forms of PAH for potential enzyme replacement therapy [Gamez et al., 2004; Kang et al., 2010].
Oral administration of tetrahydrobiopterin, the naturally occurring co-factor for the PAH reaction, reduces serum phenylalanine concentrations, especially in patients with mild hyperphenylalaninemia [Muntau et al., 2002]. However, response to tetrahydrobiopterin has also been documented in patients with classic or variant phenylketonuria [Matalon et al., 2004; van Spronsen and Enns, 2010]. These patients have mutations in the PAH gene, not in one of the genes encoding enzymes involved in tetrahydrobiopterin biosynthesis (see the section on biopterin disorders below). It has been suggested that the PAH mutations in such patients affect the structure of domains that are involved in the binding of tetrahydrobiopterin to the PAH enzyme. Tetrahydrobiopterin also may act as a chemical chaperone, preventing the PAH enzyme from misfolding or protecting PAH from inactivation [Pey et al., 2004]. If these observations and hypotheses are borne out, it may be possible to define by mutation analysis a subset of patients who would predictably benefit from co-factor supplementation. Tetrahydrobiopterin may prove useful in the treatment of maternal phenylketonuria [Trefz and Blau, 2003]. These, and other, novel therapies are under close investigation, especially given recent findings of suboptimal outcomes in phenylketonuria patients who have been continuously treated from the neonatal period [Enns et al., 2010].
Biopterin Disorders
Neonatal hyperphenylalaninemia may rarely be caused by defects in the synthesis or recycling of tetrahydrobiopterin, an essential co-factor in the PAH reaction (see Figure 32-1). Worldwide, it has been estimated that approximately 2 percent of patients with hyperphenylalaninemia have a defect in one of the four enzymes responsible for maintaining tetrahydrobiopterin levels [Blau et al., 1996]. Guanosine triphosphate cyclohydrolase (GTPCH) I and 6-pyruvoyltetrahydro-biopterin synthase (PTPS) are essential enzymes for tetrahydrobiopterin biosynthesis, whereas pterin-4α-carbinolamine dehydratase (PCD) and dihydropteridine reductase (DHPR) are responsible for tetrahydrobiopterin recycling [Blau et al., 2001]. All forms of tetrahydrobiopterin disorders that cause hyperphenylalaninemia are inherited as autosomal-recessive traits. An autosomal-dominant form of GTPCH deficiency (e.g., dopa-responsive dystonia, Segawa’s disease, hereditary progressive dystonia) manifests with dystonia, but it is not associated with elevated phenylalanine levels (see Chapter 39).
More than 600 patients with tetrahydrobiopterin disorders have been identified (BIODEF database, http://www.bh4.org/). PTPS deficiency is the most common of these disorders, occurring in 54 percent of cases of reported tetrahydrobiopterin disorders. DHPR deficiency is seen in 32 percent, whereas PCD and GTPCH deficiencies are rare, each reported in 4 percent of cases (BIODEF database, http://www.bh4.org/).
Clinical Manifestations
Most patients with GTPCH, DHPR, and PTPS deficiencies have severe forms of disease, although mild forms of DHPR and PTPS deficiencies exist, and some forms of PTPS deficiency may be transient [Blau et al., 2001]. PCD deficiency is usually not associated with significant abnormalities other than transient tone abnormalities. Untreated patients with typical severe disorders of tetrahydrobiopterin synthesis or recycling usually develop neurologic manifestations by 4 months, although symptoms can appear in the neonatal period. Clinical manifestations include microcephaly, progressive neurologic deterioration, movement disorders, delayed motor development, seizures, tone disturbances, oculogyric spasms, swallowing difficulties, hypersalivation, and hyperthermia [Blau et al., 1996, 2001; Dhondt, 1993]. Diurnal fluctuation of dystonia may occur. The clinical course is similar in severe untreated tetrahydrobiopterin deficiency, regardless of the enzymatic defect. Head MRI findings have only rarely been reported [Pietz et al., 1996]. However, in DHPR deficiency, brain abnormalities such as diffuse demyelination, atrophy, spongy vacuolation of brainstem long tracts, and basal ganglia calcification may occur. Abnormal vascular proliferation in the cortex, white matter, and basal ganglia may also be detected [Gudinchet et al., 1992; Schmidt et al., 1988].
Diagnosis
Patients with tetrahydrobiopterin defects are often identified by mandatory newborn screening programs because of hyperphenylalaninemia. All children with persistent hyperphenylalaninemia must be screened for aberrations in the levels of pterin metabolites (i.e., neopterin and biopterin). Patients with GTPCH deficiency have decreased urinary excretion of neopterin and biopterin. In PTPS deficiency, neopterin is increased and biopterin decreased, resulting in a greatly elevated neopterin to biopterin ratio (normally, the ratio is about 1:1). The neopterin to biopterin ratio in PCD deficiency is also increased but not to the same extent as in PTPS deficiency. In PCD deficiency, the characteristic feature is the presence of primapterin (7-biopterin) in the urine [Ayling et al., 2000]. In DHPR deficiency, the percentage of biopterin is elevated (>80 percent in most cases), but urine screening may miss it in some patients [Dhondt, 1984]. The measurement of DHPR activity in neonatal dried blood spots using a spectrophotometric assay is an effective method for diagnosis of DHPR deficiency. Urine pterin analysis and DHPR activity screening should be performed early in the management of a new patient with persistent hyperphenylalaninemia, or these disorders may be missed. Mutation analysis has yet to identify clear genotype-phenotype correlations [Blau et al., 1996].
Treatment
The goals of therapy are to decrease the level of phenylalanine to an acceptable range (120–360 μM) and correct the neurotransmitter deficiencies with exogenous supplementation. The diet is similar to that used to treat classic phenylketonuria, but patients tend to have a higher phenylalanine tolerance (300–700 mg/day) [Blau et al., 2001]. Tetrahydrobiopterin supplementation (2–20 mg/kg/day) is also used to help control blood phenylalanine levels. Lower tetrahydrobiopterin doses (2–5 mg/kg/day) may be effective in GTPCH and PTPS deficiencies, whereas higher doses (up to 20 mg/kg/day) may be required in DHPR deficiency. l-DOPA and 5-hydroxytryptophan are administered in a dose of 1–10 mg/kg/day. Carbidopa, an inhibitor of peripheral aromatic amino acid decarboxylase, decreases the conversion rates of l-DOPA to dopamine and 5-hydroxytryptophan to serotonin, allowing for the use of lower doses of these compounds; these therapeutic adjuncts may be especially helpful in severe forms of tetrahydrobiopterin deficiency. The optimal dose of each medication must be determined for each patient. Mild forms of disease may respond to tetrahydrobiopterin supplementation alone. Measuring levels of cerebrospinal fluid neurotransmitter metabolites (i.e., homovanillic acid and 5-hydroxyindolacetic acid) is useful in monitoring the efficacy of treatment [Blau et al., 2001; Shintaku, 2002].
Side effects of therapy include choreoathetosis, dystonia, and on-off phenomena, which are also features of the underlying disorders [Tanaka et al., 1989]. Tachycardia, diarrhea, and anorexia are associated with 5-hydroxytryptophan administration [Dhondt, 1993]. l-Deprenyl, a monoamine oxidase inhibitor, has been useful in decreasing catabolism of l-DOPA and 5-hydroxytryptophan, allowing lower dosing [Schuler et al., 1995; Spada et al., 1995, 1996]. A low concentration of cerebrospinal fluid folate is typical in DHPR deficiency, and it is treated by folinic acid supplementation (10–20 mg/day) [Shintaku, 2002]. Trimethoprim-sulfamethoxazole and methotrexate are DHPR inhibitors, and they may cause serious side effects in patients with tetrahydrobiopterin deficiency [Millot et al., 1995; Woody and Brewster, 1990]. Neurologic function may improve with therapy, but the overall prognosis for these disorders is largely unknown.
Hepatorenal Tyrosinemia
Hepatorenal tyrosinemia (i.e., tyrosinemia type I) is characterized principally by liver, kidney, and peripheral nerve involvement. The clinical spectrum ranges from severe hepatic failure in early infancy to later presentations of chronic liver disease and rickets in an older child. The overall incidence is 1 case per 100,000 births. In Quebec the incidence is quite high, at 1 in 16,700 births [Bergeron et al., 1974].
Pathophysiology
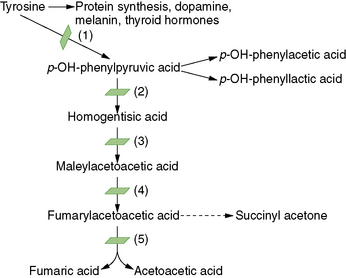
Hepatorenal tyrosinemia is caused by a deficiency of fumarylacetoacetate hydrolase, a distal enzymatic step in the processing of the amino acid tyrosine (Figure 32-2). Some investigations suggest that metabolites of tyrosine accumulating proximal to the blocked reaction step are toxic to liver and kidney, acting as alkylating agents or by disruption of sulfhydryl metabolism [Russo et al., 2001]. One of the accumulating metabolites, succinylacetone, has been implicated in the peripheral neuropathy of tyrosinemia [Sassa and Kappas, 1983; Sassa et al., 1983].
Clinical Manifestations
Neurologic involvement can include paresthesias, opisthotonic-like posture, bruxism and tongue biting, and in some cases, motor paralysis leading to respiratory failure and death [Mitchell et al., 1990]. Neurologic crises occur in up to 42 percent of individuals with tyrosinemia [Kvittingen, 1991]. These crises are biphasic, with an active period of pain, autonomic dysfunction, and sometimes paralysis lasting 1–7 days, followed by a period of recuperation. Succinylacetone blocks the heme biosynthetic pathway, and the neurologic crises – a major source of morbidity – therefore have a physiologic basis similar to those in porphyria [Russo et al., 2001].
Management
2-[2-Nitro-4-trifluoromethylbenzoyl]-1,3-cyclohexanedione (NTBC) is an inhibitor of 4-hydroxyphenylpyruvate dioxygenase (4-HPD), a proximal step in tyrosine catabolism. Inhibition thereby prevents the synthesis of succinylacetone and related metabolites, which accumulate because of the enzymatic block at fumarylacetoacetate hydrolase, a distal step in the pathway. Treatment with this compound is effective within hours and dramatically reduces the risk of neurologic and hepatic crises [Holme and Lindstedt, 2000; McKiernan, 2006]. Dietary restriction of phenylalanine and tyrosine is used in combination with NTBC. Liver transplantation is curative for hepatic and nervous system disease, and it is used in those who are refractory to nonsurgical treatment.
Acute management of neurologic crises includes analgesia, glucose (which inhibits ALA synthetase), and symptomatic treatment of hypertension. Repletion of sodium, potassium, and phosphate is necessary. The use of barbiturates and other medications that aggravate porphyria should be avoided before stabilization on NTBC [Kang and Gerald, 1970].
Other Categories of Tyrosinemia
Several causes of hypertyrosinemia exist in addition to fumarylacetoacetate hydrolase deficiency (see Figure 32-2). Deficiency of tyrosine aminotransferase causes tyrosinemia type II (oculocutaneous tyrosinemia) [Hunziker, 1980]. In type II disease, developmental delay, corneal thickening, and hyperkeratosis of palms and soles occur, but there is usually no hepatorenal involvement. Type III disease is caused by deficiency of 4-HPD and has a spectrum of manifestations, ranging from clinically normal to severe mental retardation and neurologic anomalies, including ataxia [Cerone et al., 1997; Ruetschi et al., 2000]. A 4-HPD dysfunction can also cause hawkinsinuria, a rare condition that can manifest with failure to thrive and metabolic acidosis, but it usually resolves as the patient’s metabolism matures [Borden et al., 1992].
Liver failure can lead to elevated tyrosine levels [Mitchell et al., 2001], as can postprandial testing and diseases such as vitamin C deficiency and hyperthyroidism. Premature infants may manifest transient tyrosinemia of the newborn because of temporary immaturity in the function of 4-HPD. This condition resolves spontaneously, but mild developmental delay has been reported [Nyhan, 1984].
Maple Syrup Urine Disease
In 1954, Menkes and colleagues described four siblings who died in early infancy from a cerebral degenerative disease, with onset occurring when they were 3–5 days old. Symptoms included feeding difficulty, irregular respiratory pattern, hypertonia, opisthotonus, and failure to thrive. All had urine with the smell of maple syrup [Menkes et al., 1954]. Soon thereafter, another patient with a similar history was found to have elevated levels of branched-chain amino acids in urine and blood, and the syndrome was initially referred to as maple sugar urine disease [Westall et al., 1957]. Maple syrup urine disease is caused by mitochondrial branched-chain α-ketoacid dehydrogenase complex deficiency (compared with the composite branched-chain amino acid pathways in Figure 32-6 below). The enzymatic defect leads to accumulation of branched-chain amino acids and branched-chain α-ketoacids. Five forms of maple syrup urine disease (i.e., classic, intermediate, intermittent, thiamine-responsive, and dihydrolipoyl dehydrogenase [E3] deficiency) have been delineated based on clinical presentation, level of enzyme activity, and response to thiamine administration [Chuang and Shih, 2001].
Clinical Manifestations
Classic maple syrup urine disease
In the classic form, the clinical phenotype is one of severe neonatal encephalopathy, unless presymptomatic therapy is initiated because of abnormal newborn screening, prenatal diagnosis, or positive family history. Untreated neonates typically develop symptoms by the end of the first week of life. Feeding difficulties, alternating hypertonia and hypotonia, opisthotonic posturing, abnormal movements (“fencing” or “bicycling”), and seizures commonly occur. The characteristic urine smell develops on day 5–7 of life [Strauss and Morton, 2003a]. Unless an underlying inborn error of metabolism is suspected, affected children may be misdiagnosed as having sepsis and progress to coma and death. Ketosis is often found, and hypoglycemia may occur, but severe metabolic acidosis tends not to occur. Plasma amino acid analysis reveals elevated levels of branched-chain amino acids and the diagnostic presence of alloisoleucine in plasma [Schadewaldt et al., 1999]. Urine organic acid analysis demonstrates excretion of branched-chain α-ketoacids. Hyponatremia and cerebral edema are frequent sequelae during acute metabolic decompensation [Morton et al., 2002]. Other complications include pseudotumor cerebri, pancreatitis, and eye abnormalities [Burke et al., 1991; Kahler et al., 1994]. Ocular findings in untreated or late-diagnosed patients include optic atrophy, gray optic papilla, nystagmus, ophthalmoplegia, strabismus, and cortical blindness [Burke et al., 1991]. Children who survive the initial metabolic crisis typically have significant neurodevelopmental delays and spasticity [Chuang and Shih, 2001]. Although motor, visual, and learning deficits may occur, rapid identification of affected infants and careful institution of appropriate therapy can result in normal development [Kaplan et al., 1991; Morton et al., 2002].
Neuroimaging studies (Figure 32-3) are typically abnormal in patients with untreated classic maple syrup urine disease who are in crisis. Computed tomographic (CT) scans appear normal in the first few days of life, but they reveal progression to marked generalized cerebral edema if the patient remains untreated [Brismar et al., 1990]. An unusual pattern of edema may occur, characterized by involvement of the cerebellar deep white matter, posterior brainstem, cerebral peduncles, posterior limb of the internal capsule, and posterior aspect of the centrum semiovale. Edema tends to subside in the second month of life [Brismar et al., 1990]. Patients with classic maple syrup urine disease in metabolic crisis with associated hyponatremia demonstrate a prominently increased T2 signal on brain MRI in the brainstem reticular formation, dentate nucleus, red nucleus, globus pallidus, hypothalamus, septal nuclei, and amygdala [Morton et al., 2002]. One report observed that brain MRI abnormalities were absent or only slight in sick patients with maple syrup urine disease in the absence of hyponatremia [Morton et al., 2002]. Cranial ultrasonography of neonates in acute metabolic crisis reveals symmetrically increased echogenicity of the periventricular white matter, basal ganglia, and thalami [Fariello et al., 1996]. Chronic changes, including hypomyelination of the cerebral hemispheres, cerebellum, and basal ganglia and cerebral atrophy, may supervene in poorly controlled patients. CT- and MRI-defined abnormalities and the clinical phenotype may improve after implementation of appropriate dietary therapy [Taccone et al., 1992]. Diffusion-weighted imaging and spectroscopy have also documented abnormalities during the acute phase of disease [Cavalleri et al., 2002]. Markedly restricted proton diffusion, suggestive of cytotoxic or intramyelinic sheath edema, was demonstrated in the brainstem, basal ganglia, thalami, cerebellar and periventricular white matter, and cerebral cortex in six patients with maple syrup urine disease. MR spectroscopy demonstrated abnormal elevations of branched-chain amino acids, branched-chain α-ketoacids, and lactate in the four patients. All of these changes were reversed after the institution of appropriate nutritional and antibiotic therapy to treat intercurrent illness [Jan et al., 2003].
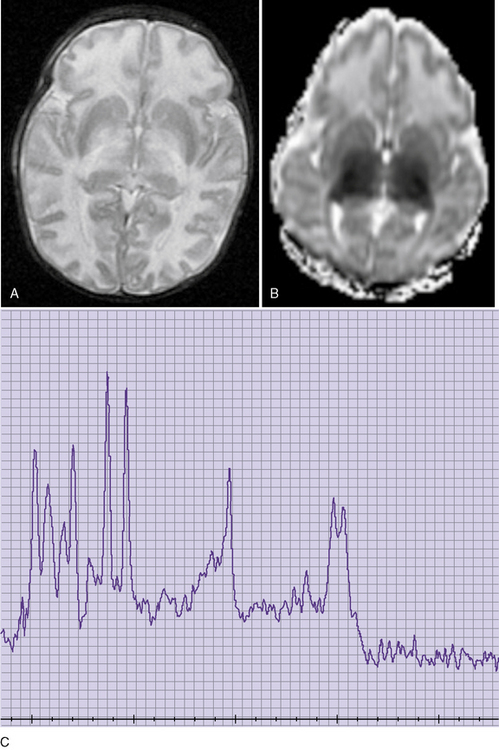
Fig. 32-3 Maple syrup urine disease.
(Courtesy of Dr. A James Barkovich, University of California, San Francisco, CA.)
A characteristic comblike EEG pattern may be demonstrated for some patients with classic maple syrup urine disease between the second and third weeks of life [Tharp, 1992]. This unusual rhythm pattern resolves with the institution of dietary therapy [Tharp, 1992].
Intermediate maple syrup urine disease
Children who have the intermediate form of maple syrup urine disease do not present in the neonatal period, despite having persistently elevated plasma levels of branched-chain amino acids. Developmental delay and failure to thrive are common. Severe neurologic impairment is absent; episodes of metabolic decompensation may occur, although severe ketoacidosis episodes are variable [Gonzalez-Rios et al., 1985a]. These children have a higher tolerance for dietary protein than those who have the classic form [Gonzalez-Rios et al., 1985a]. Rarely, patients with intermediate-type maple syrup urine disease respond to thiamine administration.
Intermittent maple syrup urine disease
Patients with intermittent maple syrup urine disease typically come to medical attention when they are 5 months to 2 years old and after stress induced by infection or high protein intake; some have been detected as late as the fifth decade of life [Chuang and Shih, 2001]. The intermittent form of maple syrup urine disease can be particularly difficult to diagnose, because affected individuals have normal levels of branched-chain amino acids and no odor between episodes of metabolic decompensation. Episodic decompensation is characterized by ataxia, disorientation, and altered behavior, which may progress to seizures, coma, and even death unless therapy is instituted. Early development and intellect are usually normal.
Thiamine-responsive maple syrup urine disease
The clinical course of patients with the thiamine-responsive variant of maple syrup urine disease is similar to that of the intermediate form of disease. Plasma levels of branched-chain amino acid and urine excretion of branched-chain α-ketoacids decline days to weeks after thiamine administration (10–1000 mg/day) is started [Scriver and Kaufman, 2001]. Patients are also treated with nutritional regimens similar to those used in other forms of maple syrup urine disease. Developmental delay may be present, but normal intelligence has also been documented [Scriver et al., 1971].
Dihydrolipoyl dehydrogenase-deficient maple syrup urine disease
The dihydrolipoyl dehydrogenase (E3)-deficient form of maple syrup urine disease is characterized by ketoacidotic crises in infancy. There is also lactic acidemia because the E3 subunit of the branched-chain α-ketoacid dehydrogenase complex is also required for catalytic function of pyruvate dehydrogenase and α-ketoglutarate dehydrogenase. In addition to the typical maple syrup urine disease metabolites, urine organic acid analysis reveals the presence of lactate, pyruvate, and α-ketoglutarate. The neonatal period is usually uneventful, but progressive neurologic deterioration, characterized by developmental delay, hypotonia or hypertonia, and dystonia, supervenes. Death in early childhood is common. Attempts at therapy have had limited success [Chuang and Shih, 2001; Sakaguchi et al., 1986].
Laboratory Tests
Maple syrup urine disease can be detected easily and accurately by tandem mass spectrometry analysis of the newborn blood spot [Chace et al., 1995]. Tandem mass spectrometry used in newborn screening is effective in identifying maple syrup urine disease, and is performed in all states in the U.S. (see http://genes-r-us.uthscsa.edu/nbsdisorders.htm) and many countries worldwide. Urine screening tests for the presence of α-ketoacids (i.e., ferric chloride and 2,4-dinitrophenylhydrazine [DNPH]) may be positive, but are nonspecific and insensitive. Plasma amino acid analysis demonstrates elevations of leucine, isoleucine, and valine (5- to 10-fold greater than normal) [Strauss and Morton, 2003a], as well as the pathognomonic finding of elevated alloisoleucine [Schadewaldt et al., 1999]. Levels of branched-chain amino acids are greatly elevated in urine and cerebrospinal fluid [Chuang and Shih, 2001]. The branched-chain α-ketoacids 2-oxoisocaproic acid, 2-oxo-3-methylvaleric acid, and 2-oxoisovaleric acid, derived from the branched-chain amino acids leucine, isoleucine, and valine, respectively, are found to be elevated on urine organic acid analysis during metabolic crises. Branched-chain amino acids levels and excretion of branched-chain α-ketoacids may be normal between episodes of decompensation in the intermittent form of disease.
The branched-chain α-ketoacid dehydrogenase complex consists of three catalytic components – a thiamine pyrophosphate-dependent carboxylase (E1) with an α2β2 structure, a transacylase (E2), and a dehydrogenase (E3) – as well as two regulatory enzymes (a kinase and a phosphatase) [Chuang and Shih, 2001]. Deficient activity of this complex leads to the accumulation of leucine, isoleucine, and valine and their corresponding α-ketoacids. The decarboxylation activity can be measured in leukocytes, lymphoblasts, or fibroblasts, and it is loosely related to the clinical phenotype: 0–2 percent of normal activity in classic maple syrup urine disease, 3–30 percent activity in intermediate, 5–20 percent in intermittent, 2–40 percent in thiamine-responsive, and 0–25 percent in E3 deficiency [Chuang and Shih, 2001; Scriver et al., 1971]. Because significant overlap exists between measured enzyme activity and clinical phenotype, enzymatic activity cannot be used to predict the clinical course with certainty.
Genetics
Maple syrup urine disease is a pan-ethnic, autosomal-recessive condition that can be caused by mutations in any of the components of the mitochondrial branched-chain α-ketoacid dehydrogenase complex. In a study of 63 individuals, E1β subunit mutations were most common (38 percent), followed by E1α (33 percent), and E2 (19 percent) mutations [Nellis and Danner, 2001]. Branched-chain α-ketoacid dehydrogenase phosphatase or kinase mutations are also predicted to cause maple syrup urine disease, but such abnormalities have not yet been detected. The overall incidence is approximately 1 case per 150,000 people in the general population, but maple syrup urine disease is more common in Old Order Mennonites in southeastern Pennsylvania (1 in 176 births) [Danner and Doering, 1998]. A novel founder mutation in the E1β subunit has been reported in the Ashkenazi Jewish population [Edelmann et al., 2001]. In general, increased residual branched-chain α-ketoacid dehydrogenase activity should convey some advantage, but there is a wide overlap between measured enzymatic activities and clinical outcome. Given the complexity of the molecular genetics, the potential for modifier gene and environmental interactions, and the multiple clinical phenotypes associated with maple syrup urine disease, a lack of definitive genotype-phenotype relationships is not surprising.
Treatment
Chronic care of the child with maple syrup urine disease includes regular visits to an integrated metabolic clinic for medical and nutritional assessment. Adequate calories (100–120 kcal/kg/day) and protein (2–3 g/kg/day) are needed for growth. Chronic valine or isoleucine deficiency may cause an exfoliative dermatitis, and supplementation of these amino acids is often needed [Koch et al., 1993]. Thiamine supplementation is administered to patients with thiamine-responsive forms of maple syrup urine disease. Because patients on restricted diets are at risk for micronutrient and essential fatty acid deficiencies, patients should be periodically monitored for such deficits and supplementation given as needed.
Because significant metabolic intoxication may occur rapidly, even in patients with apparently well-controlled disease, it is crucial to have carefully considered home and hospital emergency protocols in place for each child [Morton et al., 2002; Strauss and Morton, 2003a]. Acute metabolic decompensation (e.g., fasting or illness severe enough to cause catabolism) is a medical emergency that requires prompt intervention. Initial intervention is aimed at correcting dehydration, starting high-dose intravenous thiamine, and providing adequate calories (approximately 120–140 kcal/kg/day) to prevent further protein catabolism and higher rise in plasma leucine levels. To this end, high-dextrose intravenous fluids (to provide approximately 10 mg/kg/min) and intralipid are often administered. Branched-chain amino acid-free parenteral nutrition or enteral formula, delivered by continuous nasogastric drip, can also be used [Nyhan et al., 1998; Parini et al., 1993]. The rate of decrease of leucine is slowed in the face of valine and isoleucine levels inadequate to stimulate protein synthesis. Acute valine and isoleucine deficiency can be avoided by careful supplementation of these amino acids [Parini et al., 1993]. Leucine is reintroduced to the diet after therapeutic levels are achieved [Morton et al., 2002].
In a study of 36 maple syrup urine disease patients, plasma leucine levels fell to less than 400 μM 2–4 days after the initiation of therapy with enteral and parenteral nutrition. Initial leucine levels ranged from 233 to 778 μM in a group diagnosed on the first day of life (n = 18) and 1489 to 3359 μM in a group diagnosed between days 3 and 16 (n = 18). Over an 11-year period, neurologic examinations, gross motor development, and speech were normal in 34 of 36 children [Morton et al., 2002]. Enteral nutrition was also found to be beneficial when instituted within the first 20 days of life. Four patients receiving nasogastric drip feeding as the only treatment of neonatal classic maple syrup urine disease had normal development when 3–5 years old [Parini et al., 1993]. Hemodialysis and continuous venovenous extracorporeal removal therapies result in more rapid fall in plasma levels of branched-chain amino acids, but these modalities typically have been described in single case reports or small series with relatively short follow-up, and it is difficult to ascertain the long-term outcome of such intervention [Gouyon et al., 1996; Puliyanda et al., 2002]. Nevertheless, normal development has been reported for 8 of 12 children after continuous venovenous extracorporeal removal therapy [Jouvet et al., 2001]. Branched-chain amino acids levels often rebound after initial dialysis in cases of severe metabolic imbalance characterized by extremely high leucine levels, and dialysis may need to be repeated in such cases. Peritoneal dialysis is no longer routinely used; there is a tendency for leucine levels to plateau between 1000 and 1500 μM after 24 hours, limiting the utility of this therapeutic modality [Gortner et al., 1989]. Levels of branched-chain amino acids and branched-chain α-ketoacids tend to plateau with exchange transfusion therapy [Nyhan et al., 1998; Wendel et al., 1982].
Although enteral and intravenous therapy may be sufficient to manage many patients with maple syrup urine disease in acute crisis, various dialysis methods are commonly used and should be considered, especially when clearance of branched-chain amino acids by nutritional support is not effective or when other considerations, such as life-threatening cerebral edema, renal imbalance, or cardiovascular abnormalities, exist [Jouvet et al., 2001; Nyhan et al., 1998]. Liver transplantation has been performed rarely for maple syrup urine disease. Three patients who underwent successful transplantation were able to resume normal diets and were no longer at risk for metabolic decompensation [Wendel et al., 1999]. More recently, domino hepatic transplantion for maple syrup urine disease was successfully performed [Barshop and Khanna, 2005].
Because hyponatremia and subsequent brain edema are serious and relatively common complications, it is important to monitor serum sodium and serum and urine osmolalities closely and to replace urinary losses with saline [Morton et al., 2002]. Critical brain swelling and abnormal brainstem function may develop with only a moderate reduction in serum sodium level (by only 8–10 mEq/L) [Morton et al., 2002]. Low-dose diuretics may also be used to prevent water retention [Strauss and Morton, 2003a]. Mannitol is reserved for life-threatening episodes of increased intracranial pressure [Morton et al., 2002].
Glycine Encephalopathy
Glycine encephalopathy, also known as nonketotic hyperglycinemia, is an autosomal-recessive disorder caused by defective function of the multimeric glycine cleavage enzyme system, leading to accumulation of glycine in all body tissues, including the CNS (Figure 32-4). Gerritsen et al., 1965 described the initial patient in 1965. The glycine cleavage enzyme system has four components: the P protein (a pyridoxal phosphate-dependent glycine decarboxylase), the T protein (a tetrahydrofolate-dependent protein), the H protein (a hydrogen carrier protein), and the L protein (lipoamide dehydrogenase). Infants with classic disease present in the first week of life with apnea, lethargy, severe hypotonia, and feeding difficulties [Hoover-Fong et al., 2004]. Respiratory failure, hiccups, and intractable seizures develop, and most infants die unless assisted ventilatory support is provided. Intermittent ophthalmoplegia is a relatively frequent finding [MacDonald and Sher, 1977]. The EEG commonly has a burst suppression pattern, but hypsarrhythmia has been reported rarely [Hoover-Fong et al., 2004].
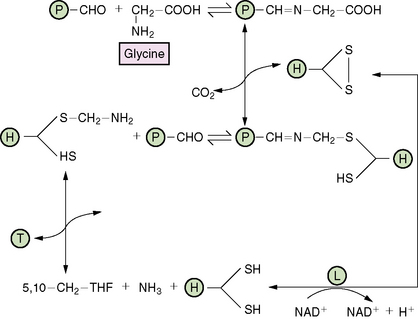
Fig. 32-4 The glycine cleavage system.
(From Scriver C, Beudet A, Sly W, Valle D, eds. The metabolic and molecular basis of inherited disease, 8th edn. New York: McGraw-Hill, 2001:2066. Reprinted with permission from The McGraw-Hill Companies.)
Brain imaging results are normal for about one-half of the neonatal-onset cases [Hoover-Fong et al., 2004]. Relatively common brain abnormalities include agenesis of the corpus callosum, progressive atrophy, and delayed myelination [Hoover-Fong et al., 2004]. Acute hydrocephalus, requiring shunting, may occur and is a poor prognostic sign [Van Hove et al., 2000]. 1H-MR spectroscopy detects increased intracerebral levels of glycine, lactate, and creatine [Viola et al., 2002]. The lethal form of glycine encephalopathy appears to be associated with elevated levels of brain myo-inositol glycine, creatine, and N-acetylaspartate. Diffusion-weighted imaging has shown high-signal-intensity lesions in the pyramidal tracts, middle cerebellar pedicles, and dentate nuclei, likely reflecting myelin spongiosis [Sener, 2003].
Atypical and transient variants of glycine encephalopathy have also been reported in patients with cerebrospinal fluid to plasma glycine ratios of more than 0.08. Atypical forms manifest in infancy or early childhood after an uneventful pregnancy and neonatal period. Clinical features include seizures (in most cases) and relatively mild developmental delay. Atypical glycine encephalopathy documented by liver enzymology has also been reported in siblings with normal cerebrospinal fluid glycine levels and cerebrospinal fluid to plasma glycine ratios [Jackson et al., 1999]. Transient glycine encephalopathy is characterized by the same initial clinical and biochemical findings as the classic form, but it has only rarely been reported. In the transient form, cerebrospinal fluid and plasma glycine levels partially or completely resolve, and most patients have normal development [Aliefendioglu et al., 2003; Korman et al., 2004; Lang et al., 2008]. Transient elevations of cerebrospinal fluid glycine and the cerebrospinal fluid to plasma glycine ratio occurred in an asphyxiated patient with pyridoxine-dependent seizures [Maeda et al., 2000].
Diagnosis of glycine encephalopathy is established by detecting an elevated cerebrospinal fluid glycine concentration, typically 15–30 times normal, in association with an increased cerebrospinal fluid to plasma glycine ratio (normal <0.02) [Applegarth and Toone, 2001; Hamosh and Johnston, 2001]. Classic neonatal-onset patients often have ratios higher than 0.2, whereas atypical patients have ratios of approximately 0.09 [Hamosh and Johnston, 2001]. A ratio higher than 0.08 is usually considered diagnostic for glycine encephalopathy. The plasma and cerebrospinal fluid samples should be obtained as closely as possible to one another, and the presence of blood in the cerebrospinal fluid invalidates the amino acid results [Applegarth and Toone, 2001]. Other causes of increased cerebrospinal fluid glycine levels include valproate therapy, brain trauma, and hypoxic-ischemic encephalopathy. Secondary elevations of plasma glycine, associated with ketosis, are often encountered in organic acidemias (e.g., methylmalonic, propionic, and isovaleric acidemias and β-ketothiolase deficiency) [Applegarth and Toone, 2001; Korman and Gutman, 2002]. Because sulfite oxidase deficiency (isolated or as part of molybdenum co-factor deficiency), folinic acid-responsive seizures, and disorders of neurotransmitters may have presentations similar to that of glycine encephalopathy, an aliquot of cerebrospinal fluid should also be frozen and saved for appropriate analyses in the event that the result of amino acid analysis is normal.
Definitive confirmation of the diagnosis may be accomplished by assaying glycine cleavage enzyme in liver. In practice, molecular testing of the genes encoding glycine cleavage system subunits is less invasive and more widely available [Hamosh et al., 2009]. Between 60 and 80 percent of patients with the classic neonatal form have defects in the P protein. T protein deficiency occurs in 5–20 percent of cases, whereas H protein and L protein defects are rarely reported [Tada and Kure, 1993]. Prenatal diagnosis by glycine cleavage enzyme system measurement in uncultured chorionic villus samples has resulted in false-negative and false-positive results in at least 1 percent of cases studied [Applegarth et al., 2000]. DNA analysis, when mutations are known, remains the most reliable form of prenatal diagnosis [Kure et al., 1999].
Comprehensive mutation analysis in 68 families with glycine encephalopathy detected GLDC (P protein gene) or AMT (T protein gene) mutations in 68 percent of neonatal and 60 percent of infantile types, respectively. No GCSH (H protein gene) mutations were identified [Kure et al., 2003]. The L protein is a component of pyruvate dehydrogenase and branched-chain ketoacid dehydrogenase, as well as the glycine cleavage enzyme system. However, L protein deficiency leads to a variant form of maple syrup urine disease or pyruvate dehydrogenase deficiency, rather than glycine encephalopathy [Applegarth and Toone, 2001]. Few glycine encephalopathy patients have been identified who carry the same mutations, making genotype-phenotype correlations problematic, although some possible correlations have been found [Applegarth and Toone, 2004]. Three of four patients with transient glycine encephalopathy and homozygosity for a novel GLDC mutation (A802V) had a normal outcome after intensive therapy as neonates. High residual activity of the mutant enzyme and therapeutic intervention during a critical period of brain sensitivity may have contributed to the good outcome in some cases of transient or mild glycine encephalopathy. The three-dimensional structures of the T protein, H protein, and L protein have been determined, which can aid in understanding the molecular mechanisms underlying the effects of missense mutations on glycine cleavage enzyme system activity [Lee et al., 2004].
In postmortem examination specimens from infants who died because of glycine encephalopathy, brain glycine concentrations are elevated 2–8-fold [Perry et al., 1977]. Neuropathology has demonstrated deficient myelination, abnormal cortical neuron morphology, and spongiosis of the white matter with associated astrocytic gliosis [Brun et al., 1979].
Treatment of glycine encephalopathy has not improved the overall dismal prognosis in the classic form of disease [Chien et al., 2004]. Therapy is focused on controlling seizures with antiepileptic drugs, decreasing tissue glycine levels, and administering N-methyl-d-aspartate (NMDA) receptor antagonists to diminish glycine-induced neuronal excitotoxicity. Valproate is contraindicated because it can inhibit the glycine cleavage enzyme system, and can cause hyperglycinemia in patients without glycine encephalopathy [Jaeken et al., 1977]. Sodium benzoate is given because of its ability to conjugate to glycine to form hippurate, which can then be excreted in the urine. A glycine-specific mitochondrial enzyme, benzoyl-coenzyme A (CoA):glycine acyltransferase, catalyzes the condensation of benzoate and glycine to form hippurate [Webster et al., 1976]. Sodium benzoate therapy can reduce plasma levels of glycine to the normal range and may have a mild effect on cerebrospinal fluid glycine levels, but it does not affect the dismal prognosis. Because high-dose sodium benzoate therapy can result in carnitine deficiency, plasma carnitine levels should be monitored closely and appropriate supplementation provided [Van Hove et al., 1995]. Dextromethorphan, an antagonist of the NMDA receptor, is also commonly used in therapy. Treatment with dextromethorphan may lead to improved seizure control and level of interaction in some patients. Ketamine has been used rarely, but it may provide benefit in controlling seizures and improving overall level of interaction [Boneh et al., 1996]. A low-protein diet has no proven efficacy and may result in severe protein malnutrition, micronutrient deficiency, and exfoliative dermatitis if not monitored carefully [Samady et al., 2000]. Sodium benzoate, alone or combined with imipramine, has been effective in improving clinical manifestations in milder forms of glycine encephalopathy [Neuberger et al., 2000; Wiltshire et al., 2000].
Sulfur Amino Acid Metabolism and the Homocystinurias
Homocysteine lies at a critical juncture between the trans-sulfuration and remethylation pathways of methionine metabolism, at which point homocysteine can be converted to cystathionine or methionine. The malfunctioning of three enzymes is known to cause homocystinuria: cystathionine β-synthase, methylene tetrahydrofolate reductase, and methionine synthase. The β-synthase enzyme is involved in the trans-sulfuration pathway (Figure 32-5), and the latter two enzymes are involved in the sulfur conservation pathway. Several of the mutations that cause methylmalonic acidemia also cause homocystinuria, and these are discussed in the section on cobalamin complementation groups. A transport abnormality, selective intestinal malabsorption of vitamin B12, can also cause homocystinuria. The overall incidence of homocystinuria is approximately 1 case in 335,000 persons, but it varies from 1 in 65,000 in Ireland to 1 in 900,000 in Japan [Naughten et al., 1998].
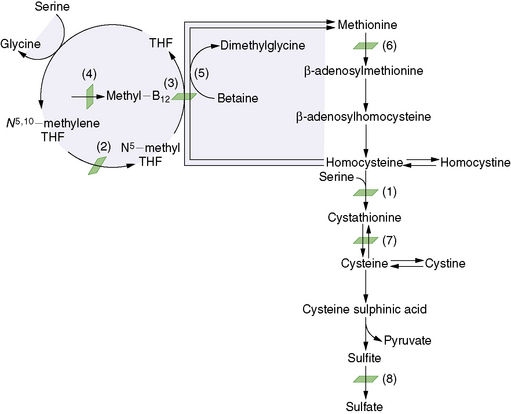
Fig. 32-5 Abbreviated diagram for the trans-sulfuration pathway.
(From Wilcox WR, Cederbaum SD. Amino acid metabolism. In: Rimoin D, Connor J, Pyeritz R, Korf B, eds. Principles and practice of medical genetics, 4th edn. Philadelphia: Churchill Livingstone, 2002:2419.)
Cystathionine β-Synthase Deficiency
Pathophysiology
The trans-sulfuration pathway converts methionine into cysteine. In cystathionine β-synthase deficiency, methionine, homocysteine, and homocystine accumulate in the blood and are excreted in large quantities in the urine [Christensen et al., 1991; Mudd et al., 1964]. Two major categories of cystathionine β-synthase deficiency have been described, one responsive to pyridoxine and the other nonresponsive [Barber and Spaeth, 1969]. One-half of patients with cystathionine β-synthase deficiency respond to pyridoxine therapy, and only modest restriction of methionine in their diet is required [Mudd et al., 1985].
Biochemical abnormalities in the CNS are thought to account for the occurrence of mental retardation and neurologic abnormalities. Synthesis of cystathionine, an important free amino acid in the brain [Brenton et al., 1965], is impaired, and this compound is virtually absent from the brain in affected individuals. Abnormalities in homocysteine, methionine, and other metabolites probably contribute to central nervous disease as well.
Arterial and venous thromboses are prominent in many organs, including the brain. Large and medium-sized blood vessels (i.e., arteries, veins, and dural sinuses) are compromised. Fibrous thickening of the intima occurs, the vessel lumen may be compromised, and the media is also involved, with increased deposition of collagen and frayed, split smooth muscle fibers. A clotting diathesis is also involved [D’Angelo and Selhub, 1997]. An increased tendency of intravascular thrombosis may be related to abnormal adhesiveness of platelets. However, no single mechanism has been demonstrated to cause the vascular complications. Homocysteine elevation is an independent risk factor for arteriosclerotic vascular disease. This factor has been demonstrated for affected patients and for individuals in the general population who may have modest elevations in homocysteine concentrations.
Degenerative changes in the fibers that hold the optic lens likely are caused by interference with fibrillin function [Sakai et al., 1986]. The bone disease likely results from abnormalities in connective tissue, such as defective fibrillin function or perhaps abnormal collagen cross-linking [Harris and Sjoerdsma, 1966].
Clinical manifestations
Four organ systems demonstrate major involvement: the central nervous, skeletal, ocular, and vascular systems [Mudd et al., 1985]. Other organs, such as the liver, hair, muscles, blood, and skin (e.g., hypopigmentation), may be involved.
When the CNS is involved, the most frequent finding is mental retardation, which can manifest as developmental delay during the first year of life. There is a spectrum of cognitive function in untreated patients, with IQ scores ranging from 10 to 138. Intelligence in B6-responsive patients tends to be higher than in B6 nonresponders. Psychiatric disturbances are common, and seizures and extrapyramidal signs occasionally are seen. Focal neurologic signs point to a cerebrovascular occlusion. Patients identified on newborn screening who receive early treatment have normal cognitive function [Yap et al., 2001].
Thromboembolic events (arterial and venous), livedo reticularis, and malar flush are some of the vascular findings. Patients have suffered from pulmonary, cerebral, and renal infarction. Cerebral venous sinus thrombosis has been documented with scanning studies. An increased risk of myocardial infarction exists in patients with homocystinuria and in hyperhomocysteinemic members of the general population [Stampfer et al., 1992]. One-half of patients will have a vascular event before age 30 years.
Laboratory tests
Homocystinuria occurs in all untreated patients [Isherwood, 1996], but this is not sufficient to establish the diagnosis because it may occur in patients with other conditions. Serum amino acid analysis reveals elevated methionine and free homocysteine, with variably low levels of cysteine. Plasma total homocysteine levels are abnormally elevated. Direct assays are used to confirm the enzymatic deficiency, and these can be performed on skin fibroblasts, liver, or leukocytes.
Treatment
The goals of treatment are to reduce the severe hyperhomocysteinemia and other biochemical abnormalities. Supportive treatment of complications is essential. Treatment of patients who are B12-responsive consists of pyridoxine in combination with folic acid and vitamin B12 [Wilcken and Wilcken, 1997]. For vitamin B12 nonresponders, treatment is achieved with a methionine-restricted, cystine-supplemented diet [Komrower et al., 1966; Perry et al., 1966]. Pyridoxine, folic acid, and vitamin B12 have been used in pyridoxine nonresponders as co-factors of methionine metabolism to promote homocysteine conversions partially to other metabolites. Betaine, a methyl donor that remethylates homocysteine to methionine, is also an effective component of treatment [Wilcken et al., 1985].
Methionine Synthase Deficiency
Clinical manifestations
Most patients present in early infancy with poor feeding, emesis, lethargy, hypotonia, seizures, and developmental delay [Watkins and Rosenblatt, 1989]. There is usually a neurologic presentation, but this can vary and can include gait disturbances and multiple sclerosis-like features [Carmel et al., 1988]. There is a strong association with megaloblastic anemia.
Laboratory tests
Serum cobalamin and folate levels are normal or high, and methylmalonic aciduria is generally not seen [Tuchman et al., 1988]. Homocystinuria is a consistent feature, and CblE and CblG can be differentiated from other causes of homocystinuria by biochemical studies of cultured cells.
Treatment
Administration of hydroxocobalamin in pharmacologic doses (with intramuscular administration at initial treatment stages) should begin as soon as the diagnosis is made. This typically leads to rapid biochemical improvement. Some patients also have improvement of anemia on folinic acid [Harding et al., 1997]. Prenatal therapy has been used for early diagnoses with success [Rosenblatt et al., 1985].
Methylene Tetrahydrofolate Reductase Deficiency
Pathophysiology
Major findings are demyelination and vascular changes, such as those seen in cystathionine β-synthase deficiency [Beckman et al., 1987]. Other changes include dilated cerebral ventricles, hydrocephalus, and microgyria [Kanwar et al., 1976; Wong et al., 1977]. Methylene tetrahydrofolate reductase deficiency is thought to result in low levels of brain folate [Levitt et al., 1971].
Clinical manifestations
Clinical findings vary with enzyme function and include developmental delay, motor and gait abnormalities, seizures, and psychiatric disease such as psychosis and schizophrenia [Haan et al., 1985; Mudd et al., 1972]. Patients rarely have megaloblastic anemia. Severe disease is often lethal. Age at presentation ranges from birth in severe cases to adulthood in milder ones.
Laboratory tests
Moderate hyperhomocysteinemia and homocystinuria are seen, with low or normal methionine levels. The homocysteine excretion is significantly lower than in cystathionine β-synthase deficiency [Fowler and Jakobs, 1998].
Treatment
Severe disease is refractory to treatment, and, although several treatments have been tried (i.e., folate, methionine, pyridoxine, cobalamin, and carnitine), none has been particularly effective [Fowler, 1998]. Betaine has somewhat improved the prognosis [Al Tawari et al., 2002; Sakura et al., 1998].
Sulfite Oxidase Deficiency
Deficiency of sulfite oxidase function may occur as an isolated enzyme defect or as part of a combined deficiency (see Figure 32-5). This rare disorder results in abnormalities of metabolism of sulfated amino acids. The cardinal feature of this condition is severe seizures in the neonatal period. Although some therapies lead to mild improvements, there is no effective treatment.
Pathophysiology
The molybdenum co-factor is composed of the metal and a small pterin group [Johnson et al., 1980a]. This prosthetic group is required for the function of three enzymes: sulfite oxidase, xanthine dehydrogenase, and aldehyde oxidase [Johnson et al., 1980b]. Most patients have mutations in one of several enzymes of the co-factor synthetic chain, whereas a minority has mutations in the sulfite oxidase gene. Postmortem examination of patients with sulfite oxidase or co-factor deficiencies has found encephalopathy with loss of neurons and myelin, attributable largely to deficiency of sulfite oxidase function and accumulation of sulfite in the brain [Roth et al., 1985]. Absence of sulfite oxidase leads to alternate metabolic pathways for sulfites, including formation of S-sulfocysteine and thiosulfate [Mudd et al., 1967]. S-sulfocysteine may substitute for cysteine in connective tissue, leading to ocular lens dislocation.
Clinical Manifestations
The clinical picture includes severe neurologic abnormalities, dislocated ocular lenses, and mental retardation [Mudd et al., 1967]. This presentation is similar in isolated sulfite oxidase deficiency cases and in patients in whom the co-factor is absent. Although some variability is seen among patients, the key feature is neonatal seizures. It is recommended that all patients with neonatal seizures have metabolic testing. Other neurologic signs can include opisthotonus, axial hypotonia with peripheral hypertonia, regression, and loss of milestones. Ophthalmologic abnormalities can include nystagmus, coloboma, and cortical blindness [Lueder and Steiner, 1995]. Craniofacial dysmorphology includes bitemporal narrowing with deep-set eyes, long palpebral fissures, thick lips, elongated philtrum, and a small nose. Heterozygote carriers are unaffected.
Laboratory Tests
Positive sulfite test results are usually observed on urine dipstick, although false-negative results can occur [van der Klei-van Moorsel et al., 1991]. The cysteine metabolite S-sulfocysteine is characteristically elevated in urine and plasma, and plasma total homocysteine may be low [Johnson and Rajagopalan, 1995; Tan et al., 2005]. Low levels of urinary urothion, a degradation product of molybdopterin, are essentially diagnostic of molybdenum co-factor deficiency, and an elevated urinary thiosulfate level is diagnostic of sulfite oxidase deficiency or molybdenum co-factor deficiency.
Treatment
Management for this condition is largely supportive because therapies are often ineffective. Molybdenum co-factor is too unstable to be used therapeutically. Patients with milder forms of isolated sulfite oxidase deficiency may respond to a diet low in sulfur-containing amino acids [Touati et al., 2000].
Hartnup’s Disease
Hartnup’s disease is an aminoaciduria that is usually clinically silent, but it can manifest with episodes of cerebellar ataxia and a pellagra-like rash. Incidence has been estimated at 1 case in 33,000 persons [Wilcken et al., 1977].
Pathophysiology
The metabolic aberration in Hartnup’s disease results from an error in the transport of monoamino-monocarboxylic (neutral) amino acids that affects renal tubular reabsorption and intestinal absorption [Baron et al., 1956; Mahon and Levy, 1986; Scriver et al., 1987]. There is deficient transport of neutral amino acids, including glutamine, histidine, valine, phenylalanine, tyrosine, tryptophan, alanine, asparagine, citrulline, isoleucine, leucine, serine, and threonine. Large amounts of the amino acids and of the amides of glutamate and aspartate are excreted in the urine [Bonetti and Dent, 1954]. The disorder arises from mutations in SLC6A19, the gene encoding the amino acid transporter B0AT1, involved in co-transport of Na+ and neutral amino acids from the luminal compartment into the cells [Kleta et al., 2004; Seow et al., 2004; Cheon et al., 2010]. Two tissue-specific forms of Hartnup’s disease are recognized: renal plus intestinal involvement, and renal involvement alone.
Amino acids, such as tryptophan, remain in the intestinal lumen [Milne et al., 1960], where they are converted to indolic compounds by bacteria and then absorbed [Asatoor et al., 1963]; these compounds are toxic to the CNS. Urinary excretion of large amounts of indican (a tryptophan metabolite) is characteristic of the disease. Large quantities of neutral amino acids are excreted in the feces [Scriver, 1965]. The lack of tryptophan absorption leads to niacin deficiency, which results in the pellagra-like symptoms and photosensitivity.
Clinical Manifestations
Patients who have been detected by population screening usually have been clinically normal, and Hartnup’s disease is usually benign [Scriver et al., 1987]. Nevertheless, some patients are affected, suggesting a monogenic trait with strong polygenic influence [Scriver, 1988]. The most prominent neurologic feature in affected individuals is attacks of cerebellar ataxia, which vary in intensity. These episodes may persist for as long as 2 weeks before improvement is evident. Less common neurologic disabilities include spasticity, wide-based gait, double vision, nystagmus, dystonia, and tremulousness. Psychiatric disturbances consisting of fear, anxiety, and mood swings may occur, as may constant headaches.
Clinical involvement varies considerably among symptomatic individuals. Neurologic symptoms are often accompanied by a rash on areas of sun-exposed skin [Baron et al., 1956]. The skin is dry and reddened, resembling pellagra (i.e., nicotinamide deficiency). Sun exposure usually exacerbates the lesions, as does sulfonamide administration. Skin may become hyperpigmented.
Laboratory Tests
Urine has strikingly elevated levels of neutral amino acids, but basic and acidic amino acids in urine are relatively normal [Tada et al., 1967]. Serum levels of amino acids are normal or low [Cusworth and Dent, 1960].
Treatment
In the rare symptomatic patient, increasing amino acid intake in the form of a high-protein diet can overcome the deficient transport and loss of neutral amino acids [Scriver et al., 1987]. Conversely, poor nutrition can lead to attacks in patients who would otherwise be asymptomatic. Symptomatic patients should protect themselves from sunlight and other aggravating agents, such as photosensitizing drugs. Administration of nicotinic acid offsets deficiency in nicotinamide synthesis [Halvorsen and Halvorsen, 1963].
Histidinemia
Histidinemia is benign in almost all affected individuals, although in rare cases, this condition may lead to CNS disease. The benign nature makes treatment unnecessary in almost all cases. Overall incidence has been estimated at between 1 in 8000 in Japan and 1 in 37,000 in Sweden [Virmani and Widhalm, 1993].
Pathophysiology
Histidinemia results from deficiency of l-histidase (l-histidine ammonia lyase) [Auerbach et al., 1962], resulting in accumulation of histidine and its metabolites in blood, urine, and cerebrospinal fluid, and in a deficiency of urocanic acid [Levy et al., 1974]. Enzyme activity is essentially undetectable in liver and other tissues of patients, but may be normal in skin [Woody et al., 1965].
Clinical Manifestations
The initial patients identified with histidinemia were mentally retarded and had speech abnormalities; the group identified was likely a result of ascertainment bias. Prospective follow-up of newborns with histidinemia detected by newborn screening did not provide evidence of disease in affected individuals [Levy et al., 1974; Tada et al., 1982]. Although it is clear that histidinemia does not produce severe disease, there is still debate about possible minor effects on speech and intelligence.
Treatment
Low-histidine diets have been successfully used to reduce blood histidine concentration; however, studies of mean IQ and general level of functioning revealed no statistical advantage of therapy. Therapy is not indicated for most patients who remain asymptomatic [Scriver and Levy, 1983]. If concern exists that elevated histidine is contributing to findings, consultation with a biochemical specialist is recommended.
Organic Acidemias
Propionic Acidemia
Propionic acidemia is caused by deficiency of propionyl-CoA carboxylase (PCC), a biotin-requiring enzyme that catalyzes the conversion of propionyl-CoA to methylmalonyl-CoA in the metabolic pathways of valine, isoleucine, methionine, threonine, and odd-chain fatty acids (Figure 32-6). This condition is estimated to occur in 1 of 50,000 to 100,000 livebirths.
Fig. 32-6 Pathways in the metabolism of the branched-chain amino acids, biotin, and vitamin B12 (cobalamin).
(From Rezvani I. Defects in metabolism of amino acids. In: Behrman R, Kliegman R, Jenson H, eds. Nelson textbook of pediatrics, 16th edn. Philadelphia: WB Saunders, 2000:355.)
Propionyl-CoA carboxylase is a multimer consisting of six α subunits (PCCA) and six β subunits (PCCB), encoded by genes on chromosomes 13q22 and 3q21–q22, respectively. Biotin, the required co-factor, is bound to the α subunit to form the fully functional enzyme. More than 40 mutations in each of the PCC-subunit genes have been described in propionic acidemia patients. There is growing evidence for genotype-phenotype correlations, with null mutations often associated with the most severe clinical outcomes [Clavero et al., 2002; Perez-Cerda et al., 2003].
Clinical Manifestations
The clinical presentation of propionic acidemia resembles that of the other so-called ketotic hyperglycinemias, including methylmalonic acidemia and isovaleric acidemia. More than 80 percent of propionic acidemia patients experience acute onset of symptoms within the first 3 months of life [Fenton et al., 2001; Sass et al., 2004a]. Symptoms of severe metabolic decompensation include vomiting, lethargy, and coma, accompanied by severe ketoacidosis, hyperammonemia, hyperglycinemia, and decreased serum carnitine. The disease is often fatal, particularly if diagnosis and implementation of treatment are delayed. Patients with onset in childhood or in adulthood have been described with episodes of neurologic decompensation associated with periods of ketoacidosis. Even for patients presenting later in life, the metabolic decompensation can be fatal [Lucke et al., 2004]. Patients with late-onset disease have been described with developmental delay or mental retardation but without a history of episodic ketoacidosis or hyperammonemia.
Diagnosis
Because abnormal elevations of propionylcarnitine are readily detected by tandem mass spectrometry, presymptomatic patients with propionic acidemia are identified in states in which expanded newborn screening by tandem mass spectrometry has been implemented. Early diagnosis of these patients, ideally before the onset of symptoms, allows initiation of specific therapies designed to reduce the risk of metabolic decompensation. The long-term outcome of propionic acidemia patients identified in this way has not been established [Leonard et al., 2003]. Some patients with propionic acidemia, particularly those with milder variant forms, have been missed by newborn screening. It is therefore important to maintain a high index of suspicion for patients presenting with symptoms compatible with this or any other organic acidemia, even in the face of normal newborn screening results.
Treatment
Long-term management of propionic acidemia consists of carnitine supplementation and a protein-restricted diet that includes supplementation with a special formula. Biotin supplementation has not been demonstrated to be effective [Fenton et al., 2001]. Liver transplantation has been performed in a small number of propionic acidemia patients, with preliminary results indicating a dramatic reduction in the risk for metabolic decompensation with less strict dietary restrictions. The long-term outcome of these patients is unknown [Yorifuji et al., 2004].
Methylmalonic Acidemias
Several genetic defects can lead to methylmalonic acidemia, in which methylmalonic acid and its derivatives accumulate in physiologic fluids. These conditions are caused by deficiencies in transport and metabolism of vitamin B12, or cobalamin (Cbl), and by mutations in enzymes requiring this co-factor (see Figure 32-6). Because of the variety of causative defects, this group of conditions has significant clinical heterogeneity and differences in response to therapy. Incidence is estimated at 1 case per 50,000 persons [Coulombe et al., 1981].
Pathophysiology
The methylmalonic acidemias can be caused by defects in adenosylcobalamin-dependent methylmalonyl-CoA mutase, intracellular cobalamin metabolism, transcobalamin II deficiency, and intrinsic factor, or by dietary cobalamin deficiency [Fowler, 1998]. Methylmalonyl-CoA mutase isomerizes methylmalonyl-CoA to succinyl-CoA, which then enters the tricarboxylic acid cycle and is converted to pyruvate. Methylmalonyl-CoA is a derivative of propionyl-CoA, which is a metabolite derived from isoleucine, valine, threonine, methionine, thymine, uracil, cholesterol, and odd-chain fatty acids.
The major causes of methylmalonic academia are divided into several different complementation groups: mut0, mut-, CblA, CblB, CblC, CblD, CblF, and CblH [Rosenblatt and Cooper, 1990]. Mutase activity is completely and partially abolished in the mut0 and mut- groups, respectively. Defective adenosylcobalamin synthesis is responsible for CblA, CblB, and CblH. CblC and CblD cause methylmalonic acidemia and homocystinuria because of cobalamin reduction abnormalities that interfere with formation of adenosylcobalamin and methylcobalamin biosynthesis. Abnormal lysosomal transport of cobalamin leads to CblF disease, which also leads to combined methylmalonic acidemia and homocystinuria. CblE and CblG diseases involve defects in homocysteine metabolism; they are discussed in the section on homocystinuria (see “Sulfur Amino Acid Metabolism and the Homocystinurias”).
Clinical Manifestations
There is significant variability in presentation, depending on the particular enzymatic deficiency, although several complementation groups share the general characteristics of failure to thrive, developmental delay, megaloblastic anemia, and neurologic dysfunction [Cooper and Rosenblatt, 1987]. Mut0, mut-, CblA, and CblB patients often present in the first few weeks of life with poor feeding, dehydration, increasing lethargy, emesis, and hypotonia [Lindblad et al., 1968; Matsui et al., 1983; Rosenberg et al., 1968]. These patients exhibit a short interval of apparent good health before presentation with symptoms. Metabolic acidosis may be catastrophic. Mild mut- or other forms of methylmalonic acidemia decompensate later in infancy or in childhood with hypoglycemia, acidosis, seizures, and lethargy. A CblC patient can present early in infancy or in later childhood with myopathy, lower-extremity paresthesias, and thrombosis due to homocystinuria [Enns et al., 1999a]. Severely affected children with CblC methylmalonic acidemia can also have ocular abnormalities, such as retinopathy, nystagmus, and worsening vision, as well as hydrocephalus and microcephaly [Rosenblatt et al., 1997]. Cranial imaging usually reveals pathology of the basal ganglia and white matter. The two initial cases reported with CblD presented in later childhood with mental retardation and behavioral problems, although subsequent reports have documented infantile onset with hypotonia and seizures, and early childhood presentations with ataxia and gait abnormalities [Goodman et al., 1970; Suormala et al., 2004]. CblF patients have been reported to have minor facial anomalies and hematologic defects [Shih et al., 1989]. Transcobalamin II deficiency can manifest as failure to thrive in the first months of life, with neurologic disease and mental retardation [Hall, 1992]. A benign form of methylmalonic acidemia has been reported in otherwise healthy children [Ledley et al., 1984].
Laboratory Tests
Methylmalonic acidemia can clinically resemble other organic acidemias, necessitating analysis of urine organic acids for diagnosis. This test reveals large amounts of methylmalonic acid, as well as methylcitrate, propionic acid, and 3-hydroxypropionic acid [Barness et al., 1963; Rosenberg et al., 1968]. Serum amino acids sometimes demonstrate elevation of glycine and hyperhomocysteinemia in the groups in which methylcobalamin metabolism is affected. Ketosis and hyperammonemia are common. Total plasma homocysteine levels are elevated in CblC, CblD, and CblF diseases. Total and free carnitine levels tend to be low, and acylcarnitine profiles reveal increased propionylcarnitine. The cobalamin transport deficiencies are assessed by measuring serum cobalamin levels and absorption by the Schilling test [Cooper and Rosenblatt, 1987]. Determination of complementation group may be approached by complementation studies in fibroblasts or, in some cases, by direct DNA analysis of genes involved in intracellular cobalamin or methylmalonate metabolism [Adams and Venditti, 2008].
Treatment
During acute metabolic crises, treatment is directed toward limiting catabolism and restricting protein intake. The usual protein intake is stopped, and fluid and glucose are given intravenously. Pharmacologic treatment includes carnitine, intramuscular hydroxocobalamin, metronidazole, or neomycin to decrease intestinal propionate production, betaine, and folate [Ogier de Baulny et al., 1998; Roe and Bohan, 1982; Rosenblatt and Cooper, 1987; Thompson et al., 1990]. Improved growth and enhanced nutrition status are seen in patients with methylmalonic acidemia fed an elemental medical food [Yannicelli et al., 2003]. Patients should consume a diet low in the micronutrient precursors proximal to the metabolic block and with adequate calories and total protein to enable growth. Plasma methylmalonic acid levels are followed for metabolic control [Nyhan et al., 1973].
Several transplantations of liver and kidneys in infants and children with methylmalonic acidemia have been reported. Although this approach seems to protect against metabolic crises, it does not lead to complete biochemical correction [Leonard et al., 2001].
Isovaleric Acidemia
Pathophysiology
Deficiency of the mitochondrial enzyme isovaleryl-CoA dehydrogenase, part of the leucine catabolic pathway (see Figure 32-6), leads to accumulation of several metabolites [Rhead and Tanaka, 1980]. Although the toxicity of individual metabolites is not well understood, isovaleric acid is an inhibitor of the tricarboxylic acid cycle and of hematopoiesis in cultured cells [Bergen et al., 1982; Hutchinson et al., 1985].
Clinical Manifestations
Historically, approximately one-half of reported patients present with an acute neonatal metabolic crisis, and the remainder have chronic, intermittent illness. Both forms result from the same inborn error, and the differences in presentation may reflect the timing of catabolic insults or genetic background. The phenotypes of isovaleric acidemia have more recently been described along a continuum of severity ranging from severely affected to mild or even asymptomatic [Vockley and Ensenauer, 2006].
The acute form occurs in infants who are clinically well at birth but who, within the first several days of life, begin to vomit, refuse feeding, and become listless, dehydrated, and lethargic [Mendiola et al., 1984]. They may have seizures, and the odor of sweaty feet often is detected. Untreated patients progress to coma and often to death, which can be caused by metabolic acidosis or attendant features such as infection [Wilson et al., 1984]. Patients who survive have the chronic, intermittent form of the disease, and they may have normal development. In the chronic form, the first episode occurs during the first year of life after a trigger such as a mild infection or protein load. Recurrent episodes involve vomiting, acidosis with ketonuria, lethargy that can progress to coma, and the cheesy or sweaty feet odor. Most patients with isovaleric acidemia who survive crises intact have normal development, but some have mild delay or even severe mental retardation.
Laboratory Tests
Because clinical features of isovaleric academia can resemble several other conditions, analysis of urine organic acids is necessary. During crises, significant elevations of isovalerylglycine and 3-hydroxyisovaleric acid are seen in urine [Tanaka et al., 1968]. Other laboratory abnormalities include metabolic acidosis with mild to moderate ketonuria and lactic acidemia, and sometimes include hyperammonemia. Hematologic abnormalities, such as thrombocytopenia, neutropenia, and pancytopenia, are common [Kelleher et al., 1980]. Hypocalcemia and hyperglycemia often occur.
Treatment
During crises, treatment should ideally be administered by a metabolic specialist. It is similar to that for other organic acidemias: protein restriction and glucose infusion to decrease protein catabolism. Management during intervening episodes includes protein restriction and supplementation with leucine-free foods. Two targeted treatment options, glycine and carnitine, conjugate with isovaleryl-CoA and lead to excretion of the nontoxic products [Cohn et al., 1978; Mayatepek et al., 1991].
3-Methylcrotonyl-CoA Carboxylase Deficiency
3-Methylcrotonyl-CoA carboxylase (MCC) deficiency, also known as 3-methylcrotonylglycinuria, is an autosomal-recessive disorder in the catabolic pathway of leucine (see Figure 32-6). MCC is a biotin-requiring enzyme consisting of two nonidentical subunits, α (MCCA) and β (MCCB), with the biotin-binding site located on the α subunit. Mutations have been identified in both subunits in patients with MCC deficiency, defining two complementation groups for this disorder [Baumgartner et al., 2001]. Although isolated MCC deficiency was once thought to be rare, an unexpectedly large number of patients have been identified in tandem mass spectrometry-based newborn screening programs, and the frequency is estimated at 1 case per 50,000 livebirths [Baumgartner et al., 2001]. Like propionyl-CoA carboxylase deficiency, MCC deficiency can arise from defects in the enzymes of the biotin use and recycling pathway: holocarboxylase synthase and biotinidase (see the section on multiple carboxylase deficiencies).
Clinical Manifestations
The clinical presentation of MCC deficiency is highly variable and ranges from apparently benign to severe and life-threatening. The classic presentation is in infancy or childhood, with rare cases occurring in the neonatal period [Bannwart et al., 1992]. Episodes are often associated with a mild infection or other stress, and they involve vomiting, feeding difficulties, lethargy, hypotonia, hyperreflexia, hypoglycemia, metabolic acidosis, and ketosis. Investigations of family members of affected patients have revealed a number of individuals with MCC deficiency with a normal clinical phenotype. MCC deficiency has been described in healthy mothers of infants identified by expanded newborn screening programs with elevated metabolites suggestive of MCC deficiency [Gibson et al., 1998]. Clinical severity, or lack thereof, is not predicted by the severity of MCC mutation or amount of residual MCC activity, in that most mutations identified in patients are associated with absent or severely diminished MCC activity [Desviat et al., 2003].
Diagnosis
Asymptomatic or presymptomatic patients with MCC deficiency are also detected in expanded newborn screening programs as having elevated 3-hydroxyisovalerylcarnitine in dried blood spots [Koeberl et al., 2003]. This finding should be followed with urine organic acid and acylcarnitine studies for the patient and the mother [Gibson et al., 1998].
Treatment
Treatment of acute episodes is aimed at restoring glucose homeostasis and correcting acidosis [Arnold et al., 2008]. Long-term treatment with carnitine supplementation and dietary restriction of leucine has been found to result in normal growth and development [Sweetman and Williams, 2001]. Responsiveness to biotin has been demonstrated in a small number of cases [Baumgartner et al., 2004].
Multiple Carboxylase Deficiencies
The biotin-responsive multiple carboxylase deficiencies include at least two autosomal-recessive disorders with somewhat distinct but overlapping clinical manifestations, referable to enzymopathies affecting the biotinylation of the four mammalian carboxylases. The carboxylases all require biotin for catalytic activity, and include cytosolic acetyl-CoA carboxylase, the rate-limiting step for the de novo synthesis of fatty acids; mitochondrial pyruvate carboxylase, catalyzing the conversion of pyruvate to oxaloacetate, a tricarboxylic acid cycle intermediate and a precursor for gluconeogenesis; mitochondrial methylcrotonyl-CoA carboxylase, catalyzing an intermediate reaction step in leucine catabolism; and mitochondrial propionyl-CoA carboxylase, which catalyzes an intermediate step in the conversion of isoleucine, valine, and other precursors to succinic acid (see Figure 32-6). In each of these enzymes, the biotin is linked to the epsilon amino group of a lysine residue, situated within the highly conserved amino acid sequence of the biotin-binding site. The clinical presentations and metabolic derangements in the multiple carboxylase deficiencies mirror those of primary deficiencies of each of the carboxylases and the clinical features of acquired nutritional biotin deficiency.
The first patient was described in 1971 and was originally reported as having biotin-responsive β-methylcrotonylglycinuria [Gompertz et al., 1971]. A little more than a decade later, two different disorders – biotinidase deficiency, a defect in the recycling of biotin, and holocarboxylase synthase deficiency, a defect in the biotinylation of carboxylases – were characterized.
Biotinidase Deficiency
Clinical manifestations
Patients with biotinidase deficiency usually present in early to middle infancy, but they may come to medical attention in very early infancy or in later childhood. Because of the usual age of presentation, biotinidase deficiency was delineated as late-onset or infantile-onset multiple carboxylase deficiency [Packman et al., 1981b; Thoene et al., 1981]. Symptoms may be insidious or acute, and can include lethargy, hypotonia, anorexia or vomiting, developmental delays, ataxia, seizures, and coma. Dermatologic manifestations are a frequent feature and can include alopecia, rashes, and mucocutaneous candidiasis. The rash bears a resemblance to that seen in essential fatty acid deficiency or acrodermatitis enteropathica [Williams et al., 1983]. Hearing loss and optic atrophy can be significant long-term complications that do not uniformly respond to therapy. Patients presenting in later childhood or adolescence can come to medical attention because of motor weakness and spastic paresis or because of loss of visual acuity [Wolf, 2007]. When studied, neuropathologic findings in biotinidase deficiency have included changes suggestive of Leigh’s disease and Wernicke’s encephalopathy [Sander et al., 1980]. Such findings are consonant with the notion that neurologic sequelae may result from abnormal intracerebral lactate concentrations [Schurmann et al., 1997].
Etiology
The defect in the infantile-onset form of biotin-responsive multiple carboxylase deficiency was determined by Wolf et al., 1983 to reside in a deficiency of biotinidase activity. Biotinidase is required in the recycling of biotin from carboxylases after the proteolytic degradative turnover of these enzymes (see Figure 32-6). When a carboxylase is hydrolyzed, biotinyl lysine is released, with the biotin remaining bound to the lysine as a compound called biocytin. The release of biotin from biocytin requires the extracellular catalytic activity of biotinidase. In the absence of biotinidase, the cleavage of biocytin does not occur, and biotin is not released for reuse. Biotinidase may also be required for the release of biotin bound to dietary protein, resulting in reduced availability of dietary biotin. The failure to recycle biotin gradually leads to biotin deficiency, a secondary deficiency of biotin-dependent carboxylase activity, and the symptoms of biotinidase deficiency.
Although the focus has been on the consequences of deficiency of the mitochondrial carboxylases, the contribution of acetyl-CoA carboxylase to the clinical manifestations also has been investigated [Gonzalez-Rios et al., 1985b; Packman et al., 1984, 1989; Proud et al., 1990]. Cells deficient in acetyl-CoA carboxylase cannot synthesize malonyl-CoA, required for de novo fatty acid synthesis and fatty acid elongation reactions. In such cells, the fatty acid pattern and composition of membranes and structural elements is abnormal, leading to speculation that such alterations may contribute to the dermatitis [Munnich et al., 1980], to the immunologic derangements reported in a number of patients [Cowan et al., 1979], and to some components of the neurologic manifestations.
Diagnosis
All states in the U.S. and many countries worldwide have incorporated biotinidase testing on filter paper blood spots in state-sponsored newborn screening programs. Ascertainment of biotinidase deficiency in such newborn screening programs has led to the identification of two broad clinical cohorts of patients: those with profound biotinidase deficiency (less than 10 percent of control subjects) and children with partial biotinidase deficiency (10–30 percent of control subjects) [Wolf, 1991]. The combined incidence of profound and partial deficiency is estimated at 1 in 60,000, with the two forms each having approximately equal incidences. The natural history of partial deficiency is not entirely known. Because there are reports of patients with partial deficiency who have become symptomatic, it has been suggested that treatment for partial deficiency might be indicated [McVoy et al., 1990; Wolf, 2010].
Holocarboxylase Synthase Deficiency
Clinical manifestations
Children with holocarboxylase synthase deficiency generally present in the first several days or weeks of life with severe and life-threatening metabolic encephalopathy. The clinical course is variably marked by irritability, lethargy, feeding problems, vomiting, tachypnea, hypertonia or hypotonia, and progression to seizures and coma. The timing of the acute presentation led to the initial characterization of patients as representing a neonatal-onset form of multiple carboxylase deficiency [Packman et al., 1981a, 1984]. Patients with milder forms of holocarboxylase synthase deficiency present later or have a more protracted clinical course [Packman et al., 1984]. These children may have alopecia, rash, ataxia, and developmental delays, a clinical presentation that overlaps that of biotinidase deficiency.
Etiology
The basic defect is in the activity of the enzyme holocarboxylase synthase [Burri et al., 1981], which catalyzes a two-step reaction that results in the covalent binding of biotin to apocarboxylases. All patients with deficient holocarboxylase synthase activity have shown detectable residual activity. Multiple mutations have been identified, with most resulting in the synthesis of an enzyme with an elevated Michaelis–Menten dissociation constant (KM) for biotin. Some patients have mutations that result in a normal KM for biotin but a reduced value for maximum velocity of enzyme-catalyzed reactions (Vmax). These observations have led to speculation that a complete absence of holocarboxylase synthase activity might be lethal [Wolf, 2007].
Diagnosis
Holocarboxylase synthase assay can be performed in cultured fibroblasts or lymphocytes and in amniotic fluid cells [Packman et al., 1982]. A more widespread clinical approach involves measurements of individual carboxylase activities in fibroblasts cultured in low-biotin medium and in high-biotin medium. If the cells are grown in low-biotin medium, individual carboxylase activities will be low, but the activities will be normal or approach normal levels if the cells are grown in high-biotin medium. This assay result contrasts with that for biotinidase deficiency fibroblasts, which demonstrates normal activities, whether grown in high- or low-biotin culture medium [Packman et al., 1984].
Prenatal diagnosis is performed by assay of carboxylases in amniocytes or chorionic villi [Packman et al., 1982; Thuy et al., 1999a], or by molecular identification of mutations in the HLCS gene [Malvagia et al., 2005]. As an adjunct to enzymatic assay and to achieve a rapid result, it is possible to measure the concentration of accumulating analytes (e.g., 3-hydroxyisovalerate, methylcitrate) in amniotic fluid, by stable isotope dilution analysis [Jakobs et al., 1984], but such studies may not be conclusive [Thuy et al., 1999b]. In two pregnancies, prenatal treatment by administration of biotin to the mother during pregnancy [Packman et al., 1982; Roth et al., 1982) was successfully performed in paradigmatic studies of the application and efficacy of co-factor therapy in such settings.
Treatment
Patients respond well to oral biotin in pharmacologic doses. Generally, the starting dose is 10 mg/day. However, the KM for biotin can range from 3 to 70 times normal [Wolf, 2007]. Some patients may require a higher dose, because the mutant enzyme has a higher KM for biotin. There is also a suggestion that patients with mutant holocarboxylase synthase enzymes exhibiting a reduced Vmax may have a poorer response to biotin therapy than those with KM values for biotin mutations [Sakamoto et al., 1999]. Accordingly, careful monitoring of clinical status (including neurologic and cutaneous manifestations), of organic acid levels (including lactate), and of conventional clinical chemistries (i.e., ammonia, electrolytes, and ketones) is mandatory to arrive at optimal dosing of biotin. After treatment levels have been achieved, no dietary restrictions are required.
3-Methylglutaconic Aciduria
Type I: Primary 3-Methylglutaconyl-CoA Hydratase Deficiency
Primary 3-methylglutaconyl-CoA hydratase deficiency is a rare, autosomal-recessive disorder caused by mutations in the 3-methylglutaconyl-CoA hydratase gene (AUH) and characterized by abnormal urinary excretion of 3-methylglutaconic and 3-hydroxyisovaleric acids. The 3-hydroxyisovaleric acid levels are not elevated in the other three types of 3-methylglutaconylaciduria. In the small number of patients reported with this disorder, the spectrum of clinical abnormalities ranges from mild to extremely severe. Although the presentation varies, many patients have had retardation, acidosis, hypoglycemia, hypotonia, and seizures. Suspicion of the diagnosis is raised by the characteristic pattern of urine organic acid excretion, and it is confirmed by assays for the 3-methylglutaconyl-CoA hydratase activity in cultured fibroblasts. Molecular studies to identify mutations in the AUH gene are available in only a limited number of laboratories. Treatment with leucine restriction or l-carnitine supplementation may be effective in some patients [Sweetman and Williams, 2001].
Type II: Barth Syndrome
Barth’s syndrome, also known as X-linked cardioskeletal myopathy and neutropenia, is characterized by mitochondrial abnormalities and abnormal excretion of 3-methylglutaconic acid. Barth’s syndrome is caused by mutations in the G4.5 gene (TAZ), which produces, through alternate splicing, a class of proteins called the tafazzins. Tafazzins are mitochondrial proteins thought to play a role in the structure and function of cardiolipin, a membrane lipid required for proper functioning of the mitochondrial electron transport chain. Clinically, cardiomyopathy and skeletal muscle weakness are usually apparent in affected males in the first year of life. Neutropenia can vary from mild to severe, even within the same patient. Many patients die before they are 4 years old, although some reports suggest an improvement in survival rates due to increased awareness of the diagnosis and more aggressive management of cardiomyopathy and infections [Barth et al., 2004]. No specific symptoms have been identified in heterozygous females, and there is no effective treatment for Barth’s syndrome.
Type III: Costeff Optic Atrophy Syndrome
Costeff’s syndrome is a rare, autosomal-recessive disorder characterized by early-onset bilateral optic atrophy and choreiform movement disorder, and by variable later-onset spasticity and mental retardation. Excretion of 3-methylglutaconic and 3-methylglutaric acids is moderately elevated, and the activity of 3-methylglutaconyl-CoA hydratase is normal. This disorder was once thought to be confined to patients of Iraqi Jewish background, but it has been described in non-Iraqi Jews [Kleta et al., 2002]. Linkage studies of large Iraqi Jewish families have identified mutations in the OPA3 gene, which encodes a protein of unknown function, as being the underlying cause of the disorder [Anikster et al., 2001]. Suspicion of the diagnosis is raised on clinical grounds and confirmed by urine organic acid analysis. No effective treatment is available [Sweetman and Williams, 2001].
Type IV: Unclassified
The type IV 3-methyglutaconic acidurias constitute a heterogeneous group of disorders. Patients share the common features of abnormal excretion and normal 3-methylglutaconyl-CoA hydratase activity. The abnormal 3-methylglutaconic aciduria may result from a variety of causes, possibly affecting energy metabolism. The varied clinical features include severe psychomotor retardation, cerebellar dysgenesis, neonatal hypotonia, absent reflexes, and optic atrophy. No specific treatment is available [Sweetman and Williams, 2001].
Beta-Ketothiolase Deficiency
Inherited β-ketothiolase deficiency, also known as mitochondrial acetoacetyl-CoA thiolase deficiency, is a rare autosomal-recessive disorder caused by defects in mitochondrial acetoacetyl-CoA thiolase, also known as β-ketothiolase, T2, or 3-oxothiolase (see Figure 32-6). This enzyme participates in the pathways of ketone body and isoleucine metabolism. It is related to, but distinct from, four other β-ketothiolase enzymes that participate in reactions of mitochondrial β-oxidation of short- and medium-chain fatty acids, cholesterol synthesis, and peroxisomal β-oxidation. Numerous mutations in the mitochondrial acetoacetyl-CoA thiolase gene (ACAT1) have been described in patients, with some completely abolishing β-ketothiolase activity and others associated with significant residual activity. The degree of clinical severity does not appear to be associated with the molecular severity of mutation nor the degree of residual activity [Fukao et al., 2001, 2003].
Clinical Manifestations
There is wide clinical variability associated with β-ketothiolase deficiency, with most patients presenting with acute ketoacidosis between 5 and 24 months. Attacks are often accompanied by moderate hyperammonemia and hyperglycinemia. Patients are typically well between episodes, with attacks often precipitated by an intercurrent illness or other stress. A small number of patients have been reported with dystonia, which reflects basal ganglia damage, and with mental retardation [Mitchell and Fukao, 2001]. A β-ketothiolase deficiency has been described in asymptomatic siblings of affected patients.
Diagnosis
Urine organic acid analysis of patients with β-ketothiolase deficiency reveals a characteristic pattern of 2-methyl-3-hydroxybutyric and 2-methylacetoacetic acids, with variable excretion of 2-butanone and tiglylglycine. The diagnosis is confirmed by assays for β-ketothiolase activity in lymphocytes or cultured fibroblasts [Mitchell and Fukao, 2001].
Treatment
Treatment of acute episodes is aimed at correcting ketoacidosis with fluids, glucose, and carnitine supplementation, and includes careful monitoring of plasma ammonia, glucose, and electrolytes. Patients are managed long-term with normal or slightly reduced protein intake and with careful monitoring of urine ketones during mild intercurrent illnesses. With appropriate treatment, the long-term prognosis is excellent for those patients in whom neurologic damage has not occurred before diagnosis [Mitchell and Fukao, 2001].
Canavan’s Disease
Canavan’s disease, also known as spongy degeneration of the brain, is an autosomal-recessive disorder caused by a deficiency of aspartoacylase (or N-acyl-l-aspartate amidohydrolase). This enzyme catalyzes the hydrolysis of N-acetyl-l-aspartic acid (NAA) to aspartic acid and acetic acid, and is particularly abundant in white matter of the brain. NAA itself is found only in the CNS, and its function is largely unknown [Beaudet, 2001]. Although the disorder is pan-ethnic, it occurs more frequently among patients of Ashkenazi Jewish descent, for whom the carrier frequency is estimated at 1 case per 40 persons. Two specific aspartoacylase mutations, E285A and Y231X, account for most of the disease alleles.
Clinical Manifestations
Patients with Canavan’s disease are typically normal for the first 1–2 months of life, but then develop hypotonia, poor head control, poor contact, and seizures, as well as macrocephaly and loss of early milestones. In most patients, neurologic abnormalities and macrocephaly are apparent by 6 months of age. Imaging studies demonstrate severe changes in the subcortical white matter and the lower layers of the gray matter, with relative sparing of the central white matter. Later features of the disorder may include spasticity, opisthotonus, and decerebrate or decorticate posturing. Most patients die within the first 3 years of life, although a small number of cases have been reported with later onset and a milder or more variable course [Beaudet, 2001].
Diagnosis
The diagnosis of Canavan’s disease is suspected on the basis of clinical evaluation and neuroimaging studies (Figure 32-7), and confirmed by urine organic acid analysis and the demonstration of abnormal excretion of NAA. Enzyme assays for aspartoacylase activity are available in only a small number of laboratories worldwide and are typically unnecessary to establish a diagnosis. Molecular analysis for common aspartoacylase mutations in the Jewish and European populations can be used for confirmation of the diagnosis and carrier testing of family members. Prenatal diagnosis can be performed by DNA analysis in cases where parental mutations are known. Prenatal diagnosis has also been performed through assays of aspartoacylase activity in chorionic villi or amniocytes, or NAA levels in amniotic fluid, but these approaches have been shown to be unreliable in a number of cases [Beaudet, 2001].
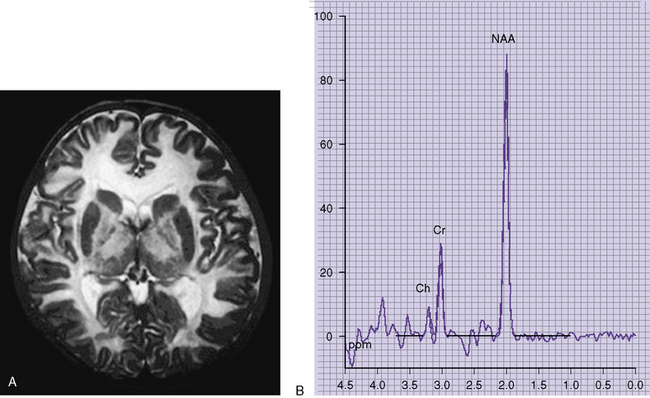
Treatment
There is no known treatment for Canavan’s disease. The American College of Obstetrics and Gynecology (ACOG) has recommended that molecular carrier testing for Canavan’s disease be offered to all Ashkenazi Jewish patients [American College of Obstetrics and Gynecology, 2004]. Such carrier screening programs are aimed at disease prevention through the identification of couples at risk of having an affected child. As with similar screening programs for Tay–Sachs disease, this is expected to reduce the occurrence of infants with Canavan’s disease born in this population dramatically.
Glutaric Aciduria Type 1
In 1975, Goodman et al., 1975 described glutaric acidemia and aciduria in siblings with a neurodegenerative disorder beginning in infancy and characterized by opisthotonus, dystonia, and athetosis. Glutaric acidemia type 1, also known as glutaryl-CoA dehydrogenase deficiency, is an autosomal-recessive condition caused by deficiency of glutaryl-CoA dehydrogenase activity and has an estimated prevalence of approximately 1 case per 100,000 persons [Lindner et al., 2004]. Glutaric acidemia type 1 is relatively common in the Old Order Amish in Lancaster County, Pennsylvania. Glutaric acidemia type 2 (i.e., multiple acyl-CoA dehydrogenase deficiency) is associated with defects in mitochondrial electron transfer flavoprotein or electron transfer flavoprotein dehydrogenase, and is discussed further in Chapter 37. Glutaryl-CoA dehydrogenase is a key enzyme in the degradation pathway for lysine, hydroxylysine, and tryptophan. Deficiency results in accumulation of glutarate and, to a lesser extent, of 3-hydroxyglutarate and glutaconate in body tissues, blood, cerebrospinal fluid, and urine.
Glutaric acidemia type 1 is characterized by irreversible focal striatal necrosis after an acute illness, most often between the ages of 3 and 18 months. Affected children often have macrocephaly, dystonia, and dyskinesia [Strauss et al., 2003]. Macrocephaly may not be present at birth, but head growth velocity is increased [Hoffmann et al., 1996]. Intraretinal hemorrhages and subdural effusions may be mistaken for nonaccidental injury [Morris et al., 1999]. In some cases, the presentation is insidious, with gradual appearance of symptoms [Hoffmann et al., 1996]. Systemic manifestations typical of many other organic acidemias, such as pronounced metabolic ketoacidosis, hypoglycemia, and hyperammonemia, do not occur [Hoffmann et al., 1996; Kyllerman et al., 2004; Strauss et al., 2003]. There appears to be a window of neurologic susceptibility to damage during the first years of life. Striatal injury is rarely encountered after age 3 years, although initial presentation as late as age 19 years has been described [Bahr et al., 2002; Strauss and Morton, 2003b].
The most characteristic head MRI finding in symptomatic children is symmetric widening of the sylvian fissure with poor operculization (“bat wing” appearance) caused by frontotemporal atrophy or hypoplasia (Figure 32-8). Other features include basal ganglia injury, subdural effusions, ventriculomegaly, and delayed myelination [Neumaier-Probst et al., 2004]. Diffusion-weighted imaging may be more sensitive in demonstrating brain lesions than CT or MRI [Elster, 2004].
Fig. 32-8 MRI in glutaric acidemia.
(Courtesy of Dr. A James Barkovich, University of California, San Francisco, CA.)
Urine organic acid analysis often documents highly elevated glutarate and lesser elevations of 3-hydroxyglutarate and glutaconate, but some children with a classic phenotype have low or undetectable levels of these metabolites (so-called low excretors) [Baric et al., 1999]. Newborn screening using tandem mass spectrometry has the potential for presymptomatic detection of glutaric acidemia type 1, although the existence of a low-excretor phenotype will undoubtedly result in missed cases [Lindner et al., 2004; Gallagher et al., 2005]]. In a study of 215 patients, complete absence of glutaryl-CoA dehydrogenase activity was found in more than 50 percent, whereas 34 percent had residual activity up to 5 percent, and a few had residual activity between 5 and 15 percent [Christensen et al., 2004]. Some correlations between severity of mutation and biochemical parameters have been established (e.g., patients with at least one mild mutation frequently have a low-excretor phenotype). However, no correlation between clinical outcome and genotype or biochemical phenotype has been established [Christensen et al., 2004].
Increased glutarate and 3-hydroxyglutarate levels may induce an imbalance in glutamatergic and GABAergic neurotransmission by inhibiting glutamate decarboxylase, the key enzyme in gamma-aminobutyric acid (GABA) synthesis, or through direct damage to striatal GABAergic neurons [Kolker et al., 2004]. 3-Hydroxyglutarate may mimic the excitatory neurotransmitter glutamate and thereby cause excitotoxic cell damage mediated through activation of NMDA receptors [Kolker et al., 2004]. Glutarate inhibits synaptosomal uptake of glutamic acid and produces striatal lesions when injected directly into rat brain [Goodman, 2004]. Other potential contributors to neurotoxicity include cytokine-induced cell damage, mitochondrial dysfunction, increased production of reactive oxygen species, and production of toxic quinolinic acid, an intermediate in tryptophan metabolism in brain [Goodman, 2004; Strauss and Morton, 2003b]. Other reports have emphasized the relatively weak neurotoxicity of glutarate and 3-hydroxyglutarate in animal models and primary neuronal cell cultures. The pathogenesis of striatal necrosis and brain lesions in glutaric acidemia type 1 remains the subject of intensive investigation [Freudenberg et al., 2004; Lund et al., 2004]. Animal models may help resolve these conflicting results.
Treatment of glutaric acidemia type 1 is based on restriction of protein and lysine intake, carnitine and riboflavin supplementation, and rapid intervention in times of intercurrent illness [Muhlhausen et al., 2004]. Antioxidants and creatine have theoretical therapeutic advantages, but such compounds are not routinely used [Muhlhausen et al., 2004; Strauss et al., 2003]. Despite early identification and aggressive management, permanent neurologic damage occurs in approximately one-third of patients [Strauss and Morton, 2003b]. Anticholinergic drugs, such as trihexyphenidyl, and botulinum toxin are beneficial in treating generalized or focal dystonia in patients with glutaric acidemia type 1 [Burlina et al., 2004]. Stereotactic pallidotomy has been performed in three children, with one improving only slightly [Muhlhausen et al., 2004; Rakocevic et al., 2004].
5-Oxoprolinuria
Glutathione (l-γ-glutamyl-l-cysteinylglycine) is a key component in cellular protection from oxidant damage. 5-Oxoprolinuria (i.e., pyroglutamic aciduria) may be caused by defects in enzymes involved in the synthesis and degradation of glutathione, including glutathione synthetase and 5-oxoprolinase, or as a secondary finding in a variety of conditions that are associated with glutathione depletion, including inborn errors of metabolism unrelated to the glutathione cycle (e.g., methylmalonic acidemia, propionic acidemia, urea cycle defects, hepatorenal tyrosinemia, mitochondrial disorders), certain medication exposure (e.g., vigabatrin, acetaminophen, antibiotics), severe burns, multi-organ failure, and malnutrition [Mayatepek, 1999].
Glutathione synthetase deficiency is an autosomal-recessive condition that features massive urine excretion of 5-oxoproline. Clinical manifestations include metabolic acidosis, mental retardation, ataxia, spasticity, seizures, psychiatric disturbances, recurrent bacterial infections, and hemolytic anemia [Ristoff et al., 2001]. Ophthalmologic findings, consisting of minor lens opacities, granular retinal hyperpigmentation, and abnormal electroretinography, were found in two sisters with glutathione synthetase deficiency [Larsson et al., 1985]. Head imaging may reveal brain atrophy [Al-Jishi et al., 1999]. Mild, moderate, and severe phenotypes have been described [Ristoff et al., 2001]. Postmortem examination has revealed generalized cerebellar atrophy, caused by a diffuse loss of granule cells, and ventriculomegaly, especially of the fourth ventricle [Skullerud et al., 1980]. Although patients with 5-oxoprolinase deficiency also excrete large amounts of 5-oxoproline, they are typically asymptomatic. The detection of developmental delay in some patients with 5-oxoprolinase deficiency may reflect ascertainment bias.
The excessive urinary excretion of 5-oxoproline should be detected easily on urine organic acid analysis. 5-Oxoproline is derived from increased synthesis of γ-glutamylcysteine, and it is present in amounts that exceed the normal capacity of 5-oxoprolinase to convert it to glutamate [Wellner et al., 1974]. Definitive diagnosis is made by measuring erythrocyte glutathione synthetase activity or DNA analysis, or both. Varieties of missense and splice site mutations have been found in the GSS gene [Shi et al., 1996].
Treatment of glutathione synthetase deficiency includes correction of metabolic acidosis and high-dose supplementation with vitamins C and E [Ristoff et al., 2001]. Supplementation with N-acetylcysteine may also increase levels of plasma and leukocyte glutathione and may be of therapeutic benefit [Jain et al., 1994; Martensson et al., 1989]. Patients with glutathione synthetase deficiency should avoid medications associated with hemolytic crises in glucose-6-phosphate dehydrogenase deficiency.
Isobutyryl-CoA Dehydrogenase Deficiency
Isobutyryl-CoA dehydrogenase deficiency is an autosomal-recessive disorder of valine metabolism that was first described by Roe et al., 1998. Defective enzyme activity results in impairment of the conversion of isobutyryl-CoA to methylacrylyl-CoA. The initial patient was a developmentally normal 2-year-old girl who developed a dilated cardiomyopathy at age 11 months. She had profound carnitine deficiency, and after carnitine supplementation, acylcarnitine analysis detected markedly increased levels of butyrylcarnitine and isobutyrylcarnitine. Urine organic acids were normal [Roe et al., 1998]. Mutations in the ACAD8 gene were later identified in this patient [Nguyen et al., 2002]. Tandem mass spectrometry newborn screening has identified asymptomatic children [Sass et al., 2004b]. It is uncertain whether children are at risk for serious clinical sequelae or whether therapy is needed.
3-Hydroxyisobutyric Aciduria
3-Hydroxyisobutyric aciduria is a rare autosomal-recessive disorder of valine metabolism; the deficient enzyme has not been identified. The initial patient had repeated episodes of ketoacidosis, failure to thrive, and chronic lactic acidemia [Ko et al., 1991]. Microcephaly, dysmorphic facial features (i.e., short, sloped forehead; telecanthus; down-slanting palpebral fissures; long, prominent philtrum; and micrognathia), seizures, cerebral dysgenesis, brain atrophy, focal white matter abnormalities, and intracerebral calcification have also been described [Chitayat et al., 1992; Sasaki et al., 2001]. This condition has been misdiagnosed as cerebral palsy [Sasaki et al., 2001]. Although most patients have had prominent mental retardation, mild disease and normal intelligence have been reported [Boulat et al., 1995]. Treatment with carnitine and dietary valine and protein restriction appear to be beneficial [Ko et al., 1991; Sasaki et al., 2001].
2-Methylbutyryl-CoA Dehydrogenase Deficiency
2-Methylbutyryl-CoA dehydrogenase deficiency is also known as short- or branched-chain acyl-CoA dehydrogenase deficiency. 2-Methylbutyryl-CoA dehydrogenase catalyzes the conversion of 2-methylbutyryl-CoA to tiglyl-CoA in the isoleucine degradative pathway [Korman, 2006]. Deficiency of this enzyme leads to increased 2-methylbutyrylglycine and 2-methylbutyrlycarnitine in physiologic fluids. A neonate with metabolic acidosis, hypoglycemia, and apnea was the initial reported case. MRI demonstrates ischemic changes bilaterally in parietal and occipital lobes, with increased signal intensity in the lentiform nuclei [Gibson et al., 2000]. Motor delay, strabismus, and generalized muscular atrophy developed during the second year of life in another patient [Andresen et al., 2000]. Tandem mass spectrometry newborn screening identified elevated 5-carbon-saturated acylcarnitine species in eight children of Hmong ancestry. All were treated with a low-protein diet and carnitine supplementation. All patients remained asymptomatic at ages 3–14 months, except for one who developed mild hypotonia [Matern et al., 2003].
Mevalonate Kinase Deficiency
Both disorders are inherited as autosomal-recessive traits. As abnormal isoprenoid synthesis can lead to fever and inflammation, these conditions are classified as autoinflammatory disorders [Houten et al., 2003].
Mevalonic aciduria is characterized by psychomotor retardation, failure to thrive, dysmorphic features (i.e., dolichocephaly, frontal bossing, down-slanting palpebral fissures, and low-set, posteriorly rotated ears), and intercurrent episodes of crisis that feature high fever, vomiting, and diarrhea. Severe cerebellar atrophy and associated ataxia are common. Ocular involvement (e.g., cataracts, retinal dystrophy), hypotonia, mild hepatosplenomegaly, and cholestatic liver disease also occur [Hoffmann et al., 1993]. Recurrent febrile crises tend to decrease with advancing age and are not present in all cases.
Patients with HIDS have lifelong recurrent febrile episodes, lymphadenopathy, splenomegaly, arthralgias, and rashes that begin in infancy. HIDS is not associated with neurologic impairment, and patients are typically asymptomatic in between episodes of fever. Because phenotypic overlap between mevalonic aciduria and HIDS exists, these conditions most likely represent a continuous spectrum of disease, rather than two distinct entities [Simon et al., 2004].
Mevalonic aciduria is diagnosed by detecting elevated mevalonic acid on urine organic acid analysis. Standard clinical chemical investigations find normal or slightly reduced cholesterol and elevated creatine kinase levels, especially during crises [Hoffmann et al., 1993]. Although the level of serum immunoglobulin D may be elevated in mevalonic aciduria, this finding is not invariable. An elevated serum IgD level is a marker for HIDS. Mild elevations of urine mevalonic acid may occur in HIDS during attacks [Cuisset et al., 2001]. Analyzing mevalonate kinase activity in lymphoblasts or using direct DNA analysis can confirm the diagnosis. Most patients are compound heterozygotes for mutant alleles. Some mutations are common to mevalonic aciduria and HIDS [Cuisset et al., 2001].
Treatment for both conditions is largely supportive. Oral cholesterol supplementation, alone or with various combinations of ursodeoxycholic acid, ubiquinone, and vitamin E, did not result in improvement of clinical or biochemical parameters. Lovastatin administration has resulted in the development of severe clinical crises, but some patients have responded dramatically to corticosteroid therapy during acute clinical crises [Hoffmann et al., 1993].
References
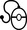 The complete list of references for this chapter is available online at www.expertconsult.com.
The complete list of references for this chapter is available online at www.expertconsult.com.
Adams D., Venditti C.P. Disorders of Intracellular Cobalamin Metabolism In. In: Pagon R.A., Bird T.C., Dolan C.R., et al, editors. GeneReviews [Internet]. Seattle (WA): University of Washington, Seattle, 1993–2008. Feb 25 [updated 2009 Aug 11]
Aliefendioglu D., Tana Aslan A., Coskun T., et al. Transient nonketotic hyperglycinemia: Two case reports and literature review. Pediatr Neurol. 2003;28:151.
Al-Jishi E., Meyer B.F., Rashed M.S., et al. Clinical, biochemical, and molecular characterization of patients with glutathione synthetase deficiency. Clin Genet. 1999;55:444.
Al Tawari A.A., Ramadan D.G., Neubauer D., et al. An early onset form of methylenetetrahydrofolate reductase deficiency: A report of a family from Kuwait. Brain Dev. 2002;24:304.
American College of Obstetrics and Gynecology. The ACOG committee opinion, no. 298, August 2004: Prenatal and preconceptional carrier screening for genetic diseases in individuals of Eastern European Jewish descent. Obstet Gynecol. 2004;104:425.
American College of Obstetricians and Gynecologists Committee on Genetics. ACOG Committee Opinion no. 449: Maternal phenylketonuria. Obstet Gynecol. 2009;114(6):1432-1433.
Andresen B.S., Christensen E., Corydon T.J., et al. Isolated 2-methylbutyrylglycinuria caused by short/branched-chain acyl-coa dehydrogenase deficiency: Identification of a new enzyme defect, resolution of its molecular basis, and evidence for distinct acyl-coa dehydrogenases in isoleucine and valine metabolism. Am J Hum Genet. 2000;67:1095.
Anikster Y., Kleta R., Shaag A., et al. Type III 3-methylglutaconic aciduria (optic atrophy plus syndrome, or Costeff optic atrophy syndrome): Identification of the OPA3 gene and its founder mutation in Iraqi Jews. Am J Hum Genet. 2001;69:1218.
Applegarth D.A., Toone J.R. Nonketotic hyperglycinemia (glycine encephalopathy): Laboratory diagnosis. Mol Genet Metab. 2001;74:139.
Applegarth D.A., Toone J.R. Glycine encephalopathy (nonketotic hyperglycinaemia): Review and update. J Inherit Metab Dis. 2004;27:417.
Applegarth D.A., Toone J.R., Rolland M.O., et al. Non-concordance of CVS and liver glycine cleavage enzyme in three families with non-ketotic hyperglycinaemia (NKH) leading to false negative prenatal diagnoses. Prenat Diagn. 2000;20:367.
Arnold G.L., Koeberl D.D., Matern D., et al. A Delphi-based consensus clinical practice protocol for the diagnosis and management of 3-methylcrotonyl coa carboxylase deficiency. Mol Genet Metab. 2008;93(4):363-370.
Asatoor A.M., Craske J., London D.R., et al. Indole production in Hartnup disease. Lancet. 1963;1:126.
Auerbach V.H., Digeorge A.M., Baldridge R.C., et al. Histidinemia. A deficiency in histidase resulting in the urinary excretion of histidine and of imidazolepyruvic acid. J Pediatr. 1962;60:487.
Ayling J.E., Bailey S.W., Boerth S.R., et al. Hyperphenylalaninemia and 7-pterin excretion associated with mutations in 4a-hydroxy-tetrahydrobiopterin dehydratase/dcoh: Analysis of enzyme activity in intestinal biopsies. Mol Genet Metab. 2000;70:179.
Bahr O., Mader I., Zschocke J., et al. Adult onset glutaric aciduria type I presenting with a leukoencephalopathy. Neurology. 2002;59:1802.
Bannwart C., Wermuth B., Baumgartner R., et al. Isolated biotin-resistant deficiency of 3-methylcrotonyl-coa carboxylase presenting as a clinically severe form in a newborn with fatal outcome. J Inherit Metab Dis. 1992;15:863.
Barber G.W., Spaeth G.L. The successful treatment of homocystinuria with pyridoxine. J Pediatr. 1969;75:463.
Baric I., Wagner L., Feyh P., et al. Sensitivity and specificity of free and total glutaric acid and 3-hydroxyglutaric acid measurements by stable-isotope dilution assays for the diagnosis of glutaric aciduria type I. J Inherit Metab Dis. 1999;22:867.
Barness L.A., Young D., Mellman W.J., et al. Methylmalonate excretion in a patient with pernicious anemia. N Engl J Med. 1963;268:144.
Baron D.N., Dent C.E., Harris H., et al. Hereditary pellagra-like skin rash with temporary cerebellar ataxia, constant renal amino-aciduria, and other bizarre biochemical features. Lancet. 1956;271:421.
Barshop B.A., Khanna A. Domino hepatic transplantation in maple syrup urine disease. N Engl J Med. 2005;353(22):2410-2411.
Barth P.G., Valianpour F., Bowen V.M., et al. X-linked cardioskeletal myopathy and neutropenia (Barth syndrome): An update. Am J Med Genet A. 2004;126:349.
Bartholome K., Lutz P., Bickel H. Determination of phenylalanine hydroxylase activity in patients with phenylketonuria and hyperphenylalaninemia. Pediatr Res. 1975;9:899.
Bauman M.L., Kemper T.L. Morphologic and histoanatomic observations of the brain in untreated human phenylketonuria. Acta Neuropathol (Berl). 1982;58:55.
Baumgartner M.R., Almashanu S., Suormala T., et al. The molecular basis of human 3-methylcrotonyl-coa carboxylase deficiency. J Clin Invest. 2001;107:495.
Baumgartner M.R., Dantas M.F., Suormala T., et al. Isolated 3-methylcrotonyl-coa carboxylase deficiency: Evidence for an allele-specific dominant negative effect and responsiveness to biotin therapy. Am J Hum Genet. 2004;75:790.
Beaudet A.L. Aspartoacylase deficiency: Canavan disease. In: Scriver C.R., Beaudet A.L., Sly W.S., et al, editors. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill; 2001:5799.
Beblo S., Reinhardt H., Muntau A.C., et al. Fish oil supplementation improves visual evoked potentials in children with phenylketonuria. Neurology. 2001;57:1488.
Beckman D.R., Hoganson G., Berlow S., et al. Pathological findings in 5,10-methylene tetrahydrofolate reductase deficiency. Birth Defects Orig Artic Ser. 1987;23:47.
Bergen B.J., Stumpf D.A., Haas R., et al. A mechanism of toxicity of isovaleric acid in rat liver mitochondria. Biochem Med. 1982;27:154.
Bergeron P., Laberge C., Grenier A. Hereditary tyrosinemia in the province of Quebec: Prevalence at birth and geographic distribution. Clin Genet. 1974;5:157.
Bickel H., Gerrard J., Hickmans E.M. Influence of phenylalanine intake on phenylketonuria. Lancet. 1953;265:812.
Blau N., Barnes I., Dhondt J.L. International database of tetrahydrobiopterin deficiencies. J Inherit Metab Dis. 1996;19:8.
Blau N., Thöny B., Cotton R.G.H., et al. Disorders of tetrahydrobiopterin and related biogenic amines. Scriver C.R., Beaudet A.L., Sly W.S., et al, editors. The metabolic and molecular bases of inherited disease. vol. II. New York: McGraw-Hill; 2001:1725.
Boneh A., Degani Y., Harari M. Prognostic clues and outcome of early treatment of nonketotic hyperglycinemia. Pediatr Neurol. 1996;15:137.
Bonetti E., Dent C.E. The determination of the optical configuration of naturally occurring amino acids using specific enzymes and paper chromatography. Biochem J. 1954;57:77.
Borden M., Holm J., Leslie J., et al. Hawkinsinuria in two families. Am J Med Genet. 1992;44:52.
Boulat O., Benador N., Girardin E., et al. 3-Hydroxyisobutyric aciduria with a mild clinical course. J Inherit Metab Dis. 1995;18:204.
Bourget L., Chang T.M. Phenylalanine ammonia-lyase immobilized in microcapsules for the depletion of phenylalanine in plasma in phenylketonuric rat model. Biochim Biophys Acta. 1986;883:432.
Brenton D.P., Cusworth D.C., Gaull G.E. Homocystinuria. Biochemical studies of tissues including a comparison with cystathioninuria. Pediatrics. 1965;35:50.
Brismar J., Aqeel A., Brismar G., et al. Maple syrup urine disease: Findings on CT and MR scans of the brain in 10 infants. Am J Neuroradiol. 1990;11:1219.
Brun A., Borjeson M., Hultberg B., et al. Neonatal non-ketotic hyperglycinemia: A clinical, biochemical and neuropathological study including electronmicroscopic findings. Neuropaediatrie. 1979;10:195.
Buck P.S. The child who never grew. New York: John Day; 1950.
Burke J.P., O’Keefe M., Bowell R., et al. Ophthalmic findings in maple syrup urine disease. Metab Pediatr Syst Ophthalmol. 1991;14:12.
Burlina A.P., Zara G., Hoffmann G.F., et al. Management of movement disorders in glutaryl-coa dehydrogenase deficiency: Anticholinergic drugs and botulinum toxin as additional therapeutic options. J Inherit Metab Dis. 2004;27:911.
Burri B.J., Sweetman L., Nyhan W.L. Mutant holocarboxylase synthetase: Evidence for the enzyme defect in early infantile biotin-responsive multiple carboxylase deficiency. J Clin Invest. 1981;68:1491.
Camfield C.S., Joseph M., Hurley T., et al. Optimal management of phenylketonuria: A centralized expert team is more successful than a decentralized model of care. J Pediatr. 2004;145:53.
Carmel R., Watkins D., Goodman S.I., et al. Hereditary defect of cobalamin metabolism (cblg mutation) presenting as a neurologic disorder in adulthood. N Engl J Med. 1988;318:1738.
Cavalleri F., Berardi A., Burlina A.B., et al. Diffusion-weighted MRI of maple syrup urine disease encephalopathy. Neuroradiology. 2002;44:499.
Cerone R., Holme E., Schiaffino M.C., et al. Tyrosinemia type III: Diagnosis and ten-year follow-up. Acta Paediatr. 1997;86:1013.
Chace D.H., Hillman S.L., Millington D.S., et al. Rapid diagnosis of maple syrup urine disease in blood spots from newborns by tandem mass spectrometry. Clin Chem. 1995;41:62.
Chace D.H., Millington D.S., Terada N., et al. Rapid diagnosis of phenylketonuria by quantitative analysis for phenylalanine and tyrosine in neonatal blood spots by tandem mass spectrometry. Clin Chem. 1993;39:66.
Cheon C.K., Lee B.H., Ko J.M., et al. Novel mutation in SLC6A19 causing late-onset seizures in Hartnup disorder. Pediatr Neurol. 2010;42(5):369-371.
Chien Y.H., Hsu C.C., Huang A., et al. Poor outcome for neonatal-type nonketotic hyperglycinemia treated with high-dose sodium benzoate and dextromethorphan. J Child Neurol. 2004;19:39.
Chitayat D., Meagher-Villemure K., Mamer O.A., et al. Brain dysgenesis and congenital intracerebral calcification associated with 3-hydroxyisobutyric aciduria. J Pediatr. 1992;121:86.
Christensen B., Refsum H., Vintermyr O., et al. Homocysteine export from cells cultured in the presence of physiological or superfluous levels of methionine: Methionine loading of non-transformed, transformed, proliferating, and quiescent cells in culture. J Cell Physiol. 1991;146:52.
Christensen E., Ribes A., Merinero B., et al. Correlation of genotype and phenotype in glutaryl-coa dehydrogenase deficiency. J Inherit Metab Dis. 2004;27:861.
Chuang D.T., Shih V.E.. Maple syrup urine disease (branched-chain ketoaciduria). Scriver C.R., Beaudet A.L., Sly W.S., et al, editors. The metabolic and molecular bases of inherited disease. vol. II. New York: McGraw-Hill; 2001:1971.
Clavero S., Martinez M.A., Perez B., et al. Functional characterization of PCCA mutations causing propionic acidemia. Biochim Biophys Acta. 2002;1588:119.
Cleary M.A., Walter J.H., Wraith J.E., et al. Magnetic resonance imaging in phenylketonuria: Reversal of cerebral white matter change. J Pediatr. 1995;127:251.
Cockburn F., Clark B.J. Recommendations for protein and amino acid intake in phenylketonuric patients. Eur J Pediatr. 1996;155(Suppl 1):S125.
Cohn R.M., Yudkoff M., Rothman R., et al. Isovaleric acidemia: Use of glycine therapy in neonates. N Engl J Med. 1978;299:996.
Cooper B.A., Rosenblatt D.S. Inherited defects of vitamin B12 metabolism. Annu Rev Nutr. 1987;7:291.
Coulombe J.T., Shih V.E., Levy H.L. Massachusetts Metabolic Disorders Screening Program. II. Methylmalonic aciduria. Pediatrics. 1981;67:26.
Cowan M.J., Wara D.W., Packman S., et al. Multiple biotin-dependent carboxylase deficiencies associated with defects in T-cell and B-cell immunity. Lancet. 1979;2:115.
Cuisset L., Drenth J.P., Simon A., et al. Molecular analysis of MVK mutations and enzymatic activity in hyper-igd and periodic fever syndrome. Eur J Hum Genet. 2001;9:260.
Cusworth D.C., Dent C.E. Renal clearances of amino acids in normal adults and in patients with aminoaciduria. Biochem J. 1960;74:550.
D’Angelo A., Selhub J. Homocysteine and thrombotic disease. Blood. 1997;90:1.
Danner D.J., Doering C.B. Human mutations affecting branched chain alpha-ketoacid dehydrogenase. Front Biosci. 1998;3:D517.
Desviat L.R., Perez-Cerda C., Perez B., et al. Functional analysis of MCCA and MCCB mutations causing methylcrotonylglycinuria. Mol Genet Metab. 2003;80:315.
Dhondt J.L. Tetrahydrobiopterin deficiencies: Preliminary analysis from an international survey. J Pediatr. 1984;104:501.
Dhondt J.L. Tetrahydrobiopterin deficiencies: Lessons from the compilation of 200 patients. Dev Brain Dysfunct. 1993;6:139.
Dipple K.M., McCabe E.R. Phenotypes of patients with “simple” Mendelian disorders are complex traits: Thresholds, modifiers, and systems dynamics. Am J Hum Genet. 2000;66:1729.
DiSilvestre D., Koch R., Groffen J. Different clinical manifestations of hyperphenylalaninemia in three siblings with identical phenylalanine hydroxylase genes. Am J Hum Genet. 1991;48:1014.
Dyer C.A., Kendler A., Philibotte T., et al. Evidence for central nervous system glial cell plasticity in phenylketonuria. J Neuropathol Exp Neurol. 1996;55:795.
Edelmann L., Wasserstein M.P., Kornreich R., et al. Maple syrup urine disease: Identification and carrier-frequency determination of a novel founder mutation in the Ashkenazi Jewish population. Am J Hum Genet. 2001;69:863.
Eisensmith R.C., Martinez D.R., Kuzmin A.I., et al. Molecular basis of phenylketonuria and a correlation between genotype and phenotype in a heterogeneous southeastern US population. Pediatrics. 1996;97:512.
Eisensmith R.C., Woo S.L. Population genetics of phenylketonuria. Acta Paediatr Suppl. 1994;407:19.
Elster A.W. Glutaric aciduria type I: Value of diffusion-weighted magnetic resonance imaging for diagnosing acute striatal necrosis. J Comput Assist Tomogr. 2004;28:98.
Enns G.M., Barkovich A.J., Rosenblatt D.S., et al. Progressive neurological deterioration and MRI changes in cblc methylmalonic acidaemia treated with hydroxocobalamin. J Inherit Metab Dis. 1999;22:599.
Enns G.M., Martinez D.R., Kuzmin A.I., et al. Molecular correlations in phenylketonuria: Mutation patterns and corresponding biochemical and clinical phenotypes in a heterogeneous California population. Pediatr Res. 1999;46:594.
Enns G.M., Koch R., Brumm V., et al. Suboptimal outcomes in patients with PKU treated early with diet alone: revisiting the evidence. Mol Genet Metab. 2010. in press
Fariello G., Dionisi-Vici C., Orazi C., et al. Cranial ultrasonography in maple syrup urine disease. Am J Neuroradiol. 1996;17:311.
Fenton W.A., Gravel R.A., Rosenblatt D.S. Disorders of propionate and methylmalonate metabolism. In: Scriver C.R., Beaudet A.L., Sly W.S., et al, editors. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill; 2001:2165.
Fishler K., Azen C.G., Henderson R., et al. Psychoeducational findings among children treated for phenylketonuria. Am J Ment Defic. 1987;92:65.
Følling I. The discovery of phenylketonuria. Acta Paediatr Suppl. 1994;407:4.
Fowler B. Genetic defects of folate and cobalamin metabolism. Eur J Pediatr. 1998;157(Suppl 2):S60.
Fowler B., Jakobs C. Post- and prenatal diagnostic methods for the homocystinurias. Eur J Pediatr. 1998;157(Suppl 2):S88.
Freudenberg F., Lukacs Z., Ullrich K. 3-Hydroxyglutaric acid fails to affect the viability of primary neuronal rat cells. Neurobiol Dis. 2004;16:581.
Fukao T., Scriver C.R., Kondo N. The clinical phenotype and outcome of mitochondrial acetoacetyl-coa thiolase deficiency (beta-ketothiolase or T2 deficiency) in 26 enzymatically proved and mutation-defined patients. Mol Genet Metab. 2001;72:109.
Fukao T., Zhang G.X., Sakura N., et al. The mitochondrial acetoacetyl-coa thiolase (T2) deficiency in Japanese patients: Urinary organic acid and blood acylcarnitine profiles under stable conditions have subtle abnormalities in T2-deficient patients with some residual T2 activity. J Inherit Metab Dis. 2003;26:423.
Gallagher R.C., Cowan T.M., Goodman S.I., et al. Glutaryl-coa dehydrogenase deficiency and newborn screening: retrospective analysis of a low excretor provides further evidence that some cases may be missed. Mol Genet Metab. 2005;86(3):417-420.
Gamez A., Wang L., Straub M., et al. Toward PKU enzyme replacement therapy: pegylation with activity retention for three forms of recombinant phenylalanine hydroxylase. Mol Ther. 2004;9:124.
Gerritsen T., Kaveggia E., Waisman H.A. A new type of idiopathic hyperglycinemia with hypo-oxaluria. Pediatrics. 1965;36:882.
Gibson K.M., Bennett M.J., Naylor E.W., et al. 3-Methylcrotonyl-coenzyme A carboxylase deficiency in Amish/Mennonite adults identified by detection of increased acylcarnitines in blood spots of their children. J Pediatr. 1998;132:519.
Gibson K.M., Burlingame T.G., Hogema B., et al. 2-Methylbutyryl-coenzyme A dehydrogenase deficiency: A new inborn error of L-isoleucine metabolism. Pediatr Res. 2000;47:830.
Gompertz D., Draffan G.H., Watts J.L., et al. Biotin-responsive beta-methylcrotonylglycinuria. Lancet. 1971;2:22.
Gonzalez-Rios M.C., Chuang D.T., Cox R.P., et al. A distinct variant of intermediate maple syrup urine disease. Clin Genet. 1985;27:153.
Gonzalez-Rios M.C., Whitney S.C., Williams M.L., et al. Lipid metabolism in biotin-responsive multiple carboxylase deficiency. J Inherit Metab Dis. 1985;8:184.
Goodman S.I. Development of pathogenic concepts in glutaryl-coa dehydrogenase deficiency: The challenge. J Inherit Metab Dis. 2004;27:801.
Goodman S.I., Markey S.P., Moe P.G., et al. Glutaric aciduria: A “new” disorder of amino acid metabolism. Biochem Med. 1975;12:12.
Goodman S.I., Moe P.G., Hammond K.B., et al. Homocystinuria with methylmalonic aciduria: Two cases in a sibship. Biochem Med. 1970;4:500.
Gortner L., Leupold D., Pohlandt F., et al. Peritoneal dialysis in the treatment of metabolic crises caused by inherited disorders of organic and amino acid metabolism. Acta Paediatr Scand. 1989;78:706.
Gouyon J.B., Semama D., Prevot A., et al. Removal of branched-chain amino acids and alpha-ketoisocaproate by haemofiltration and haemodiafiltration. J Inherit Metab Dis. 1996;19:610.
Gudinchet F., Maeder P., Meuli R.A., et al. Cranial CT and MRI in malignant phenylketonuria. Pediatr Radiol. 1992;22:223.
Guthrie R., Susi A. A simple phenylalanine method for detecting phenylketonuria in large populations of newborn infants. Pediatrics. 1963;32:338.
Güttler F., Henriksen K.F., Guldberg P. Phenylalanine tolerance and loading with relation to genotype in phenylalanine hydroxylase deficiency. Int Pediatr. 1996;11:50.
Haan E.A., Rogers J.G., Lewis G.P., et al. 5,10-Methylenetetrahydrofolate reductase deficiency. Clinical and biochemical features of a further case. J Inherit Metab Dis. 1985;8:53.
Hall C.A. The neurologic aspects of transcobalamin II deficiency. Br J Haematol. 1992;80:117.
Halvorsen K., Halvorsen S. Hartnup disease. Pediatrics. 1963;1:29.
Hamosh A., Johnston M.V.. Nonketotic hyperglycinemia. Scriver C.R., Beaudet A.L., Sly W.S., et al, editors. The metabolic and molecular bases of inherited disease. vol. II. New York: McGraw-Hill; 2001:2065.
Hamosh A., Scharer G., Van Hove J. Glycine encephalopathy. In: Pagon R.A., Bird T.C., Dolan C.R., et al, editors. GeneReviews [Internet]. Seattle (WA): University of Washington, Seattle, 1993–2002. updated 2009 Nov 24
Hanley W.B., Demshar H., Preston M.A., et al. Newborn phenylketonuria (PKU) Guthrie (BIA) screening and early hospital discharge. Early Hum Dev. 1997;47:87.
Hanley W.B., Feigenbaum A.S., Clarke J.T., et al. Vitamin B12 deficiency in adolescents and young adults with phenylketonuria. Eur J Pediatr. 1996;155(Suppl 1):S145.
Harding C.O., Arnold G., Barness L.A., et al. Functional methionine synthase deficiency due to cblg disorder: A report of two patients and a review. Am J Med Genet. 1997;71:384.
Harris E.D.Jr, Sjoerdsma A. Collagen profile in various clinical conditions. Lancet. 1966;2:707.
Hoffmann G.F., Athanassopoulos S., Burlina A.B., et al. Clinical course, early diagnosis, treatment, and prevention of disease in glutaryl-coa dehydrogenase deficiency. Neuropediatrics. 1996;27:115.
Hoffmann G.F., Charpentier C., Mayatepek E., et al. Clinical and biochemical phenotype in 11 patients with mevalonic aciduria. Pediatrics. 1993;91:915.
Holme E., Lindstedt S. Nontransplant treatment of tyrosinemia. Clin Liver Dis. 2000;4:805.
Hommes F.A., Moss L. Myelin turnover in hyperphenylalaninaemia. A re-evaluation with the HPH-5 mouse. J Inherit Metab Dis. 1992;15:243.
Hoover-Fong J.E., Shah S., Van Hove J.L., et al. Natural history of nonketotic hyperglycinemia in 65 patients. Neurology. 2004;63:1847.
Houten S.M., Frenkel J., Waterham H.R. Isoprenoid biosynthesis in hereditary periodic fever syndromes and inflammation. Cell Mol Life Sci. 2003;60:1118.
Huether G., Kaus R., Neuhoff V. Brain development in experimental hyperphenylalaninaemia: Myelination. Neuropediatrics. 1982;13:177.
Huijbregts S.C., de Sonneville L.M., van Spronsen F.J., et al. The neuropsychological profile of early and continuously treated phenylketonuria: Orienting, vigilance, and maintenance versus manipulation-functions of working memory. Neurosci Biobehav Rev. 2002;26:697.
Hunziker N. Richner-Hanhart syndrome and tyrosinemia type II. Dermatologica. 1980;160:180.
Hutchinson R.J., Bunnell K., Thoene J.G. Suppression of granulopoietic progenitor cell proliferation by metabolites of the branched-chain amino acids. J Pediatr. 1985;106:62.
Huttenlocher P.R. The neuropathology of phenylketonuria: Human and animal studies. Eur J Pediatr. 2000;159(Suppl 2):S102.
Infante J.P., Huszagh V.A. Impaired arachidonic (20:4n-6) and docosahexaenoic (22:6n-3) acid synthesis by phenylalanine metabolites as etiological factors in the neuropathology of phenylketonuria. Mol Genet Metab. 2001;72:185.
Isherwood D.M. Homocystinuria. BMJ. 1996;313:1025.
Jackson A.H., Applegarth D.A., Toone J.R., et al. Atypical nonketotic hyperglycinemia with normal cerebrospinal fluid to plasma glycine ratio. J Child Neurol. 1999;14:464.
Jaeken J., Corbeel L., Casaer P., et al. Dipropylacetate (valproate) and glycine metabolism. Lancet. 1977;2:617.
Jain A., Buist N.R., Kennaway N.G., et al. Effect of ascorbate or N-acetylcysteine treatment in a patient with hereditary glutathione synthetase deficiency. J Pediatr. 1994;124:229.
Jakobs C., Sweetman L., Nyhan W.L., et al. Stable isotope dilution analysis of 3-hydroxyisovaleric acid in amniotic fluid: Contribution to the prenatal diagnosis of inherited disorders of leucine catabolism. J Inherit Metab Dis. 1984;7:15.
Jan W., Zimmerman R.A., Wang Z.J., et al. MR diffusion imaging and MR spectroscopy of maple syrup urine disease during acute metabolic decompensation. Neuroradiology. 2003;45:393.
Johnson J.L., Hainline B.E., Rajagopalan K.V. Characterization of the molybdenum cofactor of sulfite oxidase, xanthine, oxidase, and nitrate reductase. Identification of a pteridine as a structural component. J Biol Chem. 1980;255:1783.
Johnson J.L., Rajagopalan K.V. An HPLC assay for detection of elevated urinary S-sulphocysteine, a metabolic marker of sulphite oxidase deficiency. J Inherit Metab Dis. 1995;18:40.
Johnson J.L., Waud W.R., Rajagopalan K.V., et al. Inborn errors of molybdenum metabolism: Combined deficiencies of sulfite oxidase and xanthine dehydrogenase in a patient lacking the molybdenum cofactor. Proc Natl Acad Sci USA. 1980;77:3715.
Jouvet P., Jugie M., Rabier D., et al. Combined nutritional support and continuous extracorporeal removal therapy in the severe acute phase of maple syrup urine disease. Intensive Care Med. 2001;27:1798.
Kahler S.G., Sherwood W.G., Woolf D., et al. Pancreatitis in patients with organic acidemias. J Pediatr. 1994;124:239.
Kang E.S., Gerald P.S. Hereditary tyrosinemia and abnormal pyrrole metabolism. J Pediatr. 1970;77:397.
Kang T.S., Wang L., Sarkissian C.N., et al. Converting an injectable protein therapeutic into an oral form: phenylalanine ammonia lyase for phenylketonuria. Mol Genet Metab. 2010;99(1):4-9.
Kanwar Y.S., Manaligod J.R., Wong P.W. Morphologic studies in a patient with homocystinuria due to 5,10-methylenetetrahydrofolate reductase deficiency. Pediatr Res. 1976;10:598.
Kaplan P., Mazur A., Field M., et al. Intellectual outcome in children with maple syrup urine disease. J Pediatr. 1991;119:46.
Kayaalp E., Treacy E., Waters P.J., et al. Human phenylalanine hydroxylase mutations and hyperphenylalaninemia phenotypes: A metanalysis of genotype-phenotype correlations. Am J Hum Genet. 1997;61:1309.
Kelleher J.F.Jr, Yudkoff M., Hutchinson R., et al. The pancytopenia of isovaleric acidemia. Pediatrics. 1980;65:1023.
Kleta R., Skovby F., Christensen E., et al. 3-Methylglutaconic aciduria type III in a non-Iraqi-Jewish kindred: Clinical and molecular findings. Mol Genet Metab. 2002;76:201.
Kleta R., Romeo E., Ristic Z., et al. Mutations in SLC6A19, encoding B0AT1, cause Hartnup disorder. Nat Genet. 2004;36:999-1002.
Ko F.J., Nyhan W.L., Wolff J., et al. 3-Hydroxyisobutyric aciduria: An inborn error of valine metabolism. Pediatr Res. 1991;30:322.
Koch J.H. Robert Guthrie: The PKU story. Pasadena: Hope Publishing House; 1997.
Koch R., Moseley K.D., Yano S., et al. Large neutral amino acid therapy and phenylketonuria: A promising approach to treatment. Mol Genet Metab. 2003;79:110.
Koch S.E., Packman S., Koch T.K., et al. Dermatitis in treated maple syrup urine disease. J Am Acad Dermatol. 1993;28:289.
Koeberl D.D., Millington D.S., Smith W.E., et al. Evaluation of 3-methylcrotonyl-coa carboxylase deficiency detected by tandem mass spectrometry newborn screening. J Inherit Metab Dis. 2003;26:25.
Kolker S., Koeller D.M., Okun J.G., et al. Pathomechanisms of neurodegeneration in glutaryl-coa dehydrogenase deficiency. Ann Neurol. 2004;55:7.
Komrower G.M., Lambert A.M., Cusworth D.C., et al. Dietary treatment of homocystinuria. Arch Dis Child. 1966;41:666.
Korman S.H. Inborn errors of isoleucine degradation: a review. Mol Genet Metab. 2006;89(4):289-299.
Korman S.H., Boneh A., Ichinohe A., et al. Persistent NKH with transient or absent symptoms and a homozygous GLDC mutation. Ann Neurol. 2004;56:139.
Korman S.H., Gutman A. Pitfalls in the diagnosis of glycine encephalopathy (non-ketotic hyperglycinemia). Dev Med Child Neurol. 2002;44:712.
Kure S., Kojima K., Ichinohe A., et al. A comprehensive mutation analysis of GLDC, AMT and GCSH in glycine encephalopathy. J Inherit Metab Dis. 2003;26:66.
Kure S., Rolland M.O., Leisti J., et al. Prenatal diagnosis of non-ketotic hyperglycinaemia: Enzymatic diagnosis in 28 families and DNA diagnosis detecting prevalent Finnish and Israeli-Arab mutations. Prenat Diagn. 1999;19:717.
Kvittingen E.A. Tyrosinaemia type I – an update. J Inherit Metab Dis. 1991;14:554.
Kwok S.C., Ledley F.D., DiLella A.G., et al. Nucleotide sequence of a full-length complementary DNA clone and amino acid sequence of human phenylalanine hydroxylase. Biochemistry. 1985;24:556.
Kyllerman M., Skjeldal O., Christensen E., et al. Long-term follow-up, neurological outcome and survival rate in 28 Nordic patients with glutaric aciduria type 1. Eur J Paediatr Neurol. 2004;8:121.
Lang T.F., Parr J.R., Matthews E.E., et al. Practical difficulties in the diagnosis of transient non-ketotic hyperglycinaemia. Dev Med Child Neurol. 2008;50(2):157-159.
Larsson A., Wachtmeister L., von Wendt L., et al. Ophthalmological, psychometric and therapeutic investigation in two sisters with hereditary glutathione synthetase deficiency (5-oxoprolinuria). Neuropediatrics. 1985;16:131.
Ledley F.D., Levy H.L., Shih V.E., et al. Benign methylmalonic aciduria. N Engl J Med. 1984;311:1015.
Lee H.H., Kim do J., Ahn H.J., et al. Crystal structure of T-protein of the glycine cleavage system: Cofactor binding, insights into H-protein recognition, and molecular basis for understanding nonketotic hyperglycinemia. J Biol Chem. 2004;279:50514.
Lenke R.R., Levy H.L. Maternal phenylketonuria and hyperphenylalaninemia. An international survey of the outcome of untreated and treated pregnancies. N Engl J Med. 1980;303:1202.
Leonard J.V., Vijayaraghavan S., Walter J.H. The impact of screening for propionic and methylmalonic acidaemia. Eur J Pediatr. 2003;162(Suppl 1):S21.
Leonard J.V., Walter J.H., McKiernan P.J. The management of organic acidaemias: The role of transplantation. J Inherit Metab Dis. 2001;24:309.
Levitt M., Nixon P.F., Pincus J.H., et al. Transport characteristics of folates in cerebrospinal fluid: A study utilizing doubly labeled 5-methyltetrahydrofolate and 5-formyltetrahydrofolate. J Clin Invest. 1971;50:1301.
Levy H.L., Shih V.E., Madigan P.M. Routine newborn screening for histidinemia. Clinical and biochemical results. N Engl J Med. 1974;291:1214.
Lindblad B., Lindblad B.S., Olin P., et al. Methylmalonic acidemia. A disorder associated with acidosis, hyperglycinemia, and hyperlactatemia. Acta Paediatr Scand. 1968;57:417.
Lindner M., Kolker S., Schulze A., et al. Neonatal screening for glutaryl-coa dehydrogenase deficiency. J Inherit Metab Dis. 2004;27:851.
Lucke T., Perez-Cerda C., Baumgartner M., et al. Propionic acidemia: Unusual course with late onset and fatal outcome. Metabolism. 2004;53:809.
Lueder G.T., Steiner R.D. Ophthalmic abnormalities in molybdenum cofactor deficiency and isolated sulfite oxidase deficiency. J Pediatr Ophthalmol Strabismus. 1995;32:334.
Lund T.M., Christensen E., Kristensen A.S., et al. On the neurotoxicity of glutaric, 3-hydroxyglutaric, and trans-glutaconic acids in glutaric acidemia type 1. J Neurosci Res. 2004;77:143.
MacDonald J.T., Sher P.K. Ophthalmoplegia as a sign of metabolic disease in the newborn. Neurology. 1977;27:971.
MacLeod E.L., Gleason S.T., van Calcar S.C., et al. Reassessment of phenylalanine tolerance in adults with phenylketonuria is needed as body mass changes. Mol Genet Metab. 2009;98(4):331-337.
Maeda T., Inutsuka M., Goto K., et al. Transient nonketotic hyperglycinemia in an asphyxiated patient with pyridoxine-dependent seizures. Pediatr Neurol. 2000;22:225.
Mahon B.E., Levy H.L. Maternal Hartnup disorder. Am J Med Genet. 1986;24:513.
Malvagia S., Morrone A., Pasquini E., et al. First prenatal molecular diagnosis in a family with holocarboxylase synthetase deficiency. Prenat Diagn. 2005;25(12):1117-1119.
Martensson J., Gustafsson J., Larsson A. A therapeutic trial with N-acetylcysteine in subjects with hereditary glutathione synthetase deficiency (5-oxoprolinuria). J Inherit Metab Dis. 1989;12:120.
Matalon R., Koch R., Michals-Matalon K., et al. Biopterin responsive phenylalanine hydroxylase deficiency. Genet Med. 2004;6:27.
Matalon R., Surendran S., Matalon K.M., et al. Future role of large neutral amino acids in transport of phenylalanine into the brain. Pediatrics. 2003;112:1570.
Matern D., He M., Berry S.A., et al. Prospective diagnosis of 2-methylbutyryl-coa dehydrogenase deficiency in the Hmong population by newborn screening using tandem mass spectrometry. Pediatrics. 2003;112:74.
Matsui S.M., Mahoney M.J., Rosenberg L.E. The natural history of the inherited methylmalonic acidemias. N Engl J Med. 1983;308:857.
Mayatepek E. 5-Oxoprolinuria in patients with and without defects in the gamma-glutamyl cycle. Eur J Pediatr. 1999;158:221.
Mayatepek E., Kurczynski T.W., Hoppel C.L. Long-term L-carnitine treatment in isovaleric acidemia. Pediatr Neurol. 1991;7:137.
McKiernan P.J. Nitisinone in the treatment of hereditary tyrosinaemia type 1. Drugs. 2006;66(6):743-750.
McVoy J.R., Levy H.L., Lawler M., et al. Partial biotinidase deficiency: Clinical and biochemical features. J Pediatr. 1990;116:78.
Mendiola J.Jr, Robotham J.L., Liehr J.G., et al. Neonatal lethargy due to isovaleric acidemia and hyperammonemia. Tex Med. 1984;80:52.
Menkes J.H., Hurst P.L., Craig J.M. A new syndrome: Progressive familial infantile cerebral dysfunction associated with an unusual urinary substance. Pediatrics. 1954;14:462.
Millot F., Dhondt J.L., Mazingue F., et al. Changes of cerebral biopterin and biogenic amine metabolism in leukemic children receiving 5 g/m2 intravenous methotrexate. Pediatr Res. 1995;37:151.
Milne M.D., Crawford M.A., Girao C.B., et al. The metabolic disorder in Hartnup disease. Q J Med. 1960;29:407.
Mitchell G., Larochelle J., Lambert M., et al. Neurologic crises in hereditary tyrosinemia. N Engl J Med. 1990;322:432.
Mitchell G.A., Fukao T. Inborn errors of ketone body metabolism. In: Scriver C.R., Beaudet A.L., Sly W.S., et al, editors. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill; 2001:2327.
Mitchell G.A., Russo P., Dubois J., et al. Tyrosinemia. In: Suchy F.J., editor. Liver disease in children. St Louis: Mosby–Year Book; 2001:667.
Moller H.E., Weglage J., Bick U., et al. Brain imaging and proton magnetic resonance spectroscopy in patients with phenylketonuria. Pediatrics. 2003;112:1580.
Morris A.A., Hoffmann G.F., Naughten E.R., et al. Glutaric aciduria and suspected child abuse. Arch Dis Child. 1999;80:404.
Morton D.H., Strauss K.A., Robinson D.L., et al. Diagnosis and treatment of maple syrup disease: A study of 36 patients. Pediatrics. 2002;109:999.
Mudd S.H., Finkelstein J.D., Irreverre F., et al. Homocystinuria: An enzymatic defect. Science. 1964;143:1443.
Mudd S.H., Irreverre F., Laster L. Sulfite oxidase deficiency in man: Demonstration of the enzymatic defect. Science. 1967;156:1599.
Mudd S.H., Skovby F., Levy H.L., et al. The natural history of homocystinuria due to cystathionine beta-synthase deficiency. Am J Hum Genet. 1985;37:1.
Mudd S.H., Uhlendorf B.W., Freeman J.M., et al. Homocystinuria associated with decreased methylenetetrahydrofolate reductase activity. Biochem Biophys Res Commun. 1972;46:905.
Muhlhausen C., Hoffmann G.F., Strauss K.A., et al. Maintenance treatment of glutaryl-coa dehydrogenase deficiency. J Inherit Metab Dis. 2004;27:885.
Munnich A., Saudubray J.M., Coude F.X., et al. Fatty-acid-responsive alopecia in multiple carboxylase deficiency. Lancet. 1980;1:1080.
Muntau A.C., Roschinger W., Habich M., et al. Tetrahydrobiopterin as an alternative treatment for mild phenylketonuria. N Engl J Med. 2002;347:2122.
Naughten E.R., Yap S., Mayne P.D. Newborn screening for homocystinuria: Irish and world experience. Eur J Pediatr. 1998;157(Suppl 2):S84.
Nellis M.M., Danner D.J. Gene preference in maple syrup urine disease. Am J Hum Genet. 2001;68:232.
Neuberger J.M., Schweitzer S., Rolland M.O., et al. Effect of sodium benzoate in the treatment of atypical nonketotic hyperglycinaemia. J Inherit Metab Dis. 2000;23:22.
Neumaier-Probst E., Harting I., Seitz A., et al. Neuroradiological findings in glutaric aciduria type I (glutaryl-coa dehydrogenase deficiency). J Inherit Metab Dis. 2004;27:869.
Nguyen T.V., Andresen B.S., Corydon T.J., et al. Identification of isobutyryl-coa dehydrogenase and its deficiency in humans. Mol Genet Metab. 2002;77:68.
Nyhan W. Abnormalities in amino acid metabolism in clinical medicine. Norwalk, CT: Appleton-Century-Crofts; 1984.
Nyhan W.L., Fawcett N., Ando T., et al. Response to dietary therapy in B12 unresponsive methylmalonic acidemia. Pediatrics. 1973;51:539.
Nyhan W.L., Rice-Kelts M., Klein J., et al. Treatment of the acute crisis in maple syrup urine disease. Arch Pediatr Adolesc Med. 1998;152:593.
Ogier de Baulny H., Gerard M., Saudubray J.M., et al. Remethylation defects: Guidelines for clinical diagnosis and treatment. Eur J Pediatr. 1998;157(Suppl 2):S77.
Okano Y., Eisensmith R.C., Guttler F., et al. Molecular basis of phenotypic heterogeneity in phenylketonuria. N Engl J Med. 1991;324:1232.
Paans A.M., Pruim J., Smit G.P., et al. Neurotransmitter positron emission tomographic-studies in adults with phenylketonuria, a pilot study. Eur J Pediatr. 1996;155(Suppl 1):S78.
Packman S., Caswell N., Gonzalez-Rios M.C., et al. Acetyl coa carboxylase in cultured fibroblasts: Differential biotin dependence in the two types of biotin-responsive multiple carboxylase deficiency. Am J Hum Genet. 1984;36:80.
Packman S., Cowan M.J., Golbus M.S., et al. Prenatal treatment of biotin responsive multiple carboxylase deficiency. Lancet. 1982;1:1435.
Packman S., Sweetman L., Baker H., et al. The neonatal form of biotin-responsive multiple carboxylase deficiency. J Pediatr. 1981;99:418.
Packman S., Sweetman L., Yoshino M., et al. Biotin-responsive multiple carboxylase deficiency of infantile onset. J Pediatr. 1981;99:421.
Packman S., Whitney S.C., Fitch M., et al. Abnormal fatty acid composition of biotin-responsive multiple carboxylase deficiency fibroblasts. J Inherit Metab Dis. 1989;12:47.
Parini R., Sereni L.P., Bagozzi D.C., et al. Nasogastric drip feeding as the only treatment of neonatal maple syrup urine disease. Pediatrics. 1993;92:280.
Perez-Cerda C., Clavero S., Perez B., et al. Functional analysis of PCCB mutations causing propionic acidemia based on expression studies in deficient human skin fibroblasts. Biochim Biophys Acta. 2003;1638:43.
Perry T.L., Dunn H.G., Hansen S., et al. Early diagnosis and treatment of homocystinuria. Pediatrics. 1966;37:502.
Perry T.L., Urquhart N., Hansen S. Studies of the glycine cleavage enzyme system in brain from infants with glycine encephalopathy. Pediatr Res. 1977;11:1192.
Pey A.L., Perez B., Desviat L.R., et al. Mechanisms underlying responsiveness to tetrahydrobiopterin in mild phenylketonuria mutations. Hum Mutat. 2004;24:388.
Phenylketonuria (PKU). Screening and management. NIH Consens Statement. 2000;17:1.
Phillips M.D., McGraw P., Lowe M.J., et al. Diffusion-weighted imaging of white matter abnormalities in patients with phenylketonuria. Am J Neuroradiol. 2001;22:1583.
Pietz J., Benninger C., Schmidt H., et al. Long-term development of intelligence (IQ) and EEG in 34 children with phenylketonuria treated early. Eur J Pediatr. 1988;147:361.
Pietz J., Fatkenheuer B., Burgard P., et al. Psychiatric disorders in adult patients with early-treated phenylketonuria. Pediatrics. 1997;99:345.
Pietz J., Meyding-Lamade U.K., Schmidt H. Magnetic resonance imaging of the brain in adolescents with phenylketonuria and in one case of 6-pyruvoyl tetrahydropteridine synthase deficiency. Eur J Pediatr. 1996;155(Suppl 1):S69.
Proud V.K., Rizzo W.B., Patterson J.W., et al. Fatty acid alterations and carboxylase deficiencies in the skin of biotin-deficient rats. Am J Clin Nutr. 1990;51:853.
Puliyanda D.P., Harmon W.E., Peterschmitt M.J., et al. Utility of hemodialysis in maple syrup urine disease. Pediatr Nephrol. 2002;17:239.
Rakocevic G., Lyons K.E., Wilkinson S.B., et al. Bilateral pallidotomy for severe dystonia in an 18-month-old child with glutaric aciduria. Stereotact Funct Neurosurg. 2004;82:80.
Rhead W.J., Tanaka K. Demonstration of a specific mitochondrial isovaleryl-coa dehydrogenase deficiency in fibroblasts from patients with isovaleric acidemia. Proc Natl Acad Sci USA. 1980;77:580.
Ristoff E., Mayatepek E., Larsson A. Long-term clinical outcome in patients with glutathione synthetase deficiency. J Pediatr. 2001;139:79.
Roe C.R., Bohan T.P. L-Carnitine therapy in propionicacidaemia. Lancet. 1982;1:1411.
Roe C.R., Cederbaum S.D., Roe D.S., et al. lsolated isobutyryl-coa dehydrogenase deficiency: An unrecognized defect in human valine metabolism. Mol Genet Metab. 1998;65:264.
Rosenberg L.E., Lilljeqvist A.C., Hsia Y.E. Methylmalonic aciduria. An inborn error leading to metabolic acidosis, long-chain ketonuria and intermittent hyperglycinemia. N Engl J Med. 1968;278:1319.
Rosenblatt D.S., Aspler A.L., Shevell M.I., et al. Clinical heterogeneity and prognosis in combined methylmalonic aciduria and homocystinuria (cblC). J Inherit Metab Dis. 1997;20:528.
Rosenblatt D.S., Cooper B.A. Inherited disorders of vitamin B12 metabolism. Blood Rev. 1987;1:177.
Rosenblatt D.S., Cooper B.A. Inherited disorders of vitamin B12 utilization. Bioessays. 1990;12:331.
Rosenblatt D.S., Cooper B.A., Schmutz S.M., et al. Prenatal vitamin B12 therapy of a fetus with methylcobalamin deficiency (cobalamin E disease). Lancet. 1985;1:1127.
Roth A., Nogues C., Monnet J.P., et al. Anatomo-pathological findings in a case of combined deficiency of sulphite oxidase and xanthine oxidase with a defect of molybdenum cofactor. Virchows Arch A Pathol Anat Histopathol. 1985;405:379.
Roth K.S., Yang W., Allan L., et al. Prenatal administration of biotin in biotin responsive multiple carboxylase deficiency. Pediatr Res. 1982;16:126.
Ruetschi U., Cerone R., Perez-Cerda C., et al. Mutations in the 4-hydroxyphenylpyruvate dioxygenase gene (HPD) in patients with tyrosinemia type III. Hum Genet. 2000;106:654.
Russo P.A., Mitchell G.A., Tanguay R.M. Tyrosinemia: A review. Pediatr Dev Pathol. 2001;4:212.
Sakaguchi Y., Yoshino M., Aramaki S., et al. Dihydrolipoyl dehydrogenase deficiency: A therapeutic trial with branched-chain amino acid restriction. Eur J Pediatr. 1986;145:271.
Sakai L.Y., Keene D.R., Engvall E. Fibrillin, a new 350-kd glycoprotein, is a component of extracellular microfibrils. J Cell Biol. 1986;103:2499.
Sakamoto O., Suzuki Y., Li X., et al. Relationship between kinetic properties of mutant enzyme and biochemical and clinical responsiveness to biotin in holocarboxylase synthetase deficiency. Pediatr Res. 1999;46:671.
Sakura N., Ono H., Nomura S., et al. Betaine dose and treatment intervals in therapy for homocystinuria due to 5,10-methylenetetrahydrofolate reductase deficiency. J Inherit Metab Dis. 1998;21:84.
Samady J.A., Schwartz R.A., Shih L.Y., et al. Acrodermatitis enteropathica-like eruption in an infant with nonketotic hyperglycinemia. J Dermatol. 2000;27:604.
Sander J.E., Malamud N., Cowan M.J., et al. Intermittent ataxia and immunodeficiency with multiple carboxylase deficiencies: A biotin-responsive disorder. Ann Neurol. 1980;8:544.
Sarkissian C.N., Shao Z., Blain F., et al. A different approach to treatment of phenylketonuria: Phenylalanine degradation with recombinant phenylalanine ammonia lyase. Proc Natl Acad Sci USA. 1999;96:2339.
Sasaki M., Iwata H., Sugai K., et al. A severely brain-damaged case of 3-hydroxyisobutyric aciduria. Brain Deve. 2001;23:243.
Sass J.O., Hofmann M., Skladal D., et al. Propionic acidemia revisited: A workshop report. Clin Pediatr (Phila). 2004;43:837.
Sass J.O., Sander S., Zschocke J. Isobutyryl-coa dehydrogenase deficiency: Isobutyrylglycinuria and ACAD8 gene mutations in two infants. J Inherit Metab Dis. 2004;27:741.
Sassa S., Kappas A. Hereditary tyrosinemia and the heme biosynthetic pathway. Profound inhibition of delta-aminolevulinic acid dehydratase activity by succinylacetone. J Clin Invest. 1983;71:625.
Sassa S., Kawakami M., Cerami A. Inhibition of the growth and differentiation of erythroid precursor cells by an endotoxin-induced mediator from peritoneal macrophages. Proc Natl Acad Sci USA. 1983;80:1717.
Saudubray J.M., Charpentier C. Clinical phenotypes: Diagnosis/algorithms. In: Scriver C.R., Beaudet A.L., Sly W.S., et al, editors. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill; 2001:1327.
Schadewaldt P., Bodner-Leidecker A., Hammen H.W., et al. Significance of L-alloisoleucine in plasma for diagnosis of maple syrup urine disease. Clin Chem. 1999;45:1734.
Schmidt H., Ullrich K., Korinthenberg R., et al. Basal ganglion calcification in hyperphenylalaninemia due to deficiency of dihydropteridine reductase. Pediatr Radiol. 1988;19:54.
Schuler A., Blau N., Ponzone A. Monoamine oxidase inhibitors in tetrahydrobiopterin deficiency. Eur J Pediatr. 1995;154:997.
Schurmann M., Engelbrecht V., Lohmeier K., et al. Cerebral metabolic changes in biotinidase deficiency. J Inherit Metab Dis. 1997;20:755.
Scriver C.R. Hartnup disease: A genetic modification of intestinal and renal transport of certain neutral alpha-amino acids. N Engl J Med. 1965;273:530.
Scriver C.R. Nutrient-gene interactions: The gene is not the disease and vice versa. Am J Clin Nutr. 1988;48:1505.
Scriver C.R., Kaufman S.. Hyperphenylalaninemia: Phenylalanine hydroxylase deficiency. Scriver C.R., Beaudet A.L., Sly W.S., et al, editors. The metabolic and molecular bases of inherited disease. vol. II. New York: McGraw-Hill; 2001:1667.
Scriver C.R., Levy H.L. Histidinaemia. Part I. Reconciling retrospective and prospective findings. J Inherit Metab Dis. 1983;6:51.
Scriver C.R., Mackenzie S., Clow C.L., et al. Thiamine-responsive maple-syrup-urine disease. Lancet. 1971;1:310.
Scriver C.R., Mahon B., Levy H.L., et al. The Hartnup phenotype: Mendelian transport disorder, multifactorial disease. Am J Hum Genet. 1987;40:401.
Scriver C.R., Waters P.J. Monogenic traits are not simple: Lessons from phenylketonuria. Trends Genet. 1999;15:267.
Sener R.N. Nonketotic hyperglycinemia: Diffusion magnetic resonance imaging findings. J Comput Assist Tomogr. 2003;27:538.
Seow H.F., Bröer S., Bröer A., et al. Hartnup disorder is caused by mutations in the gene encoding the neutral amino acid transporter SLC6A19. Nat Genet. 2004;36:1003-1007.
Shi Z.Z., Habib G.M., Rhead W.J., et al. Mutations in the glutathione synthetase gene cause 5-oxoprolinuria. Nat Genet. 1996;14:361.
Shih V.E., Axel S.M., Tewksbury J.C., et al. Defective lysosomal release of vitamin B12 (cb1f): A hereditary cobalamin metabolic disorder associated with sudden death. Am J Med Genet. 1989;33:555.
Shintaku H. Disorders of tetrahydrobiopterin metabolism and their treatment. Curr Drug Metab. 2002;3:123.
Simon A., Kremer H.P., Wevers R.A., et al. Mevalonate kinase deficiency: Evidence for a phenotypic continuum. Neurology. 2004;62:994.
Skullerud K., Marstein S., Schrader H., et al. The cerebral lesions in a patient with generalized glutathione deficiency and pyroglutamic aciduria (5-oxoprolinuria). Acta Neuropathol (Berl). 1980;52:235.
Smith I., Beasley M.G., Ades A.E. Effect on intelligence of relaxing the low phenylalanine diet in phenylketonuria. Arch Dis Child. 1991;66:311.
Smith I., Knowles J. Behaviour in early treated phenylketonuria: A systematic review. Eur J Pediatr. 2000;159(Suppl 2):S89.
Spada M., Blau N., Meli C., et al. Different strategies in the treatment of dihydropteridine reductase deficiency. Pteridines. 1996;7:107.
Spada M., Schuler A., Blau N., et al. Deprenyl in 6-pyruvoyl tetrahydropterin synthase deficiency (BH4 deficiency). Pteridines. 1995;5:144.
Stampfer M.J., Malinow M.R., Willett W.C., et al. A prospective study of plasma homocyst(e)ine and risk of myocardial infarction in US physicians. JAMA. 1992;268:877.
Strauss K.A., Morton D.H. Branched-chain ketoacyl dehydrogenase deficiency: Maple syrup disease. Curr Treat Options Neurol. 2003;5:329.
Strauss K.A., Morton D.H. Type I glutaric aciduria. Part 2. A model of acute striatal necrosis. Am Med Genet. 2003;121C:53.
Strauss K.A., Puffenberger E.G., Robinson D.L., et al. Type I glutaric aciduria, part 1: Natural history of 77 patients. Am J Med Genet. 2003;121C:38.
Suormala T., Baumgartner M.R., Coelho D., et al. The cbld defect causes either isolated or combined deficiency of methylcobalamin and adenosylcobalamin synthesis. J Biol Chem. 2004;279:42742.
Sweetman L., Williams J.C. Branched chain organic acidurias. In: Scriver C.R., Beaudet A.L., Sly W.S., et al, editors. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill; 2001:2125.
Taccone A., Schiaffino M.C., Cerone R., et al. Computed tomography in maple syrup urine disease. Eur J Radiol. 1992;14:207.
Tada K., Hirono H., Arakawa T. Endogenous renal clearance rates of free amino acids in prolinuric and Hartnup patients. Tohoku J Exp Med. 1967;93:57.
Tada K., Kure S. Non-ketotic hyperglycinaemia: Molecular lesion, diagnosis and pathophysiology. J Inherit Metab Dis. 1993;16:691.
Tada K., Tateda H., Arashima S., et al. Intellectual development in patients with untreated histidinemia. A collaborative study group of neonatal screening for inborn errors of metabolism in Japan. J Pediatr. 1982;101:562.
Tan W.H., Eichler F.S., Hoda S., et al. Isolated sulfite oxidase deficiency: a case report with a novel mutation and review of the literature. Pediatrics. 2005;116(3):757-766.
Tanaka K., Orr J.C., Isselbacher K.J. Identification of beta-hydroxyisovaleric acid in the urine of a patient with isovaleric acidemia. Biochim Biophys Acta. 1968;152:638.
Tanaka Y., Matsuo N., Tsuzaki S., et al. On-off phenomenon in a child with tetrahydrobiopterin deficiency due to 6-pyruvoyl tetrahydropterin synthase deficiency (BH4 deficiency). Eur J Pediatr. 1989;148:450.
Tharp B.R. Unique EEG pattern (comb-like rhythm) in neonatal maple syrup urine disease. Pediatr Neurol. 1992;8:65.
Thoene J., Baker H., Yoshino M., et al. Biotin-responsive carboxylase deficiency associated with subnormal plasma and urinary biotin. N Engl J Med. 1981;304:817.
Thompson G.N., Chalmers R.A., Walter J.H., et al. The use of metronidazole in management of methylmalonic and propionic acidaemias. Eur J Pediatr. 1990;149:792.
Thuy L.P., Jurecki E., Nemzer L., et al. Prenatal diagnosis of holocarboxylase synthetase deficiency by assay of the enzyme in chorionic villus material followed by prenatal treatment. Clin Chim Acta. 1999;284(1):59-68.
Thuy L.P., Belmont J., Nyhan W.L. Prenatal diagnosis and treatment of holocarboxylase synthetase deficiency. Prenat Diagn. 1999;19:108-112.
Touati G., Rusthoven E., Depondt E., et al. Dietary therapy in two patients with a mild form of sulphite oxidase deficiency. Evidence for clinical and biological improvement. J Inherit Metab Dis. 2000;23:45.
Treacy E.P., Delente J.J., Elkas G., et al. Analysis of phenylalanine hydroxylase genotypes and hyperphenylalaninemia phenotypes using L-[1-13C]phenylalanine oxidation rates in vivo: A pilot study. Pediatr Res. 1997;42:430.
Trefz F.K., Blau N. Potential role of tetrahydrobiopterin in the treatment of maternal phenylketonuria. Pediatrics. 2003;112:1566.
Trefz F.K., Burgard P., Konig T., et al. Genotype-phenotype correlations in phenylketonuria. Clin Chim Acta. 1993;217:15.
Tuchman M., Kelly P., Watkins D., et al. Vitamin B12-responsive megaloblastic anemia, homocystinuria, and transient methylmalonic aciduria in cble disease. J Pediatr. 1988;113:1052.
Van der, Klei-van Moorsel J.M., Smit L.M., et al. Infantile isolated sulphite oxidase deficiency: Report of a case with negative sulphite test and normal sulphate excretion. Eur J Pediatr. 1991;150:196.
Van Hove J.L., Kishnani P., Muenzer J., et al. Benzoate therapy and carnitine deficiency in non-ketotic hyperglycinemia. Am J Med Genet. 1995;59:444.
Van Hove J.L., Kishnani P.S., Demaerel P., et al. Acute hydrocephalus in nonketotic hyperglycemia. Neurology. 2000;54:754.
van Spronsen F.J., Enns G.M. Future treatment strategies in phenylketonuria. Mol Genet Metab. 2010;99(Suppl 1):S90-S95.
Viola A., Chabrol B., Nicoli F., et al. Magnetic resonance spectroscopy study of glycine pathways in nonketotic hyperglycinemia. Pediatr Res. 2002;52:292.
Virmani K., Widhalm K. Histidinemia: A biochemical variant or a disease? J Am Coll Nutr. 1993;12:115.
Vockley J., Ensenauer R. Isovaleric acidemia: new aspects of genetic and phenotypic heterogeneity. Am J Med Genet C Semin Med Genet. 2006;142C(2):95-103.
Waisbren S.E., Mahon B.E., Schnell R.R., et al. Predictors of intelligence quotient and intelligence quotient change in persons treated for phenylketonuria early in life. Pediatrics. 1987;79:351.
Waters P.J., Scriver C.R. In vitro expression analysis of mutations in phenylalanine hydroxylase: Linking genotype to phenotype and structure to function. Hum Mutat. 1998;11:4.
Watkins D., Rosenblatt D.S. Functional methionine synthase deficiency (cble and cblg): Clinical and biochemical heterogeneity. Am J Med Genet. 1989;34:427.
Webster L.T., Siddiqui U.A., Lucas S.V., et al. Identification of separate acyl-coa:glycine and acyl-coa:L-glutamine N-acyltransferase activities in mitochondrial fractions from liver of rhesus monkey and man. J Biol Chem. 1976;251:3352.
Weglage J., Ullrich K., Pietsch M., et al. Untreated non-phenylketonuric-hyperphenylalaninaemia: Intellectual and neurological outcome. Eur J Pediatr. 1996;155(Suppl 1):S26.
Weglage J., Wiedermann D., Moller H., et al. Pathogenesis of different clinical outcomes in spite of identical genotypes and comparable blood phenylalanine concentrations in phenylketonurics. J Inherit Metab Dis. 1998;21:181.
Wellner V.P., Sekura R., Meister A., et al. Glutathione synthetase deficiency, an inborn error of metabolism involving the gamma-glutamyl cycle in patients with 5-oxoprolinuria (pyroglutamic aciduria). Proc Natl Acad Sci USA. 1974;71:2505.
Welsh M.C., Pennington B.F., Ozonoff S., et al. Neuropsychology of early-treated phenylketonuria: Specific executive function deficits. Child Dev. 1990;61:1697.
Wendel U., Langenbeck U., Lombeck I., et al. Exchange transfusion in acute episodes of maple syrup urine disease. Studies on branched-chain amino and keto acids. Eur J Pediatr. 1982;138:293.
Wendel U., Saudubray J.M., Bodner A., et al. Liver transplantation in maple syrup urine disease. Eur J Pediatr. 1999;158(Suppl 2):S60.
Westall R.G., Dancis J., Miller S. Maple sugar urine disease. Am J Dis Child. 1957;94:571.
Widaman K.F., Azen C. Relation of prenatal phenylalanine exposure to infant and childhood cognitive outcomes: Results from the International Maternal PKU Collaborative Study. Pediatrics. 2003;112:1537.
Wilcken B., Yu J.S., Brown D.A. Natural history of Hartnup disease. Arch Dis Child. 1977;52:38.
Wilcken D.E., Dudman N.P., Tyrrell P.A. Homocystinuria due to cystathionine beta-synthase deficiency – the effects of betaine treatment in pyridoxine-responsive patients. Metabolism. 1985;34:1115.
Wilcken D.E., Wilcken B. The natural history of vascular disease in homocystinuria and the effects of treatment. J Inherit Metab Dis. 1997;20:295.
Williams M.L., Packman S., Cowan M.J. Alopecia and periorificial dermatitis in biotin-responsive multiple carboxylase deficiency. J Am Acad Dermatol. 1983;9:97.
Wilson W.G., Audenaert S.M., Squillaro E.J. Hyperammonaemia in a preterm infant with isovaleric acidaemia. J Inherit Metab Dis. 1984;7:71.
Wiltshire E.J., Poplawski N.K., Harrison J.R., et al. Treatment of late-onset nonketotic hyperglycinaemia: Effectiveness of imipramine and benzoate. J Inherit Metab Dis. 2000;23:15.
Wolf B. Worldwide survey of neonatal screening for biotinidase deficiency. J Inherit Metab Dis. 1991;14:923.
Wolf B. Disorders of biotin metabolism. In: Scriver C., Beaudet A., Valle D., et al, editors. The metabolic and molecular basis of inherited disease, ed 8. NY: McGraw-Hill; 2001:3935-3964.
Wolf B. Clinical issues and frequent questions about biotinidase deficiency. Mol Genet Metab. 2010;100(1):6-13.
Wolf B., Grier R.E., Allen R.J., et al. Biotinidase deficiency: The enzymatic defect in late-onset multiple carboxylase deficiency. Clin Chim Acta. 1983;131:273.
Wong P.W., Justice P., Hruby M., et al. Folic acid nonresponsive homocystinuria due to methylenetetrahydrofolate reductase deficiency. Pediatrics. 1977;59:749.
Woody N.C., Snyder C.H., Harris J.A. Histidinemia. Am J Dis Child. 1965;110:606.
Woody R.C., Brewster M.A. Adverse effects of trimethoprim-sulfamethoxazole in a child with dihydropteridine reductase deficiency. Dev Med Child Neurol. 1990;32:639.
Yannicelli S., Acosta P.B., Velazquez A., et al. Improved growth and nutrition status in children with methylmalonic or propionic acidemia fed an elemental medical food. Mol Genet Metab. 2003;80:181.
Yap S., Rushe H., Howard P.M., et al. The intellectual abilities of early-treated individuals with pyridoxine-nonresponsive homocystinuria due to cystathionine beta-synthase deficiency. J Inherit Metab Dis. 2001;24:437.
Yorifuji T., Kawai M., Mamada M., et al. Living-donor liver transplantation for propionic acidaemia. J Inherit Metab Dis. 2004;27:205.
Zeman J., Bayer M., Stepan J. Bone mineral density in patients with phenylketonuria. Acta Paediatr. 1999;88:1348.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Chapter 32 Aminoacidemias and Organic Acidemias
Laboratory Approaches to Diagnosis
Laboratory testing for the symptomatic patient can proceed at many levels, including metabolic screening tests, quantitative metabolite profiles, specific enzyme or other functional assays, and DNA mutation analysis. The specific laboratory approach is often dictated by the clinical and family history and by results of routine laboratory investigations. An extensive description of diagnostic algorithms for the laboratory evaluation of patients suspected of having a metabolic disease has been published [Saudubray and Charpentier, 2001].
For the acutely ill patient, a comprehensive evaluation should include quantitative assessment of plasma amino acids, urine organic acid analyses, plasma carnitine (free and total) levels, and identification and quantitation of acylcarnitines in plasma or serum. These tests should be ordered in conjunction with other basic tests, including hematologic cell counts, electrolytes, blood glucose, blood gases, uric acid, liver transaminases, ammonia, and lactic and pyruvic acid levels. This approach can identify many cases of amino acid disorders and organic acidemias. The interpretation of metabolic tests is greatly enhanced when the laboratory is made aware of the clinical, medication, and dietary history of the patient because these factors can significantly influence results. Depending on the clinical evaluation and results of basic chemistry studies, additional testing may be warranted; this may include levels of urine orotic acid (e.g., elevated in certain urea cycle defects) (see Chapter 33), cerebrospinal fluid glycine (together with the plasma glycine level, which is elevated in glycine encephalopathy), cerebrospinal fluid neurotransmitters (see Chapter 39), and urine S-sulfocysteine (elevated in sulfite oxidase deficiency or molybdenum co-factor deficiency).
Inheritance and Genetic Counseling
Treatment of heritable metabolic disorders involves considerations beyond the acute phase of the illness and even beyond the prognosis of the proband. Because of the importance of genetic counseling to the family, the physician has an obligation to try to arrive at a diagnosis, however poor the prognosis for the proband. Identifying a specific entity enables the family to be counseled about recurrence risks. Most inborn errors of metabolism are inherited as autosomal-recessive traits. There are a few disorders, such as the urea cycle defect of ornithine transcarbamylase deficiency, that are inherited as X-linked disorders. In the case of an autosomal-recessive condition, the affected relative is a sibling of either gender. In X-linked disorders, the affected relative may be a maternal uncle, a brother, or a mildly affected mother or other female relative. Some disorders are caused by mitochondrial DNA mutations (see Chapter 37), and maternal transmission to all children in a sibship is observed. In all circumstances, a detailed family history may reveal an affected relative who has a similar illness, and this can be of diagnostic importance. Special attention should be given to a family history of stillbirths, unexplained deaths, and neurologic diseases or delayed development of any degree or severity.
Aminoacidemias
Phenylketonuria
Phenylketonuria, described by Asbørn Følling in 1934 [Følling, 1994], is caused by deficient activity of phenylalanine hydroxylase (PAH), a hepatic enzyme that converts phenylalanine to tyrosine (Figure 32-1). The biochemical block results in the accumulation of phenylalanine, which is then converted to phenylpyruvic acid and phenyllactic acid, phenylketones that are excreted in the urine. Tetrahydrobiopterin is a necessary co-factor in the PAH reaction, and elevated phenylalanine levels rarely may be caused by inherited disorders of tetrahydrobiopterin synthesis, including guanosine triphosphate (GTP) cyclohydrolase I, 6-pyruvoyltetrahydrobiopterin synthase, pterin-4α-carbinolamine dehydratase, and dihydropteridine reductase deficiencies (see Figure 32-1). Phenylalanine is neurotoxic, and untreated patients with classic phenylketonuria are typically mentally retarded. In the 1950s, a diet in which phenylalanine intake was restricted was shown to normalize plasma phenylalanine levels and stop urinary excretion of phenylpyruvic acid [Bickel et al., 1953]. Selective restriction of phenylalanine intake by using phenylalanine-free medical formulas and foods (and tyrosine supplementation), which provides enough additional protein and nutrients to support normal growth, remains the mainstay of phenylketonuria therapy.
Fig. 32-1 Regulation of phenylalanine hydroxylase activity.
(From Wilcox WR, Cederbaum SD. Amino acid metabolism. In: Rimoin D, Connor J, Pyeritz R, Korf B, eds. Principles and practice of medical genetics, 4th edn. Philadelphia: Churchill Livingstone, 2002:2406.)
Mandatory population newborn screening for phenylketonuria, in combination with postnatal presymptomatic therapy, was begun in the 1960s using the Guthrie bacterial inhibition assay [Guthrie and Susi, 1963; Koch, 1997]. Modern newborn screening programs have switched to techniques that directly assay phenylalanine and tyrosine levels; the most recent innovation is tandem mass spectrometry. The presymptomatic institution of and continued adherence to specific dietary therapy prevents mental retardation. Phenylketonuria is a paradigmatic and landmark success story in biochemical genetics, and it is reviewed in some detail.
Classification
A blood phenylalanine level above the normal range (30–110 μM) is referred to as hyperphenylalaninemia. Patients have been classified as having nonphenylketonuria hyperphenylalaninemia if their blood phenylalanine levels without dietary therapy are 360–600 μM. Classic phenylketonuria is characterized by untreated phenylalanine levels of more than 1000 μM [Scriver and Kaufman, 2001]. A range of reduced PAH-specific activity correlates broadly with the severity of the phenotype. When it has been measured directly (i.e., liver biopsy) or indirectly (i.e., l-[1-13C]phenylalanine breath test), residual liver PAH-specific activity is relatively high in milder hyperphenylalaninemic patients, whereas enzyme activity is zero to low in the more severe cases of classic phenylketonuria [Bartholome et al., 1975]. Measured PAH activity also correlates to some degree with tolerance for dietary protein [Güttler et al., 1996]. Patients with classic phenylketonuria can tolerate very little phenylalanine in the diet (<500 mg/day). The classification of phenylketonuria into subtypes based on blood phenylalanine levels is arbitrary, and some patients (i.e., mild, variant, or atypical phenylketonuria) fall between the two extreme biochemical phenotypes. Because environmental (phenylalanine intake) and genetic (modifier genes) factors alter the biochemical and neurodevelopmental phenotypes in this single-gene disorder, phenylketonuria in many ways behaves more like a complex trait than a monogenic disorder [Kayaalp et al., 1997].
Clinical Manifestations
Profound mental retardation is the most significant clinical finding in untreated or poorly treated phenylketonuria. Acute metabolic encephalopathy, a common feature of many inborn errors of metabolism, does not occur in phenylketonuria. Children with phenylketonuria appear normal at birth and have normal early development, even if untreated. Neurologic manifestations appear insidiously and include reduced rate of growth of head circumference, developmental delay, abnormalities in muscle tone, and hyperactive deep tendon reflexes. Affected children may have lighter pigmentation than other family members (i.e., reduced melanin synthesis) and a musty odor attributed to phenylacetic acid. Eczema and decreased bone mineral density may occur [Zeman et al., 1999]. Patients exposed to chronically elevated phenylalanine levels ultimately develop microcephaly, seizures (e.g., tonic-clonic, myoclonic, infantile spasms), tremors, athetosis, and spasticity, and they may be misdiagnosed as having cerebral palsy. Psychiatric and behavior problems, including autistic behavior and attention-deficit hyperactivity disorder, are common [Pietz et al., 1997; Smith and Knowles, 2000].
Brain magnetic resonance imaging (MRI) may detect dysmyelination, especially T2 enhancement in the periventricular white matter, a finding that is potentially reversible with the initiation of dietary therapy [Cleary et al., 1995]. Abnormal areas of white matter demonstrate restricted diffusion of water, possibly indicating increased myelin turnover [Phillips et al., 2001].
In the past, most untreated phenylketonuria patients were institutionalized, and many born before universal newborn screening remain so. An eloquent and inspiring description of the life of a child with untreated phenylketonuria is given in a short monograph by Pearl S. Buck [Buck, 1950].
As a rule, well-treated classic phenylketonuria patients have normal IQs. However, recent studies have found that children and adults with phenylketonuria may experience cognitive symptoms, such as problems in executive functioning, as well as disturbance in emotional and behavioral functioning despite early and continuous treatment [Enns et al., 2010]. Dietary control of the blood phenylalanine level appears to be the best predictor of ultimate IQ [Waisbren et al., 1987], but careful psychometric testing of well-treated individuals has detected instances and degrees of impairment in visual-motor skills, abstract reasoning, problem solving, specific aspects of executive control, attention, verbal memory, expressive naming, and verbal fluency [Fishler et al., 1987]. Such neuropsychologic impairments may be a consequence of mid-dorsolateral prefrontal cortex dysfunction caused by abnormal catecholamine levels [Huijbregts et al., 2002]. Abnormal EEG patterns, including general slowing and generalized paroxysmal activity with or without spikes, may be demonstrated for children with phenylketonuria, even if they are well treated [Pietz et al., 1988]. Emotional disorders (e.g., depression, anxiety, phobias) and hyperactive behavior are more frequently encountered in persons with classic phenylketonuria than in the general population [Smith and Knowles, 2000]. However, untreated mild hyperphenylalaninemic patients are not at risk for developing neuropsychologic impairment [Weglage et al., 1996].
Maternal Phenylketonuria Syndrome
Elevated maternal blood phenylalanine levels can cross the placenta and cause fetal birth defects, including microcephaly, dysmorphic features, and congenital heart defects. Children with the maternal phenylketonuria syndrome are typically heterozygous for the mutant PAH allele, and they are not affected with phenylketonuria. More than 90 percent of children born to women with untreated classic phenylketonuria have mental retardation; 70 percent have microcephaly, 40 percent have intrauterine growth retardation, and 12 percent have congenital heart disease [Lenke and Levy, 1980]. The risk to the fetus is greatest with increasing phenylalanine levels in maternal blood. Dietary control should ideally be achieved before 3 months prior to conception, and mothers with phenylketonuria should be monitored carefully by an experienced center throughout pregnancy [ACOG, 2009]. Optimal birth outcomes occur when blood phenylalanine levels between 120 and 360 μM are achieved by 8–10 weeks’ gestation [Widaman and Azen, 2003]. Mothers with phenylketonuria can safely breastfeed their children.
Diagnosis
The Guthrie bacterial inhibition assay was a technical breakthrough, allowing newborn screening of large populations. The growth inhibition of Bacillus subtilis by β-2-thienylalanine is prevented by phenylalanine, phenylpyruvic acid, and phenyllactic acid, and this forms the basis of the Guthrie test. Fluorometric assays or tandem mass spectrometry is used in screening and monitoring [Chace et al., 1993]. False-positive results may be seen in neonates with low birth weight or liver disease, or in infants on parenteral alimentation. False-negative results may occur if the newborn screen is performed too early (especially less than 12 hours after birth) [Hanley et al., 1997]. Confirmation of the diagnosis is made by analysis of blood phenylalanine and tyrosine concentrations by means of high-performance liquid chromatography, fluorescent methods, or tandem mass spectrometry. Without the introduction of a phenylalanine-restricted diet, maximal elevation in plasma phenylalanine is typically reached within or soon after the first week of life in patients with classic phenylketonuria. Urine phenylpyruvic acid causes the appearance of a deep green color when ferric chloride is added. This ferric chloride test is sometimes performed as part of a metabolic screening panel for the evaluation of patients suspected of having an inborn error of metabolism, but it should not be used to confirm a diagnosis of phenylketonuria because of a lack of sensitivity and specificity. All patients with confirmed hyperphenylalaninemia must have urine pterins analyzed for defects in tetrahydrobiopterin metabolism. Phenylketonuria may be suspected in a child or adult and should reasonably be included in the differential diagnosis of a given patient of any age presenting with neurodevelopmental delay of unknown origin. In such settings, diagnostic testing for phenylketonuria (i.e., serum phenylalanine levels) must be done, even if there is a history or a record of a normal newborn screen.
Genetics
Phenylketonuria is an autosomal-recessive disorder with an incidence of 1 case per 10,000 people in the general white population of northern European ancestry [Eisensmith and Woo, 1994]. Phenylketonuria is more common in Turkey, Scotland, and Czechoslovakia, and among Arabic populations and Yemenite Jews (1 case per 2500–5000 persons). It is relatively uncommon in Japan and Finland, and among African Americans (1 case per 100,000–200,000 persons) [Scriver and Kaufman, 2001]. Nonphenylketonuria hyperphenylalaninemia has an overall incidence of 1 case per 50,000 persons.
The PAH gene on chromosome 12q24.1 spans 90 kb and contains 13 exons [Kwok et al., 1985]. Almost 500 mutations have been reported throughout all exons and flanking sequences. A detailed account of PAH mutant alleles and other DNA variations is maintained at the PAH Mutation Analysis Consortium website (http://www.pahdb.mcgill.ca/) [Waters and Scriver, 1998]. Most DNA alterations are missense mutations, although splice, nonsense, and frameshift mutations and large deletions and insertions have been identified. Most patients are compound heterozygotes, carrying a different mutant allele on each chromosome. Prevalences of specific mutant alleles differ from population to population [Eisensmith and Woo, 1994]. For example, R408W, IVS12nt1, and IVS10nt11 are severe mutations that account for about 50 percent of mutant alleles in Europeans, but they are rare in Asians. Conversely, mutant alleles R243Q, R413P, and Y204C are common in Asians, but they rare in Europeans [Eisensmith and Woo, 1994].
Pathogenesis
Although PAH is a hepatic enzyme, the major effect of its deficiency is brain dysfunction. Elevated phenylalanine appears to be the cause of neurotoxicity. However, the precise cause of the mental retardation observed in untreated phenylketonuria is not understood. Defective brain myelination may be related to decreased biosynthesis of myelin proteins, because brain protein synthesis is inhibited by excessive phenylalanine [Huether et al., 1982]. CNS effects may be ascribable to more global amino acid imbalances; elevated phenylalanine may affect the CNS concentrations of neutral amino acids by competitive inhibition of a shared amino acid transporter, with relative brain deprivation of tyrosine, tryptophan, and branched-chain amino acids [Huttenlocher, 2000]. Decreased brain tyrosine and tryptophan may lead to decreased neurotransmitter synthesis. The cause of abnormal brain myelination is also unclear. In a phenylketonuria mouse model (i.e., enu2 mouse), there is evidence that oligodendrocytes overexpress glial fibrillary acid protein and become nonmyelinating [Dyer et al., 1996]. Increased myelin turnover has also been observed in the enu2 mouse [Hommes and Moss, 1992]. In phenylketonuria patients studied by positron emission tomography (PET), brain protein synthesis appears to be impaired, which could also affect the myelination process [Paans et al., 1996]. Brain pathology in untreated classic phenylketonuria includes abnormalities in width of the cortical plate, cell density and organization, dendritic arborization and number of synaptic spines, and abnormal myelination [Bauman and Kemper, 1982].
Genotype-Phenotype Correlations
Mutations in the PAH gene cause phenylketonuria and nonphenylketonuria hyperphenylalaninemia. However, the final biochemical phenotype (i.e., blood phenylalanine level and dietary phenylalanine tolerance) and clinical phenotype (i.e., IQ) depend on the severity of the mutations, and are influenced by patient adherence to a strict diet, the effects of modifying genetic factors, and other environmental factors. Potential modifier genes may encode proteins mediating interindividual rates of protein synthesis (and phenylalanine use) or basal metabolism; synthesis and degradation of the PAH enzyme protein; gastrointestinal absorption of phenylalanine; hepatic uptake of circulating phenylalanine; metabolism of the tetrahydrobiopterin co-factor; and rate of phenylalanine transport across the blood–brain barrier [Dipple and McCabe, 2000; Treacy et al., 1997]. Because patients – including sibling patients – with identical mutations can have divergent neurodevelopmental progress, mutation identification may not predict the severity of the disease with certainty in a given patient [DiSilvestre et al., 1991; Enns et al., 1999b].
Genes encoding proteins responsible for transport of amino acids across the blood–brain barrier are especially attractive candidates for modifying factors in phenylketonuria. In a study of two siblings with identical genotypes but widely different IQ, in vivo nuclear MR spectroscopy documented lower peak brain phenylalanine levels and more rapid decreases in brain phenylalanine concentration in the less severely affected sibling after a phenylalanine load [Weglage et al., 1998]. Subsequent studies have confirmed the wide interindividual variation of brain to blood phenylalanine concentrations in classic phenylketonuria patients with divergent cognitive phenotypes [Moller et al., 2003].
Despite these considerations, trends can be identified in whole populations. In general, individuals with classic phenylketonuria and poor dietary control have mental retardation, although exceptions exist. If patients with severe mutations are started on strict dietary therapy in the neonatal period and maintained on such treatment throughout life, cognition will be normal. Using in vitro expression analysis in cultured cells transfected with mutant cDNAs, specific mutant alleles (i.e., genotype) can be categorized as severe or mild; such categorization correlates with biochemical or clinical severity (i.e., phenotype) in most patients in relatively homogenous populations. In a study of German and Dutch subjects, the predicted level of PAH activity correlated strongly with neonatal pretreatment levels of blood phenylalanine and dietary phenylalanine tolerance in both populations [Okano et al., 1991]. In relatively homogeneous German, Swedish, and southeastern U.S. populations, similar genotype-phenotype correlations were observed [Eisensmith et al., 1996; Kayaalp et al., 1997; Trefz et al., 1993]. However, when populations with high ethnic diversity are studied in this way, a clear genotype-phenotype correlation may not be apparent [Enns et al., 1999b; Treacy et al., 1997], and the genotype-phenotype correlation is not straightforward in some patients. Although phenylketonuria is a single-gene mendelian disorder, the observed clinical spectrum is more in keeping with a complex multifactorial trait [Scriver and Waters, 1999].
Genetic Counseling
Patients who are diagnosed in the neonatal period and who adhere to the phenylalanine-restricted diet have normal intelligence. However, learning problems can occur in well-treated patients and include problems in basic spelling, reading, and mathematical calculation skills. Patients may also be more prone to depression, anxiety, phobic tendencies, and isolation from their peers [Smith and Knowles, 2000; Welsh et al., 1990]. Such potential adverse and unpredictable manifestations should be brought to the attention of parents and carefully explained, with care and support, during the on-going genetic counseling process.
Treatment
Significant restriction of dietary phenylalanine is required for treatment, but the exact level of daily phenylalanine intake varies from patient to patient, and varies with age in an individual patient. Because phenylalanine is an essential amino acid, detrimental effects on growth and development may occur if restriction of phenylalanine intake is too severe and the blood level drops to below normal. Although there is no worldwide consensus about optimal plasma phenylalanine levels, most clinics in the United States strive to maintain levels between 120 and 360 μM in children younger than 12 years and between 120 and 600 μM in individuals older than 12 years [Phenylketonuria, 2000]. Phenylalanine levels in unaffected individuals are usually below 120 μM. In general, phenylketonuria patients who harbor severe mutations require a greater limitation of phenylalanine intake to maintain acceptable blood phenylalanine levels. However, individual variations of phenylalanine tolerance may occur, even in patients with identical genotypes. Blood phenylalanine levels therefore are monitored frequently, especially in the first year of life, and the diet is adjusted with care for each individual patient. The regimen must be initiated and overseen by experts in phenylketonuria at a specialized center, and referral of the patient to such a specialized center is mandatory. An expert, coordinated team approach is clearly the most effective way of managing phenylketonuria; stricter management improves developmental outcome [Camfield et al., 2004].
In earlier therapeutic protocols, phenylalanine restriction was continued only through the first few years of life, theoretically corresponding to the age at which brain myelination is complete. As developmental data accumulated, it became evident that treatment throughout childhood and adolescence was the best course to preserve IQ [Smith et al., 1991]. In later studies, it has been found that characteristic periventricular T2 white matter signal abnormalities on conventional MRI, restricted white matter diffusion in diffusion-weighted imaging, and electrophysiologic testing abnormalities referable to the CNS are observed in adults who are on unrestricted phenylalanine intake or poorly compliant with dietary therapy [Phillips et al., 2001]. There is evidence that MRI changes in cases of phenylketonuria are at least partially reversible if patients return to a low-phenylalanine diet [Cleary et al., 1995]. Accordingly, it is reasonable to continue therapy into adulthood, and most centers recommend lifelong treatment. Reassessment of adult phenylalanine tolerance may be necessary as body mass changes with age [MacLeod et al., 2009].
A variety of medical food products is available as special formulas for the treatment of phenylketonuria. These special formulas are low in phenylalanine or do not contain any phenylalanine, and they typically contain supplemental tyrosine and a balanced mixture of the additional amino acids, carbohydrates, essential fatty acids, vitamins, and minerals, including zinc, selenium, and molybdenum. The metabolic medical foods provide a variable amount of calories (up to 70 percent of daily requirement) in the form of starch (e.g., dextrose, cornstarch, dextromaltose, Polycose) and fat (i.e., corn or other oils), and they constitute a major source of nutrition for the lifetime of the patient. The special formula or medical food, containing no phenylalanine, is continued even after solid food is introduced. The special formula or medical food is ingested together with regular food during the same meal, providing the phenylalanine in food plus the amino acids, vitamins, and nutrients in the special formula in a complementary, beneficial manner. The medical and regular foods therefore should be given in a calculated proportion together in intervals throughout the day. Overall, the targeted total amino acid intake for children younger than 2 years is approximately 3 g/kg/day, and it is about 2 g/kg/day for older children [Cockburn and Clark, 1996]. If the medical food is ingested in a single sitting, the supply of amino acids may induce hyperinsulinism and hypoglycemia.
Medical food products continue to be modified to increase palatability and optimize the treatment of phenylketonuria. Amino acid powders and gels with added carbohydrate and with or without fats, vitamins, and minerals are examples of commonly used protein substitutes. Newer protein substitutes include amino acid tablets and capsules, which do not contain carbohydrate, vitamins, or minerals. Special amino acid bars and a protein that is almost phenylalanine-free (glycomacropeptide) are also available [van Spronsen and Enns, 2010]. Phenylketonuria dietary research has focused on making the medical food products more palatable. Although low-phenylalanine flour, pastas, cookies, and nutrition bars are available, the phenylketonuria diet remains very bland, and poor dietary compliance can be a major problem, especially after childhood. Early efforts to make a more palatable amino acid mixture have met with preliminary success, with some patients preferring the new products to traditional medical foods.
It is important to monitor complete blood cell counts and serum vitamin B12 levels periodically, because clinical and subclinical B12 deficiency has been reported in adolescents and adults with classic phenylketonuria, even in those poorly compliant with the restricted diet [Hanley et al., 1996].
In contrast to the strict dietary control required in the treatment of classic phenylketonuria, patients with nonphenylketonuria hyperphenylalaninemia (i.e., untreated blood phenylalanine levels of 360–600 μM) are not necessarily placed on the special diet as long as their phenylalanine levels are in treatment range; many of these patients are able to maintain acceptable blood phenylalanine levels with protein restriction alone. These patients have normal intelligence, and they do not have the psychologic findings or head MRI changes that have been documented in classic phenylketonuria [Weglage et al., 1996]. Dietary therapy may be recommended in some instances for pregnant women with nonphenylketonuria hyperphenylalaninemia to minimize the risk of maternal phenylketonuria syndrome.
Additional and Novel Therapies
A complementary therapeutic approach has received U.S. Food and Drug Administration approval: administration of dietary supplementation of large, neutral amino acids. Large, neutral amino acids compete with phenylalanine for transport across the blood–brain barrier by the L-type amino acid carrier and consequently decrease the level of phenylalanine in the CNS [Matalon et al., 2003; van Spronsen and Enns, 2010]. In a study of six adult subjects with classic phenylketonuria, large, neutral amino acid supplementation resulted in increased blood concentrations of tyrosine and tryptophan (the respective precursors for dopamine and serotonin) and decreased brain phenylalanine concentration, as measured by 1H-MR spectroscopy, toward the carrier range. All patients reported improvements in well-being and energy levels [Koch et al., 2003].
Deficiencies of carnitine and long-chain polyunsaturated fatty acids (i.e., arachidonic and docosahexaenoic acids) may contribute to CNS toxicity in uncontrolled phenylketonuria. Dietary supplementation of these fatty acids and of carnitine may benefit phenylketonuria patients who have low plasma levels of these essential metabolites [Infante and Huszagh, 2001]. Supplementation with omega-3, long-chain, polyunsaturated fatty acids resulted in improvement in visual-evoked potential latencies in 36 children with early-treated phenylketonuria [Beblo et al., 2001].
A novel therapeutic approach uses the nonmammalian enzyme phenylalanine lyase [van Spronsen and Enns, 2010]. This enzyme converts phenylalanine to trans-cinnamic acid, a harmless compound, and it has been found to reduce hyperphenylalaninemia in phenylketonuria rat and mouse models [Bourget and Chang, 1986; Sarkissian et al., 1999]. Enteral phenylalanine lyase therapy has the theoretic potential to increase dietary phenylalanine tolerance substantially, but significant practical hurdles need to be overcome; phenylalanine lyase is destroyed by gastric acidic pH and intestinal proteolysis. Alternative approaches being considered include the use of polyethylene glycol derivatization to produce protected forms of PAH for potential enzyme replacement therapy [Gamez et al., 2004; Kang et al., 2010].
Oral administration of tetrahydrobiopterin, the naturally occurring co-factor for the PAH reaction, reduces serum phenylalanine concentrations, especially in patients with mild hyperphenylalaninemia [Muntau et al., 2002]. However, response to tetrahydrobiopterin has also been documented in patients with classic or variant phenylketonuria [Matalon et al., 2004; van Spronsen and Enns, 2010]. These patients have mutations in the PAH gene, not in one of the genes encoding enzymes involved in tetrahydrobiopterin biosynthesis (see the section on biopterin disorders below). It has been suggested that the PAH mutations in such patients affect the structure of domains that are involved in the binding of tetrahydrobiopterin to the PAH enzyme. Tetrahydrobiopterin also may act as a chemical chaperone, preventing the PAH enzyme from misfolding or protecting PAH from inactivation [Pey et al., 2004]. If these observations and hypotheses are borne out, it may be possible to define by mutation analysis a subset of patients who would predictably benefit from co-factor supplementation. Tetrahydrobiopterin may prove useful in the treatment of maternal phenylketonuria [Trefz and Blau, 2003]. These, and other, novel therapies are under close investigation, especially given recent findings of suboptimal outcomes in phenylketonuria patients who have been continuously treated from the neonatal period [Enns et al., 2010].
Biopterin Disorders
Neonatal hyperphenylalaninemia may rarely be caused by defects in the synthesis or recycling of tetrahydrobiopterin, an essential co-factor in the PAH reaction (see Figure 32-1). Worldwide, it has been estimated that approximately 2 percent of patients with hyperphenylalaninemia have a defect in one of the four enzymes responsible for maintaining tetrahydrobiopterin levels [Blau et al., 1996]. Guanosine triphosphate cyclohydrolase (GTPCH) I and 6-pyruvoyltetrahydro-biopterin synthase (PTPS) are essential enzymes for tetrahydrobiopterin biosynthesis, whereas pterin-4α-carbinolamine dehydratase (PCD) and dihydropteridine reductase (DHPR) are responsible for tetrahydrobiopterin recycling [Blau et al., 2001]. All forms of tetrahydrobiopterin disorders that cause hyperphenylalaninemia are inherited as autosomal-recessive traits. An autosomal-dominant form of GTPCH deficiency (e.g., dopa-responsive dystonia, Segawa’s disease, hereditary progressive dystonia) manifests with dystonia, but it is not associated with elevated phenylalanine levels (see Chapter 39).
More than 600 patients with tetrahydrobiopterin disorders have been identified (BIODEF database, http://www.bh4.org/). PTPS deficiency is the most common of these disorders, occurring in 54 percent of cases of reported tetrahydrobiopterin disorders. DHPR deficiency is seen in 32 percent, whereas PCD and GTPCH deficiencies are rare, each reported in 4 percent of cases (BIODEF database, http://www.bh4.org/).
Clinical Manifestations
Most patients with GTPCH, DHPR, and PTPS deficiencies have severe forms of disease, although mild forms of DHPR and PTPS deficiencies exist, and some forms of PTPS deficiency may be transient [Blau et al., 2001]. PCD deficiency is usually not associated with significant abnormalities other than transient tone abnormalities. Untreated patients with typical severe disorders of tetrahydrobiopterin synthesis or recycling usually develop neurologic manifestations by 4 months, although symptoms can appear in the neonatal period. Clinical manifestations include microcephaly, progressive neurologic deterioration, movement disorders, delayed motor development, seizures, tone disturbances, oculogyric spasms, swallowing difficulties, hypersalivation, and hyperthermia [Blau et al., 1996, 2001; Dhondt, 1993]. Diurnal fluctuation of dystonia may occur. The clinical course is similar in severe untreated tetrahydrobiopterin deficiency, regardless of the enzymatic defect. Head MRI findings have only rarely been reported [Pietz et al., 1996]. However, in DHPR deficiency, brain abnormalities such as diffuse demyelination, atrophy, spongy vacuolation of brainstem long tracts, and basal ganglia calcification may occur. Abnormal vascular proliferation in the cortex, white matter, and basal ganglia may also be detected [Gudinchet et al., 1992; Schmidt et al., 1988].
Diagnosis
Patients with tetrahydrobiopterin defects are often identified by mandatory newborn screening programs because of hyperphenylalaninemia. All children with persistent hyperphenylalaninemia must be screened for aberrations in the levels of pterin metabolites (i.e., neopterin and biopterin). Patients with GTPCH deficiency have decreased urinary excretion of neopterin and biopterin. In PTPS deficiency, neopterin is increased and biopterin decreased, resulting in a greatly elevated neopterin to biopterin ratio (normally, the ratio is about 1:1). The neopterin to biopterin ratio in PCD deficiency is also increased but not to the same extent as in PTPS deficiency. In PCD deficiency, the characteristic feature is the presence of primapterin (7-biopterin) in the urine [Ayling et al., 2000]. In DHPR deficiency, the percentage of biopterin is elevated (>80 percent in most cases), but urine screening may miss it in some patients [Dhondt, 1984]. The measurement of DHPR activity in neonatal dried blood spots using a spectrophotometric assay is an effective method for diagnosis of DHPR deficiency. Urine pterin analysis and DHPR activity screening should be performed early in the management of a new patient with persistent hyperphenylalaninemia, or these disorders may be missed. Mutation analysis has yet to identify clear genotype-phenotype correlations [Blau et al., 1996].
Treatment
The goals of therapy are to decrease the level of phenylalanine to an acceptable range (120–360 μM) and correct the neurotransmitter deficiencies with exogenous supplementation. The diet is similar to that used to treat classic phenylketonuria, but patients tend to have a higher phenylalanine tolerance (300–700 mg/day) [Blau et al., 2001]. Tetrahydrobiopterin supplementation (2–20 mg/kg/day) is also used to help control blood phenylalanine levels. Lower tetrahydrobiopterin doses (2–5 mg/kg/day) may be effective in GTPCH and PTPS deficiencies, whereas higher doses (up to 20 mg/kg/day) may be required in DHPR deficiency. l-DOPA and 5-hydroxytryptophan are administered in a dose of 1–10 mg/kg/day. Carbidopa, an inhibitor of peripheral aromatic amino acid decarboxylase, decreases the conversion rates of l-DOPA to dopamine and 5-hydroxytryptophan to serotonin, allowing for the use of lower doses of these compounds; these therapeutic adjuncts may be especially helpful in severe forms of tetrahydrobiopterin deficiency. The optimal dose of each medication must be determined for each patient. Mild forms of disease may respond to tetrahydrobiopterin supplementation alone. Measuring levels of cerebrospinal fluid neurotransmitter metabolites (i.e., homovanillic acid and 5-hydroxyindolacetic acid) is useful in monitoring the efficacy of treatment [Blau et al., 2001; Shintaku, 2002].
Side effects of therapy include choreoathetosis, dystonia, and on-off phenomena, which are also features of the underlying disorders [Tanaka et al., 1989]. Tachycardia, diarrhea, and anorexia are associated with 5-hydroxytryptophan administration [Dhondt, 1993]. l-Deprenyl, a monoamine oxidase inhibitor, has been useful in decreasing catabolism of l-DOPA and 5-hydroxytryptophan, allowing lower dosing [Schuler et al., 1995; Spada et al., 1995, 1996]. A low concentration of cerebrospinal fluid folate is typical in DHPR deficiency, and it is treated by folinic acid supplementation (10–20 mg/day) [Shintaku, 2002]. Trimethoprim-sulfamethoxazole and methotrexate are DHPR inhibitors, and they may cause serious side effects in patients with tetrahydrobiopterin deficiency [Millot et al., 1995; Woody and Brewster, 1990]. Neurologic function may improve with therapy, but the overall prognosis for these disorders is largely unknown.
Hepatorenal Tyrosinemia
Hepatorenal tyrosinemia (i.e., tyrosinemia type I) is characterized principally by liver, kidney, and peripheral nerve involvement. The clinical spectrum ranges from severe hepatic failure in early infancy to later presentations of chronic liver disease and rickets in an older child. The overall incidence is 1 case per 100,000 births. In Quebec the incidence is quite high, at 1 in 16,700 births [Bergeron et al., 1974].
Pathophysiology
Hepatorenal tyrosinemia is caused by a deficiency of fumarylacetoacetate hydrolase, a distal enzymatic step in the processing of the amino acid tyrosine (Figure 32-2). Some investigations suggest that metabolites of tyrosine accumulating proximal to the blocked reaction step are toxic to liver and kidney, acting as alkylating agents or by disruption of sulfhydryl metabolism [Russo et al., 2001]. One of the accumulating metabolites, succinylacetone, has been implicated in the peripheral neuropathy of tyrosinemia [Sassa and Kappas, 1983; Sassa et al., 1983].
Clinical Manifestations
Neurologic involvement can include paresthesias, opisthotonic-like posture, bruxism and tongue biting, and in some cases, motor paralysis leading to respiratory failure and death [Mitchell et al., 1990]. Neurologic crises occur in up to 42 percent of individuals with tyrosinemia [Kvittingen, 1991]. These crises are biphasic, with an active period of pain, autonomic dysfunction, and sometimes paralysis lasting 1–7 days, followed by a period of recuperation. Succinylacetone blocks the heme biosynthetic pathway, and the neurologic crises – a major source of morbidity – therefore have a physiologic basis similar to those in porphyria [Russo et al., 2001].
Management
2-[2-Nitro-4-trifluoromethylbenzoyl]-1,3-cyclohexanedione (NTBC) is an inhibitor of 4-hydroxyphenylpyruvate dioxygenase (4-HPD), a proximal step in tyrosine catabolism. Inhibition thereby prevents the synthesis of succinylacetone and related metabolites, which accumulate because of the enzymatic block at fumarylacetoacetate hydrolase, a distal step in the pathway. Treatment with this compound is effective within hours and dramatically reduces the risk of neurologic and hepatic crises [Holme and Lindstedt, 2000; McKiernan, 2006]. Dietary restriction of phenylalanine and tyrosine is used in combination with NTBC. Liver transplantation is curative for hepatic and nervous system disease, and it is used in those who are refractory to nonsurgical treatment.
Acute management of neurologic crises includes analgesia, glucose (which inhibits ALA synthetase), and symptomatic treatment of hypertension. Repletion of sodium, potassium, and phosphate is necessary. The use of barbiturates and other medications that aggravate porphyria should be avoided before stabilization on NTBC [Kang and Gerald, 1970].
Other Categories of Tyrosinemia
Several causes of hypertyrosinemia exist in addition to fumarylacetoacetate hydrolase deficiency (see Figure 32-2). Deficiency of tyrosine aminotransferase causes tyrosinemia type II (oculocutaneous tyrosinemia) [Hunziker, 1980]. In type II disease, developmental delay, corneal thickening, and hyperkeratosis of palms and soles occur, but there is usually no hepatorenal involvement. Type III disease is caused by deficiency of 4-HPD and has a spectrum of manifestations, ranging from clinically normal to severe mental retardation and neurologic anomalies, including ataxia [Cerone et al., 1997; Ruetschi et al., 2000]. A 4-HPD dysfunction can also cause hawkinsinuria, a rare condition that can manifest with failure to thrive and metabolic acidosis, but it usually resolves as the patient’s metabolism matures [Borden et al., 1992].
Liver failure can lead to elevated tyrosine levels [Mitchell et al., 2001], as can postprandial testing and diseases such as vitamin C deficiency and hyperthyroidism. Premature infants may manifest transient tyrosinemia of the newborn because of temporary immaturity in the function of 4-HPD. This condition resolves spontaneously, but mild developmental delay has been reported [Nyhan, 1984].
Maple Syrup Urine Disease
In 1954, Menkes and colleagues described four siblings who died in early infancy from a cerebral degenerative disease, with onset occurring when they were 3–5 days old. Symptoms included feeding difficulty, irregular respiratory pattern, hypertonia, opisthotonus, and failure to thrive. All had urine with the smell of maple syrup [Menkes et al., 1954]. Soon thereafter, another patient with a similar history was found to have elevated levels of branched-chain amino acids in urine and blood, and the syndrome was initially referred to as maple sugar urine disease [Westall et al., 1957]. Maple syrup urine disease is caused by mitochondrial branched-chain α-ketoacid dehydrogenase complex deficiency (compared with the composite branched-chain amino acid pathways in Figure 32-6 below). The enzymatic defect leads to accumulation of branched-chain amino acids and branched-chain α-ketoacids. Five forms of maple syrup urine disease (i.e., classic, intermediate, intermittent, thiamine-responsive, and dihydrolipoyl dehydrogenase [E3] deficiency) have been delineated based on clinical presentation, level of enzyme activity, and response to thiamine administration [Chuang and Shih, 2001].
Clinical Manifestations
Classic maple syrup urine disease
In the classic form, the clinical phenotype is one of severe neonatal encephalopathy, unless presymptomatic therapy is initiated because of abnormal newborn screening, prenatal diagnosis, or positive family history. Untreated neonates typically develop symptoms by the end of the first week of life. Feeding difficulties, alternating hypertonia and hypotonia, opisthotonic posturing, abnormal movements (“fencing” or “bicycling”), and seizures commonly occur. The characteristic urine smell develops on day 5–7 of life [Strauss and Morton, 2003a]. Unless an underlying inborn error of metabolism is suspected, affected children may be misdiagnosed as having sepsis and progress to coma and death. Ketosis is often found, and hypoglycemia may occur, but severe metabolic acidosis tends not to occur. Plasma amino acid analysis reveals elevated levels of branched-chain amino acids and the diagnostic presence of alloisoleucine in plasma [Schadewaldt et al., 1999]. Urine organic acid analysis demonstrates excretion of branched-chain α-ketoacids. Hyponatremia and cerebral edema are frequent sequelae during acute metabolic decompensation [Morton et al., 2002]. Other complications include pseudotumor cerebri, pancreatitis, and eye abnormalities [Burke et al., 1991; Kahler et al., 1994]. Ocular findings in untreated or late-diagnosed patients include optic atrophy, gray optic papilla, nystagmus, ophthalmoplegia, strabismus, and cortical blindness [Burke et al., 1991]. Children who survive the initial metabolic crisis typically have significant neurodevelopmental delays and spasticity [Chuang and Shih, 2001]. Although motor, visual, and learning deficits may occur, rapid identification of affected infants and careful institution of appropriate therapy can result in normal development [Kaplan et al., 1991; Morton et al., 2002].
Neuroimaging studies (Figure 32-3) are typically abnormal in patients with untreated classic maple syrup urine disease who are in crisis. Computed tomographic (CT) scans appear normal in the first few days of life, but they reveal progression to marked generalized cerebral edema if the patient remains untreated [Brismar et al., 1990]. An unusual pattern of edema may occur, characterized by involvement of the cerebellar deep white matter, posterior brainstem, cerebral peduncles, posterior limb of the internal capsule, and posterior aspect of the centrum semiovale. Edema tends to subside in the second month of life [Brismar et al., 1990]. Patients with classic maple syrup urine disease in metabolic crisis with associated hyponatremia demonstrate a prominently increased T2 signal on brain MRI in the brainstem reticular formation, dentate nucleus, red nucleus, globus pallidus, hypothalamus, septal nuclei, and amygdala [Morton et al., 2002]. One report observed that brain MRI abnormalities were absent or only slight in sick patients with maple syrup urine disease in the absence of hyponatremia [Morton et al., 2002]. Cranial ultrasonography of neonates in acute metabolic crisis reveals symmetrically increased echogenicity of the periventricular white matter, basal ganglia, and thalami [Fariello et al., 1996]. Chronic changes, including hypomyelination of the cerebral hemispheres, cerebellum, and basal ganglia and cerebral atrophy, may supervene in poorly controlled patients. CT- and MRI-defined abnormalities and the clinical phenotype may improve after implementation of appropriate dietary therapy [Taccone et al., 1992]. Diffusion-weighted imaging and spectroscopy have also documented abnormalities during the acute phase of disease [Cavalleri et al., 2002]. Markedly restricted proton diffusion, suggestive of cytotoxic or intramyelinic sheath edema, was demonstrated in the brainstem, basal ganglia, thalami, cerebellar and periventricular white matter, and cerebral cortex in six patients with maple syrup urine disease. MR spectroscopy demonstrated abnormal elevations of branched-chain amino acids, branched-chain α-ketoacids, and lactate in the four patients. All of these changes were reversed after the institution of appropriate nutritional and antibiotic therapy to treat intercurrent illness [Jan et al., 2003].
Fig. 32-3 Maple syrup urine disease.
(Courtesy of Dr. A James Barkovich, University of California, San Francisco, CA.)
A characteristic comblike EEG pattern may be demonstrated for some patients with classic maple syrup urine disease between the second and third weeks of life [Tharp, 1992]. This unusual rhythm pattern resolves with the institution of dietary therapy [Tharp, 1992].
Intermediate maple syrup urine disease
Children who have the intermediate form of maple syrup urine disease do not present in the neonatal period, despite having persistently elevated plasma levels of branched-chain amino acids. Developmental delay and failure to thrive are common. Severe neurologic impairment is absent; episodes of metabolic decompensation may occur, although severe ketoacidosis episodes are variable [Gonzalez-Rios et al., 1985a]. These children have a higher tolerance for dietary protein than those who have the classic form [Gonzalez-Rios et al., 1985a]. Rarely, patients with intermediate-type maple syrup urine disease respond to thiamine administration.
Intermittent maple syrup urine disease
Patients with intermittent maple syrup urine disease typically come to medical attention when they are 5 months to 2 years old and after stress induced by infection or high protein intake; some have been detected as late as the fifth decade of life [Chuang and Shih, 2001]. The intermittent form of maple syrup urine disease can be particularly difficult to diagnose, because affected individuals have normal levels of branched-chain amino acids and no odor between episodes of metabolic decompensation. Episodic decompensation is characterized by ataxia, disorientation, and altered behavior, which may progress to seizures, coma, and even death unless therapy is instituted. Early development and intellect are usually normal.
Thiamine-responsive maple syrup urine disease
The clinical course of patients with the thiamine-responsive variant of maple syrup urine disease is similar to that of the intermediate form of disease. Plasma levels of branched-chain amino acid and urine excretion of branched-chain α-ketoacids decline days to weeks after thiamine administration (10–1000 mg/day) is started [Scriver and Kaufman, 2001]. Patients are also treated with nutritional regimens similar to those used in other forms of maple syrup urine disease. Developmental delay may be present, but normal intelligence has also been documented [Scriver et al., 1971].
Dihydrolipoyl dehydrogenase-deficient maple syrup urine disease
The dihydrolipoyl dehydrogenase (E3)-deficient form of maple syrup urine disease is characterized by ketoacidotic crises in infancy. There is also lactic acidemia because the E3 subunit of the branched-chain α-ketoacid dehydrogenase complex is also required for catalytic function of pyruvate dehydrogenase and α-ketoglutarate dehydrogenase. In addition to the typical maple syrup urine disease metabolites, urine organic acid analysis reveals the presence of lactate, pyruvate, and α-ketoglutarate. The neonatal period is usually uneventful, but progressive neurologic deterioration, characterized by developmental delay, hypotonia or hypertonia, and dystonia, supervenes. Death in early childhood is common. Attempts at therapy have had limited success [Chuang and Shih, 2001; Sakaguchi et al., 1986].
Laboratory Tests
Maple syrup urine disease can be detected easily and accurately by tandem mass spectrometry analysis of the newborn blood spot [Chace et al., 1995]. Tandem mass spectrometry used in newborn screening is effective in identifying maple syrup urine disease, and is performed in all states in the U.S. (see http://genes-r-us.uthscsa.edu/nbsdisorders.htm) and many countries worldwide. Urine screening tests for the presence of α-ketoacids (i.e., ferric chloride and 2,4-dinitrophenylhydrazine [DNPH]) may be positive, but are nonspecific and insensitive. Plasma amino acid analysis demonstrates elevations of leucine, isoleucine, and valine (5- to 10-fold greater than normal) [Strauss and Morton, 2003a], as well as the pathognomonic finding of elevated alloisoleucine [Schadewaldt et al., 1999]. Levels of branched-chain amino acids are greatly elevated in urine and cerebrospinal fluid [Chuang and Shih, 2001]. The branched-chain α-ketoacids 2-oxoisocaproic acid, 2-oxo-3-methylvaleric acid, and 2-oxoisovaleric acid, derived from the branched-chain amino acids leucine, isoleucine, and valine, respectively, are found to be elevated on urine organic acid analysis during metabolic crises. Branched-chain amino acids levels and excretion of branched-chain α-ketoacids may be normal between episodes of decompensation in the intermittent form of disease.
The branched-chain α-ketoacid dehydrogenase complex consists of three catalytic components – a thiamine pyrophosphate-dependent carboxylase (E1) with an α2β2 structure, a transacylase (E2), and a dehydrogenase (E3) – as well as two regulatory enzymes (a kinase and a phosphatase) [Chuang and Shih, 2001]. Deficient activity of this complex leads to the accumulation of leucine, isoleucine, and valine and their corresponding α-ketoacids. The decarboxylation activity can be measured in leukocytes, lymphoblasts, or fibroblasts, and it is loosely related to the clinical phenotype: 0–2 percent of normal activity in classic maple syrup urine disease, 3–30 percent activity in intermediate, 5–20 percent in intermittent, 2–40 percent in thiamine-responsive, and 0–25 percent in E3 deficiency [Chuang and Shih, 2001; Scriver et al., 1971]. Because significant overlap exists between measured enzyme activity and clinical phenotype, enzymatic activity cannot be used to predict the clinical course with certainty.
Genetics
Maple syrup urine disease is a pan-ethnic, autosomal-recessive condition that can be caused by mutations in any of the components of the mitochondrial branched-chain α-ketoacid dehydrogenase complex. In a study of 63 individuals, E1β subunit mutations were most common (38 percent), followed by E1α (33 percent), and E2 (19 percent) mutations [Nellis and Danner, 2001]. Branched-chain α-ketoacid dehydrogenase phosphatase or kinase mutations are also predicted to cause maple syrup urine disease, but such abnormalities have not yet been detected. The overall incidence is approximately 1 case per 150,000 people in the general population, but maple syrup urine disease is more common in Old Order Mennonites in southeastern Pennsylvania (1 in 176 births) [Danner and Doering, 1998]. A novel founder mutation in the E1β subunit has been reported in the Ashkenazi Jewish population [Edelmann et al., 2001]. In general, increased residual branched-chain α-ketoacid dehydrogenase activity should convey some advantage, but there is a wide overlap between measured enzymatic activities and clinical outcome. Given the complexity of the molecular genetics, the potential for modifier gene and environmental interactions, and the multiple clinical phenotypes associated with maple syrup urine disease, a lack of definitive genotype-phenotype relationships is not surprising.
Treatment
Chronic care of the child with maple syrup urine disease includes regular visits to an integrated metabolic clinic for medical and nutritional assessment. Adequate calories (100–120 kcal/kg/day) and protein (2–3 g/kg/day) are needed for growth. Chronic valine or isoleucine deficiency may cause an exfoliative dermatitis, and supplementation of these amino acids is often needed [Koch et al., 1993]. Thiamine supplementation is administered to patients with thiamine-responsive forms of maple syrup urine disease. Because patients on restricted diets are at risk for micronutrient and essential fatty acid deficiencies, patients should be periodically monitored for such deficits and supplementation given as needed.
Because significant metabolic intoxication may occur rapidly, even in patients with apparently well-controlled disease, it is crucial to have carefully considered home and hospital emergency protocols in place for each child [Morton et al., 2002; Strauss and Morton, 2003a]. Acute metabolic decompensation (e.g., fasting or illness severe enough to cause catabolism) is a medical emergency that requires prompt intervention. Initial intervention is aimed at correcting dehydration, starting high-dose intravenous thiamine, and providing adequate calories (approximately 120–140 kcal/kg/day) to prevent further protein catabolism and higher rise in plasma leucine levels. To this end, high-dextrose intravenous fluids (to provide approximately 10 mg/kg/min) and intralipid are often administered. Branched-chain amino acid-free parenteral nutrition or enteral formula, delivered by continuous nasogastric drip, can also be used [Nyhan et al., 1998; Parini et al., 1993]. The rate of decrease of leucine is slowed in the face of valine and isoleucine levels inadequate to stimulate protein synthesis. Acute valine and isoleucine deficiency can be avoided by careful supplementation of these amino acids [Parini et al., 1993]. Leucine is reintroduced to the diet after therapeutic levels are achieved [Morton et al., 2002].
In a study of 36 maple syrup urine disease patients, plasma leucine levels fell to less than 400 μM 2–4 days after the initiation of therapy with enteral and parenteral nutrition. Initial leucine levels ranged from 233 to 778 μM in a group diagnosed on the first day of life (n = 18) and 1489 to 3359 μM in a group diagnosed between days 3 and 16 (n = 18). Over an 11-year period, neurologic examinations, gross motor development, and speech were normal in 34 of 36 children [Morton et al., 2002]. Enteral nutrition was also found to be beneficial when instituted within the first 20 days of life. Four patients receiving nasogastric drip feeding as the only treatment of neonatal classic maple syrup urine disease had normal development when 3–5 years old [Parini et al., 1993]. Hemodialysis and continuous venovenous extracorporeal removal therapies result in more rapid fall in plasma levels of branched-chain amino acids, but these modalities typically have been described in single case reports or small series with relatively short follow-up, and it is difficult to ascertain the long-term outcome of such intervention [Gouyon et al., 1996; Puliyanda et al., 2002]. Nevertheless, normal development has been reported for 8 of 12 children after continuous venovenous extracorporeal removal therapy [Jouvet et al., 2001]. Branched-chain amino acids levels often rebound after initial dialysis in cases of severe metabolic imbalance characterized by extremely high leucine levels, and dialysis may need to be repeated in such cases. Peritoneal dialysis is no longer routinely used; there is a tendency for leucine levels to plateau between 1000 and 1500 μM after 24 hours, limiting the utility of this therapeutic modality [Gortner et al., 1989]. Levels of branched-chain amino acids and branched-chain α-ketoacids tend to plateau with exchange transfusion therapy [Nyhan et al., 1998; Wendel et al., 1982].
Although enteral and intravenous therapy may be sufficient to manage many patients with maple syrup urine disease in acute crisis, various dialysis methods are commonly used and should be considered, especially when clearance of branched-chain amino acids by nutritional support is not effective or when other considerations, such as life-threatening cerebral edema, renal imbalance, or cardiovascular abnormalities, exist [Jouvet et al., 2001; Nyhan et al., 1998]. Liver transplantation has been performed rarely for maple syrup urine disease. Three patients who underwent successful transplantation were able to resume normal diets and were no longer at risk for metabolic decompensation [Wendel et al., 1999]. More recently, domino hepatic transplantion for maple syrup urine disease was successfully performed [Barshop and Khanna, 2005].
Because hyponatremia and subsequent brain edema are serious and relatively common complications, it is important to monitor serum sodium and serum and urine osmolalities closely and to replace urinary losses with saline [Morton et al., 2002]. Critical brain swelling and abnormal brainstem function may develop with only a moderate reduction in serum sodium level (by only 8–10 mEq/L) [Morton et al., 2002]. Low-dose diuretics may also be used to prevent water retention [Strauss and Morton, 2003a]. Mannitol is reserved for life-threatening episodes of increased intracranial pressure [Morton et al., 2002].
