PART 13: Disorders of the Kidney and Urinary Tract
332e |
Cellular and Molecular Biology of the Kidney |
The kidney is one of the most highly differentiated organs in the body. At the conclusion of embryologic development, nearly 30 different cell types form a multitude of filtering capillaries and segmented nephrons enveloped by a dynamic interstitium. This cellular diversity modulates a variety of complex physiologic processes. Endocrine functions, the regulation of blood pressure and intraglomerular hemodynamics, solute and water transport, acid-base balance, and removal of drug metabolites are all accomplished by intricate mechanisms of renal response. This breadth of physiology hinges on the clever ingenuity of nephron architecture that evolved as complex organisms came out of water to live on land.
EMBRYOLOGIC DEVELOPMENT
Kidneys develop from intermediate mesoderm under the timed or sequential control of a growing number of genes, described in Fig. 332e-1. The transcription of these genes is guided by morphogenic cues that invite two ureteric buds to each penetrate bilateral metanephric blastema, where they induce primary mesenchymal cells to form early nephrons. The two ureteric buds emerge from posterior nephric ducts and mature into separate collecting systems that eventually form a renal pelvis and ureter. Induced mesenchyme undergoes mesenchymal epithelial transitions to form comma-shaped bodies at the proximal end of each ureteric bud leading to the formation of S-shaped nephrons that cleft and enjoin with penetrating endothelial cells derived from sprouting angioblasts. Under the influence of vascular endothelial growth factor A (VEGF-A), these penetrating cells form capillaries with surrounding mesangial cells that differentiate into a glomerular filter for plasma water and solute. The ureteric buds branch, and each branch produce a new set of nephrons. The number of branching events ultimately determines the total number of nephrons in each kidney. There are approximately 900,000 glomeruli in each kidney in normal birth weight adults and as few as 225,000 in low-birth-weight adults, with the latter producing numerous comorbid risks.
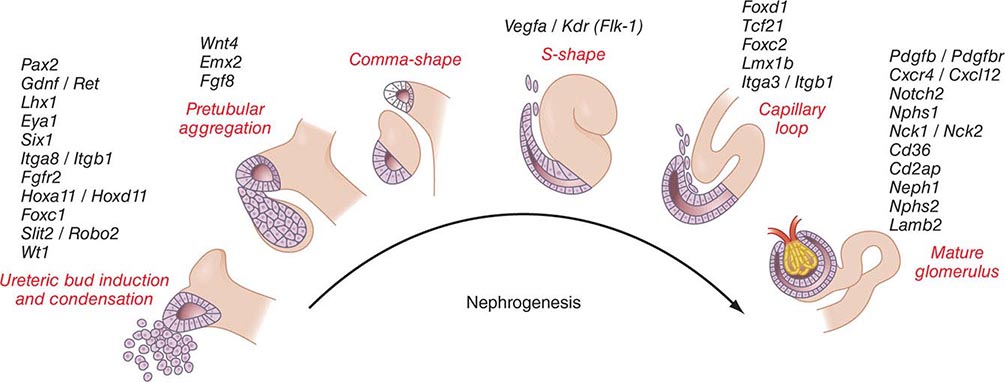
FIGURE 332e-1 Genes controlling renal nephrogenesis. A growing number of genes have been identified at various stages of glomerulotubular development in the mammalian kidney. The genes listed have been tested in various genetically modified mice, and their location corresponds to the classical stages of kidney development postulated by Saxen in 1987.
Glomeruli evolve as complex capillary filters with fenestrated endothelia under the guiding influence of VEGF-A and angiopoietin-1 secreted by adjacently developing podocytes. Epithelial podocytes facing the urinary space envelop the exterior basement membrane supporting these emerging endothelial capillaries. Podocytes are partially polarized and periodically fall off into the urinary space by epithelial-mesenchymal transition, and to a lesser extent apoptosis, only to be replenished by migrating parietal epithelia from Bowman capsule. Impaired replenishment results in heavy proteinuria. Podocytes attach to the basement membrane by special foot processes and share a slit-pore membrane with their neighbor. The slit-pore membrane forms a filter for plasma water and solute by the synthetic interaction of nephrin, annexin-4, CD2AP, FAT, ZO-1, P-cadherin, podocin, TRPC6, PLCE1, and Neph 1-3 proteins. Mutations in many of these proteins also result in heavy proteinuria. The glomerular capillaries are embedded in a mesangial matrix shrouded by parietal and proximal tubular epithelia forming Bowman capsule. Mesangial cells have an embryonic lineage consistent with arteriolar or juxtaglomerular cells and contain contractile actin-myosin fibers. These mesangial cells make contact with glomerular capillary loops, and their local matrix holds them in condensed arrangement.
Between nephrons lies the renal interstitium. This region forms a functional space surrounding glomeruli and their downstream tubules, which are home to resident and trafficking cells such as fibroblasts, dendritic cells, occasional lymphocytes, and lipid-laden macrophages. The cortical and medullary capillaries, which siphon off solute and water following tubular reclamation of glomerular filtrate, are also part of the interstitial fabric as well as a web of connective tissue that supports the kidney’s emblematic architecture of folding tubules. The relational precision of these structures determines the unique physiology of the kidney.
Each nephron is partitioned during embryologic development into a proximal tubule, descending and ascending limbs of the loop of Henle, distal tubule, and the collecting duct. These classic tubular segments build from subsegments lined by highly unique epithelia serving regional physiology. All nephrons have the same structural components, but there are two types whose structures depend on their location within the kidney. The majority of nephrons are cortical, with glomeruli located in the mid-to-outer cortex. Fewer nephrons are juxtamedullary, with glomeruli at the boundary of the cortex and outer medulla. Cortical nephrons have short loops of Henle, whereas juxtamedullary nephrons have long loops of Henle. There are critical differences in blood supply as well. The peritubular capillaries surrounding cortical nephrons are shared among adjacent nephrons. By contrast, juxtamedullary nephrons depend on individual capillaries called vasa recta. Cortical nephrons perform most of the glomerular filtration because there are more of them and because their afferent arterioles are larger than their respective efferent arterioles. The juxtamedullary nephrons, with longer loops of Henle, create an osmotic gradient for concentrating urine. How developmental instructions specify the differentiation of all these unique epithelia among various tubular segments is still unknown.
DETERMINANTS AND REGULATION OF GLOMERULAR FILTRATION
Renal blood flow normally drains approximately 20% of the cardiac output, or 1000 mL/min. Blood reaches each nephron through the afferent arteriole leading into a glomerular capillary where large amounts of fluid and solutes are filtered to form the tubular fluid. The distal ends of the glomerular capillaries coalesce to form an efferent arteriole leading to the first segment of a second capillary network (cortical peritubular capillaries or medullary vasa recta) surrounding the tubules (Fig. 332e-2A). Thus, nephrons have two capillary beds arranged in a series separated by the efferent arteriole that regulates the hydrostatic pressure in both capillary beds. The distal capillaries empty into small venous branches that coalesce into larger veins to eventually form the renal vein.
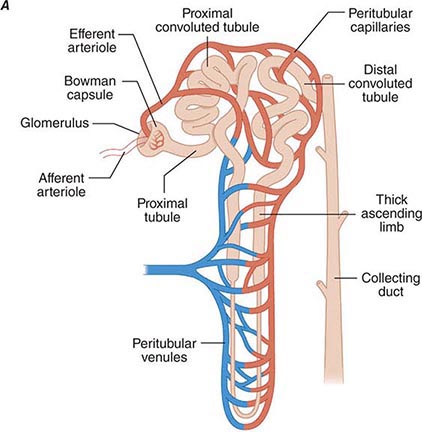
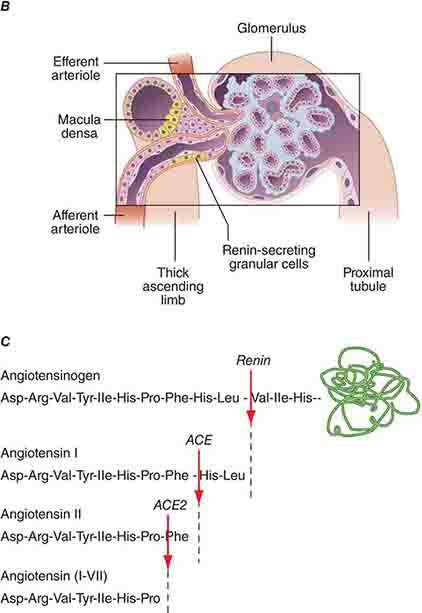
FIGURE 332e-2 Renal microcirculation and the renin-angiotensin system. A. Diagram illustrating relationships of the nephron with glomerular and peritubular capillaries. B. Expanded view of the glomerulus with its juxtaglomerular apparatus including the macula densa and adjacent afferent arteriole. C. Proteolytic processing steps in the generation of angiotensins.
The hydrostatic pressure gradient across the glomerular capillary wall is the primary driving force for glomerular filtration. Oncotic pressure within the capillary lumen, determined by the concentration of unfiltered plasma proteins, partially offsets the hydrostatic pressure gradient and opposes filtration. As the oncotic pressure rises along the length of the glomerular capillary, the driving force for filtration falls to zero on reaching the efferent arteriole. Approximately 20% of the renal plasma flow is filtered into Bowman space, and the ratio of glomerular filtration rate (GFR) to renal plasma flow determines the filtration fraction. Several factors, mostly hemodynamic, contribute to the regulation of filtration under physiologic conditions.
Although glomerular filtration is affected by renal artery pressure, this relationship is not linear across the range of physiologic blood pressures due to autoregulation of GFR. Autoregulation of glomerular filtration is the result of three major factors that modulate either afferent or efferent arteriolar tone: these include an autonomous vasoreactive (myogenic) reflex in the afferent arteriole, tubuloglomerular feedback, and angiotensin II-mediated vasoconstriction of the efferent arteriole. The myogenic reflex is a first line of defense against fluctuations in renal blood flow. Acute changes in renal perfusion pressure evoke reflex constriction or dilatation of the afferent arteriole in response to increased or decreased pressure, respectively. This phenomenon helps protect the glomerular capillary from sudden changes in systolic pressure.
Tubuloglomerular feedback (TGF) changes the rate of filtration and tubular flow by reflex vasoconstriction or dilatation of the afferent arteriole. TGF is mediated by specialized cells in the thick ascending limb of the loop of Henle called the macula densa that act as sensors of solute concentration and tubular flow rate. With high tubular flow rates, a proxy for an inappropriately high filtration rate, there is increased solute delivery to the macula densa (Fig. 332e-2B) that evokes vasoconstriction of the afferent arteriole causing GFR to return toward normal. One component of the soluble signal from the macula densa is adenosine triphosphate (ATP) released by the cells during increased NaCl reabsorption. ATP is metabolized in the extracellular space to generate adenosine, a potent vasoconstrictor of the afferent arteriole. During conditions associated with a fall in filtration rate, reduced solute delivery to the macula densa attenuates TGF, allowing afferent arteriolar dilatation and restoring glomerular filtration to normal levels. Angiotensin II and reactive oxygen species enhance, while nitric oxide (NO) blunts, TGF.
The third component underlying autoregulation of GFR involves angiotensin II. During states of reduced renal blood flow, renin is released from granular cells within the wall of the afferent arteriole near the macula densa in a region called the juxtaglomerular apparatus (Fig. 332e-2B). Renin, a proteolytic enzyme, catalyzes the conversion of angiotensinogen to angiotensin I, which is subsequently converted to angiotensin II by angiotensin-converting enzyme (ACE) (Fig. 332e-2C). Angiotensin II evokes vasoconstriction of the efferent arteriole, and the resulting increased glomerular hydrostatic pressure elevates filtration to normal levels.
MECHANISMS OF RENAL TUBULAR TRANSPORT
The renal tubules are composed of highly differentiated epithelia that vary dramatically in morphology and function along the nephron (Fig. 332e-3). The cells lining the various tubular segments form monolayers connected to one another by a specialized region of the adjacent lateral membranes called the tight junction. Tight junctions form an occlusive barrier that separates the lumen of the tubule from the interstitial spaces surrounding the tubule and also apportions the cell membrane into discrete domains: the apical membrane facing the tubular lumen and the basolateral membrane facing the interstitium. This regionalization allows cells to allocate membrane proteins and lipids asymmetrically. Owing to this feature, renal epithelial cells are said to be polarized. The asymmetric assignment of membrane proteins, especially proteins mediating transport processes, provides the machinery for directional movement of fluid and solutes by the nephron.
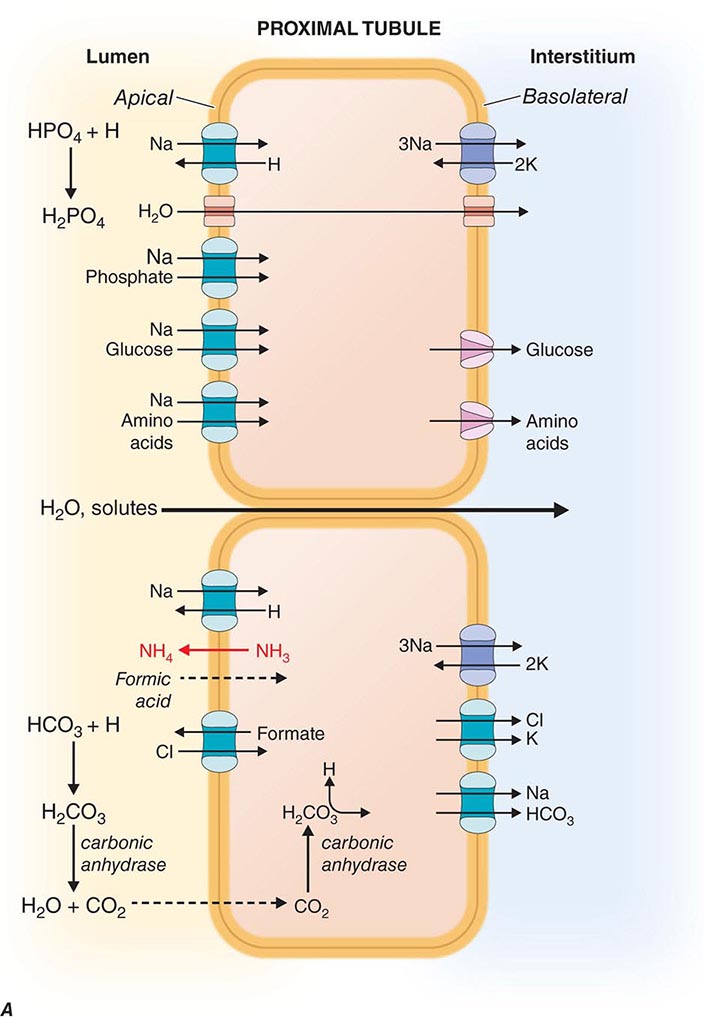
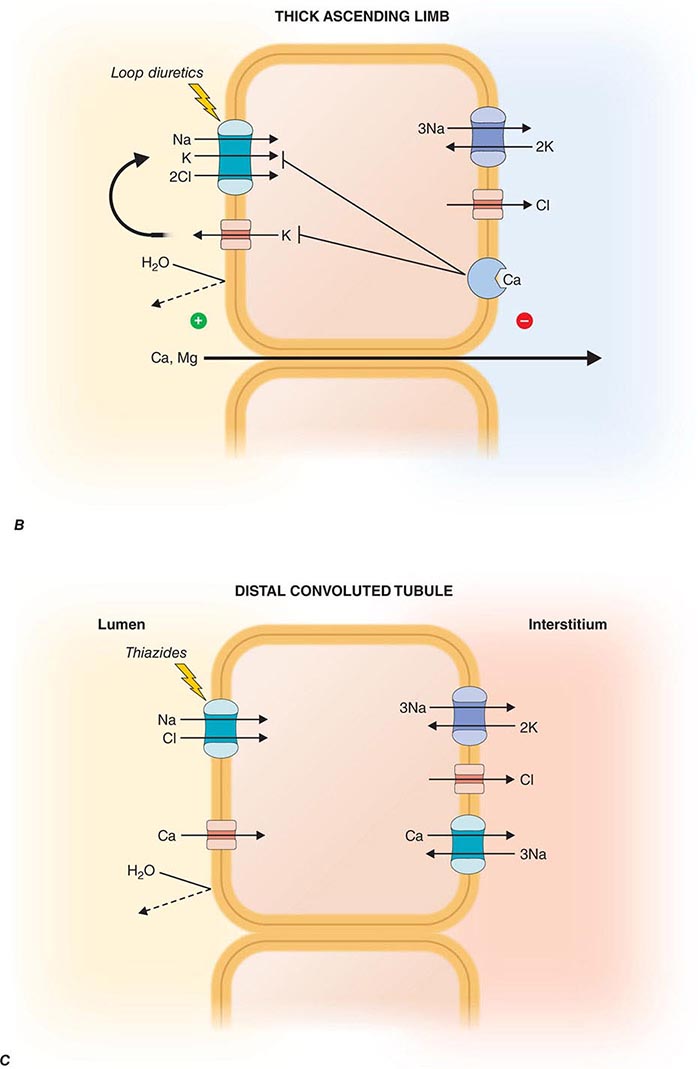
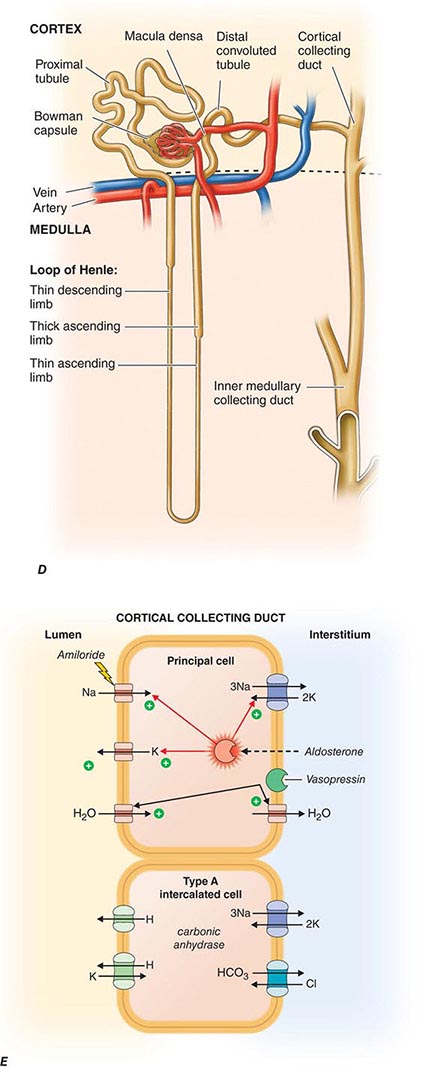
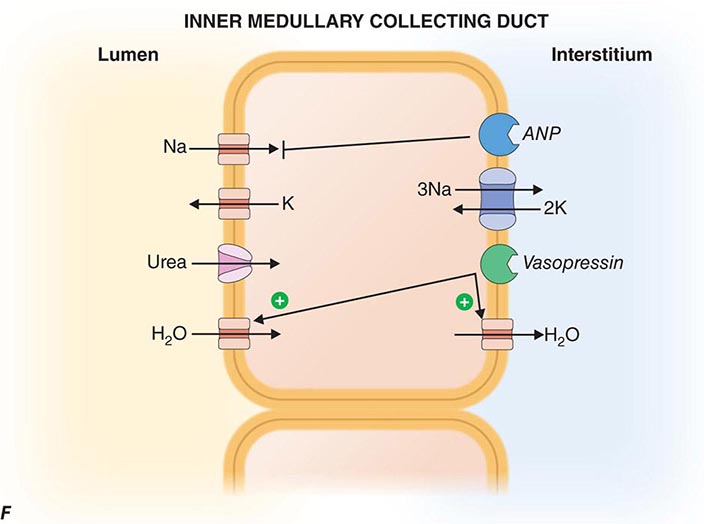
FIGURE 332e-3 Transport activities of the major nephron segments. Representative cells from five major tubular segments are illustrated with the lumen side (apical membrane) facing left and interstitial side (basolateral membrane) facing right. A. Proximal tubular cells. B. Typical cell in the thick ascending limb of the loop of Henle. C. Distal convoluted tubular cell. D. Overview of entire nephron. E. Cortical collecting duct cells. F. Typical cell in the inner medullary collecting duct. The major membrane transporters, channels, and pumps are drawn with arrows indicating the direction of solute or water movement. For some events, the stoichiometry of transport is indicated by numerals preceding the solute. Targets for major diuretic agents are labeled. The actions of hormones are illustrated by arrows with plus signs for stimulatory effects and lines with perpendicular ends for inhibitory events. Dotted lines indicate free diffusion across cell membranes. The dashed line indicates water impermeability of cell membranes in the thick ascending limb and distal convoluted tubule.
EPITHELIAL SOLUTE TRANSPORT
There are two types of epithelial transport. Movement of fluid and solutes sequentially across the apical and basolateral cell membranes (or vice versa) mediated by transporters, channels, or pumps is called cellular transport. By contrast, movement of fluid and solutes through the narrow passageway between adjacent cells is called paracellular transport. Paracellular transport occurs through tight junctions, indicating that they are not completely “tight.” Indeed, some epithelial cell layers allow rather robust paracellular transport to occur (leaky epithelia), whereas other epithelia have more effective tight junctions (tight epithelia). In addition, because the ability of ions to flow through the paracellular pathway determines the electrical resistance across the epithelial monolayer, leaky and tight epithelia are also referred to as low- or high-resistance epithelia, respectively. The proximal tubule contains leaky epithelia, whereas distal nephron segments, such as the collecting duct, contain tight epithelia. Leaky epithelia are most well suited for bulk fluid reabsorption, whereas tight epithelia allow for more refined control and regulation of transport.
MEMBRANE TRANSPORT
Cell membranes are composed of hydrophobic lipids that repel water and aqueous solutes. The movement of solutes and water across cell membranes is made possible by discrete classes of integral membrane proteins, including channels, pumps, and transporters. These different mechanisms mediate specific types of transport activities, including active transport (pumps), passive transport (channels), facilitated diffusion (transporters), and secondary active transport (cotransporters). Active transport requires metabolic energy generated by the hydrolysis of ATP. Active transport pumps are ion-translocating ATPases, including the ubiquitous Na+/K+-ATPase, the H+-ATPases, and Ca2+-ATPases. Active transport creates asymmetric ion concentrations across a cell membrane and can move ions against a chemical gradient. The potential energy stored in a concentration gradient of an ion such as Na+ can be used to drive transport through other mechanisms (secondary active transport). Pumps are often electrogenic, meaning they can create an asymmetric distribution of electrostatic charges across the membrane and establish a voltage or membrane potential. The movement of solutes through a membrane protein by simple diffusion is called passive transport. This activity is mediated by channels created by selectively permeable membrane proteins, and it allows solute or water to move across a membrane driven by favorable concentration gradients or electrochemical potential. Facilitated diffusion is a specialized type of passive transport mediated by simple transporters called carriers or uniporters. For example, hexose transporters such as GLUT2 mediate glucose transport by tubular cells. These transporters are driven by the concentration gradient for glucose that is highest in extracellular fluids and lowest in the cytoplasm due to rapid metabolism. Many other transporters operate by translocating two or more ions/solutes in concert either in the same direction (symporters or cotransporters) or in opposite directions (antiporters or exchangers) across the cell membrane. The movement of two or more ions/solutes may produce no net change in the balance of electrostatic charges across the membrane (electroneutral), or a transport event may alter the balance of charges (electrogenic). Several inherited disorders of renal tubular solute and water transport occur as a consequence of mutations in genes encoding a variety of channels, transporter proteins, and their regulators (Table 332e-1).
|
INHERITED DISORDERS AFFECTING RENAL TUBULAR ION AND SOLUTE TRANSPORT |
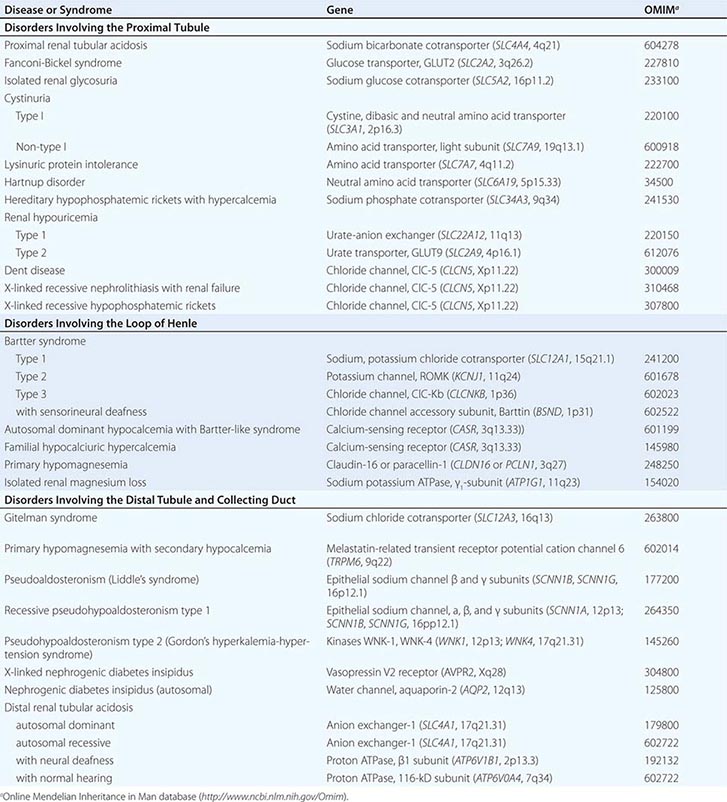
SEGMENTAL NEPHRON FUNCTIONS
Each anatomic segment of the nephron has unique characteristics and specialized functions enabling selective transport of solutes and water (Fig. 332e-3). Through sequential events of reabsorption and secretion along the nephron, tubular fluid is progressively conditioned into urine. Knowledge of the major tubular mechanisms responsible for solute and water transport is critical for understanding hormonal regulation of kidney function and the pharmacologic manipulation of renal excretion.
PROXIMAL TUBULE
The proximal tubule is responsible for reabsorbing ~60% of filtered NaCl and water, as well as ~90% of filtered bicarbonate and most critical nutrients such as glucose and amino acids. The proximal tubule uses both cellular and paracellular transport mechanisms. The apical membrane of proximal tubular cells has an expanded surface area available for reabsorptive work created by a dense array of microvilli called the brush border, and leaky tight junctions enable high-capacity fluid reabsorption.
Solute and water pass through these tight junctions to enter the lateral intercellular space where absorption by the peritubular capillaries occurs. Bulk fluid reabsorption by the proximal tubule is driven by high oncotic pressure and low hydrostatic pressure within the peritubular capillaries. Cellular transport of most solutes by the proximal tubule is coupled to the Na+ concentration gradient established by the activity of a basolateral Na+/K+-ATPase (Fig. 332e-3A). This active transport mechanism maintains a steep Na+ gradient by keeping intracellular Na+ concentrations low. Solute reabsorption is coupled to the Na+ gradient by Na+-dependent transporters such as Na+-glucose and Na+-phosphate cotransporters. In addition to the paracellular route, water reabsorption also occurs through the cellular pathway enabled by constitutively active water channels (aquaporin-1) present on both apical and basolateral membranes.
Proximal tubular cells reclaim bicarbonate by a mechanism dependent on carbonic anhydrases. Filtered bicarbonate is first titrated by protons delivered to the lumen by Na+/H+ exchange. The resulting carbonic acid (H2CO3) is metabolized by brush border carbonic anhydrase to water and carbon dioxide. Dissolved carbon dioxide then diffuses into the cell, where it is enzymatically hydrated by cytoplasmic carbonic anhydrase to re-form carbonic acid. Finally, intracellular carbonic acid dissociates into free protons and bicarbonate anions, and bicarbonate exits the cell through a basolateral Na+/HCO3– cotransporter. This process is saturable, resulting in urinary bicarbonate excretion when plasma levels exceed the physiologically normal range (24-26 meq/L). Carbonic anhydrase inhibitors such as acetazolamide, a class of weak diuretic agents, block proximal tubule reabsorption of bicarbonate and are useful for alkalinizing the urine.
The proximal tubule contributes to acid secretion by two mechanisms involving the titration of the urinary buffers ammonia (NH3) and phosphate. Renal NH3 is produced by glutamine metabolism in the proximal tubule. Subsequent diffusion of NH3 out of the proximal tubular cell enables trapping of H+ secreted by sodium-proton exchange in the lumen as ammonium ion (NH4+). Cellular K+ levels inversely modulate proximal tubular ammoniagenesis, and in the setting of high serum K+ from hypoaldosteronism, reduced ammoniagenesis facilitates the appearance of type IV renal tubular acidosis. Filtered hydrogen phosphate ion (HPO42–) is also titrated in the proximal tubule by secreted H+ to form H2PO4–, and this reaction constitutes a major component of the urinary buffer referred to as titratable acid. Most filtered phosphate ion is reabsorbed by the proximal tubule through a sodium-coupled cotransport process that is regulated by parathyroid hormone.
Chloride is poorly reabsorbed throughout the first segment of the proximal tubule, and a rise in Cl– concentration counterbalances the removal of bicarbonate anion from tubular fluid. In later proximal tubular segments, cellular Cl– reabsorption is initiated by apical exchange of cellular formate for higher luminal concentrations of Cl–. Once in the lumen, formate anions are titrated by H+ (provided by Na+/H+ exchange) to generate neutral formic acid, which can diffuse passively across the apical membrane back into the cell where it dissociates a proton and is recycled. Basolateral Cl– exit is mediated by a K+/Cl– cotransporter.
Reabsorption of glucose is nearly complete by the end of the proximal tubule. Cellular transport of glucose is mediated by apical Na+-glucose cotransport coupled with basolateral, facilitated diffusion by a glucose transporter. This process is also saturable, leading to glycosuria when plasma levels exceed 180-200 mg/dL, as seen in untreated diabetes mellitus.
The proximal tubule possesses specific transporters capable of secreting a variety of organic acids (carboxylate anions) and bases (mostly primary amine cations). Organic anions transported by these systems include urate, dicarboxylic acid anions (succinate), ketoacid anions, and several protein-bound drugs not filtered at the glomerulus (penicillins, cephalosporins, and salicylates). Probenecid inhibits renal organic anion secretion and can be clinically useful for raising plasma concentrations of certain drugs like penicillin and oseltamivir. Organic cations secreted by the proximal tubule include various biogenic amine neurotransmitters (dopamine, acetylcholine, epinephrine, norepinephrine, and histamine) and creatinine. The ATP-dependent transporter P-glycoprotein is highly expressed in brush border membranes and secretes several medically important drugs, including cyclosporine, digoxin, tacrolimus, and various cancer chemotherapeutic agents. Certain drugs like cimetidine and trimethoprim compete with endogenous compounds for transport by the organic cation pathways. Although these drugs elevate serum creatinine levels, there is no change in the actual GFR.
The proximal tubule, through distinct classes of Na+-dependent and Na+-independent transport systems, reabsorbs amino acids efficiently. These transporters are specific for different groups of amino acids. For example, cystine, lysine, arginine, and ornithine are transported by a system comprising two proteins encoded by the SLC3A1 and SLC7A9 genes. Mutations in either SLC3A1 or SLC7A9 impair reabsorption of these amino acids and cause the disease cystinuria. Peptide hormones, such as insulin and growth hormone, β2-microglobulin, albumin, and other small proteins, are taken up by the proximal tubule through a process of absorptive endocytosis and are degraded in acidified endocytic lysosomes. Acidification of these vesicles depends on a vacuolar H+-ATPase and Cl– channel. Impaired acidification of endocytic vesicles because of mutations in a Cl– channel gene (CLCN5) causes low-molecular-weight proteinuria in Dent disease.
LOOP OF HENLE
The loop of Henle consists of three major segments: descending thin limb, ascending thin limb, and ascending thick limb. These divisions are based on cellular morphology and anatomic location, but also correlate with specialization of function. Approximately 15–25% of filtered NaCl is reabsorbed in the loop of Henle, mainly by the thick ascending limb. The loop of Henle has an important role in urinary concentration by contributing to the generation of a hypertonic medullary interstitium in a process called countercurrent multiplication. The loop of Henle is the site of action for the most potent class of diuretic agents (loop diuretics) and also contributes to reabsorption of calcium and magnesium ions.
The descending thin limb is highly water permeable owing to dense expression of constitutively active aquaporin-1 water channels. By contrast, water permeability is negligible in the ascending limb. In the thick ascending limb, there is a high level of secondary active salt transport enabled by the Na+/K+/2Cl– cotransporter on the apical membrane in series with basolateral Cl– channels and Na+/K+-ATPase (Fig. 332e-3B). The Na+/K+/2Cl– cotransporter is the primary target for loop diuretics. Tubular fluid K+ is the limiting substrate for this cotransporter (tubular concentration of K+ is similar to plasma, about 4 meq/L), but transporter activity is maintained by K+ recycling through an apical potassium channel. The cotransporter also enables reabsorption of NH4+ in lieu of K+, and this leads to accumulation of both NH4+ and NH3 in the medullary interstitium. An inherited disorder of the thick ascending limb, Bartter syndrome, also results in a salt-wasting renal disease associated with hypokalemia and metabolic alkalosis; loss-of-function mutations in one of five distinct genes encoding components of the Na+/K+/2Cl– cotransporter (NKCC2), apical K+ channel (KCNJ1), basolateral Cl– channel (CLCNKB, BSND), or calcium-sensing receptor (CASR) can cause Bartter syndrome.
Potassium recycling also contributes to a positive electrostatic charge in the lumen relative to the interstitium that promotes divalent cation (Mg2+ and Ca2+) reabsorption through a paracellular pathway. A Ca2+-sensing, G-protein-coupled receptor (CaSR) on basolateral membranes regulates NaCl reabsorption in the thick ascending limb through dual signaling mechanisms using either cyclic AMP or eicosanoids. This receptor enables a steep relationship between plasma Ca2+ levels and renal Ca2+ excretion. Loss-of-function mutations in CaSR cause familial hypercalcemic hypocalciuria because of a blunted response of the thick ascending limb to extracellular Ca2+. Mutations in CLDN16 encoding paracellin-1, a transmembrane protein located within the tight junction complex, leads to familial hypomagnesemia with hypercalciuria and nephrocalcinosis, suggesting that the ion conductance of the paracellular pathway in the thick limb is regulated.
The loop of Henle contributes to urine-concentrating ability by establishing a hypertonic medullary interstitium that promotes water reabsorption by the downstream inner medullary collecting duct. Countercurrent multiplication produces a hypertonic medullary interstitium using two countercurrent systems: the loop of Henle (opposing descending and ascending limbs) and the vasa recta (medullary peritubular capillaries enveloping the loop). The countercurrent flow in these two systems helps maintain the hypertonic environment of the inner medulla, but NaCl reabsorption by the thick ascending limb is the primary initiating event. Reabsorption of NaCl without water dilutes the tubular fluid and adds new osmoles to medullary interstitial fluid. Because the descending thin limb is highly water permeable, osmotic equilibrium occurs between the descending limb tubular fluid and the interstitial space, leading to progressive solute trapping in the inner medulla. Maximum medullary interstitial osmolality also requires partial recycling of urea from the collecting duct.
DISTAL CONVOLUTED TUBULE
The distal convoluted tubule reabsorbs ~5% of the filtered NaCl. This segment is composed of a tight epithelium with little water permeability. The major NaCl-transporting pathway uses an apical membrane, electroneutral thiazide-sensitive Na+/Cl– cotransporter in tandem with basolateral Na+/K+-ATPase and Cl– channels (Fig. 332e-3C). Apical Ca2+-selective channels (TRPV5) and basolateral Na+/Ca2+ exchange mediate calcium reabsorption in the distal convoluted tubule. Ca2+ reabsorption is inversely related to Na+ reabsorption and is stimulated by parathyroid hormone. Blocking apical Na+/Cl– cotransport will reduce intracellular Na+, favoring increased basolateral Na+/Ca2+ exchange and passive apical Ca2+ entry. Loss-of-function mutations of SLC12A3 encoding the apical Na+/Cl– cotransporter cause Gitelman syndrome, a salt-wasting disorder associated with hypokalemic alkalosis and hypocalciuria. Mutations in genes encoding WNK kinases, WNK-1 and WNK-4, cause pseudohypoaldosteronism type II or Gordon syndrome characterized by familial hypertension with hyperkalemia. WNK kinases influence the activity of several tubular ion transporters. Mutations in this disorder lead to overactivity of the apical Na+/Cl– cotransporter in the distal convoluted tubule as the primary stimulus for increased salt reabsorption, extracellular volume expansion, and hypertension. Hyperkalemia may be caused by diminished activity of apical K+ channels in the collecting duct, a primary route for K+ secretion. Mutations in TRPM6 encoding Mg2+ permeable ion channels also cause familial hypomagnesemia with hypocalcemia. A molecular complex of TRPM6 and TRPM7 proteins is critical for Mg2+ reabsorption in the distal convoluted tubule.
COLLECTING DUCT
The collecting duct modulates the final composition of urine. The two major divisions, the cortical collecting duct and inner medullary collecting duct, contribute to reabsorbing ~4-5% of filtered Na+ and are important for hormonal regulation of salt and water balance. The cortical collecting duct contains high-resistance epithelia with two cell types. Principal cells are the main water, Na+-reabsorbing, and K+-secreting cells, and the site of action of aldosterone, K+-sparing diuretics, and mineralocorticoid receptor antagonists such as spironolactone. The other cells are type A and B intercalated cells. Type A intercalated cells mediate acid secretion and bicarbonate reabsorption also under the influence of aldosterone. Type B intercalated cells mediate bicarbonate secretion and acid reabsorption.
Virtually all transport is mediated through the cellular pathway for both principal cells and intercalated cells. In principal cells, passive apical Na+ entry occurs through the amiloride-sensitive, epithelial Na+ channel (ENaC) with basolateral exit via the Na+/K+-ATPase (Fig. 332e-3E). This Na+ reabsorptive process is tightly regulated by aldosterone and is physiologically activated by a variety of proteolytic enzymes that cleave extracellular domains of ENaC; plasmin in the tubular fluid of nephrotic patients, for example, activates ENaC, leading to sodium retention. Aldosterone enters the cell across the basolateral membrane, binds to a cytoplasmic mineralocorticoid receptor, and then translocates into the nucleus, where it modulates gene transcription, resulting in increased Na+ reabsorption and K+ secretion. Activating mutations in ENaC increase Na+ reclamation and produce hypokalemia, hypertension, and metabolic alkalosis (Liddle’s syndrome). The potassium-sparing diuretics amiloride and triamterene block ENaC, causing reduced Na+ reabsorption.
Principal cells secrete K+ through an apical membrane potassium channel. Several forces govern the secretion of K+. Most importantly, the high intracellular K+ concentration generated by Na+/K+-ATPase creates a favorable concentration gradient for K+ secretion into tubular fluid. With reabsorption of Na+ without an accompanying anion, the tubular lumen becomes negative relative to the cell interior, creating a favorable electrical gradient for secretion of potassium. When Na+ reabsorption is blocked, the electrical component of the driving force for K+ secretion is blunted, and this explains lack of excess urinary K+ loss during treatment with potassium-sparing diuretics or mineralocorticoid receptor antagonists. K+ secretion is also promoted by aldosterone actions that increase regional Na+ transport favoring more electronegativity and by increasing the number and activity of potassium channels. Fast tubular fluid flow rates that occur during volume expansion or diuretics acting “upstream” of the cortical collecting duct also increase K+ secretion, as does the presence of relatively nonreabsorbable anions (including bicarbonate and semisynthetic penicillins) that contribute to the lumen-negative potential. Off-target effects of certain antibiotics, such as trimethoprim and pentamidine, block ENaCs and predispose to hyperkalemia, especially when renal K+ handling is impaired for other reasons. Principal cells, as described below, also participate in water reabsorption by increased water permeability in response to vasopressin.
Intercalated cells do not participate in Na+ reabsorption but, instead, mediate acid-base secretion. These cells perform two types of transport: active H+ transport mediated by H+-ATPase (proton pump), and Cl–/HCO3– exchange. Intercalated cells arrange the two transport mechanisms on opposite membranes to enable either acid or base secretion. Type A intercalated cells have an apical proton pump that mediates acid secretion and a basolateral Cl–/HCO3– anion exchanger for bicarbonate reabsorption (Fig. 332e-3E); aldosterone increases the number of H+-ATPase pumps, sometimes contributing to the development of metabolic alkalosis. Secreted H+ is buffered by NH3 that has diffused into the collecting duct lumen from the surrounding interstitium. By contrast, type B intercalated cells have the anion exchanger on the apical membrane to mediate bicarbonate secretion while the proton pump resides on the basolateral membrane to enable acid reabsorption. Under conditions of acidemia, the kidney preferentially uses type A intercalated cells to secrete the excess H+ and generate more HCO3–. The opposite is true in states of bicarbonate excess with alkalemia where the type B intercalated cells predominate. An extracellular protein called hensin mediates this adaptation.
Inner medullary collecting duct cells share many similarities with principal cells of the cortical collecting duct. They have apical Na+ and K+ channels that mediate Na+ reabsorption and K+ secretion, respectively (Fig. 332e-3F). Inner medullary collecting duct cells also have vasopressin-regulated water channels (aquaporin-2 on the apical membrane, aquaporin-3 and -4 on the basolateral membrane). The antidiuretic hormone vasopressin binds to the V2 receptor on the basolateral membrane and triggers an intracellular signaling cascade through G-protein-mediated activation of adenylyl cyclase, resulting in an increase in the cellular levels of cyclic AMP. This signaling cascade stimulates the insertion of water channels into the apical membrane of the inner medullary collecting duct cells to promote increased water permeability. This increase in permeability enables water reabsorption and production of concentrated urine. In the absence of vasopressin, inner medullary collecting duct cells are water impermeable, and urine remains dilute.
Sodium reabsorption by inner medullary collecting duct cells is also inhibited by the natriuretic peptides called atrial natriuretic peptide or renal natriuretic peptide (urodilatin); the same gene encodes both peptides but uses different posttranslational processing of a common preprohormone to generate different proteins. Atrial natriuretic peptides are secreted by atrial myocytes in response to volume expansion, whereas urodilatin is secreted by renal tubular epithelia. Natriuretic peptides interact with either apical (urodilatin) or basolateral (atrial natriuretic peptides) receptors on inner medullary collecting duct cells to stimulate guanylyl cyclase and increase levels of cytoplasmic cGMP. This effect in turn reduces the activity of the apical Na+ channel in these cells and attenuates net Na+ reabsorption, producing natriuresis.
The inner medullary collecting duct transports urea out of the lumen, returning urea to the interstitium, where it contributes to the hypertonicity of the medullary interstitium. Urea is recycled by diffusing from the interstitium into the descending and ascending limbs of the loop of Henle.
HORMONAL REGULATION OF SODIUM AND WATER BALANCE
The balance of solute and water in the body is determined by the amounts ingested, distributed to various fluid compartments, and excreted by skin, bowel, and kidneys. Tonicity, the osmolar state determining the volume behavior of cells in a solution, is regulated by water balance (Fig. 332e-4A), and extracellular blood volume is regulated by Na+ balance (Fig. 332e-4B). The kidney is a critical modulator of both physiologic processes.
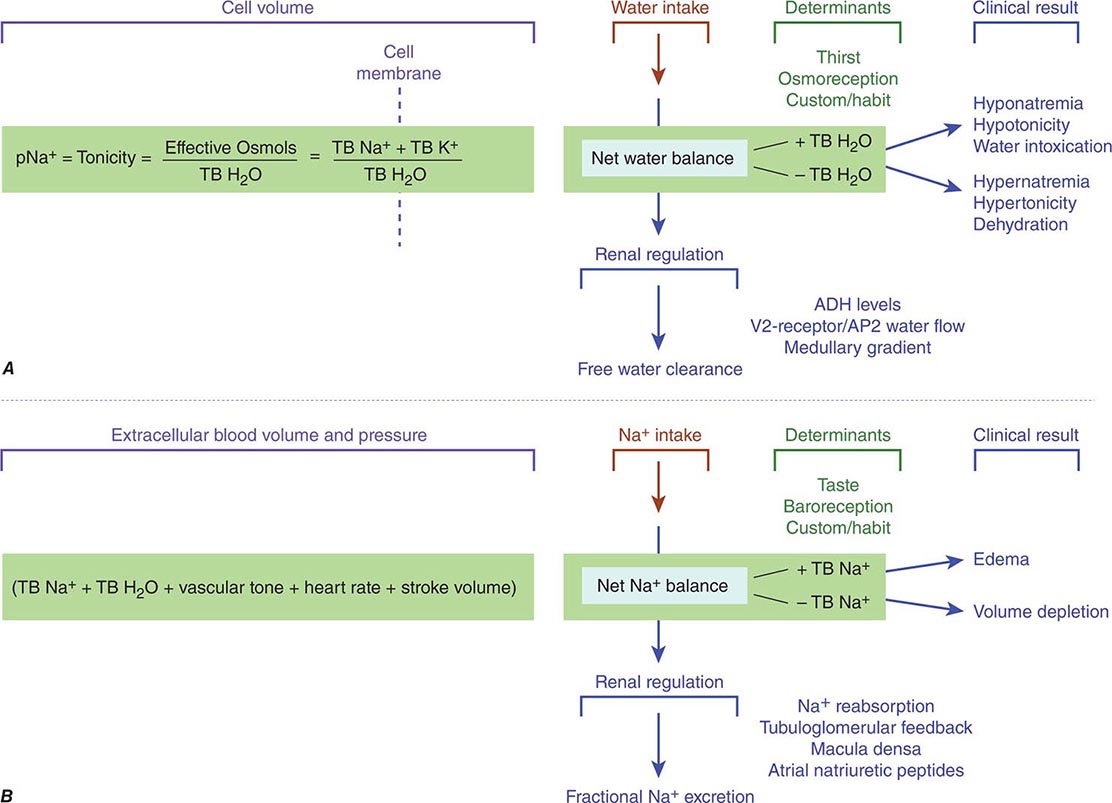
FIGURE 332e-4 Determinants of sodium and water balance. A. Plasma Na+ concentration is a surrogate marker for plasma tonicity, the volume behavior of cells in a solution. Tonicity is determined by the number of effective osmoles in the body divided by the total body H2O (TB H2O), which translates simply into the total body Na (TB Na+) and anions outside the cell separated from the total body K (TB K+) inside the cell by the cell membrane. Net water balance is determined by the integrated functions of thirst, osmoreception, Na reabsorption, vasopressin release, and the strength of the medullary gradient in the kidney, keeping tonicity within a narrow range of osmolality around 280 mosmol/L. When water metabolism is disturbed and total body water increases, hyponatremia, hypotonicity, and water intoxication occur; when total body water decreases, hypernatremia, hypertonicity, and dehydration occur. B. Extracellular blood volume and pressure are an integrated function of total body Na+ (TB Na+), total body H2O (TB H2O), vascular tone, heart rate, and stroke volume that modulates volume and pressure in the vascular tree of the body. This extracellular blood volume is determined by net Na balance under the control of taste, baroreception, habit, Na+ reabsorption, macula densa/tubuloglomerular feedback, and natriuretic peptides. When Na+ metabolism is disturbed and total body Na+ increases, edema occurs; when total body Na+ is decreased, volume depletion occurs. ADH, antidiuretic hormone; AQP2, aquaporin-2.
WATER BALANCE
Tonicity depends on the variable concentration of effective osmoles inside and outside the cell causing water to move in either direction across its membrane. Classic effective osmoles, like Na+, K+, and their anions, are solutes trapped on either side of a cell membrane, where they collectively partition and obligate water to move and find equilibrium in proportion to retained solute; Na+/K+-ATPase keeps most K+ inside cells and most Na+ outside. Normal tonicity (~280 mosmol/L) is rigorously defended by osmoregulatory mechanisms that control water balance to protect tissues from inadvertent dehydration (cell shrinkage) or water intoxication (cell swelling), both of which are deleterious to cell function (Fig. 332e-4A).
The mechanisms that control osmoregulation are distinct from those governing extracellular volume, although there is some shared physiology in both processes. While cellular concentrations of K+ have a determinant role in any level of tonicity, the routine surrogate marker for assessing clinical tonicity is the concentration of serum Na+. Any reduction in total body water, which raises the Na+ concentration, triggers a brisk sense of thirst and conservation of water by decreasing renal water excretion mediated by release of vasopressin from the posterior pituitary. Conversely, a decrease in plasma Na+ concentration triggers an increase in renal water excretion by suppressing the secretion of vasopressin. Whereas all cells expressing mechanosensitive TRPV1, 2, or 4 channels, among potentially other sensors, respond to changes in tonicity by altering their volume and Ca2+ concentration, only TRPV+ neuronal cells connected to the organum vasculosum of the lamina terminalis are osmoreceptive. Only these cells, because of their neural connectivity and adjacency to a minimal blood-brain barrier, modulate the downstream release of vasopressin by the posterior lobe of the pituitary gland. Secretion is stimulated primarily by changing tonicity and secondarily by other nonosmotic signals such as variable blood volume, stress, pain, nausea, and some drugs. The release of vasopressin by the posterior pituitary increases linearly as plasma tonicity rises above normal, although this varies, depending on the perception of extracellular volume (one form of cross-talk between mechanisms that adjudicate blood volume and osmoregulation). Changing the intake or excretion of water provides a means for adjusting plasma tonicity; thus, osmoregulation governs water balance.
The kidneys play a vital role in maintaining water balance through the regulation of renal water excretion. The ability to concentrate urine to an osmolality exceeding that of plasma enables water conservation, whereas the ability to produce urine more dilute than plasma promotes excretion of excess water. For water to enter or exit a cell, the cell membrane must express aquaporins. In the kidney, aquaporin-1 is constitutively active in all water-permeable segments of the proximal and distal tubules, whereas vasopressin-regulated aquaporin-2, -3, and -4 in the inner medullary collecting duct promote rapid water permeability. Net water reabsorption is ultimately driven by the osmotic gradient between dilute tubular fluid and a hypertonic medullary interstitium.
SODIUM BALANCE
The perception of extracellular blood volume is determined, in part, by the integration of arterial tone, cardiac stroke volume, heart rate, and the water and solute content of extracellular fluid. Na+ and accompanying anions are the most abundant extracellular effective osmoles and together support a blood volume around which pressure is generated. Under normal conditions, this volume is regulated by sodium balance (Fig. 332e-4B), and the balance between daily Na+ intake and excretion is under the influence of baroreceptors in regional blood vessels and vascular hormone sensors modulated by atrial natriuretic peptides, the renin-angiotensin-aldosterone system, Ca2+ signaling, adenosine, vasopressin, and the neural adrenergic axis. If Na+ intake exceeds Na+ excretion (positive Na+ balance), then an increase in blood volume will trigger a proportional increase in urinary Na+ excretion. Conversely, when Na+ intake is less than urinary excretion (negative Na+ balance), blood volume will decrease and trigger enhanced renal Na+ reabsorption, leading to decreased urinary Na+ excretion.
The renin-angiotensin-aldosterone system is the best-understood hormonal system modulating renal Na+ excretion. Renin is synthesized and secreted by granular cells in the wall of the afferent arteriole. Its secretion is controlled by several factors, including β1-adrenergic stimulation to the afferent arteriole, input from the macula densa, and prostaglandins. Renin and ACE activity eventually produce angiotensin II that directly or indirectly promotes renal Na+ and water reabsorption. Stimulation of proximal tubular Na+/H+ exchange by angiotensin II directly increases Na+ reabsorption. Angiotensin II also promotes Na+ reabsorption along the collecting duct by stimulating aldosterone secretion by the adrenal cortex. Constriction of the efferent glomerular arteriole by angiotensin II indirectly increases the filtration fraction and raises peritubular capillary oncotic pressure to promote tubular Na+ reabsorption. Finally, angiotensin II inhibits renin secretion through a negative feedback loop. Alternative metabolism of angiotensin by ACE2 generates the vasodilatory peptide angiotensin 1-7 that acts through Mas receptors to counterbalance several actions of angiotensin II on blood pressure and renal function (Fig. 332e-2C).
Aldosterone is synthesized and secreted by granulosa cells in the adrenal cortex. It binds to cytoplasmic mineralocorticoid receptors in the collecting duct principal cells that increase activity of ENaC, apical membrane K+ channel, and basolateral Na+/K+-ATPase. These effects are mediated in part by aldosterone-stimulated transcription of the gene encoding serum/glucocorticoid-induced kinase 1 (SGK1). The activity of ENaC is increased by SGK1-mediated phosphorylation of Nedd4-2, a protein that promotes recycling of the Na+ channel from the plasma membrane. Phosphorylated Nedd4-2 has impaired interactions with ENaC, leading to increased channel density at the plasma membrane and increased capacity for Na+ reabsorption by the collecting duct.
Chronic exposure to aldosterone causes a decrease in urinary Na+ excretion lasting only a few days, after which Na+ excretion returns to previous levels. This phenomenon, called aldosterone escape, is explained by decreased proximal tubular Na+ reabsorption following blood volume expansion. Excess Na+ that is not reabsorbed by the proximal tubule overwhelms the reabsorptive capacity of more distal nephron segments. This escape may be facilitated by atrial natriuretic peptides that lose their effectiveness in the clinical settings of heart failure, nephrotic syndrome, and cirrhosis, leading to severe Na+ retention and volume overload.
333e |
Adaptation of the Kidney to Injury |
Many years ago Claude Bernard (1878) introduced the concepts of milieu extérieur (the environment where an organism lives) and a milieu intérieur (the environment in which the tissues of that organism live). He argued that the milieu intérieur varied very little and that there were vital mechanisms that functioned to maintain this internal environment constant. Walter B. Cannon later extended these concepts by recognizing that the constancy of the internal state, which he termed the homeostatic state, was evidence of physiologic mechanisms that act to maintain this minimal variability. In higher animals, the plasma is maintained remarkably constant in composition both within an individual and among individuals. The kidney plays a vital role in this constancy. The kidney changes the composition of the urine to maintain electrolyte and acid-base balance and can produce hormones that can maintain constancy of blood hemoglobin and mineral metabolism. When the kidney is injured, the remaining functional mass responds and attempts to continue to maintain the milieu intérieur. It is remarkable how well the residual nephrons can perform in this task so that in many cases homeostasis is maintained until the glomerular filtration rate (GFR) drops to very low levels. At this point, the functional tissue can no longer compensate. In this chapter, we will discuss a number of these compensatory adaptations that the kidney makes in response to injury in an attempt to protect itself and protect the milieu intérieur. A theme that permeates, however, is that these adaptive processes can often be maladaptive and contribute to enhanced renal dysfunction, facilitating a positive feedback process that is inherently unstable.
RESPONSES OF THE KIDNEY TO REDUCED NUMBERS OF NEPHRONS DURING DEVELOPMENT
Renal disease is associated with a reduction in functional nephrons. The rest of the kidney adapts to this reduction by increasing blood flow to and the size of the remaining glomeruli and increasing size and function of the remaining tubules. Robert Platt, in 1936, argued that “…a high glomerular pressure, together with loss of nephrons (destroyed by disease) [is] an explanation of the peculiarities of renal function in this stage of kidney disease.” The raised glomerular pressure will increase the amount of filtrate produced by each nephron and thus compensate for a time for the destruction of part of the kidney. But eventually there are too few nephrons remaining to produce an adequate filtrate, even though they may work under the highest possible pressure, associated with a high systemic blood pressure. The responses to kidney injury can be both adaptive and maladaptive, and in many cases, the early adaptive responses can become maladaptive over time, leading to progressive decline in the anatomic and functional integrity of the kidney. As described previously, the early responses are likely in many cases motivated by attempts to maintain the constancy of the milieu intérieur for the survival of the organism (Claude Bernard).
Barry Brenner in the 1960s and 1970s carried out micropuncture experiments to define the pressures in glomerular capillaries as well as afferent and efferent resistances and modeled the behavior of the factors that governed glomerular filtration in health and disease. According to the Brenner Hyperfiltration Hypothesis, a reduction in the number of nephrons results in glomerular hypertension, hyperfiltration, and enlargement of glomeruli and this hyperfiltration results in damage to those glomeruli over time and ultimately decreased kidney function. According to this hypothesis, a positive feedback process is set into motion whereby injury to the glomeruli will result in further hyperfiltration to other glomeruli and hence more accelerated injury to those glomeruli. Since nephrons are not generated after 34–36 weeks of gestation or after birth (if earlier than 34–36 weeks) in humans, this hypothesis implies a deterministic effect of low nephron numbers at birth. There is over a 10-fold variation in the number of nephrons per kidney in the population (200,000 to over 2.5 million). This variation is not explained by kidney size in the adult. Children born with low birth weights would be more prone to kidney disease as adults. There are many reasons why there might be reduced nephron numbers at birth: developmental abnormalities, genetic predisposition, and environmental factors, such as malnutrition. There are thought to be interactions between these various factors. Reduced nephron mass can also occur with chronic kidney disease (CKD) in the adult, and the response of the kidney is similar qualitatively with hyperfiltration of the remaining nephrons.
Developmental Abnormalities There are many congenital abnormalities of the kidney and urinary tract (CAKUT). Dysplastic kidneys have varying degrees of abnormalities that interfere with their function. Anatomically abnormal kidneys can be associated with abnormalities of the lower urinary tract. Urinary tract abnormalities resulting in obstruction or vesicoureteric reflux can dramatically alter the normal development of the kidney nephrons. Dysplastic or hypoplastic kidneys can be cystic in patterns that are distinct from polycystic kidney disease. Of course, autosomal recessive kidney disease can result in widespread cyst formation.
Hypoplastic kidneys are characterized by a reduced number of functional nephrons. One definition of hypoplastic kidneys is as follows: “Kidney mass below two standard deviations of that of age-matched normal [individuals] or a combined kidney mass of less than half normal for the patient’s age.” Renal agenesis and cystic dysplasia often affects only one kidney. This results in hypertrophy of the other kidney if it is unaffected by any congenital abnormality itself. Although there is hypertrophy in size, it is not clear if this is associated with an increase in the number of nephrons on the contralateral side.
The prevalence of CAKUT has been generally found to be between 0.003 and 0.2%, depending on the population studied. This excludes fetuses with transient upper renal tract dilatation likely related to the high rate of fetal urine flow rate. In the adult U.S. Renal Data System (USRDS) of patients with end-stage kidney disease, approximately 0.6% are listed as having dysplastic or hypoplastic kidneys as a primary cause of the disease. This is likely an underestimate, however, because many patients with “small kidneys” may be misdiagnosed with chronic glomerulonephritis or chronic pyelonephritis.
Environmental Contributions to Reduced Nephron Mass The most important environmental factor responsible for reduced nephron number is growth restriction within the uterus. This has been associated with disease processes such as diabetes mellitus in the mother, but there also is a strong genetic disposition. Low-birth-weight children are more likely to be born to mothers who, themselves, were born with low birth weight. There are clearly other environmental factors. Caloric restriction during pregnancy in humans has been associated with altered glucose as adults and increased risk for hypertension. In one study, it was found that if women were calorie restricted in midgestation, the time of most rapid nephrogenesis, there was a threefold incidence of albuminuria in their children when they were tested as adults. Factors such as deficiency in vitamin A, sodium, zinc, or iron have been implicated as predisposing to abnormal kidney development. Other environmental factors that can influence kidney development are medications taken by the mother, such as dexamethasone, angiotensin-converting enzyme inhibitors, and angiotensin receptor antagonists (Table 333e-1). Protein restriction in mice during pregnancy can reduce lifespan of the offspring by 200 days. Obesity may play an important role in determining kidney outcome long term in patients with reduced kidney mass. It has been shown in mice fed a high-fat diet that the rodents that had reduced nephron number had a greater incidence of hypertension and renal fibrosis.
|
DRUGS THAT INHIBIT NEPHROGENESIS |
Implications of Low Nephron Number at Birth David Barker was the first to describe the association between low birth weight and later cardiovascular death. This was followed by studies relating low birth weight to risk for diabetes, stroke, hypertension, and CKD. It has been found that there is an inverse relationship between nephron number and blood pressure in adults. This relationship was found in Caucasians but not in African Americans. Approximately one-third of children with a single functioning kidney at the age of 10 years had signs of renal injury as determined by the presence of hypertension, albuminuria, or the use of renoprotective drugs. Another study revealed that 20–40% of patients born with a single functional kidney had renal failure requiring dialysis by 30 years of age.
ADAPTIVE RESPONSES OF THE KIDNEY TO REDUCED KIDNEY MASS THAT CHARACTERIZES CHRONIC KIDNEY DISEASE
In the early stages of CKD, there are many adaptations structurally and functionally that limit the consequences of the loss of nephrons on total-body homeostasis. In later stages of disease, however, these adaptations are insufficient to counteract the consequences of nephron loss and in fact often become maladaptive.
Counterbalance Renal counterbalance was defined by Hinman in 1923 as “an attempt on the part of the less injured or uninjured portion (of the kidney) to take over the work of the more injured portion.” Hinman defined “renal reserve” to be of two types: “native reserve, which is the normal physiological response to stimulation … and acquired reserve, which involves growth or compensation due to overstimulation.” It was known that removal of one kidney results in an increase in size of the contralateral kidney. If, instead of nephrectomy, one kidney is rendered ischemic and the other left intact, there is a resultant atrophy of the postischemic kidney. If the contralateral kidney is removed, however, before the atrophy becomes too severe, then the postischemic kidney increases markedly in size. With the contralateral kidney in place, there is vasoconstriction and reduced renal blood flow to the postischemic kidney. This is rapidly reversed, however, when the contralateral normal kidney is removed. The factors responsible for the persistent initial (prenephrectomy) vasoconstriction and those responsible for the rapid vasodilation and enhanced growth after contralateral nephrectomy are unknown.
Hypertrophy Because nephrons of mammals, in contrast to those of fish, cannot regenerate, the loss of functional units of the kidney, either due to disease or surgery, results in anatomic and functional changes in the remaining nephrons. As described above, there is increased blood flow to remaining glomeruli with potentially adverse effects over time of the resultant increased size of the remaining glomeruli and hyperfiltration (Fig. 333e-1). In addition, there is hypertrophy of the tubules. Some of the mediators of this hypertrophy of the remaining functional tubules are listed in Table 333e-2. In the adult, within a few weeks after unilateral nephrectomy for donation of a kidney, the GFR is approximately 70% of the prenephrectomy value. It then remains relatively stable for most patients over 15–20 years. The hyperfiltration is related to an increase in renal blood flow likely secondary to dilatation of the afferent arterioles potentially due to increases in nitric oxide (NO) production. The rate of increase in GFR is slower in the adult than it is in the young after nephrectomy. There are a number of factors that have been implicated at the cellular and nephron level to account for the compensatory hypertrophy that ensues after removal of functional nephrons (Table 333e-2).
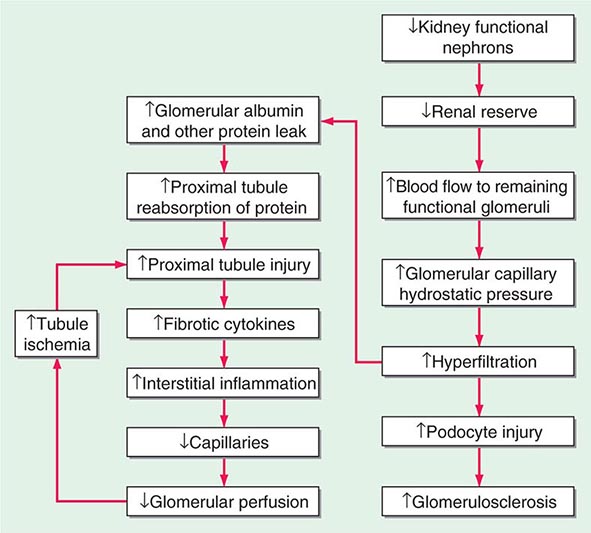
FIGURE 333e-1 Some of the pathophysiologic mechanisms involved with the maladaptive response to a reduction in the number of functional nephrons due to prenatal factors or postnatal disease processes.
|
FACTORS IMPLICATED IN COMPENSATORY RENAL GROWTH AFTER NEPHRON LOSS |
With increased blood flow to the kidney, there is glomerular hypertension (i.e., an increase in glomerular capillary pressure). There is increased wall tension and force on the capillary wall that is counteracted by contractile properties of the endothelium and elastic properties of the glomerular basement membrane. The force is conveyed to podocytes, which adapt by reinforcing cell cycle arrest and increasing cell adhesion in an adaptive attempt to maintain the delicate architecture of the interdigitating foot processes. Over time, however, these increased forces due to glomerular hypertension lead to podocyte damage and glomerulosclerosis.
Other Systemic and Renal Adaptations to Reduced Nephron Function With reduced functional nephrons, as is seen in CKD, there are many other systemic adaptations that occur to preserve the milieu intérieur because the kidney is involved in so many regulatory networks that are then stressed when there is dysfunction. In the 1960s, Neil Bricker introduced the “intact nephron hypothesis.” According to his concept, with decreases in the number of functioning nephrons, each remaining nephron has to adapt to carry a larger burden of transport, synthetic function, and regulatory function.
POTASSIUM Under normal and abnormal conditions, most of the filtered potassium is reabsorbed in the proximal tubule so that excretion is determined by secretion by the distal nephron. Potassium handling is altered in CKD protecting the organism somewhat from lethal hyperkalemia. Hyperkalemia is a common feature of individuals with CKD. Hyperkalemia (if not severe and dangerous) is adaptive in that it promotes potassium secretion by the principal cells of the collecting duct. When patients with CKD are given a potassium load, they can excrete it at the same rate as patients with normal renal function except that they do so at a higher serum potassium, consistent with the view that the hyperkalemia facilitates potassium excretion. The direct effect of hyperkalemia on potassium secretion by the distal nephron is independent of changes in aldosterone levels, but “normal” levels of aldosterone are necessary to see the effect of hyperkalemia on potassium excretion. Elevated potassium stimulates the production of aldosterone, and this effect is also seen in patients with CKD. Aldosterone increases the density and activity of the basolateral Na+-K+ ATPase and the number of Na+ channels in the apical membrane of the collecting duct. In CKD, the excretion of the dietary load of potassium occurs at the expense of an elevation in serum potassium concentrations.
SODIUM As renal function is reduced with CKD, there is a reduced ability to excrete sodium. Thus, patients with advanced kidney disease are often fluid overloaded. In early disease, however, there are functional adaptations that the kidney assumes to help to maintain the milieu intérieur. With loss of functional nephrons, the remaining nephrons are hyperperfused and are hyperfiltering in a manner that can be influenced by dietary protein intake. Although protein restriction can decrease this compensatory hyperperfusion, there is generally more sodium and water filtered and delivered to the remaining nephrons. There is some preservation of glomerulotubular balance with increased proximal tubule sodium and water reabsorption associated with increased levels of the Na/H exchanger in apical membranes of the tubule. The tubuloglomerular feedback (TGF) of the remaining nephrons is sensitive to sodium intake. With high sodium intake in normal renal function, a negative feedback process occurs by which increased distal delivery results in reduced GFR and hence filtration of sodium. In CKD, the TGF becomes a positive feedback process by which increased distal delivery results in increased filtration so that the need to excrete an increased amount of sodium per nephron is achieved. This conversion from a negative feedback process to a positive feedback process may be due to conversion of an adenosine-dominated vasoconstrictive feedback on the afferent arteriole of the glomerulus to a NO-dominated vasodilatory feedback. Like so many of these adaptive responses, this one may turn maladaptive, resulting in higher intraglomerular hydrostatic pressures with increased mechanical strain on the glomerular capillary wall and podocytes and increased glomerulosclerosis as a consequence.
ACID-BASE HOMEOSTASIS The kidneys excrete approximately 1 mEq/kg per day of dietary acid load under normal dietary conditions. With decreased kidney functional mass, there is an adaptive response to increase H+ excretion by the remaining functional nephrons. This takes the form of enhanced nephron ammoniagenesis and increased distal nephron H+ ion secretion, which is mediated by the renin-angiotensin system and endothelin-1. NH3 is produced by deamidization of glutamine in the proximal tubule. NH3 is converted to NH4+ in the collecting duct, where it buffers the secreted H+. It has been argued, however, that these mechanistic attempts to enhance H+ secretion can be maladaptive in that they can contribute to kidney inflammation and fibrosis and hence facilitate the progression of CKD.
MINERAL METABOLISM In CKD, there is a decrease in the ability of the kidney to excrete phosphate and produce 1,25-dihydroxyvitamin D3 [1,25(OH)2D3]. There is a resultant increase in serum phosphate and reduction in serum calcium (Fig. 333e-2). In response, the body adapts by increasing production of parathyroid hormone (PTH) and fibroblast growth factor-23 (FGF-23) in an attempt to increase phosphaturia. The elevated levels of PTH act on bone to increase bone resorption and on osteocytes to increase FGF-23 expression. Elevated levels of PTH increase FGF-23 expression by activating protein kinase A and wnt signaling in osteoblast-like cells. There are a number of other factors that increase bone FGF-23 production in CKD including systemic acidosis, altered hydroxyapatite metabolism, changes in bone matrix, and release of low-molecular-weight FGFs. Although the production of PTH and FGF-23 initially are adaptive attempts to maintain body phosphate levels by enhancing excretion by the kidney, they become maladaptive due to systemic effects on the cardiovascular system and bone, as renal function continues to deteriorate. PTH and FGF-23 decrease the kidney’s ability to reabsorb phosphate by decreasing the levels of the sodium-phosphate cotransporters NaPi2a and NaPi2c on the apical and basolateral membranes of the renal tubule. FGF-23 also reduces the ability of the kidney to generate 1,25(OH)2D3. In the parathyroid gland, the FGF-23 receptor, the klotho-fibroblast growth factor 1 complex, is downregulated with a consequent loss of the normal action of FGF-23 to downregulate PTH production. PTH and FGF-23 have been implicated in the cardiovascular disease that is so characteristic of patients with CKD. With CKD, there is less klotho expression in the kidney and the parathyroid glands. Klotho deficiency contributes to soft tissue calcifications in CKD. FGF-23 has been associated with increased mortality in CKD and has been reported to be involved causally in the development of left ventricular hypertrophy. PTH also has been reported to directly affect rat myocardial cells, increasing calcium entry into the cells and contributing to death of the cells.
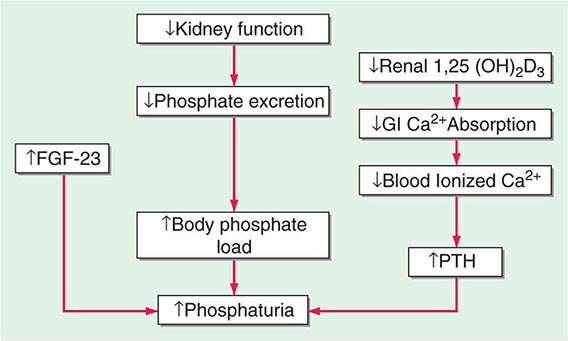
FIGURE 333e-2 Modification of the trade-off hypothesis of Slatopolsky and Bricker as it relates to the adaptation of the body to decreased functional renal mass in an attempt to maintain calcium and phosphate stores and serum levels. 1,25(OH)2D3, 1,25-dihydroxy-vitamin D3; FGF-23, fibroblast growth factor-23; GI, gastrointestinal; PTH, parathyroid hormone.
THE EFFECTS OF ACUTE KIDNEY INJURY ON SUSCEPTIBILITY TO SUBSEQUENT INJURY (PRECONDITIONING)
Preconditioning represents activation by the organism of intrinsic defense mechanisms to cope with pathologic conditions. Ischemic preconditioning is the phenomenon whereby a prior ischemic insult renders the organ resistant to a subsequent ischemic insult. Renal protection afforded by prior renal injury was described approximately 100 years ago, in 1912, by Suzuki, who noted that the kidney became resistant to uranium nephrotoxicity if the animal had previously been exposed to a sublethal dose of uranium. This resistance of the renal epithelium to recurrent toxic injury was proposed to be a defense mechanism of the kidney. There have been a number of studies over the years demonstrating that preconditioning with a number of renal toxicants leads to protection against injury associated with a second exposure to the same toxicant or to another nephrotoxicant. It is not, however, a universal finding that toxins confer resistance to subsequent insults.
Kidney ischemic preconditioning is the conveyance of protection against ischemia due to prior exposure of the kidney to sublethal episodes of ischemia. In some experiments in rodents, these prior exposures were short (e.g., 5 min) and repeated or longer. Subsequent protection was generally found at 1–2 h or up to 48 h, but there has been a report of protection in the mouse for up to 12 weeks after the preconditioning exposures. Unilateral ischemia, with the contralateral kidney left alone, was also protective against a subsequent ischemic insult to the postischemic kidney, revealing that systemic uremia was not necessary for protection.
Remote Ischemic Preconditioning Remote ischemic preconditioning is a therapeutic strategy by which protection can be afforded in one vascular bed by ischemia to another vascular bed in the same organ or a different organ. A large number of studies have demonstrated that ischemia to one organ protects against ischemia to another. There are very few mechanistic studies of remote preconditioning in the kidney. In one study, naloxone blocked preconditioning in the kidney, implicating opiates as effectors. Remote preconditioning induced by ischemia to the muscle of the arm induced by a blood pressure cuff can result in protection of the kidney against a subsequent insult, such as one related to contrast agents in humans. Some of the cellular processes and signaling mechanisms proposed to explain preconditioning in the kidney and other organs are listed in Table 333e-3. These protective processes, most of which have been identified in the heart, involve multiple signaling pathways that affect decreased apoptosis, inhibition of mitochondrial permeability transition pores, activation of survival pathways, autophagy, and other pathways involved in reducing energy consumption or reactive oxygen production. In a study from our laboratory, inducible NO synthase was found to be an important contributor to the adaptive response to kidney injury, which results in protection against a subsequent insult. Identification of the responsible protective factor(s) mediating the advantageous adaptive response to remote ischemic preconditioning would provide a therapeutic approach for prevention of acute kidney injury or facilitation of a protective adaptation to kidney injury.
|
FACTORS AND PROCESSES IMPLICATED AS PROTECTIVE MEDIATORS OF ISCHEMIC PRECONDITIONING |
ADAPTIVE RESPONSE OF THE KIDNEY TO ACUTE INJURY
Adaptive Response to Hypoxic Injury Hypoxia plays a role in ischemic, septic, and toxic acute kidney injury. Many conditions result in a global or regional impairment of oxygen delivery. This is particularly important in the outer medulla where there is baseline reduced oxygen tension and a complex capillary network that, by its nature, is susceptible to interruption. In addition, the S3 segment of the proximal tubule is very dependent on oxidative metabolism, whereas the medullary thick ascending limb of the nephron that also traverses the outer medulla can adapt to hypoxia by converting to glycolysis as a primary energy source.
One proposed adaptive response to hypoxia is a reduction in glomerular filtration with consequent reduction in “work” requirement for reabsorption of solutes by the tubule. This was termed acute renal success by Thurau many years ago. The importance of this has been questioned, however, because there is no significant reduction in renal oxygen consumption in post–cardiac surgery patients with acute kidney injury in the setting of reduced GFR and renal blood flow.
If hypoxia or other influences, such as toxins, damage the proximal tubule and interfere with reabsorption of sodium and water, it is important that the kidney adapt in such a way so that there is not a large natriuresis that might compromise intravascular volume and blood pressure. This is accomplished, at least in part, by tubuloglomerular feedback (TGF). The increased distal delivery of salt and water results in a homeostatic adaptation to decrease glomerular filtration and hence decrease tubular delivery of salt and water through the glomerulus and reduce the delivery to the distal nephron. This adaptive response to acute injury is different from the role of TGF in CKD, as we have discussed previously in this chapter. In chronic disease with reduced nephron function, there is a steady-state need to increase excretion of sodium, whereas with acute injury, excretion of sodium is reduced.
Many genes are activated by hypoxia that are adaptive in serving to protect the cell and organ. With hypoxia, hypoxia-inducible factor (HIF) 1α rapidly accumulates due to the inhibition of the HIF prolyl-hydroxylases, which normally promote HIF1α proteasomal degradation. HIF1α then dimerizes with HIF1β and the dimer moves to the nucleus, where it upregulates a number of genes whose protein products are involved in energy metabolism, angiogenesis, and apoptosis, enhancing oxygen delivery and metabolic adaptation to hypoxia. This takes the form of a complex interplay among factors that regulate perfusion, cellular redox state, and mitochondrial function. For example, upregulation of NO production by sepsis results in vasodilatation and reduction in mitochondrial respiration and oxygen consumption. In addition, HIF1 activation in endothelial cells may be important for adaptive preservation of the microvasculature during and after hypoxia. Better understanding of the role that the HIFs play in protective adaptation has led to an aggressive development of HIF prolyl-hydroxylase inhibitors by biotechnology and pharmaceutical companies for clinical use.
Adaptive Response to Toxic Injury Specific to the Proximal Tubule One can model an acute kidney injury by genetically inserting a Simian diphtheria toxin (DT) receptor into the proximal tubule and then adding either a single dose of DT or multiple doses of the toxin. Repair of the kidney after a single dose of DT can be shown to be adaptive with few longer term sequelae. There is a very robust proliferative response of the proximal tubule cells to replace the cells that die as a result of the DT. Ultimately the inflammation resolves, and there is little, if any, residual interstitial inflammation, expansion, or matrix deposition.
Maladaptive Response of the Kidney to Acute Injury By contrast to the above adaptive repair that occurs after a single insult, after three doses of DT administered at weekly intervals, there is maladaptive repair with development over time of a chronic interstitial infiltrate, increased myofibroblast proliferation, tubulointerstitial fibrosis, and tubular atrophy, as well as an increase in serum creatinine (0.6 ± 0.1 mg/dL vs 0.18 ± 0.02 mg/dL in control mice) by week 5, 2 weeks after the last dose in the thrice-treated animals. There is a dramatic increase in the number of interstitial cells that expressed the platelet-derived growth factor receptor β (pericytes/perivascular fibroblasts), αSMA (myofibroblasts), FSP-1/S100A4 (fibroblast specific protein-1), and F4/80 (macrophages). In addition, there is loss of endothelial cells, interstitial capillaries, and development of focal global and segmental glomerulosclerosis.
It has become increasingly recognized as a result of large epidemiologic studies that even mild forms of acute kidney injury are associated with adverse short- and long-term outcomes including onset or progression of CKD and more rapid progression to end-stage kidney disease. Experimental models in animals, such as the DT model described above, provide pathophysiologic explanations for how the effects of acute injury can lead to chronic inflammation, vascular rarefaction, tubular cell atrophy, interstitial fibrosis, and glomerulosclerosis. Recurrent specific tubular injury leads to a pattern very typical of CKD in humans: tubular atrophy, interstitial chronic inflammation and fibrosis, vascular rarefaction, and glomerulosclerosis. The mechanisms involved in the development of glomerulosclerosis evoked by primary tubular injury may be multifactorial. Damage to nephron segments may lead to sluffing of cells into the lumen and to tubular obstruction. Progressive narrowing of the early proximal tubule near the glomerular tuft can lead to a sclerotic atubular glomerulus like those that are seen with ureteral obstruction. There may be paracrine signaling from injured and regenerating/undifferentiated epithelium to directly impact the glomerulus. Alternatively, a progressive tubulointerstitial reaction originating around atrophic and undifferentiated tubules may directly encroach upon the glomerular tuft. The loss of interstitial capillaries may lead to a progressive reduction of glomerular blood flow with ischemia to the glomerulus and to the kidney regions perfused by the postglomerular capillaries. This speaks to the fact that primary tubular injury can trigger a response that adversely affects multiple compartments of the kidney and leads to a positive feedback process, involving loss of capillaries, glomerulosclerosis, persistent ischemia, tubular atrophy, increased fibrosis, and ultimately kidney failure.
334 |
Acute Kidney Injury |
Acute kidney injury (AKI), previously known as acute renal failure, is characterized by the sudden impairment of kidney function resulting in the retention of nitrogenous and other waste products normally cleared by the kidneys. AKI is not a single disease but, rather, a designation for a heterogeneous group of conditions that share common diagnostic features: specifically, an increase in the blood urea nitrogen (BUN) concentration and/or an increase in the plasma or serum creatinine (SCr) concentration, often associated with a reduction in urine volume. It is important to recognize that AKI is a clinical diagnosis and not a structural one. A patient may have AKI without injury to the kidney parenchyma. AKI can range in severity from asymptomatic and transient changes in laboratory parameters of glomerular filtration rate (GFR), to overwhelming and rapidly fatal derangements in effective circulating volume regulation and electrolyte and acid-base composition of the plasma.
EPIDEMIOLOGY
AKI complicates 5–7% of acute care hospital admissions and up to 30% of admissions to the intensive care unit, particularly in the setting of diarrheal illnesses, infectious diseases like malaria and leptospirosis, and natural disasters such as earthquakes. The incidence of AKI has grown by more than fourfold in the United States since 1988 and is estimated to have a yearly incidence of 500 per 100,000 population, higher than the yearly incidence of stroke. AKI is associated with a markedly increased risk of death in hospitalized individuals, particularly in those admitted to the ICU where in-hospital mortality rates may exceed 50%. AKI increases the risk for the development or worsening of chronic kidney disease. Patients who survive and recover from an episode of severe AKI requiring dialysis are at increased risk for the later development of dialysis-requiring end-stage kidney disease. AKI may be community-acquired or hospital-acquired. Common causes of community-acquired AKI include volume depletion, adverse effects of medications, and obstruction of the urinary tract. The most common clinical settings for hospital-acquired AKI are sepsis, major surgical procedures, critical illness involving heart or liver failure, intravenous iodinated contrast administration, and nephrotoxic medication administration.
AKI IN THE DEVELOPING WORLD
![]() AKI is also a major medical complication in the developing world, where the epidemiology differs from that in developed countries due to differences in demographics, economics, geography, and comorbid disease burden. While certain features of AKI are common to both—particularly since urban centers of some developing countries increasingly resemble those in the developed world—many etiologies for AKI are region-specific such as envenomations from snakes, spiders, caterpillars, and bees; infectious causes such as malaria and leptospirosis; and crush injuries and resultant rhabdomyolysis from earthquakes.
AKI is also a major medical complication in the developing world, where the epidemiology differs from that in developed countries due to differences in demographics, economics, geography, and comorbid disease burden. While certain features of AKI are common to both—particularly since urban centers of some developing countries increasingly resemble those in the developed world—many etiologies for AKI are region-specific such as envenomations from snakes, spiders, caterpillars, and bees; infectious causes such as malaria and leptospirosis; and crush injuries and resultant rhabdomyolysis from earthquakes.
ETIOLOGY AND PATHOPHYSIOLOGY
The causes of AKI have traditionally been divided into three broad categories: prerenal azotemia, intrinsic renal parenchymal disease, and postrenal obstruction (Fig. 334-1).
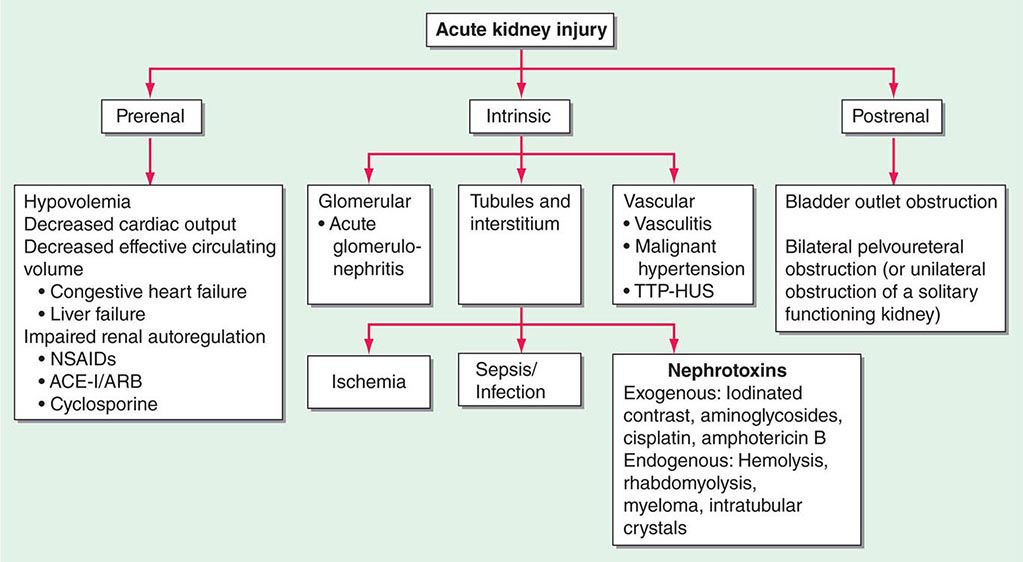
FIGURE 334-1 Classification of the major causes of acute kidney injury. ACE-I, angiotensin-converting enzyme inhibitor-I; ARB, angiotensin receptor blocker; NSAIDs, nonsteroidal anti-inflammatory drugs; TTP-HUS, thrombotic thrombocytopenic purpura–hemolytic-uremic syndrome.
PRERENAL AZOTEMIA
Prerenal azotemia (from “azo,” meaning nitrogen, and “-emia”) is the most common form of AKI. It is the designation for a rise in SCr or BUN concentration due to inadequate renal plasma flow and intraglomerular hydrostatic pressure to support normal glomerular filtration. The most common clinical conditions associated with prerenal azotemia are hypovolemia, decreased cardiac output, and medications that interfere with renal autoregulatory responses such as nonsteroidal anti-inflammatory drugs (NSAIDs) and inhibitors of angiotensin II (Fig. 334-2). Prerenal azotemia may coexist with other forms of intrinsic AKI associated with processes acting directly on the renal parenchyma. Prolonged periods of prerenal azotemia may lead to ischemic injury, often termed acute tubular necrosis (ATN). By definition, prerenal azotemia involves no parenchymal damage to the kidney and is rapidly reversible once intraglomerular hemodynamics are restored.
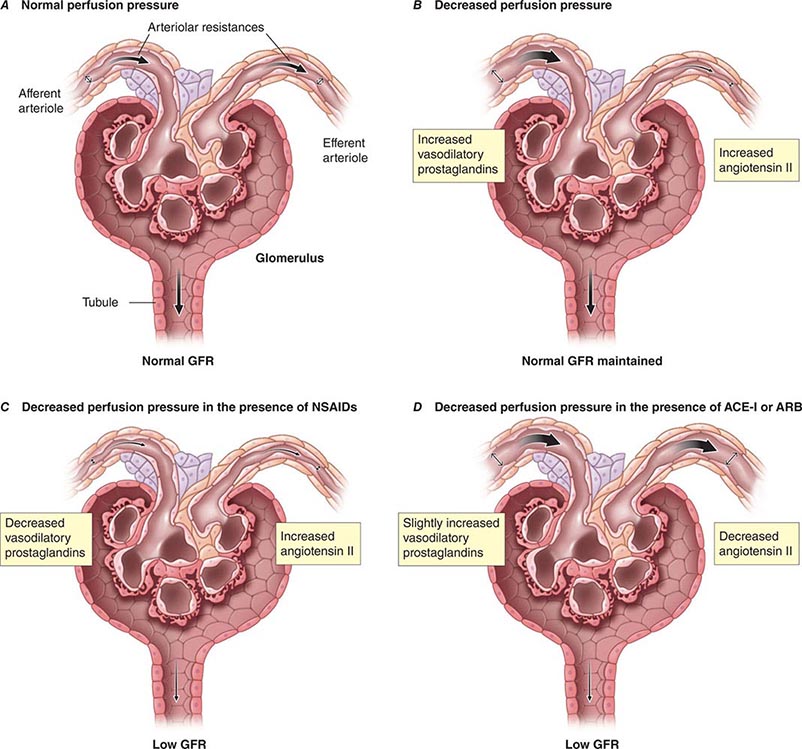
FIGURE 334-2 Intrarenal mechanisms for autoregulation of the glomerular filtration rate (GFR) under decreased perfusion pressure and reduction of the GFR by drugs. A. Normal conditions and a normal GFR. B. Reduced perfusion pressure within the autoregulatory range. Normal glomerular capillary pressure is maintained by afferent vasodilatation and efferent vasoconstriction. C. Reduced perfusion pressure with a nonsteroidal anti-inflammatory drug (NSAID). Loss of vasodilatory prostaglandins increases afferent resistance; this causes the glomerular capillary pressure to drop below normal values and the GFR to decrease. D. Reduced perfusion pressure with an angiotensin-converting enzyme inhibitor (ACE-I) or an angiotensin receptor blocker (ARB). Loss of angiotensin II action reduces efferent resistance; this causes the glomerular capillary pressure to drop below normal values and the GFR to decrease. (From JG Abuelo: N Engl J Med 357:797-805, 2007; with permission.)
Normal GFR is maintained in part by the relative resistances of the afferent and efferent renal arterioles, which determine the glomerular plasma flow and the transcapillary hydraulic pressure gradient that drive glomerular ultrafiltration. Mild degrees of hypovolemia and reductions in cardiac output elicit compensatory renal physiologic changes. Because renal blood flow accounts for 20% of the cardiac output, renal vasoconstriction and salt and water reabsorption occur as homeostatic responses to decreased effective circulating volume or cardiac output in order to maintain blood pressure and increase intravascular volume to sustain perfusion to the cerebral and coronary vessels. Mediators of this response include angiotensin II, norepinephrine, and vasopressin (also termed antidiuretic hormone). Glomerular filtration can be maintained despite reduced renal blood flow by angiotensin II–mediated renal efferent vasoconstriction, which maintains glomerular capillary hydrostatic pressure closer to normal and thereby prevents marked reductions in GFR if renal blood flow reduction is not excessive.
In addition, a myogenic reflex within the afferent arteriole leads to dilation in the setting of low perfusion pressure, thereby maintaining glomerular perfusion. Intrarenal biosynthesis of vasodilator prostaglandins (prostacyclin, prostaglandin E2), kallikrein and kinins, and possibly nitric oxide (NO) also increase in response to low renal perfusion pressure. Autoregulation is also accomplished by tubuloglomerular feedback, in which decreases in solute delivery to the macula densa (specialized cells within the distal tubule) elicit dilation of the juxtaposed afferent arteriole in order to maintain glomerular perfusion, a mechanism mediated, in part, by NO. There is a limit, however, to the ability of these counterregulatory mechanisms to maintain GFR in the face of systemic hypotension. Even in healthy adults, renal autoregulation usually fails once the systolic blood pressure falls below 80 mmHg.
A number of factors determine the robustness of the autoregulatory response and the risk of prerenal azotemia. Atherosclerosis, long-standing hypertension, and older age can lead to hyalinosis and myointimal hyperplasia, causing structural narrowing of the intrarenal arterioles and impaired capacity for renal afferent vasodilation. In chronic kidney disease, renal afferent vasodilation may be operating at maximal capacity in order to maximize GFR in response to reduced functional renal mass. Drugs can affect the compensatory changes evoked to maintain GFR. NSAIDs inhibit renal prostaglandin production, limiting renal afferent vasodilation. Angiotensin-converting enzyme (ACE) inhibitors and angiotensin receptor blockers (ARBs) limit renal efferent vasoconstriction; this effect is particularly pronounced in patients with bilateral renal artery stenosis or unilateral renal artery stenosis (in the case of a solitary functioning kidney) because renal efferent vasoconstriction is needed to maintain GFR due to low renal perfusion. The combined use of NSAIDs with ACE inhibitors or ARBs poses a particularly high risk for developing prerenal azotemia.
Many individuals with advanced cirrhosis exhibit a unique hemodynamic profile that resembles prerenal azotemia despite total-body volume overload. Systemic vascular resistance is markedly reduced due to primary arterial vasodilation in the splanchnic circulation, resulting ultimately in activation of vasoconstrictor responses similar to those seen in hypovolemia. AKI is a common complication in this setting, and it can be triggered by volume depletion and spontaneous bacterial peritonitis. A particularly poor prognosis is seen in the case of type 1 hepatorenal syndrome, in which AKI without an alternate cause (e.g., shock and nephrotoxic drugs) persists despite volume administration and withholding of diuretics. Type 2 hepatorenal syndrome is a less severe form characterized mainly by refractory ascites.
INTRINSIC AKI
The most common causes of intrinsic AKI are sepsis, ischemia, and nephrotoxins, both endogenous and exogenous (Fig. 334-3). In many cases, prerenal azotemia advances to tubular injury. Although classically termed “acute tubular necrosis,” human biopsy confirmation of tubular necrosis is, in general, often lacking in cases of sepsis and ischemia; indeed, processes such as inflammation, apoptosis, and altered regional perfusion may be important contributors pathophysiologically. Other causes of intrinsic AKI are less common and can be conceptualized anatomically according to the major site of renal parenchymal damage: glomeruli, tubulointerstitium, and vessels.
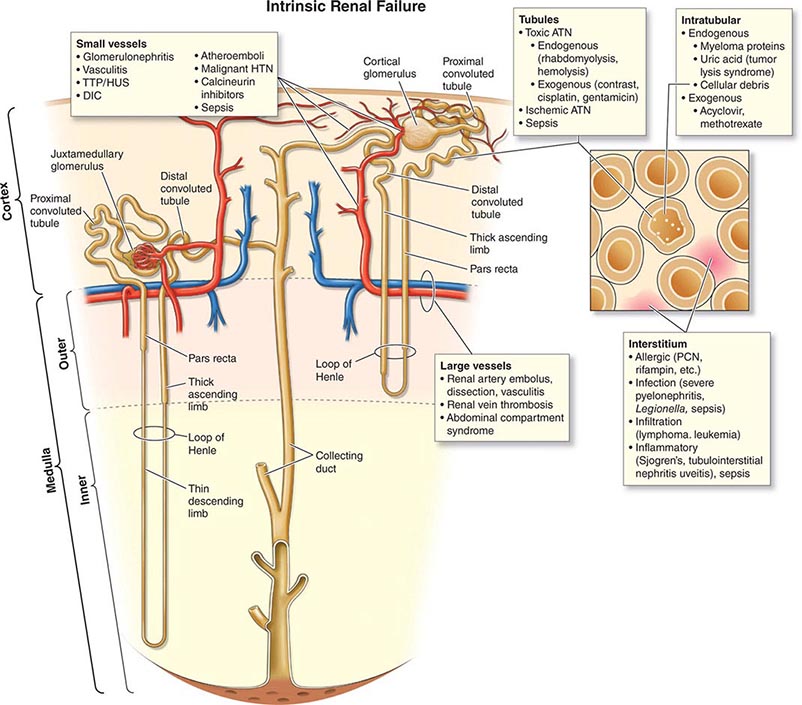
FIGURE 334-3 Major causes of intrinsic acute kidney injury. ATN, acute tubular necrosis; DIC, disseminated intravascular coagulation; HTN, hypertension; PCN, penicillin; TTP/HUS, thrombotic thrombocytopenic purpura/hemolytic-uremic syndrome; TINU, tubulointerstitial nephritis-uveitis.
SEPSIS-ASSOCIATED AKI
In the United States, more than 700,000 cases of sepsis occur each year. AKI complicates more than 50% of cases of severe sepsis and greatly increases the risk of death. Sepsis is also a very important cause of AKI in the developing world. Decreases in GFR with sepsis can occur even in the absence of overt hypotension, although most cases of severe AKI typically occur in the setting of hemodynamic collapse requiring vasopressor support. While there is clearly tubular injury associated with AKI in sepsis as manifest by the presence of tubular debris and casts in the urine, postmortem examinations of kidneys from individuals with severe sepsis suggest that other factors, perhaps related to inflammation, mitochondrial dysfunction, and interstitial edema, must be considered in the pathophysiology of sepsis-induced AKI.
The hemodynamic effects of sepsis—arising from generalized arterial vasodilation, mediated in part by cytokines that upregulate the expression of inducible NO synthase in the vasculature—can lead to a reduction in GFR. The operative mechanisms may be excessive efferent arteriole vasodilation, particularly early in the course of sepsis, or renal vasoconstriction from activation of the sympathetic nervous system, the renin-angiotensin-aldosterone system, vasopressin, and endothelin. Sepsis may lead to endothelial damage, which results in microvascular thrombosis, activation of reactive oxygen species, and leukocyte adhesion and migration, all of which may injure renal tubular cells.
ISCHEMIA-ASSOCIATED AKI
Healthy kidneys receive 20% of the cardiac output and account for 10% of resting oxygen consumption, despite constituting only 0.5% of the human body mass. The kidneys are also the site of one of the most hypoxic regions in the body, the renal medulla. The outer medulla is particularly vulnerable to ischemic damage because of the architecture of the blood vessels that supply oxygen and nutrients to the tubules. Enhanced leukocyte-endothelial interactions in the small vessels lead to inflammation and reduced local blood flow to the metabolically very active S3 segment of the proximal tubule, which depends on oxidative metabolism for survival. Ischemia alone in a normal kidney is usually not sufficient to cause severe AKI, as evidenced by the relatively low risk of severe AKI even after total interruption of renal blood flow during suprarenal aortic clamping or cardiac arrest. Clinically, AKI more commonly develops when ischemia occurs in the context of limited renal reserve (e.g., chronic kidney disease or older age) or coexisting insults such as sepsis, vasoactive or nephrotoxic drugs, rhabdomyolysis, or the systemic inflammatory states associated with burns and pancreatitis. Prerenal azotemia and ischemia-associated AKI represent a continuum of the manifestations of renal hypoperfusion. Persistent preglomerular vasoconstriction may be a common underlying cause of the reduction in GFR seen in AKI; implicated factors for vasoconstriction include activation of tubuloglomerular feedback from enhanced delivery of solute to the macula densa following proximal tubule injury, increased basal vascular tone and reactivity to vasoconstrictive agents, and decreased vasodilator responsiveness. Other contributors to low GFR include backleak of filtrate across damaged and denuded tubular epithelium and mechanical obstruction of tubules from necrotic debris (Fig. 334-4).
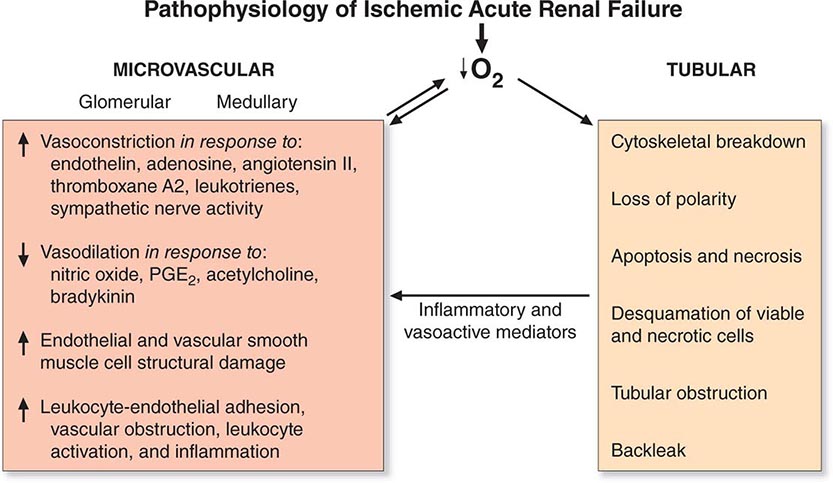
FIGURE 334-4 Interacting microvascular and tubular events contributing to the pathophysiology of ischemic acute kidney injury. PGE2, prostaglandin E2. (From JV Bonventre, JM Weinberg: J Am Soc Nephrol 14:2199, 2003.)
Postoperative AKI Ischemia-associated AKI is a serious complication in the postoperative period, especially after major operations involving significant blood loss and intraoperative hypotension. The procedures most commonly associated with AKI are cardiac surgery with cardiopulmonary bypass (particularly for combined valve and bypass procedures), vascular procedures with aortic cross clamping, and intraperitoneal procedures. Severe AKI requiring dialysis occurs in approximately 1% of cardiac and vascular surgery procedures. The risk of severe AKI has been less well studied for major intraperitoneal procedures but appears to be of comparable magnitude. Common risk factors for postoperative AKI include underlying chronic kidney disease, older age, diabetes mellitus, congestive heart failure, and emergency procedures. The pathophysiology of AKI following cardiac surgery is multifactorial. Major AKI risk factors are common in the population undergoing cardiac surgery. The use of nephrotoxic agents including iodinated contrast for cardiac imaging prior to surgery may increase the risk of AKI. Cardiopulmonary bypass is a unique hemodynamic state characterized by nonpulsatile flow and exposure of the circulation to extracorporeal circuits. Longer duration of cardiopulmonary bypass is a risk factor for AKI. In addition to ischemic injury from sustained hypoperfusion, cardiopulmonary bypass may cause AKI through a number of mechanisms including extracorporeal circuit activation of leukocytes and inflammatory processes, hemolysis with resultant pigment nephropathy (see below), and aortic injury with resultant atheroemboli. AKI from atheroembolic disease, which can also occur following percutaneous catheterization of the aorta, or spontaneously, is due to cholesterol crystal embolization resulting in partial or total occlusion of multiple small arteries within the kidney. Over time, a foreign body reaction can result in intimal proliferation, giant cell formation, and further narrowing of the vascular lumen, accounting for the generally subacute (over a period of weeks rather than days) decline in renal function.
Burns and Acute Pancreatitis Extensive fluid losses into the extravascular compartments of the body frequently accompany severe burns and acute pancreatitis. AKI is an ominous complication of burns, affecting 25% of individuals with more than 10% total body surface area involvement. In addition to severe hypovolemia resulting in decreased cardiac output and increased neurohormonal activation, burns and acute pancreatitis both lead to dysregulated inflammation and an increased risk of sepsis and acute lung injury, all of which may facilitate the development and progression of AKI. Individuals undergoing massive fluid resuscitation for trauma, burns, and acute pancreatitis can also develop the abdominal compartment syndrome, where markedly elevated intraabdominal pressures, usually higher than 20 mmHg, lead to renal vein compression and reduced GFR.
Diseases of the Microvasculature Leading to Ischemia Microvascular causes of AKI include the thrombotic microangiopathies (antiphospholipid antibody syndrome, radiation nephritis, malignant nephrosclerosis, and thrombotic thrombocytopenic purpura/hemolytic-uremic syndrome [TTP-HUS]), scleroderma, and atheroembolic disease. Large-vessel diseases associated with AKI include renal artery dissection, thromboembolism, thrombosis, and renal vein compression or thrombosis.
NEPHROTOXIN-ASSOCIATED AKI
The kidney has very high susceptibility to nephrotoxicity due to extremely high blood perfusion and concentration of circulating substances along the nephron where water is reabsorbed and in the medullary interstitium; this results in high-concentration exposure of toxins to tubular, interstitial, and endothelial cells. Nephrotoxic injury occurs in response to a number of pharmacologic compounds with diverse structures, endogenous substances, and environmental exposures. All structures of the kidney are vulnerable to toxic injury, including the tubules, interstitium, vasculature, and collecting system. As with other forms of AKI, risk factors for nephrotoxicity include older age, chronic kidney disease (CKD), and prerenal azotemia. Hypoalbuminemia may increase the risk of some forms of nephrotoxin-associated AKI due to increased free circulating drug concentrations.
Contrast Agents Iodinated contrast agents used for cardiovascular and computed tomography (CT) imaging are a leading cause of AKI. The risk of AKI, or “contrast nephropathy,” is negligible in those with normal renal function but increases markedly in the setting of CKD, particularly diabetic nephropathy. The most common clinical course of contrast nephropathy is characterized by a rise in SCr beginning 24–48 h following exposure, peaking within 3–5 days, and resolving within 1 week. More severe, dialysis-requiring AKI is uncommon except in the setting of significant preexisting CKD, often in association with congestive heart failure or other coexisting causes for ischemia-associated AKI. Patients with multiple myeloma and renal disease are particularly susceptible. Low fractional excretion of sodium and relatively benign urinary sediment without features of tubular necrosis (see below) are common findings. Contrast nephropathy is thought to occur from a combination of factors, including (1) hypoxia in the renal outer medulla due to perturbations in renal microcirculation and occlusion of small vessels; (2) cytotoxic damage to the tubules directly or via the generation of oxygen free radicals, especially because the concentration of the agent within the tubule is markedly increased; and (3) transient tubule obstruction with precipitated contrast material. Other diagnostic agents implicated as a cause of AKI are high-dose gadolinium used for magnetic resonance imaging (MRI) and oral sodium phosphate solutions used as bowel purgatives.
Antibiotics Several antimicrobial agents are commonly associated with AKI. Aminoglycosides and amphotericin B both cause tubular necrosis. Nonoliguric AKI (i.e., without a significant reduction in urine volume) accompanies 10–30% of courses of aminoglycoside antibiotics, even when plasma levels are in the therapeutic range. Aminoglycosides are freely filtered across the glomerulus and then accumulate within the renal cortex, where concentrations can greatly exceed those of the plasma. AKI typically manifests after 5–7 days of therapy and can present even after the drug has been discontinued. Hypomagnesemia is a common finding.
Amphotericin B causes renal vasoconstriction from an increase in tubuloglomerular feedback as well as direct tubular toxicity mediated by reactive oxygen species. Nephrotoxicity from amphotericin B is dose and duration dependent. This drug binds to tubular membrane cholesterol and introduces pores. Clinical features of amphotericin B nephrotoxicity include polyuria, hypomagnesemia, hypocalcemia, and nongap metabolic acidosis.
Vancomycin may be associated with AKI, particularly when trough levels are high, but a causal relationship with AKI has not been definitively established. Acyclovir can precipitate in tubules and cause AKI by tubular obstruction, particularly when given as an intravenous bolus at high doses (500 mg/m2) or in the setting of hypovolemia. Foscarnet, pentamidine, tenofovir, and cidofovir are also frequently associated with AKI due to tubular toxicity. AKI secondary to acute interstitial nephritis can occur as a consequence of exposure to many antibiotics, including penicillins, cephalosporins, quinolones, sulfonamides, and rifampin.
Chemotherapeutic Agents Cisplatin and carboplatin are accumulated by proximal tubular cells and cause necrosis and apoptosis. Intensive hydration regimens have reduced the incidence of cisplatin nephrotoxicity, but it remains a dose-limiting toxicity. Ifosfamide may cause hemorrhagic cystitis and tubular toxicity, manifested as type II renal tubular acidosis (Fanconi’s syndrome), polyuria, hypokalemia, and a modest decline in GFR. Antiangiogenesis agents, such as bevacizumab, can cause proteinuria and hypertension via injury to the glomerular microvasculature (thrombotic microangiopathy). Other antineoplastic agents such as mitomycin C and gemcitabine may cause thrombotic microangiopathy with resultant AKI.
Toxic Ingestions Ethylene glycol, present in automobile antifreeze, is metabolized to oxalic acid, glycolaldehyde, and glyoxylate, which may cause AKI through direct tubular injury. Diethylene glycol is an industrial agent that has been the cause of outbreaks of severe AKI around the world due to adulteration of pharmaceutical preparations. The metabolite 2-hydroxyethoxyacetic acid (HEAA) is thought to be responsible for tubular injury. Melamine contamination of foodstuffs has led to nephrolithiasis and AKI, either through intratubular obstruction or possibly direct tubular toxicity. Aristolochic acid was found to be the cause of “Chinese herb nephropathy” and “Balkan nephropathy” due to contamination of medicinal herbs or farming. The list of environmental toxins is likely to grow and contribute to a better understanding of previously catalogued “idiopathic” chronic tubular interstitial disease, a common diagnosis in both the developed and developing world.
Endogenous Toxins AKI may be caused by a number of endogenous compounds, including myoglobin, hemoglobin, uric acid, and myeloma light chains. Myoglobin can be released by injured muscle cells, and hemoglobin can be released during massive hemolysis leading to pigment nephropathy. Rhabdomyolysis may result from traumatic crush injuries, muscle ischemia during vascular or orthopedic surgery, compression during coma or immobilization, prolonged seizure activity, excessive exercise, heat stroke or malignant hyperthermia, infections, metabolic disorders (e.g., hypophosphatemia, severe hypothyroidism), and myopathies (drug-induced, metabolic, or inflammatory). Pathogenic factors for AKI include intrarenal vasoconstriction, direct proximal tubular toxicity, and mechanical obstruction of the distal nephron lumen when myoglobin or hemoglobin precipitates with Tamm-Horsfall protein (uromodulin, the most common protein in urine and produced in the thick ascending limb of the loop of Henle), a process favored by acidic urine. Tumor lysis syndrome may follow initiation of cytotoxic therapy in patients with high-grade lymphomas and acute lymphoblastic leukemia; massive release of uric acid (with serum levels often exceeding 15 mg/dL) leads to precipitation of uric acid in the renal tubules and AKI (Chap. 331). Other features of tumor lysis syndrome include hyperkalemia and hyperphosphatemia. The tumor lysis syndrome can also occasionally occur spontaneously or with treatment for solid tumors or multiple myeloma. Myeloma light chains can also cause AKI by direct tubular toxicity and by binding to Tamm-Horsfall protein to form obstructing intratubular casts. Hypercalcemia, which can also be seen in multiple myeloma, may cause AKI by intense renal vasoconstriction and volume depletion.
Allergic Acute Tubulointerstitial Disease and Other Causes of Intrinsic AKI While many of the ischemic and toxic causes of AKI previously described result in tubulointerstitial disease, many drugs are also associated with the development of an allergic response characterized by an inflammatory infiltrate and often peripheral and urinary eosinophilia. AKI may be caused by severe infections and infiltrative diseases. Diseases of the glomeruli or vasculature can lead to AKI by compromising blood flow within the renal circulation. Glomerulonephritis and vasculitis are less common causes of AKI. It is particularly important to recognize these diseases early because they require timely treatment with immunosuppressive agents or therapeutic plasma exchange.
POSTRENAL ACUTE KIDNEY INJURY
(See also Chap. 343) Postrenal AKI occurs when the normally unidirectional flow of urine is acutely blocked either partially or totally, leading to increased retrograde hydrostatic pressure and interference with glomerular filtration. Obstruction to urinary flow may be caused by functional or structural derangements anywhere from the renal pelvis to the tip of the urethra (Fig. 334-5). Normal urinary flow rate does not rule out the presence of partial obstruction, because the GFR is normally two orders of magnitude higher than the urinary flow rate. For AKI to occur in healthy individuals, obstruction must affect both kidneys unless only one kidney is functional, in which case unilateral obstruction can cause AKI. Unilateral obstruction may cause AKI in the setting of significant underlying CKD or, in rare cases, from reflex vasospasm of the contralateral kidney. Bladder neck obstruction is a common cause of postrenal AKI and can be due to prostate disease (benign prostatic hypertrophy or prostate cancer), neurogenic bladder, or therapy with anticholinergic drugs. Obstructed Foley catheters can cause postrenal AKI if not recognized and relieved. Other causes of lower tract obstruction are blood clots, calculi, and urethral strictures. Ureteric obstruction can occur from intraluminal obstruction (e.g., calculi, blood clots, sloughed renal papillae), infiltration of the ureteric wall (e.g., neoplasia), or external compression (e.g., retroperitoneal fibrosis, neoplasia, abscess, or inadvertent surgical damage). The pathophysiology of postrenal AKI involves hemodynamic alterations triggered by an abrupt increase in intratubular pressures. An initial period of hyperemia from afferent arteriolar dilation is followed by intrarenal vasoconstriction from the generation of angiotensin II, thromboxane A2, and vasopressin, and a reduction in NO production. Reduced GFR is due to underperfusion of glomeruli and, possibly, changes in the glomerular ultrafiltration coefficient.
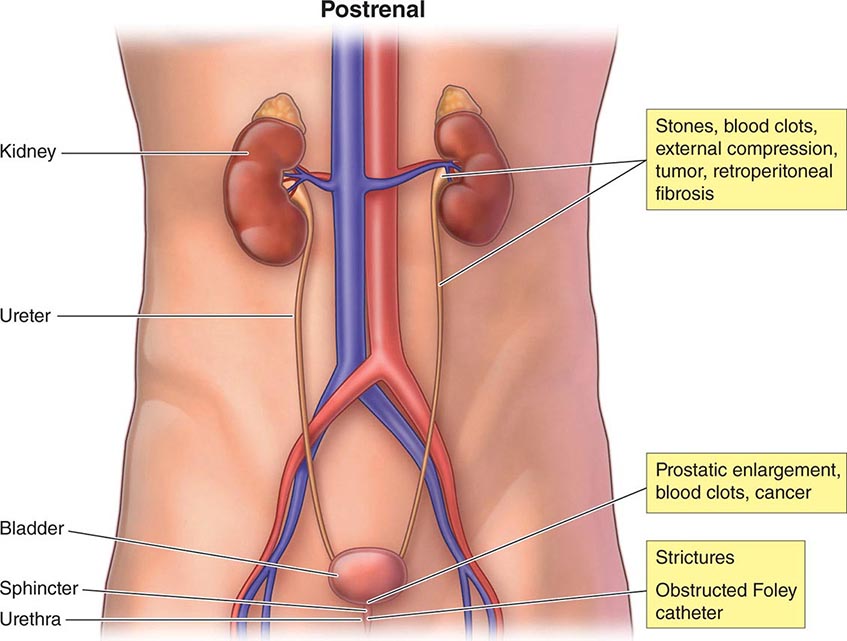
FIGURE 334-5 Anatomic sites and causes of obstruction leading to postrenal acute kidney injury.
DIAGNOSTIC EVALUATION (TABLE 334-1)
|
MAJOR CAUSES, CLINICAL FEATURES, AND DIAGNOSTIC STUDIES FOR PRERENAL AND INTRINSIC ACUTE KIDNEY INJURY |
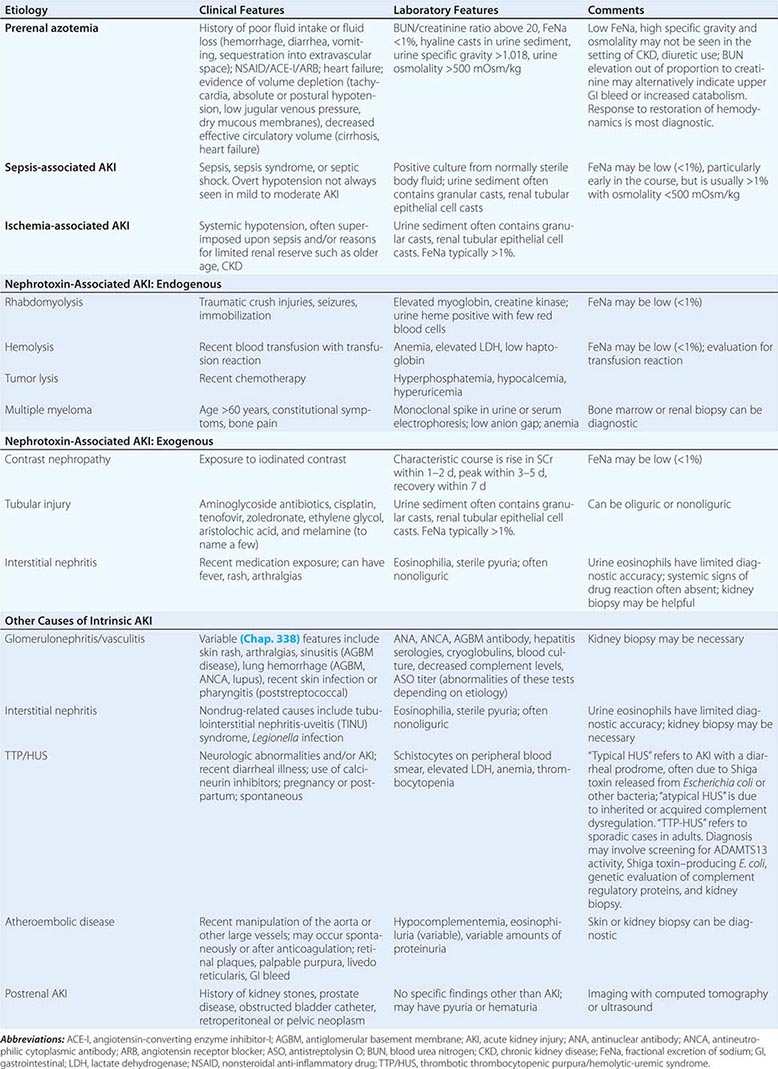
The presence of AKI is usually inferred by an elevation in the SCr concentration. AKI is currently defined by a rise from baseline of at least 0.3 mg/dL within 48 h or at least 50% higher than baseline within 1 week, or a reduction in urine output to less than 0.5 mL/kg per hour for longer than 6 h. It is important to recognize that given this definition, some patients with AKI will not have tubular or glomerular damage (e.g., prerenal azotemia). The distinction between AKI and CKD is important for proper diagnosis and treatment. The distinction is straightforward when a recent baseline SCr concentration is available, but more difficult in the many instances in which the baseline is unknown. In such cases, clues suggestive of CKD can come from radiologic studies (e.g., small, shrunken kidneys with cortical thinning on renal ultrasound, or evidence of renal osteodystrophy) or laboratory tests such as normocytic anemia in the absence of blood loss or secondary hyperparathyroidism with hyperphosphatemia and hypocalcemia, consistent with CKD. No set of tests, however, can rule out AKI superimposed on CKD because AKI is a frequent complication in patients with CKD, further complicating the distinction. Serial blood tests showing continued substantial rise of SCr represents clear evidence of AKI. Once the diagnosis of AKI is established, its cause needs to be determined.
HISTORY AND PHYSICAL EXAMINATION
The clinical context, careful history taking, and physical examination often narrow the differential diagnosis for the cause of AKI. Prerenal azotemia should be suspected in the setting of vomiting, diarrhea, glycosuria causing polyuria, and several medications including diuretics, NSAIDs, ACE inhibitors, and ARBs. Physical signs of orthostatic hypotension, tachycardia, reduced jugular venous pressure, decreased skin turgor, and dry mucous membranes are often present in prerenal azotemia. A history of prostatic disease, nephrolithiasis, or pelvic or paraaortic malignancy would suggest the possibility of postrenal AKI. Whether or not symptoms are present early during obstruction of the urinary tract depends on the location of obstruction. Colicky flank pain radiating to the groin suggests acute ureteric obstruction. Nocturia and urinary frequency or hesitancy can be seen in prostatic disease. Abdominal fullness and suprapubic pain can accompany massive bladder enlargement. Definitive diagnosis of obstruction requires radiologic investigations.
A careful review of all medications is imperative in the evaluation of an individual with AKI. Not only are medications frequently a cause of AKI, but doses of administered medications must be adjusted for estimated GFR. Idiosyncratic reactions to a wide variety of medications can lead to allergic interstitial nephritis, which may be accompanied by fever, arthralgias, and a pruritic erythematous rash. The absence of systemic features of hypersensitivity, however, does not exclude the diagnosis of interstitial nephritis.
AKI accompanied by palpable purpura, pulmonary hemorrhage, or sinusitis raises the possibility of systemic vasculitis with glomerulonephritis. Atheroembolic disease can be associated with livedo reticularis and other signs of emboli to the legs. A tense abdomen should prompt consideration of acute abdominal compartment syndrome, which requires measurement of bladder pressure. Signs of limb ischemia may be clues to the diagnosis of rhabdomyolysis.
URINE FINDINGS
Complete anuria early in the course of AKI is uncommon except in the following situations: complete urinary tract obstruction, renal artery occlusion, overwhelming septic shock, severe ischemia (often with cortical necrosis), or severe proliferative glomerulonephritis or vasculitis. A reduction in urine output (oliguria, defined as <400 mL/24 h) usually denotes more severe AKI (i.e., lower GFR) than when urine output is preserved. Oliguria is associated with worse clinical outcomes. Preserved urine output can be seen in nephrogenic diabetes insipidus characteristic of longstanding urinary tract obstruction, tubulointerstitial disease, or nephrotoxicity from cisplatin or aminoglycosides, among other causes. Red or brown urine may be seen with or without gross hematuria; if the color persists in the supernatant after centrifugation, then pigment nephropathy from rhabdomyolysis or hemolysis should be suspected.
The urinalysis and urine sediment examination are invaluable tools, but they require clinical correlation because of generally limited sensitivity and specificity (Fig. 334-6) (Chap. 62e). In the absence of preexisting proteinuria from CKD, AKI from ischemia or nephrotoxins leads to mild proteinuria (<1 g/d). Greater proteinuria in AKI suggests damage to the glomerular ultrafiltration barrier or excretion of myeloma light chains; the latter are not detected with conventional urine dipsticks (which detect albumin) and require the sulfosalicylic acid test or immunoelectrophoresis. Atheroemboli can cause a variable degree of proteinuria. Extremely heavy proteinuria (“nephrotic range,” >3.5 g/d) can occasionally be seen in glomerulonephritis, vasculitis, or interstitial nephritis (particularly from NSAIDs). AKI can also complicate cases of minimal change disease, a cause of the nephrotic syndrome (Chap. 332e). If the dipstick is positive for hemoglobin but few red blood cells are evident in the urine sediment, then rhabdomyolysis or hemolysis should be suspected.
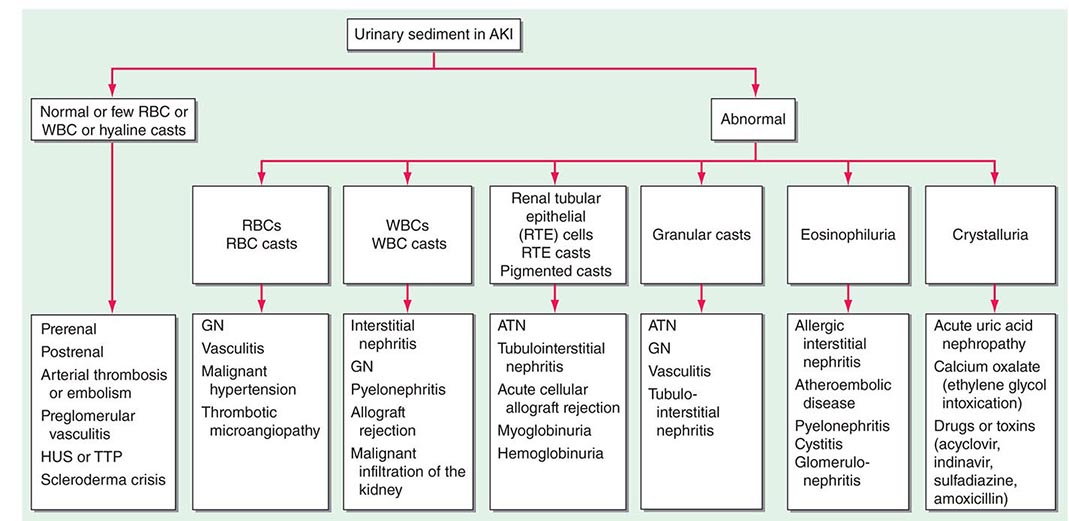
FIGURE 334-6 Interpretation of urinary sediment findings in acute kidney injury (AKI). ATN, acute tubular necrosis; GN, glomerulonephritis; HUS, hemolytic-uremic syndrome; RBCs, red blood cells; RTE, renal tubular epithelial; TTP, thrombotic thrombocytopenic purpura; WBCs, white blood cells. (Adapted from L Yang, JV Bonventre: Diagnosis and clinical evaluation of acute kidney injury. In Comprehensive Nephrology, 4th ed. J Floege et al [eds]. Philadelphia, Elsevier, 2010.)
Prerenal azotemia may present with hyaline casts or an unremarkable urine sediment exam. Postrenal AKI may also lead to an unremarkable sediment, but hematuria and pyuria may be seen depending on the cause of obstruction. AKI from ATN due to ischemic injury, sepsis, or certain nephrotoxins has characteristic urine sediment findings: pigmented “muddy brown” granular casts and tubular epithelial cell casts. These findings may be absent in more than 20% of cases, however. Glomerulonephritis may lead to dysmorphic red blood cells or red blood cell casts. Interstitial nephritis may lead to white blood cell casts. The urine sediment findings overlap somewhat in glomerulonephritis and interstitial nephritis, and a diagnosis is not always possible on the basis of the urine sediment alone. Urine eosinophils have a limited role in differential diagnosis; they can be seen in interstitial nephritis, pyelonephritis, cystitis, atheroembolic disease, or glomerulonephritis. Crystalluria may be important diagnostically. The finding of oxalate crystals in AKI should prompt an evaluation for ethylene glycol toxicity. Abundant uric acid crystals may be seen in the tumor lysis syndrome.
BLOOD LABORATORY FINDINGS
Certain forms of AKI are associated with characteristic patterns in the rise and fall of SCr. Prerenal azotemia typically leads to modest rises in SCr that return to baseline with improvement in hemodynamic status. Contrast nephropathy leads to a rise in SCr within 24–48 h, peak within 3–5 days, and resolution within 5–7 days. In comparison, atheroembolic disease usually manifests with more subacute rises in SCr, although severe AKI with rapid increases in SCr can occur in this setting. With many of the epithelial cell toxins such as aminoglycoside antibiotics and cisplatin, the rise in SCr is characteristically delayed for 3–5 days to 2 weeks after initial exposure.
A complete blood count may provide diagnostic clues. Anemia is common in AKI and is usually multifactorial in origin. It is not related to an effect of AKI solely on production of red blood cells because this effect in isolation takes longer to manifest. Peripheral eosinophilia can accompany interstitial nephritis, atheroembolic disease, polyarteritis nodosa, and Churg-Strauss vasculitis. Severe anemia in the absence of bleeding may reflect hemolysis, multiple myeloma, or thrombotic microangiopathy (e.g., HUS or TTP). Other laboratory findings of thrombotic microangiopathy include thrombocytopenia, schistocytes on peripheral blood smear, elevated lactate dehydrogenase level, and low haptoglobin content. Evaluation of patients suspected of having TTP-HUS includes measurement of levels of the von Willebrand factor cleaving protease (ADAMTS13) and testing for Shiga toxin–producing Escherichia coli. “Atypical HUS” constitutes the majority of adult cases of HUS; genetic testing is important because it is estimated that 60–70% of atypical HUS patients have mutations in genes encoding proteins that regulate the alternative complement pathway.
AKI often leads to hyperkalemia, hyperphosphatemia, and hypocalcemia. Marked hyperphosphatemia with accompanying hypocalcemia, however, suggests rhabdomyolysis or the tumor lysis syndrome. Creatine phosphokinase levels and serum uric acid are elevated in rhabdomyolysis, while tumor lysis syndrome shows normal or marginally elevated creatine kinase and markedly elevated serum uric acid. The anion gap may be increased with any cause of uremia due to retention of anions such as phosphate, hippurate, sulfate, and urate. The co-occurrence of an increased anion gap and an osmolal gap may suggest ethylene glycol poisoning, which may also cause oxalate crystalluria. Low anion gap may provide a clue to the diagnosis of multiple myeloma due to the presence of unmeasured cationic proteins. Laboratory blood tests helpful for the diagnosis of glomerulonephritis and vasculitis include depressed complement levels and high titers of antinuclear antibodies (ANAs), antineutrophilic cytoplasmic antibodies (ANCAs), antiglomerular basement membrane (AGBM) antibodies, and cryoglobulins.
RENAL FAILURE INDICES
Several indices have been used to help differentiate prerenal azotemia from intrinsic AKI when the tubules are malfunctioning. The low tubular flow rate and increased renal medullary recycling of urea seen in prerenal azotemia may cause a disproportionate elevation of the BUN compared to creatinine. Other causes of disproportionate BUN elevation need to be kept in mind, however, including upper gastrointestinal bleeding, hyperalimentation, increased tissue catabolism, and glucocorticoid use.
The fractional excretion of sodium (FeNa) is the fraction of the filtered sodium load that is reabsorbed by the tubules, and is a measure of both the kidney’s ability to reabsorb sodium as well as endogenously and exogenously administered factors that affect tubular reabsorption. As such, it depends on sodium intake, effective intravascular volume, GFR, diuretic intake, and intact tubular reabsorptive mechanisms. With prerenal azotemia, the FeNa may be below 1%, suggesting avid tubular sodium reabsorption. In patients with CKD, a FeNa significantly above 1% can be present despite a superimposed prerenal state. The FeNa may also be above 1% despite hypovolemia due to treatment with diuretics. Low FeNa is often seen early in glomerulonephritis and other disorders and, hence, should not be taken as prima facie evidence of prerenal azotemia. Low FeNa is therefore suggestive, but not synonymous, with effective intravascular volume depletion, and should not be used as the sole guide for volume management. The response of urine output to crystalloid or colloid fluid administration may be both diagnostic and therapeutic in prerenal azotemia. In ischemic AKI, the FeNa is frequently above 1% because of tubular injury and resultant inability to reabsorb sodium. Several causes of ischemia-associated and nephrotoxin-associated AKI can present with FeNa below 1%, however, including sepsis (often early in the course), rhabdomyolysis, and contrast nephropathy.
The ability of the kidney to produce a concentrated urine is dependent upon many factors and reliant on good tubular function in multiple regions of the kidney. In the patient not taking diuretics and with good baseline kidney function, urine osmolality may be above 500 mOsm/kg in prerenal azotemia, consistent with an intact medullary gradient and elevated serum vasopressin levels causing water reabsorption resulting in concentrated urine. In elderly patients and those with CKD, however, baseline concentrating defects may exist, making urinary osmolality unreliable in many instances. Loss of concentrating ability is common in septic or ischemic AKI, resulting in urine osmolality below 350 mOsm/kg, but the finding is not specific.
RADIOLOGIC EVALUATION
Postrenal AKI should always be considered in the differential diagnosis of AKI because treatment is usually successful if instituted early. Simple bladder catheterization can rule out urethral obstruction. Imaging of the urinary tract with renal ultrasound or CT should be undertaken to investigate obstruction in individuals with AKI unless an alternate diagnosis is apparent. Findings of obstruction include dilation of the collecting system and hydroureteronephrosis. Obstruction can be present without radiologic abnormalities in the setting of volume depletion, retroperitoneal fibrosis, encasement with tumor, and also early in the course of obstruction. If a high clinical index of suspicion for obstruction persists despite normal imaging, antegrade or retrograde pyelography should be performed. Imaging may also provide additional helpful information about kidney size and echogenicity to assist in the distinction between acute versus CKD. In CKD, kidneys are usually smaller unless the patient has diabetic nephropathy, HIV-associated nephropathy, or infiltrative diseases. Normal sized kidneys are expected in AKI. Enlarged kidneys in a patient with AKI suggests the possibility of acute interstitial nephritis. Vascular imaging may be useful if venous or arterial obstruction is suspected, but the risks of contrast administration should be kept in mind. MRI with gadolinium-based contrast agents should be avoided if possible in severe AKI due to the possibility of inducing nephrogenic system fibrosis, a rare but serious complication seen most commonly in patients with end-stage renal disease.
KIDNEY BIOPSY
If the cause of AKI is not apparent based on the clinical context, physical examination, laboratory studies, and radiologic evaluation, kidney biopsy should be considered. The kidney biopsy can provide definitive diagnostic and prognostic information about acute kidney disease and CKD. The procedure is most often used in AKI when prerenal azotemia, postrenal AKI, and ischemic or nephrotoxic AKI have been deemed unlikely, and other possible diagnoses are being considered such as glomerulonephritis, vasculitis, interstitial nephritis, myeloma kidney, HUS and TTP, and allograft dysfunction. Kidney biopsy is associated with a risk of bleeding, which can be severe and organ- or life-threatening in patients with thrombocytopenia or coagulopathy.
NOVEL BIOMARKERS
BUN and creatinine are functional biomarkers of glomerular filtration rather than tissue injury biomarkers and, therefore, may be suboptimal for the diagnosis of actual parenchymal kidney damage. BUN and creatinine are also relatively slow to rise after kidney injury. Several novel kidney injury biomarkers have been investigated and show promise for earlier and accurate diagnosis of AKI. Kidney injury molecule-1 (KIM-1) is a type 1 transmembrane protein that is abundantly expressed in proximal tubular cells injured by ischemia or nephrotoxins such as cisplatin. KIM-1 is not expressed in appreciable quantities in the absence of tubular injury or in extrarenal tissues. KIM-1’s functional role may be to confer phagocytic properties to tubular cells, enabling them to clear debris from the tubular lumen after kidney injury. KIM-1 can be detected shortly after ischemic or nephrotoxic injury in the urine and, therefore, may be an easily tested biomarker in the clinical setting. Neutrophil gelatinase associated lipocalin (NGAL, also known as lipocalin-2 or siderocalin) is another novel biomarker of AKI. NGAL was first discovered as a protein in granules of human neutrophils. NGAL can bind to iron siderophore complexes and may have tissue-protective effects in the proximal tubule. NGAL is highly upregulated after inflammation and kidney injury and can be detected in the plasma and urine within 2 h of cardiopulmonary bypass–associated AKI. Other candidate biomarkers of AKI include interleukin (IL) 18, a proinflammatory cytokine of the IL-1 superfamily that may mediate ischemic proximal tubular injury, and L-type fatty acid binding protein, which is expressed in ischemic proximal tubule cells and may be renoprotective by binding free fatty acids and lipid peroxidation products. A number of other biomarkers are under investigation for early and accurate identification of AKI and for risk stratification to identify individuals at increased risk. The optimal use of novel AKI biomarkers in clinical settings is an area of ongoing investigation.
COMPLICATIONS
The kidney plays a central role in homeostatic control of volume status, blood pressure, plasma electrolyte composition, and acid-base balance, and for excretion of nitrogenous and other waste products. Complications associated with AKI are, therefore, protean, and depend on the severity of AKI and other associated conditions. Mild to moderate AKI may be entirely asymptomatic, particularly early in the course.
UREMIA
Buildup of nitrogenous waste products, manifested as an elevated BUN concentration, is a hallmark of AKI. BUN itself poses little direct toxicity at levels below 100 mg/dL. At higher concentrations, mental status changes and bleeding complications can arise. Other toxins normally cleared by the kidney may be responsible for the symptom complex known as uremia. Few of the many possible uremic toxins have been definitively identified. The correlation of BUN and SCr concentrations with uremic symptoms is extremely variable, due in part to differences in urea and creatinine generation rates across individuals.
HYPERVOLEMIA AND HYPOVOLEMIA
Expansion of extracellular fluid volume is a major complication of oliguric and anuric AKI, due to impaired salt and water excretion. The result can be weight gain, dependent edema, increased jugular venous pressure, and pulmonary edema; the latter can be life threatening. Pulmonary edema can also occur from volume overload and hemorrhage in pulmonary renal syndromes. AKI may also induce or exacerbate acute lung injury characterized by increased vascular permeability and inflammatory cell infiltration in lung parenchyma. Recovery from AKI can sometimes be accompanied by polyuria, which, if untreated, can lead to significant volume depletion. The polyuric phase of recovery may be due to an osmotic diuresis from retained urea and other waste products as well as delayed recovery of tubular reabsorptive functions.
HYPONATREMIA
Administration of excessive hypotonic crystalloid or isotonic dextrose solutions can result in hypoosmolality and hyponatremia, which, if severe, can cause neurologic abnormalities, including seizures.
HYPERKALEMIA
Abnormalities in plasma electrolyte composition can be mild or life threatening. Frequently the most concerning complication of AKI is hyperkalemia. Marked hyperkalemia is particularly common in rhabdomyolysis, hemolysis, and tumor lysis syndrome due to release of intracellular potassium from damaged cells. Potassium affects the cellular membrane potential of cardiac and neuromuscular tissues. Muscle weakness may be a symptom of hyperkalemia. The more serious complication of hyperkalemia is due to effects on cardiac conduction, leading to potentially fatal arrhythmias.
ACIDOSIS
Metabolic acidosis, usually accompanied by an elevation in the anion gap, is common in AKI, and can further complicate acid-base and potassium balance in individuals with other causes of acidosis, including sepsis, diabetic ketoacidosis, or respiratory acidosis.
HYPERPHOSPHATEMIA AND HYPOCALCEMIA
AKI can lead to hyperphosphatemia, particularly in highly catabolic patients or those with AKI from rhabdomyolysis, hemolysis, and tumor lysis syndrome. Metastatic deposition of calcium phosphate can lead to hypocalcemia. AKI-associated hypocalcemia may also arise from derangements in the vitamin D–parathyroid hormone–fibroblast growth factor-23 axis. Hypocalcemia is often asymptomatic but can lead to perioral paresthesias, muscle cramps, seizures, carpopedal spasms, and prolongation of the QT interval on electrocardiography. Calcium levels should be corrected for the degree of hypoalbuminemia, if present, or ionized calcium levels should be followed. Mild, asymptomatic hypocalcemia does not require treatment.
BLEEDING
Hematologic complications of AKI include anemia and bleeding, both of which are exacerbated by coexisting disease processes such as sepsis, liver disease, and disseminated intravascular coagulation. Direct hematologic effects from AKI-related uremia include decreased erythropoiesis and platelet dysfunction.
INFECTIONS
Infections are a common precipitant of AKI and also a dreaded complication of AKI. Impaired host immunity has been described in end-stage renal disease and may be operative in severe AKI.
CARDIAC COMPLICATIONS
The major cardiac complications of AKI are arrhythmias, pericarditis, and pericardial effusion.
MALNUTRITION
AKI is often a severely hypercatabolic state, and therefore, malnutrition is a major complication.
OUTCOME AND PROGNOSIS
The development of AKI is associated with a significantly increased risk of in-hospital and long-term mortality, longer length of stay, and increased costs. Prerenal azotemia, with the exception of the cardiorenal and hepatorenal syndromes, and postrenal azotemia carry a better prognosis than most cases of intrinsic AKI. The kidneys may recover even after severe, dialysis-requiring AKI. Survivors of an episode of AKI requiring temporary dialysis, however, are at extremely high risk for progressive CKD, and up to 10% may develop end-stage renal disease. Postdischarge care under the supervision of a nephrologist for aggressive secondary prevention of kidney disease is prudent. Patients with AKI are more likely to die prematurely after they leave the hospital even if their kidney function has recovered.
335 |
Chronic Kidney Disease |
Chronic kidney disease (CKD) encompasses a spectrum of different pathophysiologic processes associated with abnormal kidney function and a progressive decline in glomerular filtration rate (GFR). Figure 335-1 provides a recently updated classification, in which stages of CKD are stratified by both estimated GFR and the degree of albuminuria, in order to predict risk of progression of CKD. Previously, CKD had been staged solely by the GFR. However, the risk of worsening of kidney function is closely linked to the amount of albuminuria, and so it has been incorporated into the classification.
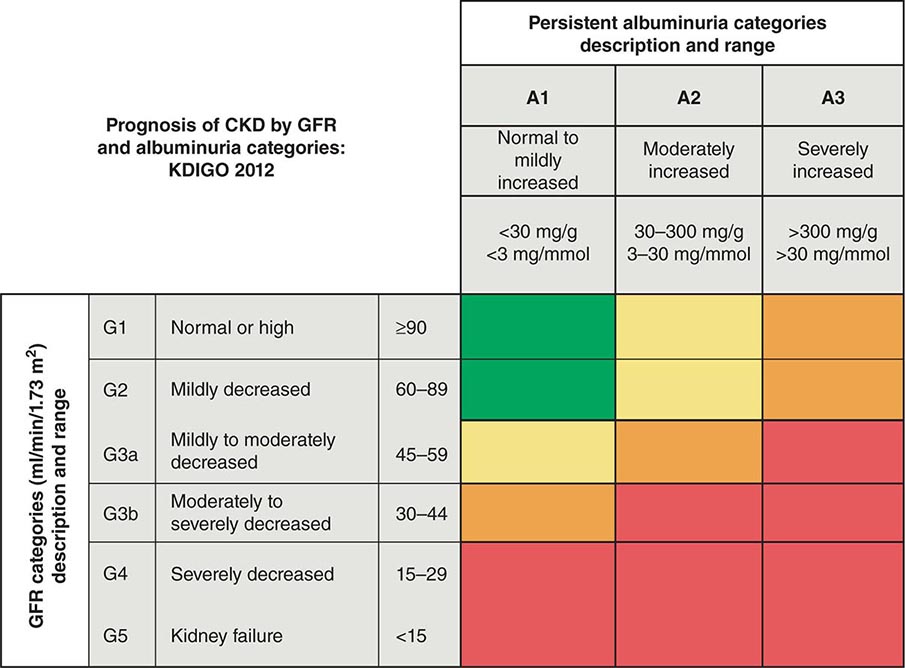
FIGURE 335-1 Kidney Disease Improving Global Outcome (KDIGO) classification of chronic kidney disease (CKD). Gradation of color from green to red corresponds to increasing risk and progression of CKD. GFR, glomerular filtration rate. (Reproduced with permission from Kidney Int Suppl 3:5-14, 2013.)
The pathophysiologic processes, adaptations, clinical presentations, assessment, and therapeutic interventions associated with CKD will be the focus of this chapter. The dispiriting term end-stage renal disease represents a stage of CKD where the accumulation of toxins, fluid, and electrolytes normally excreted by the kidneys results in the uremic syndrome. This syndrome leads to death unless the toxins are removed by renal replacement therapy, using dialysis or kidney transplantation. These interventions are discussed in Chaps. 336 and 337. End-stage renal disease will be supplanted in this chapter by the term stage 5 CKD.
PATHOPHYSIOLOGY OF CHRONIC KIDNEY DISEASE
The pathophysiology of CKD involves two broad sets of mechanisms of damage: (1) initiating mechanisms specific to the underlying etiology (e.g., genetically determined abnormalities in kidney development or integrity, immune complex deposition and inflammation in certain types of glomerulonephritis, or toxin exposure in certain diseases of the renal tubules and interstitium) and (2) a set of progressive mechanisms, involving hyperfiltration and hypertrophy of the remaining viable nephrons, that are a common consequence following long-term reduction of renal mass, irrespective of underlying etiology (Chap. 333e). The responses to reduction in nephron number are mediated by vasoactive hormones, cytokines, and growth factors. Eventually, these short-term adaptations of hypertrophy and hyperfiltration become maladaptive as the increased pressure and flow within the nephron predisposes to distortion of glomerular architecture, abnormal podocyte function, and disruption of the filtration barrier leading to sclerosis and dropout of the remaining nephrons (Fig. 335-2). Increased intrarenal activity of the renin-angiotensin system (RAS) appears to contribute both to the initial adaptive hyperfiltration and to the subsequent maladaptive hypertrophy and sclerosis. This process explains why a reduction in renal mass from an isolated insult may lead to a progressive decline in renal function over many years (Fig. 335-3).
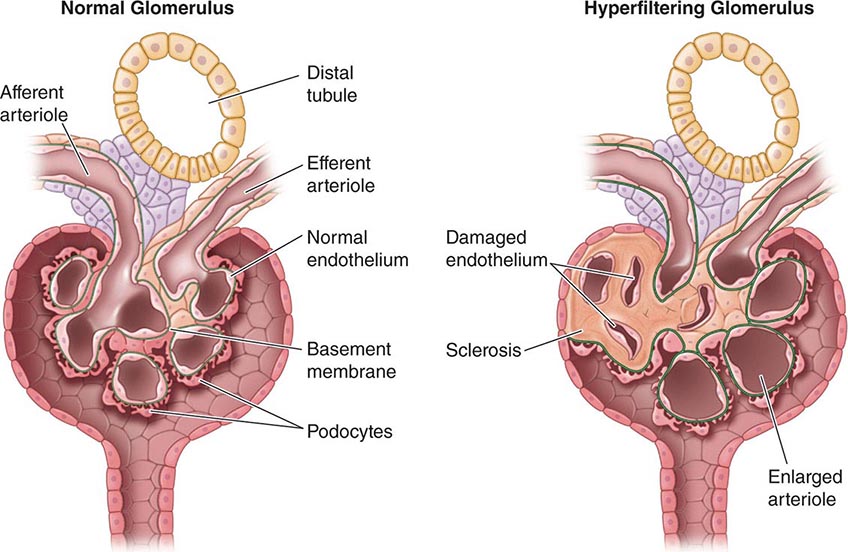
FIGURE 335-2 Left: Schema of the normal glomerular architecture. Right: Secondary glomerular changes associated with a reduction in nephron number, including enlargement of capillary lumens and focal adhesions, which are thought to occur consequent to compensatory hyperfiltration and hypertrophy in the remaining nephrons. (Modified from JR Ingelfinger: N Engl J Med 348:99, 2003.)
FIGURE 335-3 Left: Low-power photomicrograph of a normal kidney showing normal glomeruli and healthy tubulointerstitium without fibrosis. Right: Low-power photomicrograph of chronic kidney disease with sclerosis of many glomeruli and severe tubulointerstitial fibrosis (Masson trichrome, ×40 magnification).(Slides courtesy of the late Dr. Andrew Herzenberg.)
IDENTIFICATION OF RISK FACTORS AND STAGING OF CKD
It is important to identify factors that increase the risk for CKD, even in individuals with normal GFR. Risk factors include small for gestation birth weight, childhood obesity, hypertension, diabetes mellitus, autoimmune disease, advanced age, African ancestry, a family history of kidney disease, a previous episode of acute kidney injury, and the presence of proteinuria, abnormal urinary sediment, or structural abnormalities of the urinary tract.
Many rare inherited forms of CKD follow a Mendelian inheritance pattern, often as part of a systemic syndrome, with the most common in this category being autosomal dominant polycystic kidney disease. In addition, recent research in the genetics of predisposition to common complex diseases (Chap. 82) has revealed DNA sequence variants at a number of genetic loci that are associated with common forms of CKD. A striking example is the finding of allelic versions of the APOL1 gene, of West African population ancestry, which contributes to the several-fold higher frequency of certain common etiologies of nondiabetic CKD (e.g., focal segmental glomerulosclerosis) observed among African and Hispanic Americans. The prevalence in West African populations seems to have arisen as an evolutionary adaptation conferring protection from tropical pathogens. As in other common diseases with a heritable component, an environmental trigger (such as a viral pathogen) is required to transform genetic risk into disease.
To stage CKD, it is necessary to estimate the GFR rather than relying on serum creatinine concentration (Table 335-1). Many laboratories now report an estimated GFR, or eGFR, using one of these equations.
|
RECOMMENDED EQUATIONS FOR ESTIMATION OF GLOMERULAR FILTRATION RATE (GFR) USING SERUM CREATININE CONCENTRATION (SCR), AGE, SEX, RACE, AND BODY WEIGHT |
Abbreviation: CKD-EPI, Chronic Kidney Disease Epidemiology Collaboration.
The normal annual mean decline in GFR with age from the peak GFR (~120 mL/min per 1.73 m2) attained during the third decade of life is ~1 mL/min per year per 1.73 m2, reaching a mean value of 70 mL/min per 1.73 m2 at age 70. Although reduced GFR occurs with human aging, the lower GFR signifies a true loss of kidney function, with all of the implications that apply to the corresponding stage of CKD. The mean GFR is lower in women than in men. For example, a woman in her 80s with a normal serum creatinine may have a GFR of just 50 mL/min per 1.73 m2. Thus, even a mild elevation in serum creatinine concentration (e.g., 130 μmol/L [1.5 mg/dL]) often signifies a substantial reduction in GFR in most individuals.
the equations for estimating GFR are valid only if the patient is in steady state, that is, the serum creatinine is neither rising nor falling over days.
Measurement of albuminuria is also helpful for monitoring nephron injury and the response to therapy in many forms of CKD, especially chronic glomerular diseases. Although an accurate 24-h urine collection is the standard for measurement of albuminuria, the measurement of protein-to-creatinine ratio in a spot first-morning urine sample is often more practical to obtain and correlates well, but not perfectly, with 24-h urine collections. Microalbuminuria (Fig. 335-1, stage A2) refers to the excretion of amounts of albumin too small to detect by urinary dipstick or conventional measures of urine protein. It is a good screening test for early detection of renal disease, and may be a marker for the presence of microvascular disease in general. If a patient has a large amount of excreted albumin, there is no reason to test for microalbuminuria.
Stages 1 and 2 CKD are usually not associated with any symptoms arising from the decrement in GFR. If the decline in GFR progresses to stages 3 and 4, clinical and laboratory complications of CKD become more prominent. Virtually all organ systems are affected, but the most evident complications include anemia and associated easy fatigability; decreased appetite with progressive malnutrition; abnormalities in calcium, phosphorus, and mineral-regulating hormones, such as 1,25(OH)2D3 (calcitriol), parathyroid hormone (PTH), and fibroblast growth factor 23 (FGF-23); and abnormalities in sodium, potassium, water, and acid-base homeostasis. Many patients, especially the elderly, will have eGFR values compatible with stage 2 or 3 CKD. However, the majority of these patients will show no further deterioration of renal function. The primary care physician is advised to recheck kidney function, and if it is stable and not associated with proteinuria, the patient can usually be managed in this setting. However, if there is evidence of decline of GFR, uncontrolled hypertension, or proteinuria, referral to a nephrologist is appropriate. If the patient progresses to stage 5 CKD, toxins accumulate such that patients usually experience a marked disturbance in their activities of daily living, well-being, nutritional status, and water and electrolyte homeostasis, eventuating in the uremic syndrome.
ETIOLOGY AND EPIDEMIOLOGY
It has been estimated from population survey data that at least 6% of the adult population in the United States has CKD at stages 1 and 2. An additional 4.5% of the U.S. population is estimated to have stages 3 and 4 CKD. Table 335-2 lists the five most frequent categories of causes of CKD, cumulatively accounting for greater than 90% of the CKD disease burden worldwide. The relative contribution of each category varies among different geographic regions. The most frequent cause of CKD in North America and Europe is diabetic nephropathy, most often secondary to type 2 diabetes mellitus. Patients with newly diagnosed CKD often also present with hypertension. When no overt evidence for a primary glomerular or tubulointerstitial kidney disease process is present, CKD is often attributed to hypertension. However, it is now appreciated that such individuals can be considered in two categories. The first includes patients with a silent primary glomerulopathy, such as focal segmental glomerulosclerosis, without the overt nephrotic or nephritic manifestations of glomerular disease (Chap. 338). The second includes patients in whom progressive nephrosclerosis and hypertension is the renal correlate of a systemic vascular disease, often also involving large- and small-vessel cardiac and cerebral pathology. This latter combination is especially common in the elderly, in whom chronic renal ischemia as a cause of CKD may be underdiagnosed. The increasing incidence of CKD in the elderly has been ascribed, in part, to decreased mortality rate from the cardiac and cerebral complications of atherosclerotic vascular disease, enabling a greater segment of the population to eventually manifest the renal component of generalized vascular disease. Nevertheless, it should be appreciated that the vast majority of such patients with early stages of CKD will succumb to the cardiovascular and cerebrovascular consequences of the vascular disease before they can progress to the most advanced stages of CKD. Indeed, even a minor decrement in GFR or the presence of albuminuria is now recognized as a major risk factor for cardiovascular disease.
|
LEADING CATEGORIES OF ETIOLOGIES OF CKDa |
PATHOPHYSIOLOGY AND BIOCHEMISTRY OF UREMIA
Although serum urea and creatinine concentrations are used to measure the excretory capacity of the kidneys, accumulation of these two molecules themselves does not account for the many symptoms and signs that characterize the uremic syndrome in advanced renal failure. Hundreds of toxins that accumulate in renal failure have been implicated in the uremic syndrome. These include water-soluble, hydrophobic, protein-bound, charged, and uncharged compounds. Additional categories of nitrogenous excretory products include guanidino compounds, urates and hippurates, products of nucleic acid metabolism, polyamines, myoinositol, phenols, benzoates, and indoles. It is thus evident that the serum concentrations of urea and creatinine should be viewed as being readily measured, but incomplete, surrogate markers for these compounds, and monitoring the levels of urea and creatinine in the patient with impaired kidney function represents a vast oversimplification of the uremic state.
The uremic syndrome and the disease state associated with advanced renal impairment involve more than renal excretory failure. A host of metabolic and endocrine functions normally performed by the kidneys is also impaired or suppressed, and this results in anemia, malnutrition, and abnormal metabolism of carbohydrates, fats, and proteins. Furthermore, plasma levels of many hormones, including PTH, FGF-23, insulin, glucagon, steroid hormones including vitamin D and sex hormones, and prolactin, change with CKD as a result of reduced excretion, decreased degradation, or abnormal regulation. Finally, CKD is associated with worsening systemic inflammation. Elevated levels of C-reactive protein are detected along with other acute-phase reactants, whereas levels of so-called negative acute-phase reactants, such as albumin and fetuin, decline with progressive reduction in GFR. Thus, the inflammation associated with CKD is important in the malnutrition-inflammation-atherosclerosis/calcification syndrome, which contributes in turn to the acceleration of vascular disease and comorbidity associated with advanced kidney disease.
In summary, the pathophysiology of the uremic syndrome can be divided into manifestations in three spheres of dysfunction: (1) those consequent to the accumulation of toxins that normally undergo renal excretion, including products of protein metabolism; (2) those consequent to the loss of other kidney functions, such as fluid and electrolyte homeostasis and hormone regulation; and (3) progressive systemic inflammation and its vascular and nutritional consequences.
CLINICAL AND LABORATORY MANIFESTATIONS OF CHRONIC KIDNEY DISEASE AND UREMIA
Uremia leads to disturbances in the function of virtually every organ system. Chronic dialysis can reduce the incidence and severity of many of these disturbances, so that the overt and florid manifestations of uremia have largely disappeared in the modern health setting. However, even optimal dialysis therapy is not completely effective as renal replacement therapy, because some disturbances resulting from impaired kidney function fail to respond to dialysis.
FLUID, ELECTROLYTE, AND ACID-BASE DISORDERS
Sodium and Water Homeostasis In most patients with stable CKD, the total-body content of sodium and water is modestly increased, although this may not be apparent on clinical examination. With normal renal function, the tubular reabsorption of filtered sodium and water is adjusted so that urinary excretion matches intake. Many forms of kidney disease (e.g., glomerulonephritis) disrupt this balance such that dietary intake of sodium exceeds its urinary excretion, leading to sodium retention and attendant extracellular fluid volume (ECFV) expansion. This expansion may contribute to hypertension, which itself can accelerate the nephron injury. As long as water intake does not exceed the capacity for water clearance, the ECFV expansion will be isotonic and the patient will have a normal plasma sodium concentration (Chap. 333e). Hyponatremia is not commonly seen in CKD patients but, when present, often responds to water restriction. The patient with ECFV expansion (peripheral edema, sometimes hypertension poorly responsive to therapy) should be counseled regarding salt restriction. Thiazide diuretics have limited utility in stages 3–5 CKD, such that administration of loop diuretics, including furosemide, bumetanide, or torsemide, may also be needed. Resistance to loop diuretics in CKD often mandates use of higher doses than those used in patients with more normal kidney function. The combination of loop diuretics with metolazone, which inhibits the sodium chloride co-transporter of the distal convoluted tubule, can promote renal salt excretion. Diuretic resistance with intractable edema and hypertension in advanced CKD may serve as an indication to initiate dialysis.
In addition to problems with salt and water excretion, some patients with CKD may instead have impaired renal conservation of sodium and water. When an extrarenal cause for fluid loss, such as gastrointestinal (GI) loss, is present, these patients may be prone to ECFV depletion because of the inability of the failing kidney to reclaim filtered sodium adequately. Furthermore, depletion of ECFV, whether due to GI losses or overzealous diuretic therapy, can further compromise kidney function through underperfusion, or a “prerenal” basis, leading to acute-on-chronic kidney failure. In this setting, cautious volume repletion with normal saline may return the ECFV to normal and restore renal function to baseline without having to intervene with dialysis.
Potassium Homeostasis In CKD, the decline in GFR is not necessarily accompanied by a parallel decline in urinary potassium excretion, which is predominantly mediated by aldosterone-dependent secretion in the distal nephron. Another defense against potassium retention in these patients is augmented potassium excretion in the GI tract. Notwithstanding these two homeostatic responses, hyperkalemia may be precipitated in certain settings. These include increased dietary potassium intake, protein catabolism, hemolysis, hemorrhage, transfusion of stored red blood cells, and metabolic acidosis. In addition, a host of medications can inhibit renal potassium excretion and lead to hyperkalemia. The most important medications in this respect include the RAS inhibitors and spironolactone and other potassium-sparing diuretics such as amiloride, eplerenone, and triamterene.
Certain causes of CKD can be associated with earlier and more severe disruption of potassium-secretory mechanisms in the distal nephron, out of proportion to the decline in GFR. These include conditions associated with hyporeninemic hypoaldosteronism, such as diabetes, and renal diseases that preferentially affect the distal nephron, such as obstructive uropathy and sickle cell nephropathy.
Hypokalemia is not common in CKD and usually reflects markedly reduced dietary potassium intake, especially in association with excessive diuretic therapy or concurrent GI losses. The use of potassium supplements and potassium-sparing diuretics may be risky in patients with impaired renal function, and should be constantly reevaluated as GFR declines.
Metabolic Acidosis Metabolic acidosis is a common disturbance in advanced CKD. The majority of patients can still acidify the urine, but they produce less ammonia and, therefore, cannot excrete the normal quantity of protons in combination with this urinary buffer. Hyperkalemia, if present, further depresses ammonia production. The combination of hyperkalemia and hyperchloremic metabolic acidosis is often present, even at earlier stages of CKD (stages 1–3), in patients with diabetic nephropathy or in those with predominant tubulointerstitial disease or obstructive uropathy; this is a non-anion-gap metabolic acidosis.
With worsening renal function, the total urinary net daily acid excretion is usually limited to 30–40 mmol, and the anions of retained organic acids can then lead to an anion-gap metabolic acidosis. Thus, the non-anion-gap metabolic acidosis that can be seen in earlier stages of CKD may be complicated by the addition of an anion-gap metabolic acidosis as CKD progresses. In most patients, the metabolic acidosis is mild; the pH is rarely <7.35 and can usually be corrected with oral sodium bicarbonate supplementation. Animal and human studies have suggested that even modest degrees of metabolic acidosis may be associated with the development of protein catabolism. Alkali supplementation may attenuate the catabolic state and possibly slow CKD progression and accordingly is recommended when the serum bicarbonate concentration falls below 20–23 mmol/L. The concomitant sodium load mandates careful attention to volume status and the need for diuretic agents.
DISORDERS OF CALCIUM AND PHOSPHATE METABOLISM
The principal complications of abnormalities of calcium and phosphate metabolism in CKD occur in the skeleton and the vascular bed, with occasional severe involvement of extraosseous soft tissues. It is likely that disorders of bone turnover and disorders of vascular and soft tissue calcification are related to each other (Fig. 335-3).
Bone Manifestations of CKD The major disorders of bone disease can be classified into those associated with high bone turnover with increased PTH levels (including osteitis fibrosa cystica, the classic lesion of secondary hyperparathyroidism) and low bone turnover with low or normal PTH levels (adynamic bone disease and osteomalacia).
The pathophysiology of secondary hyperparathyroidism and the consequent high-turnover bone disease is related to abnormal mineral metabolism through the following events: (1) declining GFR leads to reduced excretion of phosphate and, thus, phosphate retention; (2) the retained phosphate stimulates increased synthesis of both FGF-23 by osteocytes and PTH and stimulates growth of parathyroid gland mass; and (3) decreased levels of ionized calcium, resulting from suppression of calcitriol production by FGF-23 and by the failing kidney, as well as phosphate retention, also stimulate PTH production. Low calcitriol levels contribute to hyperparathyroidism, both by leading to hypocalcemia and also by a direct effect on PTH gene transcription. These changes start to occur when the GFR falls below 60 mL/min.
FGF-23 is part of a family of phosphatonins that promotes renal phosphate excretion. Recent studies have shown that levels of this hormone, secreted by osteocytes, increase early in the course of CKD, even before phosphate retention and hyperphosphatemia. FGF-23 may defend normal serum phosphorus in at least three ways: (1) increased renal phosphate excretion; (2) stimulation of PTH, which also increases renal phosphate excretion; and (3) suppression of the formation of 1,25(OH)2D3, leading to diminished phosphorus absorption from the GI tract. Interestingly, high levels of FGF-23 are also an independent risk factor for left ventricular hypertrophy and mortality in CKD, dialysis, and renal transplant patients. Moreover, elevated levels of FGF-23 may indicate the need for therapeutic intervention (e.g., phosphate restriction), even when serum phosphate levels are within the normal range.
Hyperparathyroidism stimulates bone turnover and leads to osteitis fibrosa cystica. Bone histology shows abnormal osteoid, bone and bone marrow fibrosis, and in advanced stages, the formation of bone cysts, sometimes with hemorrhagic elements so that they appear brown in color, hence the term brown tumor. Clinical manifestations of severe hyperparathyroidism include bone pain and fragility, brown tumors, compression syndromes, and erythropoietin resistance in part related to the bone marrow fibrosis. Furthermore, PTH itself is considered a uremic toxin, and high levels are associated with muscle weakness, fibrosis of cardiac muscle, and nonspecific constitutional symptoms.
Low-turnover bone disease can be grouped into two categories—adynamic bone disease and osteomalacia. Adynamic bone disease is increasing in prevalence, especially among diabetics and the elderly. It is characterized by reduced bone volume and mineralization and may result from excessive suppression of PTH production, chronic inflammation, or both. Suppression of PTH can result from the use of vitamin D preparations or from excessive calcium exposure in the form of calcium-containing phosphate binders or high-calcium dialysis solutions. Complications of adynamic bone disease include an increased incidence of fracture and bone pain and an association with increased vascular and cardiac calcification. Occasionally the calcium will precipitate in the soft tissues into large concretions termed “tumoral calcinosis” (Fig. 335-4).
FIGURE 335-4 Tumoral calcinosis. This patient was on hemodialysis for many years and was nonadherent to dietary phosphorus restriction or the use of phosphate binders. He was chronically severely hyperphosphatemic. He developed an enlarging painful mass on his arm that was extensively calcified.
Calcium, Phosphorus, and the Cardiovascular System Recent epidemiologic evidence has shown a strong association between hyperphosphatemia and increased cardiovascular mortality rate in patients with stage 5 CKD and even in patients with earlier stages of CKD. Hyperphosphatemia and hypercalcemia are associated with increased vascular calcification, but it is unclear whether the excessive mortality rate is mediated by this mechanism. Studies using computed tomography (CT) and electron-beam CT scanning show that CKD patients have calcification of the media in coronary arteries and even heart valves that appear to be orders of magnitude greater than that in patients without renal disease. The magnitude of the calcification is proportional to age and hyperphosphatemia and is also associated with low PTH levels and low bone turnover. It is possible that in patients with advanced kidney disease, ingested calcium cannot be deposited in bones with low turnover and, therefore, is deposited at extraosseous sites, such as the vascular bed and soft tissues. It is interesting in this regard that there is also an association between osteoporosis and vascular calcification in the general population. Finally, hyperphosphatemia can induce a change in gene expression in vascular cells to an osteoblast-like profile, leading to vascular calcification and even ossification.
Other Complications of Abnormal Mineral Metabolism Calciphylaxis (calcific uremic arteriolopathy) is a devastating condition seen almost exclusively in patients with advanced CKD. It is heralded by livedo reticularis and advances to patches of ischemic necrosis, especially on the legs, thighs, abdomen, and breasts (Fig. 335-5). Pathologically, there is evidence of vascular occlusion in association with extensive vascular and soft tissue calcification. It appears that this condition is increasing in incidence. Originally it was ascribed to severe abnormalities in calcium and phosphorus control in dialysis patients, usually associated with advanced hyperparathyroidism. However, more recently, calciphylaxis has been seen with increasing frequency in the absence of severe hyperparathyroidism. Other etiologies have been suggested, including the increased use of oral calcium as a phosphate binder. Warfarin is commonly used in hemodialysis patients, and one of the effects of warfarin therapy is to decrease the vitamin K–dependent regeneration of matrix GLA protein. This latter protein is important in preventing vascular calcification. Thus, warfarin treatment is considered a risk factor for calciphylaxis, and if a patient develops this syndrome, this medication should be discontinued and replaced with alternative forms of anticoagulation.
FIGURE 335-5 Calciphylaxis. This peritoneal dialysis patient was on chronic warfarin therapy for atrial fibrillation. She noticed a small painful nodule on the abdomen that was followed by progressive skin necrosis and ulceration of the anterior abdominal wall. She was treated with hyperbaric oxygen, intravenous thiosulfate, and discontinuation of warfarin, with slow resolution of the ulceration.
CARDIOVASCULAR ABNORMALITIES
Cardiovascular disease is the leading cause of morbidity and mortality in patients at every stage of CKD. The incremental risk of cardiovascular disease in those with CKD compared to the age- and sex-matched general population ranges from 10- to 200-fold, depending on the stage of CKD. Between 30 and 45% of patients reaching stage 5 CKD already have advanced cardiovascular complications. As a result, most patients with CKD succumb to cardiovascular disease (Fig. 335-6) before ever reaching stage 5 CKD. Thus, the focus of patient care in earlier CKD stages should be directed to prevention of cardiovascular complications.
FIGURE 335-6 U.S. Renal Data System showing increased likelihood of dying rather than starting dialysis or reaching stage 5 chronic kidney disease (CKD). ![]() , Death;
, Death; ![]() , ESRD;
, ESRD; ![]() , event-free. DM; diabetes mellitus. (Adapted from RN Foley et al: J Am Soc Nephrol 16:489-495, 2005.)
, event-free. DM; diabetes mellitus. (Adapted from RN Foley et al: J Am Soc Nephrol 16:489-495, 2005.)
Ischemic Vascular Disease The presence of any stage of CKD is a major risk factor for ischemic cardiovascular disease, including occlusive coronary, cerebrovascular, and peripheral vascular disease. The increased prevalence of vascular disease in CKD patients derives from both traditional (“classic”) and nontraditional (CKD-related) risk factors. Traditional risk factors include hypertension, hypervolemia, dyslipidemia, sympathetic overactivity, and hyperhomocysteinemia. The CKD-related risk factors comprise anemia, hyperphosphatemia, hyperparathyroidism, increased FGF-23, sleep apnea, and generalized inflammation. The inflammatory state associated with a reduction in kidney function is reflected in increased circulating acute-phase reactants, such as inflammatory cytokines and C-reactive protein, with a corresponding fall in the “negative acute-phase reactants,” such as serum albumin and fetuin. The inflammatory state appears to accelerate vascular occlusive disease, and low levels of fetuin may permit more rapid vascular calcification, especially in the face of hyperphosphatemia. Other abnormalities seen in CKD may augment myocardial ischemia, including left ventricular hypertrophy and microvascular disease. In addition, hemodialysis, with its attendant episodes of hypotension and hypovolemia, may further aggravate coronary ischemia and repeatedly stun the myocardium. Interestingly, however, the largest increment in cardiovascular mortality rate in dialysis patients is not necessarily directly associated with documented acute myocardial infarction but, instead, presents with congestive heart failure and all of its manifestations and sudden death.
Cardiac troponin levels are frequently elevated in CKD without evidence of acute ischemia. The elevation complicates the diagnosis of acute myocardial infarction in this population. Serial measurements may be needed, and if the level is unchanged, it is possible that there is no acute myocardial ischemia. Therefore, the trend in levels over the hours after presentation may be more informative than a single, elevated level. Interestingly, consistently elevated levels are an independent prognostic factor for adverse cardiovascular events in this population.
Heart Failure Abnormal cardiac function secondary to myocardial ischemia, left ventricular hypertrophy, and frank cardiomyopathy, in combination with the salt and water retention that can be seen with CKD, often results in heart failure or even pulmonary edema. Heart failure can be a consequence of diastolic or systolic dysfunction, or both. A form of “low-pressure” pulmonary edema can also occur in advanced CKD, manifesting as shortness of breath and a “bat wing” distribution of alveolar edema fluid on the chest x-ray. This finding can occur even in the absence of ECFV overload and is associated with normal or mildly elevated pulmonary capillary wedge pressure. This process has been ascribed to increased permeability of alveolar capillary membranes as a manifestation of the uremic state, and it responds to dialysis. Other CKD-related risk factors, including anemia and sleep apnea, may contribute to the risk of heart failure.
Hypertension and Left Ventricular Hypertrophy Hypertension is one of the most common complications of CKD. It usually develops early during the course of CKD and is associated with adverse outcomes, including the development of ventricular hypertrophy and a more rapid loss of renal function. Many studies have shown a relationship between the level of blood pressure and the rate of progression of diabetic and nondiabetic kidney disease. Left ventricular hypertrophy and dilated cardiomyopathy are among the strongest risk factors for cardiovascular morbidity and mortality in patients with CKD and are thought to be related primarily, but not exclusively, to prolonged hypertension and ECFV overload. In addition, anemia and the placement of an arteriovenous fistula for hemodialysis can generate a high cardiac output state and consequent heart failure.
The absence of hypertension may signify poor left ventricular function. Indeed, in epidemiologic studies of dialysis patients, low blood pressure actually carries a worse prognosis than does high blood pressure. This mechanism, in part, accounts for the “reverse causation” seen in dialysis patients, wherein the presence of traditional risk factors, such as hypertension, hyperlipidemia, and obesity, appear to portend a better prognosis. Importantly, these observations derive from cross-sectional studies of late-stage CKD patients and should not be interpreted to discourage appropriate management of these risk factors in CKD patients, especially at early stages. In contrast to the general population, it is possible that in late-stage CKD, low blood pressure, reduced body mass index, and hypolipidemia indicate the presence of an advanced malnutrition-inflammation state, with poor prognosis.
The use of exogenous erythropoiesis-stimulating agents can increase blood pressure and the requirement for antihypertensive drugs. Chronic ECFV overload is also a contributor to hypertension, and improvement in blood pressure can often be seen with the use of dietary sodium restriction, diuretics, and fluid removal with dialysis. Nevertheless, because of activation of the RAS and other disturbances in the balance of vasoconstrictors and vasodilators, some patients remain hypertensive despite careful attention to ECFV status.
Pericardial Disease Chest pain with respiratory accentuation, accompanied by a friction rub, is diagnostic of pericarditis. Classic electrocardiographic abnormalities include PR-interval depression and diffuse ST-segment elevation. Pericarditis can be accompanied by pericardial effusion that is seen on echocardiography and can rarely lead to tamponade. However, the pericardial effusion can be asymptomatic, and pericarditis can be seen without significant effusion.
Pericarditis is observed in advanced uremia, and with the advent of timely initiation of dialysis, is not as common as it once was. It is now more often observed in underdialyzed, nonadherent patients than in those starting dialysis.
HEMATOLOGIC ABNORMALITIES
Anemia A normocytic, normochromic anemia is observed as early as stage 3 CKD and is almost universal by stage 4. The primary cause in patients with CKD is insufficient production of erythropoietin (EPO) by the diseased kidneys. Additional factors are reviewed in Table 335-3.
|
CAUSES OF ANEMIA IN CKD |
The anemia of CKD is associated with a number of adverse pathophysiologic consequences, including decreased tissue oxygen delivery and utilization, increased cardiac output, ventricular dilation, and ventricular hypertrophy. Clinical manifestations include fatigue and diminished exercise tolerance, angina, heart failure, decreased cognition and mental acuity, and impaired host defense against infection. In addition, anemia may play a role in growth restriction in children with CKD. Although many studies in CKD patients have found that anemia and resistance to exogenous erythropoietic-stimulating agents (ESA) are associated with a poor prognosis, the relative contribution to a poor outcome of the low hematocrit itself, versus inflammation as a cause of the anemia and ESA resistance, remains unclear.
Abnormal Hemostasis Patients with later stages of CKD may have a prolonged bleeding time, decreased activity of platelet factor III, abnormal platelet aggregation and adhesiveness, and impaired prothrombin consumption. Clinical manifestations include an increased tendency to bleeding and bruising, prolonged bleeding from surgical incisions, menorrhagia, and GI bleeding. Interestingly, CKD patients also have a greater susceptibility to thromboembolism, especially if they have renal disease that includes nephrotic-range proteinuria. The latter condition results in hypoalbuminemia and renal loss of anticoagulant factors, which can lead to a thrombophilic state.
NEUROMUSCULAR ABNORMALITIES
Central nervous system (CNS), peripheral, and autonomic neuropathy as well as abnormalities in muscle structure and function are all well-recognized complications of CKD. Subtle clinical manifestations of uremic neuromuscular disease usually become evident at stage 3 CKD. Early manifestations of CNS complications include mild disturbances in memory and concentration and sleep disturbance. Neuromuscular irritability, including hiccups, cramps, and twitching, becomes evident at later stages. In advanced untreated kidney failure, asterixis, myoclonus, seizures, and coma can be seen.
Peripheral neuropathy usually becomes clinically evident after the patient reaches stage 4 CKD, although electrophysiologic and histologic evidence occurs earlier. Initially, sensory nerves are involved more than motor, lower extremities more than upper, and distal parts of the extremities more than proximal. The “restless leg syndrome” is characterized by ill-defined sensations of sometimes debilitating discomfort in the legs and feet relieved by frequent leg movement. If dialysis is not instituted soon after onset of sensory abnormalities, motor involvement follows, including muscle weakness. Evidence of peripheral neuropathy without another cause (e.g., diabetes mellitus) is an indication for starting renal replacement therapy. Many of the complications described above will resolve with dialysis, although subtle nonspecific abnormalities may persist.
GASTROINTESTINAL AND NUTRITIONAL ABNORMALITIES
Uremic fetor, a urine-like odor on the breath, derives from the breakdown of urea to ammonia in saliva and is often associated with an unpleasant metallic taste (dysgeusia). Gastritis, peptic disease, and mucosal ulcerations at any level of the GI tract occur in uremic patients and can lead to abdominal pain, nausea, vomiting, and GI bleeding. These patients are also prone to constipation, which can be worsened by the administration of calcium and iron supplements. The retention of uremic toxins also leads to anorexia, nausea, and vomiting.
Protein restriction may be useful to decrease nausea and vomiting; however, it may put the patient at risk for malnutrition and should be carried out, if possible, in consultation with a registered dietitian specializing in the management of CKD patients. Protein-energy malnutrition, a consequence of low protein and caloric intake, is common in advanced CKD and is often an indication for initiation of renal replacement therapy. Metabolic acidosis and the activation of inflammatory cytokines can promote protein catabolism. Assessment for protein-energy malnutrition should begin at stage 3 CKD. A number of indices are useful in this assessment and include dietary history, including food diary and subjective global assessment; edema-free body weight; and measurement of urinary protein nitrogen appearance. Dual-energy x-ray absorptiometry is now widely used to estimate lean body mass versus ECFV. Adjunctive tools include clinical signs, such as skinfold thickness, mid-arm muscle circumference, and additional laboratory tests such as serum pre-albumin and cholesterol levels. Nutritional guidelines for patients with CKD are summarized in the “Treatment” section.
ENDOCRINE-METABOLIC DISTURBANCES
Glucose metabolism is impaired in CKD, as evidenced by a slowing of the rate at which blood glucose levels decline after a glucose load. However, fasting blood glucose is usually normal or only slightly elevated, and the mild glucose intolerance does not require specific therapy. Because the kidney contributes to insulin removal from the circulation, plasma levels of insulin are slightly to moderately elevated in most uremic patients, both in the fasting and postprandial states. Because of this diminished renal degradation of insulin, patients on insulin therapy may need progressive reduction in dose as their renal function worsens. Many hypoglycemic agents, including the gliptins, require dose reduction in renal failure, and some, such as metformin, are contraindicated when the GFR is less than half of normal.
In women with CKD, estrogen levels are low, and menstrual abnormalities, infertility, and inability to carry pregnancies to term are common. When the GFR has declined to ~40 mL/min, pregnancy is associated with a high rate of spontaneous abortion, with only ~20% of pregnancies leading to live births, and pregnancy may hasten the progression of the kidney disease itself. Women with CKD who are contemplating pregnancy should consult first with a nephrologist in conjunction with an obstetrician specializing in high-risk pregnancy. Men with CKD have reduced plasma testosterone levels, and sexual dysfunction and oligospermia may supervene. Sexual maturation may be delayed or impaired in adolescent children with CKD, even among those treated with dialysis. Many of these abnormalities improve or reverse with intensive dialysis or with successful renal transplantation.
DERMATOLOGIC ABNORMALITIES
Abnormalities of the skin are prevalent in progressive CKD. Pruritus is quite common and one of the most vexing manifestations of the uremic state. In advanced CKD, even on dialysis, patients may become more pigmented, and this is felt to reflect the deposition of retained pigmented metabolites, or urochromes. Although many of the cutaneous abnormalities improve with dialysis, pruritus is often tenacious. The first lines of management are to rule out unrelated skin disorders, such as scabies, and to treat hyperphosphatemia, which can cause itch. Local moisturizers, mild topical glucocorticoids, oral antihistamines, and ultraviolet radiation have been reported to be helpful.
A skin condition unique to CKD patients called nephrogenic fibrosing dermopathy consists of progressive subcutaneous induration, especially on the arms and legs. The condition is similar to scleromyxedema and is seen very rarely in patients with CKD who have been exposed to the magnetic resonance contrast agent gadolinium. Current recommendations are that patients with CKD stage 3 (GFR 30–59 mL/min) should minimize exposure to gadolinium, and those with CKD stages 4–5 (GFR <30 mL/min) should avoid the use of gadolinium agents unless it is medically necessary. Concomitant liver disease appears to be a risk factor. However, no patient should be denied an imaging investigation that is critical to management, and under such circumstances, rapid removal of gadolinium by hemodialysis (even in patients not yet receiving renal replacement therapy) shortly after the procedure may mitigate this sometimes devastating complication.
EVALUATION AND MANAGEMENT OF PATIENTS WITH CKD
INITIAL APPROACH
History and Physical Examination Symptoms and overt signs of kidney disease are often subtle or absent until renal failure supervenes. Thus, the diagnosis of kidney disease often surprises patients and may be a cause of skepticism and denial. Particular aspects of the history that are germane to renal disease include a history of hypertension (which can cause CKD or more commonly be a consequence of CKD), diabetes mellitus, abnormal urinalyses, and problems with pregnancy such as preeclampsia or early pregnancy loss. A careful drug history should be elicited: patients may not volunteer use of analgesics, for example. Other drugs to consider include nonsteroidal anti-inflammatory agents, cyclooxygenase-2 (COX-2) inhibitors, antimicrobials, chemotherapeutic agents, antiretroviral agents, proton pump inhibitors, phosphate-containing bowel cathartics, and lithium. In evaluating the uremic syndrome, questions about appetite, weight loss, nausea, hiccups, peripheral edema, muscle cramps, pruritus, and restless legs are especially helpful. A careful family history of kidney disease, together with assessment of manifestations in other organ systems such as auditory, visual, and integumentary, may lead to the diagnosis of a heritable form of CKD (e.g., Alport or Fabry disease, cystinosis) or shared environmental exposure to nephrotoxic agents (e.g., heavy metals, aristolochic acid). It should be noted that clustering of CKD, sometimes of different etiologies, is often observed within families.
The physical examination should focus on blood pressure and target organ damage from hypertension. Thus, funduscopy and precordial examination (left ventricular heave, a fourth heart sound) should be carried out. Funduscopy is important in the diabetic patient, because it may show evidence of diabetic retinopathy, which is associated with nephropathy. Other physical examination manifestations of CKD include edema and sensory polyneuropathy. The finding of asterixis or a pericardial friction rub not attributable to other causes usually signifies the presence of the uremic syndrome.
Laboratory Investigation Laboratory studies should focus on a search for clues to an underlying causative or aggravating disease process and on the degree of renal damage and its consequences. Serum and urine protein electrophoresis, looking for multiple myeloma, should be obtained in all patients >35 years with unexplained CKD, especially if there is associated anemia and elevated, or even inappropriately normal, serum calcium concentration in the face of renal insufficiency. In the presence of glomerulonephritis, autoimmune diseases such as lupus and underlying infectious etiologies such as hepatitis B and C and HIV should be tested. Serial measurements of renal function should be obtained to determine the pace of renal deterioration and ensure that the disease is truly chronic rather than acute or subacute and hence potentially reversible. Serum concentrations of calcium, phosphorus, vitamin D, and PTH should be measured to evaluate metabolic bone disease. Hemoglobin concentration, iron, vitamin B12, and folate should also be evaluated. A 24-h urine collection may be helpful, because protein excretion >300 mg may be an indication for therapy with ACE inhibitors or ARBs.
Imaging Studies The most useful imaging study is a renal ultrasound, which can verify the presence of two kidneys, determine if they are symmetric, provide an estimate of kidney size, and rule out renal masses and evidence of obstruction. Because it takes time for kidneys to shrink as a result of chronic disease, the finding of bilaterally small kidneys supports the diagnosis of CKD of long-standing duration, with an irreversible component of scarring. If the kidney size is normal, it is possible that the renal disease is acute or subacute. The exceptions are diabetic nephropathy (where kidney size is increased at the onset of diabetic nephropathy before CKD supervenes), amyloidosis, and HIV nephropathy, where kidney size may be normal in the face of CKD. Polycystic kidney disease that has reached some degree of renal failure will almost always present with enlarged kidneys with multiple cysts (Chap. 339). A discrepancy >1 cm in kidney length suggests either a unilateral developmental abnormality or disease process or renovascular disease with arterial insufficiency affecting one kidney more than the other. The diagnosis of renovascular disease can be undertaken with different techniques, including Doppler sonography, nuclear medicine studies, or CT or magnetic resonance imaging (MRI) studies. If there is a suspicion of reflux nephropathy (recurrent childhood urinary tract infection, asymmetric renal size with scars on the renal poles), a voiding cystogram may be indicated. However, in most cases, by the time the patient has CKD, the reflux has resolved, and even if still present, repair does not improve renal function. Radiographic contrast imaging studies are not particularly helpful in the investigation of CKD. Intravenous or intraarterial dye should be avoided where possible in the CKD patient, especially with diabetic nephropathy, because of the risk of radiographic contrast dye–induced renal failure. When unavoidable, appropriate precautionary measures include avoidance of hypovolemia at the time of contrast exposure, minimization of the dye load, and choice of radiographic contrast preparations with the least nephrotoxic potential. Additional measures thought to attenuate contrast-induced worsening of renal function include judicious administration of sodium bicarbonate–containing solutions and N -acetylcysteine.
Kidney Biopsy In the patient with bilaterally small kidneys, renal biopsy is not advised because (1) it is technically difficult and has a greater likelihood of causing bleeding and other adverse consequences, (2) there is usually so much scarring that the underlying disease may not be apparent, and (3) the window of opportunity to render disease-specific therapy has passed. Other contraindications to renal biopsy include uncontrolled hypertension, active urinary tract infection, bleeding diathesis (including ongoing anticoagulation), and severe obesity. Ultrasound-guided percutaneous biopsy is the favored approach, but a surgical or laparoscopic approach can be considered, especially in the patient with a single kidney where direct visualization and control of bleeding are crucial. In the CKD patient in whom a kidney biopsy is indicated (e.g., suspicion of a concomitant or superimposed active process such as interstitial nephritis or in the face of accelerated loss of GFR), the bleeding time should be measured, and if increased, desmopressin should be administered immediately prior to the procedure.
A brief run of hemodialysis (without heparin) may also be considered prior to renal biopsy to normalize the bleeding time.
ESTABLISHING THE DIAGNOSIS AND ETIOLOGY OF CKD
The most important initial diagnostic step is to distinguish newly diagnosed CKD from acute or subacute renal failure, because the latter two conditions may respond to targeted therapy. Previous measurements of serum creatinine concentration are particularly helpful in this regard. Normal values from recent months or even years suggest that the current extent of renal dysfunction could be more acute, and hence reversible, than might otherwise be appreciated. In contrast, elevated serum creatinine concentration in the past suggests that the renal disease represents a chronic process. Even if there is evidence of chronicity, there is the possibility of a superimposed acute process (e.g., ECFV depletion, urinary infection or obstruction, or nephrotoxin exposure) supervening on the chronic condition. If the history suggests multiple systemic manifestations of recent onset (e.g., fever, polyarthritis, rash), it should be assumed that renal insufficiency is part of an acute systemic illness.
Although renal biopsy can usually be performed in early CKD (stages 1–3), it is not always indicated. For example, in a patient with a history of type 1 diabetes mellitus for 15–20 years with retinopathy, nephrotic-range proteinuria, and absence of hematuria, the diagnosis of diabetic nephropathy is very likely and biopsy is usually not necessary. However, if there were some other finding not typical of diabetic nephropathy, such as hematuria or white blood cell casts, or absence of diabetic retinopathy, some other disease may be present and a biopsy may be indicated.
In the absence of a clinical diagnosis, renal biopsy may be the only recourse to establish an etiology in early-stage CKD. However, as noted above, once the CKD is advanced and the kidneys are small and scarred, there is little utility and significant risk in attempting to arrive at a specific diagnosis. Genetic testing is increasingly entering the repertoire of diagnostic tests, since the patterns of injury and kidney morphologic abnormalities often reflect overlapping causal mechanisms, whose origins can sometimes be attributed to a genetic predisposition or cause.
IMPLICATIONS FOR GLOBAL HEALTH
In contrast to the natural decline and successful eradication of many devastating infectious diseases, there is rapid growth in the prevalence of metabolic and vascular disease in developing countries. Diabetes mellitus is becoming increasingly prevalent in these countries, perhaps due in part to change in dietary habits, diminished physical activity, and weight gain. Therefore, it follows that there will be a proportionate increase in vascular and renal disease. Health care agencies must plan for improved screening for early detection, prevention, and treatment plans in these nations and must start considering options for improved availability of renal replacement therapies.