106 Allergic Disorders
 Key Points
Key Points• Allergy refers to inappropriate responses (hypersensitivity) by the host’s immune system.
• Anaphylaxis is an acute, potentially fatal, systemic allergic reaction. Deaths in patients with anaphylaxis are caused by acute respiratory failure.
• Foods, medications, and insect stings are the most common agents causing anaphylaxis.
• Epinephrine is the first-choice medication in the treatment of anaphylaxis, with H1 and H2 antihistamines and steroids being helpful adjuncts.
• Most cases of angioedema are mediated by IgE and respond to allergic therapy. Certain forms of angioedema, however, are mediated by bradykinin and may respond to new classes of medications, the kallikrein inhibitor (ecallantide) and C1 esterase inhibitor concentrates (Cinryze, Berinert).
Allergic Disease: Allergic Rhinits, Insect Stings, Drug Allergy
Epidemiology
The prevalence of allergic disorders, including the incidence of anaphylaxis, has been increasing worldwide in the last few decades and is a topic of intensive study.1,2 Features of Western lifestyles, such as changes in infant diets, widespread use of antibiotics, smaller family size, and cleaner child care, are believed to reduce stimulatory antigenic exposure in an individual’s early years. This has led to an environment in which the immune system is dominated by a persistent allergy-prone system.3
Allergic Rhinitis
It is estimated that up to 42% of Americans suffer from some form of allergic rhinitis at any one time,4 and it has been shown to have a strong correlation with asthma. In recent studies, nearly 40% of adults with allergic rhinitis were also found to have asthma, and 80% of asthmatics demonstrated signs of rhinitis.
Insect Stings
Serious systemic reactions to insect stings are rare and occur in 1% of children and 3% of adults. Anaphylactic reactions account for approximately 50 deaths annually in the United States. There is a 2 : 1 male-to-female distribution, and adult male agricultural workers are considered the most vulnerable population. Individuals who experience large local reactions are less likely to have a systemic reaction (5% to 10%).5
Drug Allergy
A drug allergy is defined as an immune-mediated adverse response to a drug. An adverse drug reaction is defined as a noxious, unintended, or undesired response to a drug taken at a normal dose for the prevention, diagnosis, or treatment of a disease,6 and a drug side effect is an expected and known (adverse) effect of taking the drug that is not the intended therapeutic outcome. Although the true frequency is unknown, drug allergy is thought to account for approximately one third of adverse drug reactions.7 Adverse drug reactions affect 10% to 20% of hospitalized patients and more than 7% of the general population.
Pathophysiology
The immune system protects the host by distinguishing self from nonself; it tolerates the former but attacks the latter.8,9 Allergy, allergic diseases, and hypersensitivity reactions arise when our immune system reacts inappropriately to allergens with resultant harm to the host.1,2,10,11 For allergic diseases to occur, predisposed individuals need to first be exposed to an allergen through a process called sensitization.
Hypersensitivity reactions are mechanistically divided into four types of reactions according to the Gell and Combs classification system (Box 106.1). The immediate hypersensitivity reaction (type I), which is mediated by IgE, serves as the classic model of the immune response to allergen. On initial exposure to allergen, T helper 2 (TH2) cells are activated, which results in the production of an array of cytokines that exert their effects on the T cells themselves, B cells, and antigen-presenting cells. IgE is elaborated and attached to the high-affinity Fc receptor on the surface of mast cells and basophils. Fixation of allergen-specific IgE leads to a series of cellular and molecular changes that prime these cells for future exposure. On reexposure, allergen cross-links the cell-bound IgE on the surface of mast cells and basophils, thereby setting in motion a complex cascade of events that lead to the release of preformed mediators such as histamine, lipid mediators, and cytokines and subsequent activation of various inflammatory pathways. These mediators and products of secondary inflammatory pathways cause adherence and chemotaxis of inflammatory cells, increased capillary permeability, vasodilation, smooth muscle contraction, and sensory nerve stimulation. A few allergen molecules can thus cause the release of a large number of mediator molecules in a designed amplification response. Examples of type I hypersensitivity reactions include allergic rhinitis, allergic asthma, urticaria, angioedema, and anaphylaxis.
See Box 106.1, Types of Hypersensitivity, online at www.expertconsult.com.
Box 106.1 Types of Hypersensitivity
Allergens are typically carbohydrate or protein molecules (or parts of a larger molecule) that elicit an immune response.10
The inflammation that occurs in allergy is divided into three temporal phases.10 Early-phase reactions occur within minutes of exposure and are considered immediate type I hypersensitivity reactions. Late-phase reactions typically occur within 2 to 6 hours and peak 6 to 9 hours after exposure. This response is thought to be due to newly synthesized cytokines, growth factors, and chemokines, which were released more slowly than the preformed mediators primarily responsible for the early-phase reaction. This reaction often involves airway narrowing and hypersecretion of mucus in the lungs, in addition to the erythema, warmth, and pain experienced in the skin. In some individuals there is no clinical distinction between the early and late phases. Chronic allergic inflammation is the final phase in the inflammatory process. It occurs after persistent or repetitive exposure to specific allergens and results in tissue remodeling and structural changes in affected cells. Chronic allergic inflammation can further increase epithelial injury, mucus production, and thickening of airway walls.
The majority of serious sting-related reactions are caused by insects belonging to the order Hymenoptera (yellow jackets, hornets, honeybees, wasps, and fire ants).5 Their venom contains histamine, dopamine, various peptides, and protein enzymes that are either vasoactive or can elicit significant allergic reactions (IgE mediated).
Presenting Signs and Symptoms
Insect Stings (Hymenoptera Venom Allergy)
Patients with systemic allergic reactions from stings may exhibit generalized urticaria, angioedema, wheezing, stridor, anxiety, or other signs of respiratory and circulatory insufficiency. Most stings, however, cause self-limited local reactions consisting of redness, pain, and itching at the site of the sting. These reactions typically develop within minutes and usually last for a few hours. Local reactions rarely put patients at risk for future systemic reactions.5 Fire ant stings tend to produce pustulelike lesions. Local stings that result in subsequent swelling near the oropharynx (either cutaneous or deep) can cause airway compromise, and affected patients should be observed in the emergency department (ED) until the symptoms have resolved.
Differential Diagnosis and Medical Decision Making
Allergic Rhinitis
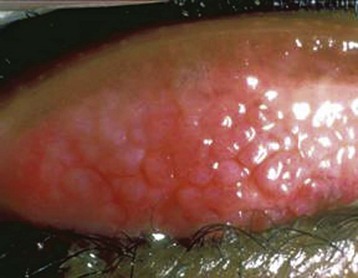
Box 106.2 lists other diagnostic considerations in patients with symptoms that may mimic allergic rhinitis.12 Patients older than 20 years should be investigated for nonallergic causes (e.g., polyps). Atopic patients with severely inflamed conjunctivae, lids, and periorbital structures should raise the possibility of atopic keratoconjunctivitis (Fig. 106.1) and vernal keratoconjunctivitis (Fig. 106.2). These two types of chronic allergic conjunctivitis have the potential to cause corneal erosions and ulcers leading to vision loss and should be managed in consultation with an ophthalmologist.
Box 106.2
Causes of Rhinorrhea and Nasal Obstruction
Modified from Greiner AN. Allergic rhinitis: impact of the disease and considerations for management. Med Clin North Am 2006;90:17–38.
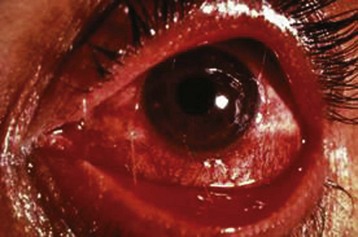
Fig. 106.1 Atopic keratoconjunctivitis.
(From Baba I. Red eye—first aid at the primary level. Community Eye Health 2005;18:70–72.)
Drug Allergy
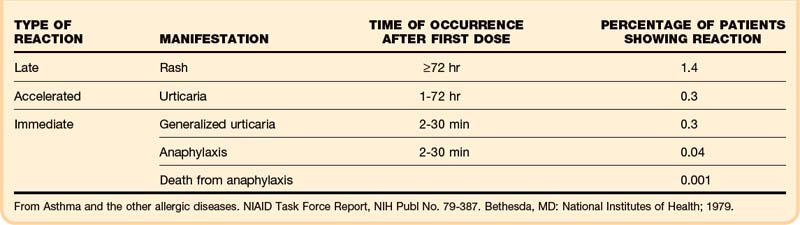
Studies show that 4.4% of patients whose penicillin allergy history was confirmed by a positive skin test experienced an allergic reaction to cephalosporins.13 Only 10% to 20% of patients who report a history of penicillin allergy are truly allergic when assessed by skin testing.14 Although the overall risk for a cross-allergic reaction to cephalosporins in patients with a history of a penicillin allergy is low, the use of cephalosporins requires weighing the risks versus benefits based on an informed discussion between the patient and treating physician. Table 106.1 shows the incidence of allergic reactions to penicillin.15 Some factors can transiently cause T cells to falsely identify the penicillin epitope as being allergic. For example, in up to half (50%) of patients with mononucleosis, a maculopapular rash develops after taking amoxicillin. These same patients often have no adverse drug reaction on subsequent challenge with amoxicillin at a later time. Penicillin allergy should not be diagnosed in such patients.
Treatment
Allergic Rhinitis
Oral second-generation H1 blockers (loratadine, fexofenadine), oral decongestants (pseudoephedrine), and nasal decongestants (oxymetazoline, phenylephrine) can be used for mild, intermittent symptoms.16 Moderate to severe and persistent nasal symptoms may require the addition of intranasal steroid (fluticasone, triamcinolone, budesonide), or chromone derivative such as cromoglycate and nedocromil. An intraocular antihistamine (olopatadine), intraocular chromone, or intraocular ketorolac can be used for ocular allergies, including conjunctivitis.17
Drug Allergy
The most prudent approach in managing possible drug allergy–related complaints in the ED is to discontinue use of the suspect medication or medications, treat the allergic symptoms, and prescribe a suitable alternative drug or drugs. Severe symptoms should be treated in the same way as anaphylaxis (see Box 106.8). Patients with Stevens-Johnson syndrome and toxic epidermal necrolysis require a multidisciplinary approach that includes an intensivist, burn surgeon, and endocrinologist or allergist. For minor allergic drug reactions, H1 antihistamines can be prescribed for itching, flushing, and rash. Steroids are reserved for serious or extensive drug reactions.
Urticaria
Epidemiology
Urticaria (hives) is a fairly common reaction that affects approximately 20% of the population at some point in their lifetime.18 It has numerous different underlying causes and consists of several different types and subtypes. Spontaneous urticaria is broadly divided into acute (<6 weeks) and chronic (≥6 weeks) forms, with the latter representing approximately 10% to 20% of cases.
Pathophysiology
The classic wheal lesions associated with urticaria are the result of edema within the mid and upper dermal layers of the skin. Dilation of lymphatic vessels and capillary venules allows extravasation of protein-rich fluid into the surrounding tissue. This complex inflammatory process involves a variable mixture of macrophages, T cells, neutrophils, eosinophils, and mast cells.19
Presenting Signs and Symptoms
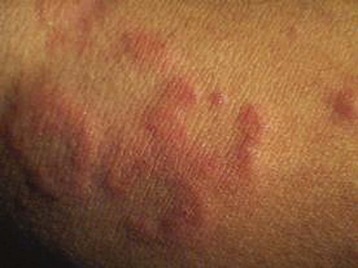
Patients with urticaria usually have hives of variable duration and location.20,21 Urticarial lesions are pruritic, erythematous, raised rashes that blanch on palpation. The lesions are typically round or oval with serpiginous borders, but they may vary in color, size, and shape (Fig. 106.3). They may be localized or appear throughout the body, but there is a slight predilection for the trunk, hands, feet, lips, tongue, and ears. Urticaria usually starts with erythema (flare) as a result of capillary vasodilation in the superficial layer of the dermis. As the protein-rich fluid extravasates into surrounding tissue, it evolves into raised wheals and may change from red to white. A history of pruritic red rash that changes in size and shape, with extension and regression over a period of hours or days, favors the diagnosis of urticaria.
Differential Diagnosis and Medical Decision Making
The first step in narrowing the differential diagnosis of urticaria is to determine whether the urticaria is acute (<6 weeks of symptoms) or chronic (≥6 weeks of symptoms).20,21 The differential diagnosis of urticaria is outlined in Box 106.3 and illustrated in Figure 106.4.

Fig. 106.4 Algorithm for the diagnosis and treatment of urticaria.
ASA, Acetylsalicylic acid; c/w, consistent with; NSAIDs, nonsteroidal antiinflammatory drugs.
See Box 106.3, Differential Diagnosis of Urticaria, online at www.expertconsult.com
Box 106.3
Differential Diagnosis of Urticaria
Foods, Additives
Seafood (fish, shellfish, scombroid), tree nuts, eggs, seeds
Foods containing allergens that are cross-reactive with latex, including bananas, avocados, kiwi fruit, chestnuts
Food preservatives such as tartrazine, benzoates, and sulfites
Other: cow’s milk, pork, strawberries, wheat, chocolate, tomatoes
Systemic Disease
Rheumatologic or immunologic: vasculitis, serum sickness, systemic lupus erythematosus, juvenile rheumatoid arthritis
Endocrinologic: thyroid (both hyperthyroid and hypothyroid) disease, progesterone dermatitis, pruritic urticarial papules and plaques of pregnancy, premenstrual flare-up
Skin disease: mastocytosis, dermatitis herpetiformis, amyloidosis, pemphigoid
Neoplastic: paraneoplastic syndrome, Hodgkin disease, leukemia
Types of Urticaria
Dermatographism: may begin suddenly after a viral illness or drug therapy
Pressure or vibratory urticaria: caused by tight clothing around the waist (may be misdiagnosed as scabies)
Heat urticaria: may be caused by a rise in core temperature after a hot bath, fever, vigorous exercise (also known as cholinergic urticaria because it can be induced by injection of a cholinergic agent)
Cold urticaria: caused by exposure to cold temperatures
Solar urticaria: occurs after exposure to intense sunlight or artificial light (antigens produced in the skin may interact with immunoglobulin E)
Water urticaria: prickling skin sensation without skin lesions within 15 minutes of contact with water
Exercise urticaria: may progress to anaphylaxis
Contact urticaria: urticarial wheals or hives develop within 30 to 60 minutes after cutaneous exposure to certain agents, including plants (nettles), animals (caterpillars, jellyfish), latex, cosmetics, and various chemicals
Modified from Dibbern Jr DA. Urticaria: selected highlights and recent advances. Med Clin North Am 2006;90:187–209.
Treatment
Therapy for acute urticaria includes avoidance of the suspected causative agent and administration of H1 antihistamines (see Fig. 106.4). The second-generation H1 antihistamines (cetirizine, loratadine, fexofenadine, desloratadine) are currently recommended as the first-line drugs.18,20
For urticaria judged difficult to control with antihistamines alone, prednisone can be added.20 If the urticarial rash is extensive and pruritus is severe, epinephrine can be administered (0.3 to 0.5 mL of a 1 : 1000 dilution intramuscularly). It may be repeated every 1 to 2 hours as needed. Caution is advised when administering epinephrine to patients at risk for coronary heart disease (>35 years of age, other risk factors for coronary artery disease). Patients with chronic urticaria can be prescribed a nonsedating H1 antihistamine (in higher dosages) as a temporizing measure and be referred to an allergist for further care.
Angioedema
Pathophysiology
Angioedema (swelling) refers to vasodilation and edema in the lower dermis and subcutaneous layers of the skin.22 In the majority of cases angioedema shares a similar allergic mechanism, but in selected cases it is the result of a reaction mediated by bradykinin. Both forms of angioedema can appear similar clinically, with bradykinin-mediated angioedema typically being manifested as angioedema without urticaria. They both commonly affect the eyelids, face, lips, and tongue and can potentially cause life-threatening respiratory compromise. It is important to distinguish the two forms because bradykinin-mediated angioedema may respond better to the new classes of medications, including recombinant C1 factor, kallikrein inhibitor, or a bradykinin receptor antagonist.
Bradykinin-mediated angioedema typically falls into one of two groups, HAE or acquired angioedema (AAE). HAE was first described by William Osler in 1888. It is a rare autosomal dominant disorder with an overall incidence of 1 in 50,000.23 Approximately 25% of cases are the result of a spontaneous mutation. HAE has three subtypes. Type I (85%) is characterized by low levels of C1 esterase inhibitor (C1 INH), type II (<15%) consists of normal but poor functioning levels of C1 INH, and type III (rare) has normal C1 INH levels and activity but probably involves another unidentified defect or gene mutation at the receptor level. Currently, bradykinin is thought to be the primary molecule responsible for angioedema in patients with HAE. Documented triggers include infection, stress, trauma, oral contraceptives, pregnancy, and menstruation.23
AAE comprises a heterogeneous group that includes drug-induced, idiopathic, and environmental agent–related angioedema and acquired C1 INH deficiency (ACID). ACID is characterized by a lack of inheritance and low-level or nonfunctional C1 INH. Drug-induced angioedema is most commonly caused by ACEIs.24 Up to 50% of those who still take an ACEI after an episode of angioedema will experience recurrent episodes. Discontinuation of the medication results in resolution of the symptoms in the majority of patients (within 24 to 48 hours). ACEIs block the degradation of kinins and thereby lead to elevated levels of bradykinin and other peptides. This results in vasodilation and tissue edema of the deeper layers of the skin, which is clinically manifested as angioedema.24
Differential Diagnosis and Medical Decision Making
When angioedema is suggested by the history and physical examination, it is important to determine whether it is associated with other allergic findings, urticaria, a family history, or other medications (ACEIs). The differential diagnosis of angioedema is outlined in Box 106.4.
Treatment
The first priority in the management of patients with acute angioedema is to secure a patent airway if needed.22,23 Endotracheal intubation should be considered early in patients with progressive laryngeal edema. As soon as the patient is situated, intravenous access should be established, oxygen administered, and the patient placed on a monitor.
Epinephrine, antihistamines, and corticosteroids are generally ineffective in the treatment of HAE and ACEI-induced angioedema, although they are commonly given in the acute setting. The use of new drugs for acute HAE attacks and ACEI-induced angioedema is still in the formative stage. Plasma-derived C1 INH concentrate (Berinert, 20 U/kg intravenously, Cinryze, 1000 units intravenously) is currently available in the United States.15 Ecallantide, a kallikrein inhibitor (30 mg subcutaneously in adults), is another recent agent approved by the Food and Drug Administration for the treatment of acute HAE. Aminocaproic acid, tranexamic acid, and anabolic androgens (danazol, stanozolol) are often prescribed for HAE prophylaxis but may be considered for the treatment of acute episodes. Additional treatment alternatives include solvent detergent–treated plasma or fresh frozen plasma, although these therapies are considered less safe.23
Follow-up, Next Steps in Care, and Patient Education
Patients with mild symptoms who improved clinically after treatment and were observed for 4 to 6 hours in the ED can be discharged home with H1 antihistamines and a short course of steroids. If the patient was taking an ACEI, its use should be stopped. Replacement candidates include calcium channel blockers and thiazides. Angiotensin receptor blockers are deemed acceptable by some authorities because the risk for angioedema from these agents is considered acceptably low.24,25 Beta-blockers are not advised in the initial setting because of the theoretic risk of exacerbating the angioedema and potential antagonistic effects with epinephrine.
Anaphylaxis
Epidemiology
Anaphylaxis is a life-threatening systemic hypersensitivity reaction that is rapid in onset and usually precipitated within minutes of exposure to an allergen. Because of lack of data the true incidence of anaphylaxis is unknown, but recent evidence suggests that it is increasing and its incidence may be as high as 2%.26,27 Risk factors for anaphylaxis include atopy, higher socioeconomic status, northern locations, female sex (adults), and route of allergen exposure. An individual with a history of asthma is more likely to experience a severe or fatal reaction.27
Triggers
Foods, predominantly peanuts, tree nuts, shellfish, fish, eggs, soy, and cow milk, are the most common cause of anaphylaxis and are responsible for up to 30% of all fatal cases. Insect stings, primarily by those belonging to the order Hymenoptera, are the second most common cause of anaphylaxis. They account for approximately 18.5% of anaphylactic cases. Medications are the third most common cause of anaphylaxis (13.7%). Although the β-lactams (penicillin) are commonly involved, several other drugs are known to cause anaphylaxis, including NSAIDs, aspirin, protamine, vancomycin, neuromuscular blocking agents, and newer biologic modifiers such as omalizumab, infliximab, and cetuximab.27,28
Although the three groups just mentioned (foods, stings, and drugs) are responsible for the majority of anaphylactic reactions, other prominent triggers of anaphylaxis include latex, seminal fluid, transfusion products, allergen immunotherapy, contrast dye, methylmethacrylate (bone cement), and physical activity (exercise). Exercise-induced anaphylaxis is noteworthy because its pathophysiology is unknown. Some authorities suspect co-triggers, such as a specific food, drug, or recent high pollen exposure as the actual culprit.29 In approximately 20% of anaphylactic cases, no identifiable triggers are found, and the reaction is termed idiopathic anaphylaxis.30
Presenting Signs and Symptoms
Anaphylaxis is a systemic reaction of rapid onset that involves multiple organ systems, principally the cutaneous, respiratory, cardiovascular, gastrointestinal, and central nervous systems (Box 106.5).31 Patients may initially experience warmth and tingling of the face, mouth, and chest, followed by nasal congestion, sneezing, and ocular itching and tearing. Urticaria, pruritus, and angioedema occur roughly 90% of the time.
Box 106.5
Common Clinical Findings in Patients with Anaphylaxis
Modified from Simons FE. Anaphylaxis. J Allergy Clin Immunol 2010;125(Suppl 2):S161–81 (with permission).
The majority of anaphylactic reactions become clinically evident within minutes after parenteral exposure (average of 5 to 30 minutes), with a longer latent time (2 hours) after the ingestion of a triggering agent. In general, the sooner the clinical syndrome is manifested after exposure to the allergen, the more severe the reaction. Most fatalities occur within the first 30 minutes after exposure. In the setting of shock, cutaneous symptoms may be absent as a result of compensatory vasoconstriction. Anaphylaxis may precipitate an acute coronary syndrome in what is known as cardiac anaphylaxis.32 More commonly in the elderly, the initial complaint may be abdominal pain and cramping.
In approximately 20% of patients, anaphylaxis may recur 1 to 72 hours after apparent clinical resolution of all signs and symptoms in what is called a biphasic reaction.33 There is currently no evidence or expert consensus on clinical predictors of the occurrence of a biphasic anaphylactic reaction, although it has been suggested that a biphasic reaction is rare in patients without hypotension and airway obstruction during the initial evaluation.34 Most authorities recommend an observation period (8 to 24 hours) after the initial manifestation to minimize the untoward effects of the biphasic reaction.33
Differential Diagnosis and Medical Decision Making
The clinical spectrum of anaphylaxis overlaps that of several other syndromes, especially those involving the skin and cardiorespiratory system (Box 106.6). Vasovagal reactions may mimic early anaphylaxis, although these patients usually have bradycardia, hypotension, diaphoresis, and pallor, as opposed to the tachycardia, hypotension, diaphoresis, and urticaria usually associated with anaphylaxis. Anaphylactic shock may also appear clinically indistinguishable from other forms of shock (distributive, septic, or cardiogenic). The acute onset and characteristic angioedematous urticarial rash favor the diagnosis of anaphylactic shock. Flushing syndromes also frequently mimic anaphylactic reactions. They may be associated with dry skin or diaphoresis and are the result of numerous different drugs, ingestants, and other physiologic syndromes.
Box 106.6 Differential Diagnosis of Anaphylaxis
Anaphylaxis is diagnosed clinically. The diagnosis is considered highly likely when any of three clinical criteria in Box 106.7 are met.35
Box 106.7
Clinical Diagnosis of Anaphylaxis
Anaphylaxis is highly likely when any of the following three criteria are fulfilled:
1. Acute onset (minutes to hours) of an illness with cutaneous or mucosal involvement AND at least one of the following:
2. Two or more of the following occurring rapidly after exposure to a probable allergen (minutes to several hours):
3. Reduced blood pressure after exposure to a known allergen for that patient (age specific, less than 90 mm Hg, or 30% decline from baseline)
Reproduced from Sampson HA, Munoz-Furlong A, Campbell RL, et al. Second symposium on the definition and management of anaphylaxis: summary report—Second National Institute of Allergy and Infectious Disease/Food Allergy and Anaphylaxis Network symposium. Ann Emerg Med 2006;47:373–80.
Currently, histamine and tryptase are the only measurable markers of anaphylaxis in clinical laboratories.31 Histamine plasma levels become elevated approximately 10 minutes after the onset of symptoms and remain so for up to an hour. Peak levels of serum tryptase occur between 60 and 90 minutes and persist for as long as 6 hours. These markers must be determined within the time frames mentioned and may be helpful when it is uncertain whether anaphylaxis has occurred. It should be noted that food-induced anaphylaxis may not be accompanied by elevated levels of tryptase. Additional laboratory tests and imaging may be necessary to help rule out other disease processes.
Treatment
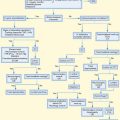
Early administration of epinephrine is the mainstay of treatment of anaphylaxis (Box 106.8). It should be administered quickly and preferably intramuscularly in the lateral aspect of the thigh (vastus lateralis). Epinephrine should be considered if anaphylaxis is suspected even when only one system (e.g., skin) is involved. Doses can be repeated every 5 to 10 minutes or more frequently as indicated clinically.27,31 Caution should be exercised in patients with risk factors for ischemic heart disease, although there are no absolute contraindications to epinephrine in those initially seen in anaphylactic shock. Intravenous infusion should be used only in patients who remain hypotensive and have failed to respond to multiple intramuscular injections.27,31 Inhaled epinephrine may be used as adjunctive therapy for laryngeal edema but should never replace the intramuscular or intravenous route (Fig. 106.5).
Box 106.8 Treatment Options for Anaphylaxis
General
Two large-bore intravenous lines (intraosseous if unable to acquire intravenous access)
Placement of the patient on a monitor
Supplement oxygen (nasal canula, non-rebreather mask)
Consideration of a tourniquet—if anaphylaxis is due to an insect sting at the distal end of an extremity (blood pressure cuff inflated to approximately 20 mm Hg greater than systolic blood pressure), release every 10 minutes for 1 minute or when the symptoms resolve
Epinephrine
Intramuscular (subcutaneous acceptable)—1 : 1000; adult: 0.3 to 0.5 mL every 5 minutes as necessary, titrated to effect; pediatric: 0.01 mL/kg every 5 minutes as necessary, titrated to effect)
Alternatively, EpiPen (0.3 mL) or EpiPen Jr (0.15 mL) can be administered into the anterolateral aspect of the thigh; removal of clothing unnecessary
Intravenous infusion—1 : 100,000; 0.1 mL of a 1 : 1000 dilution in 10 mL of normal saline (NS), 100 mcg, over a 10-minute period; equivalent to 10 mcg/min for a 10-min period, titrated to effect; repeat as necessary; continuous hemodynamic monitoring required
Continuous infusion—1 mL of a 1 : 1000 dilution of epinephrine in 250 mL of 5% dextrose in water (D5W) results in a concentration of 4 mcg/mL; can be started at 1 mcg/min and increased to 4 mcg/min if needed
Pediatric continuous infusion—rate of 0.1 mcg/kg/min (up to 30 kg) advised, with increasing increments of 0.1 mcg/kg/min to a maximum of 1.5 mcg/kg/min
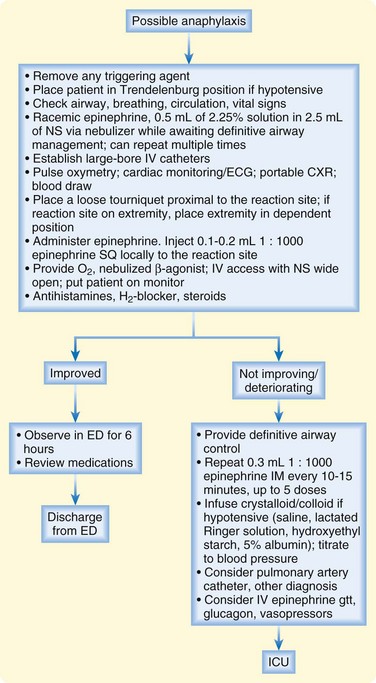
Fig. 106.5 Treatment algorithm for possible anaphylaxis (see Box 106.8 for drug dosages and indications).
Hypotension is treated aggressively with crystalloid infusion and colloid as indicated clinically. Caution should be taken in patients with a history of congestive heart failure. Patients with persistent hypotension despite appropriate doses of epinephrine and intravenous fluid resuscitation should be administered vasopressors drips (see Box 106.8).
 Red Flags
Red FlagsSpecial Considerations
Patients Taking Beta-Blockers
Patients currently taking beta-blockers may not respond adequately to epinephrine and intravenous fluids. A glucagon infusion should be strongly considered in these cases (see Box 106.8).27,31 Conversely, these patients may also demonstrate unopposed α-adrenergic effects after epinephrine administration and may require an alpha-blocker such as phentolamine.
Patients with Allergy to Intravenous Contrast Media
Pretreatment with antihistamines and steroids significantly decreases the frequency and severity of anaphylactoid reactions in patients who have sustained a previous reaction to intravenous radiocontrast media. Each hospital has its own pretreatment protocols for clinical procedures; Box 106.9 reproduces one such protocol.36
Box 106.9
Standard Pharmacologic Treatment Protocol for Patients with a History of Radiocontrast-Induced Anaphylaxis
Prednisone, 50 mg PO given 13 hours, 7 hours, and 1 hour before the procedure
Diphenhydramine, 50 mg IM given 1 hour before the procedure
Consider giving an H2 antagonist, such as ranitidine, 300 mg PO as drops 3 hours before the procedure
Modified from Lieberman P. Anaphylaxis. Med Clin North Am 2006;90:77–95.
Follow-up, Next Steps in Care, and Patient Education
Patients who experience complete resolution of symptoms and were never hypotensive can potentially be discharged home. Although the time frame for biphasic reactions varies significantly, limited literature suggests that patients be observed in the ED for up to 8 hours.33 Oral H1 antihistamines such as diphenhydramine (50 mg every 6 hours for 72 hours) and H2 blockers such as ranitidine (150 mg every 12 hours for 72 hours) along with oral prednisone may prevent possible relapse.37 These patients should be instructed to return to the ED if they experience any recurrent symptoms consistent with their previous anaphylactic reaction.
Tips and Tricks
Prevention of Anaphylaxis and Anaphylactic Death
Obtain a thorough drug allergy and atopic history.
Check all drugs for proper labeling.
Give drugs orally rather than parenterally when possible.
Give drugs in distal end of the extremity if possible when a parenteral route is necessary.
Always have resuscitation equipment available when administering antigenic compounds.
Ensure that patients remain in the emergency department for at least 30 minutes after drug administration.
Use unrelated drugs when feasible in susceptible patients.
Encourage patients to carry warning identification (Medic-Alert, wallet ID).
Educate patients on the technique of self-injection of epinephrine and urge them to carry the treatment kit at all times.
Instruct patients to avoid known antigens (stinging insects, foods, antibiotics).
Refer patients to an allergist for a skin test and possible immunotherapy.
1 Kay AB. Allergy and allergic diseases. Second of two parts. N Engl J Med. 2001;344:109–113.
2 Kay AB. Allergy and allergic diseases. First of two parts. N Engl J Med. 2001;344:30–37.
3 Weiss ST. Eat dirt—the hygiene hypothesis and allergic diseases. N Engl J Med. 2002;347:930–931.
4 Settipane RA. Demographics and epidemiology of allergic and nonallergic rhinitis. Allergy Asthma Proc. 2001;22:185–189.
5 Ellis AK, Day JH. Clinical reactivity to insect stings. Curr Opin Allergy Clin Immunol. 2005;5:349–354.
6 World Health Organization. The importance of pharmacovigilance—safety monitoring of medicinal products. Geneva: World Health Organization; 2002.
7 Gomes ER, Demoly P. Epidemiology of hypersensitivity drug reactions. Curr Opin Allergy Clin Immunol. 2005;5:309–316.
8 Chaplin DD. 1. Overview of the immune response. J Allergy Clin Immunol. 2003;111(2 Suppl):S442–S459.
9 Chaplin DD. 1. Overview of the human immune response. J Allergy Clin Immunol. 2006;117(2 Suppl Mini-Primer):S430–S435.
10 Galli SJ, Tsai M, Piliponsky AM. The development of allergic inflammation. Nature. 2008;454:445–454.
11 Gould HJ, Sutton BJ, Beavil AJ, et al. The biology of IGE and the basis of allergic disease. Annu Rev Immunol. 2003;21:579–628.
12 Greiner AN. Allergic rhinitis: impact of the disease and considerations for management. Med Clin North Am. 2006;90:17–38.
13 Kelkar PS, Li JT. Cephalosporin allergy. N Engl J Med. 2001;345:804–809.
14 Salkind AR, Cuddy PG, Foxworth JW. Is this patient allergic to penicillin? An evidence-based analysis of the likelihood of penicillin allergy. JAMA. 2001;285:2498–2505.
15 Asthma and the other allergic diseases. NIAID Task Force Report. NIH Publ No. 79–387. Bethesda, MD: National Institutes of Health; 1979.
16 Brozek JL, Bousquet J, Baena-Cagnani CE, et al. Allergic Rhinitis and its Impact on Asthma (ARIA) guidelines: 2010 revision. J Allergy Clin Immunol. 2010;126:466–476.
17 Owen CG, Shah A, Henshaw K, et al. Topical treatments for seasonal allergic conjunctivitis: systematic review and meta-analysis of efficacy and effectiveness. Br J Gen Pract. 2004;54:451–456.
18 Zuberbier T, Asero R, Bindslev-Jensen C, et al. for the Dermatology Section of the European Academy of Allergology and Clinical Immunology; Global Allergy and Asthma European Network; European Dermatology Forum; World Allergy Organization. EAACI/GA(2)LEN/EDF/WAO guideline: definition, classification and diagnosis of urticaria. Allergy. 2009;64:1417–1426.
19 Bischoff SC, Kramer S. Human mast cells, bacteria, and intestinal immunity. Immunol Rev. 2007;217:329–337.
20 Dibbern Jr, DA. Urticaria: selected highlights and recent advances. Med Clin North Am. 2006;90:187–209.
21 Maurer M, Grabbe J. Urticaria: its history-based diagnosis and etiologically oriented treatment. Dtsch Arztebl Int. 2008;105:458–465.
22 Kaplan AP, Greaves MW. Angioedema. J Am Acad Dermatol. 2005;53:373–388.
23 Bowen T, Cicardi M, Farkas H, et al. 2010 International consensus algorithm for the diagnosis, therapy and management of hereditary angioedema. Allergy Asthma Clin Immunol. 2010;6:24.
24 Hoover T, Lippmann M, Grouzmann E, et al. Angiotensin converting enzyme inhibitor induced angio-oedema: a review of the pathophysiology and risk factors. Clin Exp Allergy. 2010;40:50–61.
25 Malde B, Regalado J, Greenberger PA. Investigation of angioedema associated with the use of angiotensin-converting enzyme inhibitors and angiotensin receptor blockers. Ann Allergy Asthma Immunol. 2007;98:57–63.
26 Lieberman P. Epidemiology of anaphylaxis. Curr Opin Allergy Clin Immunol. 2008;8:316–320.
27 Lieberman P, Nicklas RA, Oppenheimer J, et al. The diagnosis and management of anaphylaxis practice parameter: 2010 update. J Allergy Clin Immunol. 2010;126:477–480.
28 Ben-Shoshan M, Clarke AE. Anaphylaxis: past, present and future. Allergy. 2011;66:1–14.
29 Schwartz LB, Delgado L, Craig T, et al. Exercise-induced hypersensitivity syndromes in recreational and competitive athletes: a PRACTALL consensus report (what the general practitioner should know about sports and allergy). Allergy. 2008;63:953–961.
30 Greenberger PA. Idiopathic anaphylaxis. Immunol Allergy Clin North Am. 2007;27:273–293. vii–viii
31 Simons FE. Anaphylaxis. J Allergy Clin Immunol. 2010;125(2 Suppl 2):S161–S181.
32 Ridella M, Bagdure S, Nugent K, et al. Kounis syndrome following beta-lactam antibiotic use: review of literature. Inflamm Allergy Drug Targets. 2009;8:11–16.
33 Tole JW, Lieberman P. Biphasic anaphylaxis: review of incidence, clinical predictors, and observation recommendations. Immunol Allergy Clin North Am. 2007;27:309–326. viii
34 Golden DB. Patterns of anaphylaxis: acute and late phase features of allergic reactions. Novartis Found Symp. 2004;257:101–110.
35 Sampson HA, Munoz-Furlong A, Campbell RL, et al. Second symposium on the definition and management of anaphylaxis: summary report—Second National Institute of Allergy and Infectious Disease/Food Allergy and Anaphylaxis Network Symposium. Ann Emerg Med. 2006;47:373–380.
36 Lieberman P. Anaphylaxis. Med Clin North Am. 2006;90:77–95. viii
37 Choo KJ, Simons E, Sheikh A. Glucocorticoids for the treatment of anaphylaxis: Cochrane systematic review. Allergy. 2010;65:1205–1211.