[level-membership-for-anesthesiology-category]
Chapter 31 Acute severe asthma
Acute severe asthma is a medical emergency associated with significant morbidity and mortality. The majority of adverse outcomes are attributed to underestimation of severity with delayed and/or inadequate treatment1–3 and are potentially preventable.
The worldwide prevalence of asthma varies widely (2–37% in children),4 but it is now clear that the overall prevalence has increased worldwide,5,6 with life-threatening episodes affecting an estimated 0.5% of asthmatics per year.7 Australia, New Zealand and the UK have amongst the highest incidences.7 In Australia, 9% have asthma as a long-term condition8 and up to 40% of children have asthma symptoms at some time.5
Although the prevalence of asthma is increasing, many countries have achieved reductions in hospital presentations and admissions,9,10 reduced intensive care admissions and reduced overall asthma mortality.9,10 Improved community management of asthma,1,2 more widespread use of inhaled corticosteroids3 and other preventive measures have been given the credit for these improvements.
Despite reducing admissions, significant and potentially preventable mortality continues to occur in those patients who do require intensive care or mechanical ventilation.11,12
CLINICAL DEFINITION
Asthma has been defined as a lung disease with the following characteristics:13
Exacerbations of asthma are characterised by increasing dyspnoea, cough, wheeze, chest tightness and decreased expiratory airflow. Status asthmaticus has had varying definitions. However, for practical purposes, any patient not responding to initial doses of nebulised bronchodilators should be considered to have status asthmaticus.14
AETIOLOGY
The pathogenesis of asthma is complex, with both genetic and environmental influences. The increase in asthma prevalence has been attributed to the ‘hygiene hypothesis’15 which suggests that reduced exposure to childhood infections as a result of antibiotics and hygienic lifestyle promotes an imbalance in T-cell phenotype leading to inflammatory cytokine overproduction. Immunoglobulin (Ig) E-dependent mechanisms appear to be particularly important in generating the characteristic state of airway inflammation and bronchial hyperreactivity with the allergens in the local environment dictating the specificity of the antibody response.16 Triggers of acute asthma can be non-specific (cold air, exercise, atmospheric pollutants), specific allergens (housemite, pollen, animal danders), modifiers of airway control (aspirin, β-blockers) or stress and emotion. No precipitant can be identified in over 30% of patients.
PATHOPHYSIOLOGY
The postmortem airway pathology of patients who die from acute asthma includes bronchial wall thickening from oedema and inflammatory cell infiltrate, hypertrophy and hyperplasia of bronchial smooth muscle and submucosal glands, deposition of collagen beneath the epithelial basement membrane and prominent intraluminal secretions. These secretions may narrow or occlude the small airways and postmortem studies frequently report extensive plugging and atelectasis.17,18 When interpreting the latter findings it is important to remember that during life in very severe asthma, the lungs are hyperinflated close to total lung capacity (TLC) by maximal inspiratory effort and to beyond TLC during mechanical ventilation. At this time radiological atelectasis is rare, suggesting that most airways are communicating at these high lung volumes, even if airways are very narrowed. Only at death does the prolonged apnoea allow lung deflation, widespread airway closure and alveolar gas absorption to give the postmortem appearance of extensive complete occlusion that was not present to the same degree during life.
In some deaths bronchial mucus is absent; in these cases airway obstruction may be mainly due to intense smooth bronchoconstriction.
This observation may account for two patterns of progression of asthma:
CLINICAL FEATURES AND ASSESSMENT OF SEVERITY
Triage and assessment of severity of the acute asthma attack are crucial. Underestimation or non-measurement of asthma severity is associated with increased mortality.26,27 Assessment has two key features: assessment of initial severity and ongoing assessment of response to treatment.
HISTORY
Any history of prior intubation and mechanical ventilation for asthma is a predictor for life-threatening asthma.26,27 A history of poor asthma control and multiple recent medical presentations for asthma are recognised risk factors. Other risk factors include a poor response to prior treatments and poor psychosocial circumstances.
PHYSICAL EXAMINATION
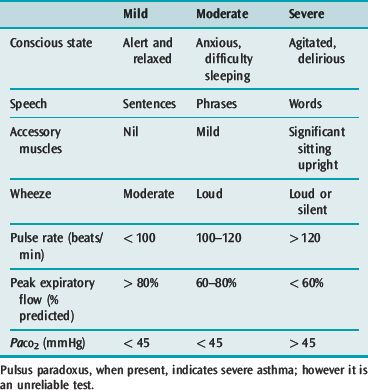
The general appearance and level of distress can be an important indicator of severity (Table 31.1). Use of accessory muscles, suprasternal retraction, markedly diminished breath sounds or a silent chest, central cyanosis, inability to speak (sentences, phrases, single words), a disturbance in the level of consciousness, upright posture and diaphoresis all suggest a severe attack.28 A respiratory rate > 30 breaths/min, pulse rate > 120 beats/min and pulsus paradoxus of > 15 mmHg are associated with severe asthma, though their absence does not preclude life-threatening asthma.
VENTILATORY FUNCTION TESTS
The patient may not be able to perform these due to breathlessness. However, forced expiratory volume in 1 second (FEV1) and peak expiratory flow rate (PEFR) are useful indicators of severity and response to treatment when done serially. An FEV1 < 1.0 litre or PEFR < 100 l/min indicates very severe asthma at significant risk of requiring mechanical ventilation. In some patients forced expiration may worsen symptoms; these measurements should cease if this occurs.29
PULSE OXIMETRY
Pulse oximetry (SpO2) is usually readily available and provides a rapid assessment of oxygenation. It is also very valuable in regulation oxygen therapy, both for avoiding hypoxia (SpO2 > 90%)30 and avoiding potential side-effects of hyperoxia (SpO2 < 95%).31 Of course, pulse oximetry cannot assess arterial PCO2 or acid–base state or lactate and hence does not replace the need for blood gases
ARTERIAL BLOOD GASES
Arterial hypoxaemia is almost invariably present in a patient with severe asthma breathing room air, though it usually responds well to low-level oxygen supplementation (28–35%).30 Blood gases should not delay initiation of treatment, and are not required in mild asthma or moderate asthma that is responding well to treatment. They are very important in severe asthma, or moderate asthma with inadequate treatment response. The arterial PaCO2 is an important measure of severity and, if hypercapnia is present, an important guide to treatment response.
Ventilation is initially increased in an acute attack, leading to hypocapnia and a respiratory alkalosis.30 As the asthmatic attack worsens, the work of breathing, V/Q mismatch and adverse cardiopulmonary interactions all increase, and the minute ventilation required to maintain the same alveolar ventilation and PaCO2 increases. Eventually the patient is incapable of meeting this demand and the PaCO2 rises. The presence of hypercapnic acidosis is associated with an FEV1 of < 20% predicted and reliably indicates that asthma is severe. A metabolic acidosis may also be present, and this is most commonly due to lactic acidosis32 associated with intravenous or continuous nebulised β-agonists.33,34
CHEST X-RAY
Although not generally helpful in assessing severity,35 a chest X-ray should be performed when asthma is severe or refractory to treatment, when barotrauma or lower respiratory tract infection is suspected or when the diagnosis is in doubt. It is not required in milder attacks that respond well to treatment.
ASSESSMENT OF TREATMENT RESPONSE
Repeated evaluation of the patient’s response to treatment is a valuable tool in assessing severity of acute asthma. The response to the first 2 hours of treatment is an important predictor of outcome.36 Adverse events are associated with underestimation of severity, inadequate observation after initial assessment and under treatment. Ongoing evaluation of response to therapy is critical. This evaluation should include repeated assessments of:
MANAGEMENT
ESTABLISHED TREATMENTS
Initial therapy of acute severe asthma should include the following.
OXYGEN
Hypoxaemia contributes to life-threatening events that complicate acute severe asthma.37 Humidified supplemental oxygen should be titrated to achieve a SpO2 > 90%. The risk of oxygen-induced increasing hypercapnia with coexisting chronic obstructive pulmonary disease or pre-existing chronic hypercapnia is a well-known reason to maintain a lower SpO2 (e.g. 90–92%). This phenomenon was not believed to be relevant to the majority of patients with acute asthma; however, there is now emerging evidence that hyperoxia may be harmful to a more widespread group38 by releasing pulmonary hypoxic vasoconstriction, worsening V/Q matching and increasing hypercapnia. A recent randomised controlled trial31 showed a decrease in PaCO2 in a group receiving 28% O2 and an increase in PaCO2 in a group receiving 100% O2.
β-AGONISTS
Short-acting β-agonists remain the first-line bronchodilator therapy of choice.6,39–41 Agents include salbutamol (albuterol), terbutaline, isoprenaline and epinephrine. Salbutamol is generally the agent of first choice as it has relative β2-selectivity, with decreased β1-mediated cardiac toxicity. Long-acting β-agonists such as salmeterol have no role in status asthmaticus due to slow onset of action and association with fatalities in this setting.14 Eformotarol is a combined long- and short-acting β-agonist which could theoretically be used for acute asthma; however it is in dry powder form and unlikely to be helpful in intubated and mechanically ventilated patients. β-agonists cause bronchodilatation by stimulation of β2-receptors on airway smooth muscle and may reduce bronchial mucosal oedema.42
β-agonists are best given by metered-dose inhaler (MDI) and a spacer device. There are data to suggest that, in non-intubated patients, MDIs combined with a spacer device are more effective than nebulisers and are cheaper to use.43,44 In intubated patients both nebulisers and MDIs have been used effectively.45 The dose per MDI inhalation is 100 μg and this can be given, in severe asthma, in repeated doses at 1–5-minute intervals.
Alternatively, β-agonists can be given by nebuliser in high and repeated doses.46 The typical adult dose of salbutamol is 5–10 mg (in 2.5–5.0 ml diluent volume) every 2–4 hours but more frequent doses with a higher total dose are often required in severe asthma. It should be noted that less than 10% of the nebulised drug reaches the lung even under ideal conditions.47 Continuous nebulisation appears to be superior to intermittent doses and is commonly used at the beginning of treatment in severe asthma.14,48 The nebuliser should be driven by oxygen with the flow at 10–12 l/min and a reservoir volume of 2–4 ml so as to produce particles in the desired 1–3 μm range.49 The total dose should be modulated by response to treatment and the level of toxic side-effects.
Two-thirds of patients presenting acutely will respond well to inhaled β-agonists,50 irrespective of the method of administration. The remaining one-third are refractory even to high doses and usually require longer periods of intense treatment, including multiple other agents.
Intravenous β-agonists remain controversial. There is no clear evidence of benefit51 and significant side-effects. Despite this, intravenous β-agonists have a theoretical benefit of additional access to lung units with severe airflow obstruction and poor nebulised drug delivery, and some studies have demonstrated improved response when intravenous β-agonist is used.52 Intravenous β-agonists continue to be considered if the patient is not responding to continuous nebulisation.53 The typical dose is 5–20 μg/min but doses > 10 μg/min should be used with caution because of side-effects, which should be monitored closely. Salbutamol 100–300 μg may also be given intravenously to non-intubated patients in extremis or delivered down an endotracheal tube if there is not time to gain intravenous access.
Side-effects of β-agonists include tachycardia, arrhythmias, hypertension, hypotension, tremor, hypokalaemia, worsening of ventilation–perfusion mismatch and hyperglycaemia,54 but the most common side-effect of parenteral β-agonist – also occasionally seen with continuous nebulised β-agonists – is lactic acidosis. This occurs in over 70% of patients, has an onset within 2–4 hours of commencing an infusion or following an intravenous statim dose, levels may reach 4–12 mmol/l and may significantly add to respiratory acidosis and respiratory distress.34,55,56 Parenteral infusions should be initially limited to 10 μg/min and statim doses should not exceed 250 μg. Serum bicarbonate and lactate should be regularly monitored. If lactic acidosis becomes significant, the salbutamol infusion should be reduced or ceased. Lactic acidosis will generally resolve within 4–6 hours of infusion cessation and is seldom a problem with infusions in place for more than 24 hours.
Long-term high-dose β-agonist use has been associated with increased mortality,57 but whether high-dose β-agonists are a marker of disease severity, an indicator of suboptimal inhaled steroids or a direct cause of death is unclear. These concerns do not apply in the treatment of the acute asthma attack.
ANTICHOLINERGICS
Anticholinergics cause bronchodilatation by decreasing parasympathetic-mediated cholinergic bronchomotor tone.58 Ipratropium bromide is the most commonly used anticholinergic for asthma and is a quaternary derivative of atropine. A number of studies and meta-analyses now suggest clear additional benefit and few side-effects when ipratropium bromide is added to the β-agonist regimen59,60 and ipratropium is now considered accepted first-line therapy for acute severe asthma in conjunction with β-agonist therapy. Preservative-induced bronchoconstriction has been reported in a few patients and can be prevented by using preservative-free solutions.61 The bronchodilatation effect of ipratropium bromide appeared to be maximal with a dose of 250 μg when studied in children between 9 and 17 years of age. The optimal dose is not known in adults; a reasonable regimen would be to add 500 μg ipratropium bromide to the salbutamol nebuliser every 2–6 hours; however, initial dose intervals as low as 10–20 minutes have been recommended.62
CORTICOSTEROIDS
The role of corticosteroids in the acute asthma attack has been well established. Systemic steroids should be considered in all but mild exacerbations of asthma.41 Their benefits include increased β-responsiveness of airway smooth muscle, decreased inflammatory cell response and decreased mucus secretion. Early treatment with corticosteroids has been shown to decrease the likelihood of hospitalisation and decrease the mortality rate from acute asthma. Systematic reviews41,63 suggest that effects commence within 6–12 hours, that oral administration is as effective as intravenous and that there is little evidence of benefit for initial daily doses exceeding 800 mg/day hydrocortisone (160 mg/day methylprednisolone) given in four divided doses.
Inhaled steroids have established long-term benefit and are believed to be a major factor in asthma mortality reduction.1–3 There is now emerging evidence that inhaled steroids may also have a role during an acute attack64,65 and it appears reasonable to use them routinely from day 1 as they may also enable more rapid dose reduction of parenteral steroids, potentially reducing side-effects.
Side-effects of corticosteroids include hyperglycaemia, hypokalaemia, hypertension, acute psychosis and myopathy,66,67 though they are usually well tolerated acutely. The immunosuppressive effects can increase the risk of infections, including Legionella, Pneumocystis carinii and varicella,68,69 especially when the patient is on long-term corticosteroids. Allergic reactions including anaphylaxis have been reported with the use of most corticosteroid preparations.
AMINOPHYLLINE
There have been conflicting reports regarding the efficacy of aminophylline in acute asthma ranging from no benefit70 to improved lung function and improved outcome.71 However it is accepted that aminophylline is an inferior bronchodilator, with a narrow therapeutic range and frequent side-effects,72 including headache, nausea, vomiting and restlessness; cardiac arrhythmias and convulsions can occur at serum levels above 200 μmol/l (40 mg/l).
As a result, aminophylline is not a first-line treatment.40,41,53 Aminophylline may be given to patients with acute asthma who are not showing a favourable response to full treatment with first-line agents. Careful administration and monitoring are required with an initial loading dose of 3 mg/kg (maximum 6 mg/kg, omitted if the patient is already taking oral theophylline) and an infusion of 0.5 mg/kg per hour. This should be reduced in patients with cirrhosis, cardiac failure or chronic obstructive pulmonary disease and in patients taking cimetidine, erythromycin or antiviral vaccines. Drug levels should be taken after a loading dose (if given), and then 24 hours later, aiming for a level of 30–80 μmol/l (5–12 mg/l). Levels should be repeated daily thereafter until stability has been achieved. The duration should be based on the response to treatment.
NON-ESTABLISHED TREATMENTS
EPINEPHRINE
Epinephrine has some theoretical advantages over pure β2-agonists in that its additional α-agonist actions of vasoconstriction and mucosal shrinkage may improve airway calibre. However, in practice, nebulised or subcutaneous epinephrine has not been shown to confer any advantage over nebulised β2-agonists and is not recommended because of its cardiac side-effects.62 Epinephrine may be tried in the patient who is failing to respond to conventional treatment. The nebulised dose is 2–4 mg in 2–4 ml (1% solution, 0.05 ml/kg) 1–4-hourly. The subcutaneous dose is 0.2–0.5 mg (0.2–0.5 ml of 1:1000 epinephrine) repeated if necessary 2–3 times at 30-minute intervals. Epinephrine by infusion may avert mechanical ventilation in very severe cases but should be used with caution with electrocardiogram monitoring and preferably with central venous access. An initial intravenous dose of 0.2–1.0 mg (2–10 ml of 1:10 000 epinephrine) is given slowly over 3–5 minutes. This may be followed by a continuous infusion of 1–20 μg/min, which is weaned when the acute attack subsides.73
MAGNESIUM SULPHATE
Magnesium sulphate is postulated to block calcium channels, and possibly acetylcholine release at the neuromuscular junction, leading to smooth-muscle relaxation and bronchodilatation. It appeared to be well tolerated in early studies, and a number of randomised, double-blind, prospective trials were performed adding intravenous or nebulised magnesium sulphate to conventional therapy in adults with acute asthma. Some showed benefit whereas others showed no benefit.
Three meta-analyses74–76did not support routine use of magnesium and it is not recommended for acute asthma. If given, recommended doses are 5–10 mmol (1.25–2.5 g, 2.5–5.0 ml of 50% solution) given slowly over 20 minutes but doses up to 40–80 mmol (10–20 g) have been given.77 Side-effects include hypotension, flushing, sedation, weakness, areflexia, respiratory depression and cardiac arrhythmias seen at higher serum levels (> 5 mmol/dl or 12 mg/dl). Serum concentrations should be measured if repeated or high doses are used.
HELIOX
Inhalation of a helium:oxygen mixture reduces gas density and turbulence with reduced airflow resistance. The most effective gas mixture is 70% helium (30% oxygen) and the minimum concentration likely to provide benefit is 60% helium. Work of breathing is decreased and pulmonary access of inhaled bronchodilators may be improved.78,79 Small case-series and reports have suggested benefit from Heliox, randomised prospective trials have both positive and negative results80,81 and a recent meta-analysis82 shows a trend towards improved lung function, but insufficient evidence to recommend use of Heliox in severe asthma. However, it otherwise appears safe and may be tried in critical asthma to avert intubation or during difficult mechanical ventilation providing the patient can tolerate 30–40% O2.
ANAESTHETIC AGENTS
Ketamine, a dissociative anaesthetic agent, has been used in severe asthma.83 It may cause bronchodilatation by both sympathomimetic potentiation and a direct effect on airway smooth muscle.84 Small case-series have suggested some benefit, although a small randomised controlled trial found no benefit with ketamine in the treatment of acute severe asthma.85 Ketamine may be a useful induction agent for endotracheal intubation (dose 1–2 mg/kg) as it may ameliorate the bronchoconstrictor response to intubation. It has been used as a continuous infusion in the dose range of 0.5–2 mg/kg per hour85,86 to treat refractory asthma. Side-effects include increased bronchial secretions, a hyperdynamic cardiovascular response and hallucinations; the hallucinations can be reduced with concomitant benzodiazepines.
The volatile inhalational agents, including halothane, isoflurane and enflurane, have been used in mechanically ventilated patients with severe asthma. Clinical data are limited to small case-series and side-effects include direct myocardial depression, arrhythmias and hypotension.87 The volatile anaesthetic agents should be used with great care and usually only as a prelude to or during invasive ventilation. An anaesthetic machine or custom-fitted ventilator is required for safe administration.
LEUKOTRIENE ANTAGONISTS
Leukotriene antagonists have shown benefit in chronic asthma88 and there is some evidence of benefit in acute asthma.89,90 However there is insufficient evidence and these benefits were of insufficient magnitude to recommend these agents for acute severe asthma.
BRONCHOALVEOLAR LAVAGE
Bronchoalveolar lavage has been used in severe refractory asthma to clear mucous plugging during mechanical ventilation.91 It can transiently worsen bronchospasm and hypoxaemia and should be used when airflow obstruction has stabilised. It may have a role in ventilated patients with resistant mucus impaction, but is rarely used.
THERAPIES NOT RECOMMENDED
Antibiotics are not indicated92 unless there is evidence of infection. Antihistamines are not effective. Inhaled mucolytics have been shown to have no benefit and may worsen airflow obstruction. Sedation is unsafe in acute asthma. There is a clear association between their use and avoidable deaths.93 Patients with severe asthma should not be sedated unless being intubated and ventilated or in carefully monitored circumstances.
VENTILATION IN ASTHMA
DYNAMIC HYPERINFLATION
In all degrees of airflow obstruction, slow expiratory airflow results in incomplete exhalation of gas during normal expiratory times. Gas is trapped in the lungs by the arrival of the next breath and the lungs are unable to return their normal passive relaxation volume (functional residual capacity, FRC). Incomplete exhalation of each successive breath causes progressive accumulation of trapped gas, called dynamic hyperinflation (DH; Figure 31.1).94 This continues until an equilibrium point is reached, where the exhaled volume increases to match the inspired volume (see Figure 31.1).94 This equilibrium occurs because increasing lung volume increases small-airway calibre and lung elastic recoil pressure, both of which improve expiratory airflow and allow the inspired tidal volume to be exhaled in the expiratory time available. There are three primary determinants of this equilibrium point. Gas trapped at the end of expiration exerts a positive pressure on the alveoli; this pressure is intrinsic positive end-expiratory pressure (PEEPi) or auto-PEEP.94,95 During expiration, sequential closure of the most severely obstructed airways occurs, with only the less obstructed airways remaining in communication with the central airway at the end of tidal expiration.96 As a consequence, measured PEEPi underestimates the true magnitude of PEEPi and is recognised as being insensitive to changes in severity.96
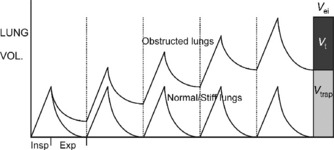
In mild airflow obstruction, this process is adaptive as it allows the desired minute ventilation, which could not be achieved at FRC, to be achieved at a higher lung volume (Figure 31.2: ‘mild AO’). In mild AO, a normal FRC can still be reached with prolonged passive expiratory period but the mildly narrowed airways undergo airway closure at lower lung volumes, reducing the expiratory reserve capacity (ER Cap) and increasing residual volume (RV; see Figure 31.2). This is in contrast to acute lung injury (ALI), where lung collapse reduces all lung volumes (see Figure 31.2: Vt, ER Cap and RV).
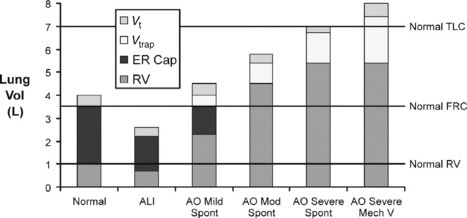
As asthma becomes more severe (see Figure 31.2: ‘mod AO’), the static FRC that would be reached with a long expiratory time is elevated above the normal FRC by airway closure (static hyperinflation), there is no ER Cap, DHI further increases lung volume to a level where work of breathing is increased by lower lung compliance and respiratory muscles become less efficient from shortening and mechanical disadvantage. At this level some degree of dyspnoea is expected.
When asthma is severe enough to risk requiring mechanical ventilation, static hyperinflation may increase FRC by 50%97 and very little DHI is required to reach total lung capacity (see Figure 31.2: ‘severe AO’). In this circumstance, the minute ventilation required to maintain normocapnia would cause hyperinflation beyond normal total lung capacity. During spontaneous ventilation, the patient with severe asthma is unable to exceed total lung capacity (see Figure 31.2), has a much lower maximum minute ventilation capacity and as a result must become hypercapnic even at maximum respiratory effort with no fatigue.14 When mechanical ventilation is commenced, increased tidal volume and rate delivery are easily able to increase minute ventilation and DHI well beyond normal total lung capacity (see Figure 31.2).94 The consequences of this are commonly hypotension (due to increased intrathoracic pressure and decreased venous return) and barotrauma (Table 31.2).22,94,97,98
NON-INVASIVE VENTILATION (NIV)
NIV has been widely used for a variety of respiratory problems.99 In severe asthma there are a number of potential benefits. Externally applied PEEP may help overcome PEEPi due to gas trapping and thus reduce the inspiratory threshold work of breathing. Augmentation of inspiration with NIV may further decrease the work ofinspiration and increase tidal volume and minute ventilation. If tidal volume is increased with a shorter inspiratory time, then increased minute ventilation can occur without a proportional increase in dynamic hyperinflation. Both inspiratory augmentation and PEEP may facilitate airspace opening, thus reducing V/Q mismatch.100,101
The role of NIV is well established in chronic obstructive pulmonary disease where a number of randomised trials have shown benefit.102,103 Although there have been no large randomised trials in acute asthma, there is increasing evidence of benefit from NIV for this indication, with all studies reporting positive outcome.104,105 Non-randomised studies have reported improvements in respiratory acidosis and respiratory rate following the introduction of NIV, and low requirements for invasive ventilation. In a small prospectively randomised study of acute asthma, Soroksky et al.105showed better lung function and decreased hospitalisation rate with NIV.
Contraindications to NIV include cardiac or respiratory arrest, a decreased conscious state, severe upper gastrointestinal bleeding, haemodynamic instability, facial trauma or surgery, inability to protect the airway and clear secretions and high risk of aspiration.100
Complications of NIV include nasal bridge ulceration, mask discomfort, nasal congestion, gastric insufflation, aspiration, hypotension and pneumothorax;101 however hypotension and pneumothorax are uncommon compared with their risk during mechanical ventilation.
INVASIVE VENTILATION
Invasive mechanical ventilation in acute severe asthma may be life-saving, but can be associated with significant morbidity and mortality.97 Institution of invasive ventilation with endotracheal intubation carries the risk of inadvertent pulmonary hyperinflation94,95,97 and potential aggravation of bronchospasm, and a significant part of the morbidity and mortality has been attributed to pulmonary hyperinflation.94,95,97 Despite these risks, the incidence of mechanical ventilation for asthma is decreasing and mortality of patients ventilated for asthma is also decreasing in some series.9,10
The decision to intubate is based primarily on the degree of respiratory distress as assessed by an experienced clinician and patients themselves. A patient with a high PaCO2 (e.g. > 70 mmHg) who is dyspnoeic but not distressed and who may respond to full treatment over a few hours needs close observation but not immediate intubation. Patients are often able to tolerate hypercapnia without requiring invasive ventilation.106 A patient with a lower PaCO2 (e.g. 50–60 mmHg) who has been unwell for days and who has a deteriorating status despite treatment is likely to need intubation. A patient who complains of respiratory exhaustion is likely to need intubation. Absolute indications for intubation include cardiac or respiratory arrest, severe hypoxia or rapid deterioration of conscious state.14,54
Once the decision to intubate has been made, a safe option is to perform rapid-sequence intubation using the orotracheal approach. The largest possible endotracheal tube should be used to reduce the work of breathing and to reduce the risk of occlusion by the tenacious secretions that often occur with asthma. Once the endotracheal tube is in place, slow hand ventilation (8–10 breaths/min) should maintain oxygenation until the ventilator can be connected.
INITIAL VENTILATOR SETTINGS
The principles of initial mechanical ventilation are to avoid excessive DHI107 and hypoventilation (Figure 31.3) by commencing with a minute ventilation < 115 ml/kg per min (< 8 l/min in a 70-kg patient) best achieved with a tidal volume of 5–7 ml/kg, a respiratory rate of 10–12 breaths/min and a short inspiratory time to ensure an expiratory time of ≥ 4 seconds.22,94,97,98,107 This degree of hypoventilation will usually result in hypercapnic acidosis and continued respiratory distress necessitating heavy sedation, and sometimes requiring 1–2 bolus doses of a neuromuscular-blocking agent (NMBA). During initial controlled hypoventilation, PEEP will further increase lung volume and should not be used.98
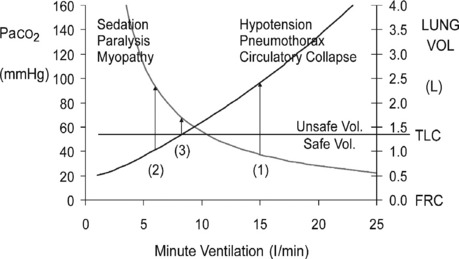
This use of volume-controlled ventilation is most established for this ventilatory pattern. In volume control, a high inspiratory flow rate (70–100 l/min) is required to achieve a short inspiratory time. This will result in a high peak airway pressure but this will lower DHI and plateau airway pressure (Pplat) and reduce barotrauma compared with lower inspiratory flow rates.94,97 Pressure-controlled or assisted modes have been used108 without adverse consequences. The theoretical advantage of a safe pressure limit in pressure modes is offset by the fact that equilibrium with the set safe pressure cannot be reached during the short inspiratory times required and thus either a higher pressure must be set or more than necessary hypoventilation may occur. It is not clear if one mode is superior to the other.
ASSESSMENT OF DYNAMIC HYPERINFLATION
Once mechanical ventilation has started, the degree of DHI should be assessed by measurement of the plateau airway pressure (Pplat). This is the airway pressure after transient expiratory occlusion at the end of inspiration. This may be achieved by an end-inspiratory hold function on most current ventilators, which delays the commencement of the next breath (Figure 31.4a) or by applying a 0.5-second ‘plateau’ which does not delay the onset of the next breath. The latter should be applied for a single breath only, as application for several breaths in a row will decrease expiratory time and progressively increase DHI. This is the most easily measured estimate of average alveolar pressure at the end of inspiration and is directly proportional to the degree of hyperinflation and should be maintained at < 25 cmH2O.94,97
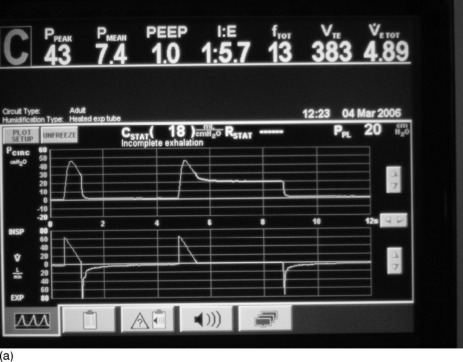
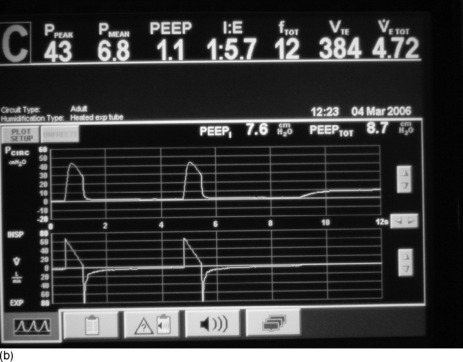
Figure 31.4 (a) The ventilator circuit pressure and flow traces versus time during controlled ventilation of a patient with severe airflow obstruction. Note the use of a low rate, low tidal volume (Vt), a high inspiratory flow rate (Vi 80 l/min) and hence a short inspiratory time (te < 1 second). This causes a high peak airway pressure but allows a long expiratory time (> 4 seconds). Expiratory flow is low throughout expiration and appears close to baseline at the onset of the next breath (second on screen), suggesting minimal gas trapping, but when an intrinsic positive end-expiratory pressure (PEEPi) manoeuvre is done (end-expiratory pause) there is a surprising degree of PEEPi present (7.6 cmH2O). (b) The ventilator circuit pressure and flow traces versus time during controlled ventilation of the same patient as in Figure 31.4b. When a Pplat manoeuvre is done (end-inspiratory pause) the Pplat is safe (20 cmH2O) with this ventilator pattern (despite the degree of PEEPi present in Figure 31.4b). Note the low Pplat despite the high peak airway pressure.
Intrinsic PEEP is the airway pressure during occlusion of expiratory flow at the end of expiration (Figure 31.4b). This also is an automated end-expiratory hold function on most current ventilators. However this measurement is known to underestimate true PEEPi, as a consequence of small-airway closure during expiration, resulting in many higher-pressure alveoli not communicating with the central airway at the end of expiration.96 Because of this, PEEPi can be used to show the presence of DHI but is not recommended to regulate the mechanical ventilation. Ideally PEEPi should be < 12 cmH2O, though the exact safe level is unknown.
End-inspiratory lung volume (VEI) (see Figures 31.1 and 31.2) < 20 ml/kg (1.4 litres in a 70-kg patient) has been shown to be a good predictor of complications during mechanical ventilation97 and to correlate with total lung volume.22 It is the total exhaled volume during a period of apnoea (30–90 seconds) in a paralysed patient. Although valuable, it is not routinely used as it requires neuromuscular blockade and a volumetric measuring device.
Assessment of the change in blood pressure and central venous pressure during a period of transient ventilatordisconnection (1–2 min) or transient ventilator rate reduction (4–6 breaths/min for 2–4 min): if DHI has been suppressing circulation then a significant increase in blood pressure and reduction in central venous pressure will occur.
ADJUSTMENT OF VENTILATION
Ventilatory patterns with an excessive minute ventilation risk hypotension and barotrauma and are associated with a high mortality. Using profound hypoventilation will guarantee avoidance of these complications but usually necessitates heavy sedation and neuromuscular blockade (see Figure 31.3). This, in association with parenteral steroids, has a high probability of myopathy,66,109,110 which may cause severe prolonged disability. To minimise the risk of both these complications, DHI should be carefully assessed and the minimum amount of hypoventilation used to achieve a safe level of DHI (see Figure 31.3) in association with less sedation and minimal or no use of NMBAs.
Hypercapnia is usually present but is well tolerated111 and does not appear to depress cardiac function. There is no evidence of benefit from sodium bicarbonate but it may reduce acidaemia-induced respiratory distress and may be given if the pH is less than 7.1.
COMPLICATIONS OF INVASIVE VENTILATION IN ASTHMA
Hypotension may be caused by sedation, DHI, pneumothoraces or arrhythmias.94,97 Hypovolaemia may be a contributory factor but is rarely a cause. Hypotension may be mild or life-threatening.112 Hypotension due to DHI may be diagnosed by recovery of blood pressure during apnoea of 60 seconds (the ‘apnoea test’)112 or by a longer period of low ventilator rate (4–6 breaths/min for 2–4 min). If this occurs, ventilation should be continued at a lower rate.
Circulatory arrest with apparent electromechanical dissociation is a recognised complication that may occur within 10 minutes of intubation and can lead to death or severe cerebral ischaemic injury if not managed correctly.112–114 Standard mechanical ventilation recommendations (minute ventilation 115 ml/kg per min) have been estimated to be safe for 80% of patients requiring mechanical ventilation for acute severe asthma, with the remaining 20% requiring a small to moderate reduction in minute ventilation to return DHI to a safe level.97 A small percentage of patients with unusually severe asthma can rapidly develop excessive DHI during initial uncontrolled mechanical ventilation, leading to electromechanical dissociation, sometimes despite ‘safe’ levels of minute ventilation. If the cause of this is not immediately recognised, it can lead to prolonged and unnecessary cardiopulmonary resuscitation, unsafe procedures such as intercostal vascular access needles or pericardial taps and risk cerebral injury and death.112–114 When this occurs, immediate disconnection from the ventilator for 60–90 seconds (the ‘apnoea test’; see above) or profound hypoventilation (2–3 breaths/min)54 will diagnose and improve this situation. An even smaller percentage of patients may remain hypotensive despite profound hypoventilation with marked hypercapnia, fluid loading and inotropes. These patients may require Heliox delivered by the mechanical ventilator115 or extracorporeal membrane oxygenation.114,116,117
Pneumothoraces were common before the advent of protective ventilatory strategies. DHI during mechanical ventilation was probably the major causative factor involved,94,97 but pneumothorax may also occur in association with subclavian central venous catheter insertion and as a consequence of intercostal needle insertion for suspected tension pneumothorax during circulatory arrest (see above). The presence of severe airflow obstruction prevents lung collapse and favours gas loss through the ruptured alveoli, with the result that tension is almost always present in the pneumothorax. Once a unilateral tension pneumothorax is present, this will necessarily reduce ventilation to that lung and redistribute ventilation to the contralateral lung, thereby further increasing DHI in the second lung, and bilateral tension pneumothoraces may result with severely adverse consequences.
Acute necrotising myopathy is a serious complication that may occur in patients who are invasively ventilated for asthma and receive NMBAs or very deep sedation.66,109,110,118,119 It is characterised by weakness with electromyographic evidence of myopathy and increased serum creatine kinase levels. Muscle biopsy reveals two patterns: myonecrosis with muscle cell vacuolisation or predominant type II fibre atrophy.66,118 Recovery can be slow with prolonged weaning from mechanical ventilation and the need for rehabilitation. Incomplete recovery after 12 months has been reported in a few patients.119,120 The aetiology of the myopathy appears to be a combination of the effects of corticosteroids and NMBAs with the duration of paralysis a strong predictor of myopathy.119,120 The type of NMBA used seems to make no difference to the incidence of myopathy.118 The relative contribution of corticosteroids versus NMBAs in the causation of myopathy is unclear; it seems wise to minimise the dose of parenteral corticosteroids with early introduction of nebulised agents and to minimise or avoid NMBAs if possible.
MORTALITY, LONG-TERM OUTCOME AND FOLLOW-UP
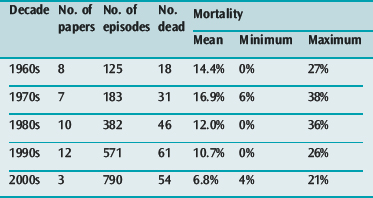
In a summary of 37 papers reporting the outcomes of 1260 patients requiring mechanical ventilation for asthma in the four decades prior to the year 200055 there was an overall mortality of 12.4% with a progressive reduction in reported mortality during that period (see Table 31.2). A selection of reports9,11,12 published since 2000 have shown continued reduction in overall mortality (see Table 31.2) but with mortalities as high as 21% still occurring in one series.11
The largest and most comprehensive recent series9 reported 1899 patients admitted to 22 Australian intensive care units over an 8-year period (1996–2003). This series reported a requirement for mechanical ventilation for 36% of patients admitted with severe asthma and a progressive reduction in annual mortality of the ventilated patients from 10% to 3% over the 8-year period.
The need for invasive ventilation is associated with an increased risk of death9,11,121 and survivors of these near-fatal episodes of asthma have an increased risk of intensive care readmission and an increased risk of death after hospital discharge.27
For these reasons, patients who have an episode of asthma severe enough to require hospitalisation, particularly intensive care admission, need careful follow-up. This should include active identification and avoidance of precipitants, aggressive bronchodilator therapy including inhaled steroids, regular medical review, regular measurement of lung function, management plans for a deteriorating status and ready access to emergency services.9
1 Abramson MJ, Bailey MJ, Couper FJ, et al. Are asthma medications and management related to deaths from asthma? Am J Respir Crit Care Med. 2001;163:12-18.
2 McCaul KA, Wakefield MA, Roder DM, et al. Trends in hospital readmission for asthma: has the Australian National Asthma Campaign had an effect? Med J Aust. 2000;172:62-66.
3 Suissa S, Ernst P, Benayoun S, et al. Low-dose inhaled corticosteroids and the prevention of death from asthma. N Engl J Med. 2000;343:332-336.
4 The International Study of Asthma and Allergies in Childhood (ISAAC) Steering Committee. Worldwide variation in prevalence of symptoms of asthma, allergic rhinoconjunctivitis, and atopic eczema: ISAAC. Lancet. 1998;351:1225-1232.
5 Pearce N, Weiland S, Keil U, et al. Self-reported prevalence of asthma symptoms in children in Australia, England, Germany and New Zealand: an international comparison using the ISAAC protocol. Eur Respir J. 1993;6:1455-1461.
6 Worldwide variations in the prevalence of asthma symptoms: the international study of asthma and allergies in childhood (ISAAC). Eur Respir J. 1998;12:315-335.
7 Sly RM. Changing prevalence of allergic rhinitis and asthma. Ann Allergy Asthma Immunol. 1999;82:233-248. quiz 248–52
8 Strong K, de Looper M, Magnus P. Asthma mortality in Australia, 1980–1996. Aust Health Rev. 1998;21:255-263.
9 Stow PJ, Pilcher DV, Wilson J, et al. Improved outcomes from acute severe asthma in Australian intensive care units (1996–2003). Thorax. 2007;62:842-847.
10 Wilson DH, Tucker G, Frith P, et al. Trends in hospital admissions and mortality from asthma and chronic obstructive pulmonary disease in Australia, 1993–2003. Med J Aust. 2007;186:408-411.
11 Afessa B, Morales I, Cury JD. Clinical course and outcome of patients admitted to an ICU for status asthmaticus. Chest. 2001;120:1616-1621.
12 Gehlbach B, Kress JP, Kahn J, et al. Correlates of prolonged hospitalization in inner-city ICU patients receiving noninvasive and invasive positive pressure ventilation for status asthmaticus. Chest. 2002;122:1709-1714.
13 US Department of Health and Human Services. Guidelines for the Diagnosis and Management of Asthma. Maryland: National Asthma Education Program, 1991.
14 Werner HA. Status asthmaticus in children: a review. Chest. 2001;119:1913-1929.
15 Mattes J, Karmaus W. The use of antibiotics in the first year of life and development of asthma: which comes first? Clin Exp Allergy. 1999;29:729-732.
16 Busse WW, Lemanske RFJr. Asthma. N Engl J Med. 2001;344:350-362.
17 Bai TR, Cooper J, Koelmeyer T, et al. The effect of age and duration of disease on airway structure in fatal asthma. Am J Respir Crit Care Med. 2000;162:663-669.
18 Jeffery PK. Bronchial biopsies and airway inflammation. Eur Respir J. 1996;9:1583-1587.
19 Wasserfallen J, Schaller M, Feihl F, et al. Sudden asphyxic asthma: a distinct entity? Am Rev Respir Dis. 1990;142:108-111.
20 Kolbe J, Fergusson W, Garrett J. Rapid onset asthma: a severe but uncommon manifestation. Thorax. 1998;53:241-247.
21 Woodruff PG, Emond SD, Singh AK, et al. Sudden-onset severe acute asthma: clinical features and response to therapy. Acad Emerg Med. 1998;5:695-701.
22 Tuxen D, Williams T, Scheinkestel C, et al. Use of a measurement of pulmonary hyperinflation to control the level of mechanical ventilation in patients with severe asthma. Am Rev Respir Dis. 1992;146:1136-1142.
23 Evans TW. International Consensus Conferences in Intensive Care Medicine: non-invasive positive pressure ventilation in acute respiratory failure. Organised jointly by the American Thoracic Society, the European Respiratory Society, the European Society of Intensive Care Medicine, and the Société de Réanimation de Langue Française, and approved by the ATS Board of Directors, December 2000. Intens Care Med, 27. 2001, 166-178.
24 Pinsky M. Cardiopulmonary interactions associated with airflow obstruction. In: Hall J, Corbridge T, Rodrigo C, et al, editors. Acute Asthma: Assessment and Management. New York: McGraw-Hill; 2000:105-123.
25 Rossi A, Ganassini A, Brusasco V. Airflow obstruction and dynamic hyperinflation. In: Hall J, Corbridge T, Rodrigo C, et al, editors. Acute Asthma: Assessment and Management. New York: McGraw-Hill; 2000:57-82.
26 McFadden ERJr. Acute severe asthma. Am J Respir Crit Care Med. 2003;168:740-759.
27 McFadden ERJr., Warren EL. Observations on asthma mortality. Ann Intern Med. 1997;127:142-147.
28 Brenner B, Abraham E, Simon R. Position and diaphoresis in acute asthma. Am J Med. 1983;74:1005-1009.
29 Lemarchrand P, Labrune S, Herer B, et al. Cardiorespiratory arrest following peak expiratory flow measurement during attack of asthma. Chest. 1991;100:1168-1169.
30 Carruthers DM, Harrison BD. Arterial blood gas analysis or oxygen saturation in the assessment of acute asthma? Thorax. 1995;50:186-188.
31 Rodrigo GJ, Rodriquez Verde M, Peregalli V, et al. Effects of short-term 28% and 100% oxygen on Paco2 and peak expiratory flow rate in acute asthma: a randomized trial. Chest. 2003;124:1312-1317.
32 Mountain R, Heffner J, Brackett N, et al. Acid–base disturbances in acute asthma. Chest. 1990;98:651-655.
33 Manthous CA. Lactic acidosis in status asthmaticus: three cases and review of the literature. Chest. 2001;119:1599-1602.
34 Prakash S, Mehta S. Lactic acidosis in asthma: report of two cases and review of the literature. Can Respir J. 2002;9:203-208.
35 White C, Cole R, Lubetsky H, et al. Acute asthma: admission chest radiography in hospitalized adult patients. Chest. 1991;100:14-16.
36 Rodrigo G, Rodrigo C. Assessment of the patient with acute asthma in the emergency department. A factor analytic study. Chest. 1993;104:1325-1328.
37 Beasley R, Pearce N, Crane J, et al. Asthma mortality and inhaled beta agonist therapy. Aust NZ J Med. 1991;21:753-763.
38 Chien JW, Ciufo R, Novak R, et al. Uncontrolled oxygen administration and respiratory failure in acute asthma. Chest. 2000;117:728-733.
39 National Asthma Campaign. Asthma Management Handbook 1998. Melbourne: National Asthma Campaign, 1998.
40 BTS guidelines on asthma management. Thorax. 2003;58(suppl. 1):i1-94.
41 Global Strategy for Asthma Management and Prevention. Global Initiative for Asthma (GINA). Eur Respir J. 2006;31:143-178.
42 Rossing T, Fanta C, Goldstein D, et al. Emergency therapy of asthma: comparison of the acute effects of parenteral and inhaled sympathomimetics and infused aminophylline. Am Rev Respir Dis. 1980;122:365-371.
43 Cates C, Rowe B, Bara A. Holding chamber versus nebulisers for beta-agonist treatment of acute asthma (Cochrane review). Oxford, UK: Cochrane Library: Update Software; 2002.
44 Newman KB, Milne S, Hamilton C, et al. A comparison of albuterol administered by metered-dose inhaler and spacer with albuterol by nebulizer in adults presenting to an urban emergency department with acute asthma. Chest. 2002;121:1036-1041.
45 Manthous CA, Chatila W, Schmidt GA, et al. Treatment of bronchospasm by metered-dose inhaler albuterol in mechanically ventilated patients. Chest. 1995;107:210-213.
46 Rodrigo G. Inhaled therapy for acute adult asthma. Curr Opin Allergy Immunol. 2003;3:169-175.
47 Bisgaard H. Delivery of inhaled medication to children. J Asthma. 1997;34:443-467.
48 Papo MC, Frank J, Thompson AE. A prospective, randomized study of continuous versus intermittent nebulized albuterol for severe status asthmaticus in children. Crit Care Med. 1993;21:1479-1486.
49 Dolovich MA. Influence of inspiratory flow rate, particle size, and airway caliber on aerosolized drug delivery to the lung. Respir Care. 2000;45:597-608.
50 Rodrigo C, Rodrigo G. Therapeutic response patterns to high and cumulative doses of salbutamol in acute severe asthma. Chest. 1998;113:593-598.
51 Travers AH, Rowe BH, Barker S, et al. The effectiveness of IV beta-agonists in treating patients with acute asthma in the emergency department: a meta-analysis. Chest. 2002;122:1200-1207.
52 Browne GJ, Penna AS, Phung X, et al. Randomised trial of intravenous salbutamol in early management of acute severe asthma in children. Lancet. 1997;349:301-305.
53 Canadian Asthma Consensus Report. Management of patients with asthma in the emergency department and in the hospital. Can Med Assoc J. 1999;161(suppl):S53-59.
54 Corbridge SJ, Corbridge TC. Severe exacerbations of asthma. Crit Care Nurs Q. 2004;27:207-228. quiz 229–30
55 Tuxen D. Mechanical ventilation in asthma. In: Evans T, Hinds C, editors. Recent Advances in Critical Care Medicine Number 4. London: Churchill Livingstone; 1996:165-189.
56 Willmot D. A 24 year old woman admitted to the critical care unit, with ‘resistant’ asthma and a metabolic acidosis. Crit Care Resusc. 2000;2:228-229.
57 Spitzer WO, Suissa S, Ernst P, et al. The use of beta-agonists and the risk of death and near death from asthma. N Engl J Med. 1992;326:501-506.
58 Beakes DE. The use of anticholinergics in asthma. J Asthma. 1997;34:357-368.
59 Stoodley RG, Aaron SD, Dales RE. The role of ipratropium bromide in the emergency management of acute asthma exacerbation: a metaanalysis of randomized clinical trials. Ann Emerg Med. 1999;34:8-18.
60 Rodrigo GJ, Rodrigo C. The role of anticholinergics in acute asthma treatment: an evidence-based evaluation. Chest. 2002;121:1977-1987.
61 Bryant D, Rogers P. Effects of ipratropium bromide nebuliser solution with and without preservatives in the treatment of acute and stable asthma. Chest. 1992;102:742-747.
62 Rodrigo GJ, Rodrigo C, Hall JB. Acute asthma in adults: a review. Chest. 2004;125:1081-1102.
63 Rowe B, Spooner C, Ducharme F, et al. Early emergency department treatment of acute asthma with systemic corticosteroids (Cochrane review). Oxford, UK: Update Software, 2002.
64 Rodrigo GJ, Rodrigo C. Triple inhaled drug protocol for the treatment of acute severe asthma. Chest. 2003;123:1908-1915.
65 Edmonds ML, Camargo CAJr., Pollack CVJr., et al. The effectiveness of inhaled corticosteroids in the emergency department treatment of acute asthma: a meta-analysis. Ann Emerg Med. 2002;40:145-154.
66 Douglass J, Tuxen D, Horne M, et al. Myopathy in severe asthma. Am Rev Respir Dis. 1992;146:517-519.
67 Klein-Gitelman MS, Pachman LM. Intravenous corticosteroids: adverse reactions are more variable than expected in children. J Rheumatol. 1998;25:1995-2002.
68 Abernathy-Carver KJ, Fan LL, Boguniewicz M, et al. Legionella and Pneumocystis pneumonias in asthmatic children on high doses of systemic steroids. Pediatr Pulmonol. 1994;18:135-138.
69 Kasper WJ, Howe PM. Fatal varicella after a single course of corticosteroids. Pediatr Infect Dis J. 1990;9:729-732.
70 Goodman DC, Littenberg B, O’Connor GT, et al. Theophylline in acute childhood asthma: a meta-analysis of its efficacy. Pediatr Pulmonol. 1996;21:211-218.
71 Yung M, South M. Randomised controlled trial of aminophylline for severe acute asthma. Arch Dis Child. 1998;79:405-410.
72 Parameswaran K, Belda J, Rowe B, et al. Addition of intravenous aminophylline to beta2-agonists in adults with acute asthma (Cochrane review). Oxford, UK: Update Software, 2002.
73 Tirot P, Bouachour G, Varache N, et al. Use of intravenous epinephrine in severe acute asthma. Rev Mal Respir. 1992;9:319-323.
74 Alter HJ, Koepsell TD, Hilty WM. Intravenous magnesium as an adjuvant in acute bronchospasm: a meta-analysis. Ann Emerg Med. 2000;36:191-197.
75 Rowe BH, Bretzlaff JA, Bourdon C, et al. Intravenous magnesium sulfate treatment for acute asthma in the emergency department: a systematic review of the literature. Ann Emerg Med. 2000;36:181-190.
76 Rodrigo G, Rodrigo C, Burschtin O. Efficacy of magnesium sulfate in acute adult asthma: a meta-analysis of randomized trials. Am J Emerg Med. 2000;18:216-221.
77 Sydow M, Crozier T, Zielman S, et al. High-dose intravenous magnesium sulfate in the management of life threatening status asthmaticus. Inten Care Med. 1993;19:467-471.
78 Hess DR, Acosta FL, Ritz RH, et al. The effect of heliox on nebulizer function using a beta-agonist bronchodilator. Chest. 1999;115:184-189.
79 Kress JP, Noth I, Gehlbach BK, et al. The utility of albuterol nebulized with heliox during acute asthma exacerbations. Am J Respir Crit Care Med. 2002;165:1317-1321.
80 Carter E. Quality of writing in manuscripts. Chest. 1999;116:1494.
81 Kass JE, Terregino CA. The effect of heliox in acute severe asthma: a randomized controlled trial. Chest. 1999;116:296-300.
82 Rodrigo GJ, Rodrigo C, Pollack CV, et al. Use of helium-oxygen mixtures in the treatment of acute asthma: a systematic review. Chest. 2003;123:891-896.
83 Sarma V. Use of ketamine in acute severe asthma. Acta Anaesthesiol Scand. 1992;36:106-107.
84 Sato N, Matsuki A, Zsigmond E, et al. Ketamine relaxes and airway smooth muscle contracted by endothelium. Anesth Analg. 1997;84:900-906.
85 Howton JC, Rose J, Duffy S, et al. Randomized, double-blind, placebo-controlled trial of intravenous ketamine in acute asthma. Ann Emerg Med. 1996;27:170-175.
86 Youssef-Ahmed MZ, Silver P, Nimkoff L, et al. Continuous infusion of ketamine in mechanically ventilated children with refractory bronchospasm. Intens Care Med. 1996;22:972-976.
87 Tobias JD, Garrett JS. Therapeutic options for severe, refractory status asthmaticus: inhalational anaesthetic agents, extracorporeal membrane oxygenation and helium/oxygen ventilation. Paediatr Anaesth. 1997;7:47-57.
88 Dockhorn RJ, Baumgartner RA, Leff JA, et al. Comparison of the effects of intravenous and oral montelukast on airway function: a double blind, placebo controlled, three period, crossover study in asthmatic patients. Thorax. 2000;55:260-265.
89 Camargo CAJr., Smithline HA, Malice MP, et al. A randomized controlled trial of intravenous montelukast in acute asthma. Am J Respir Crit Care Med. 2003;167:528-533.
90 Silverman R. Ipratropium bromide in emergency management of acute asthma exacerbation. Ann Emerg Med. 2000;35:197-198.
91 Smith D, Deshazo R. Bronchoalveolar lavage in asthma. State of the art. Am Rev Respir Dis. 1993;148:523-532.
92 Graham V, Lasserson T, Rowe B, editors. Antibiotics for Acute Asthma (Cochrane Review). Oxford, UK: Update Software, 2002.
93 Joseph KS, Blais L, Ernst P, et al. Increased morbidity and mortality related to asthma among asthmatic patients who use major tranquillisers. Br Med J. 1996;312:79-82.
94 Tuxen D, Lane S. The effects of ventilatory pattern on hyperinflation, airway pressures, and circulation in mechanical ventilation of patients with severe airflow obstruction. Am Rev Respir Dis. 1987;136:872-879.
95 Pepe P, Marini J. Occult positive end-expiratory pressure in mechanically ventilated patients with airflow obstruction. Am Rev Respir Dis. 1982;126:166-170.
96 Leatherman J, Ravenscraft S. Low measured auto-positive end-expiratory pressure during mechanical ventilation of patients with severe asthma: hidden auto-positive end-expiratory pressure. Crit Care Med. 1996;24:541-546.
97 Williams T, Tuxen D, Scheinkestel C, et al. Risk factors for morbidity in mechanically ventilated patients with acute severe asthma. Am Rev Respir Dis. 1992;146:607-615.
98 Tuxen D. Detrimental effects of positive end-expiratory pressure during controlled mechanical ventilation of patients with severe airflow obstruction. Am Rev Respir Dis. 1989;140:5-9.
99 Brochard L. Noninvasive ventilation for acute respiratory failure. JAMA. 2002;288:932-935.
100 International Consensus Conferences in Intensive Care Medicine: noninvasive positive pressure ventilation in acute respiratory failure. Am J Respir Crit Care Med. 2001;163:283-291.
101 Mehta S, Hill S. Noninvasive ventilation. Am J Respir Crit Care Med. 2001;163:540-577.
102 Brochard L, Mancebo J, Wysocki M, et al. Noninvasive ventilation for acute exacerbations of chronic obstructive pulmonary disease. N Engl J Med. 1995;333:817-822.
103 Kramer N, Meyer T, Meharg J, et al. Randomised prospective trial of noninvasive positive pressure ventilation in acute respiratory failure. Am J Respir Crit Care Med. 1995;151:1799-1806.
104 Fernandez MM, Villagra A, Blanch L, et al. Non-invasive mechanical ventilation in status asthmaticus. Intens Care Med. 2001;27:486-492.
105 Soroksky A, Stav D, Shpirer I. A pilot prospective, randomized, placebo-controlled trial of bilevel positive airway pressure in acute asthmatic attack. Chest. 2003;123:1018-1025.
106 Mountain R, Sahn S. Clinical features and outcome in patients with acute asthma presenting with hypercapnia. Am Rev Respir Dis. 1988;138:535-539.
107 Peigang Y, Marini J. Ventilation of patients with asthma and chronic pulmonary disease. Curr Opin Crit Care. 2002;8:70-78.
108 Mansel J, Stogner S, Petrini M, et al. Mechanical ventilation in patients with acute severe asthma. Am J Med. 1990;89:42-48.
109 Hansen-Flaschen J, Cowen J, Raps E. Neuromuscular blockade in the Intensive Care Unit. More than we bargained for. Am Rev Respir Dis. 1993;147:234-236.
110 Nates J, Cooper D, Tuxen D. Acute weakness syndromes in critically ill patients – a reappraisal. Anaesth Int Care. 1997;25:502-513.
111 Bellomo R, McLaughlan P, Tai E, et al. Asthma requiring mechanical ventilation. A low morbidity approach. Chest. 1994;105:891-896.
112 Rosengarten P, Tuxen D, Dziukas L, et al. Circulatory arrest induced by intermittent positive pressure ventilation in a patient with severe asthma. Anaes Int Care. 1990;19:118-121.
113 Kollef M. Lung hyperinflation caused by inappropriate ventilation resulting in electromechanical dissociation: a case report. Heart Lung. 1992;21:74-77.
114 Mabuchi N, Takasu H, Ito S, et al. Successful extracorporeal lung assist (ECLA) for a patient with severe asthma and cardiac arrest. Clin Intens Med. 1991;2:292-294.
115 Gluck E, Onorato D, Castriotta R. Helium–oxygen mixtures in intubated patients with status asthmaticus and respiratory acidosis. Chest. 1990;98:693-698.
116 King D, Smales C, Arnold A, et al. Extracorporeal membrane oxygenation as emergency treatment for life threatening acute severe asthma. Postgrad Med J. 1986;62:555-557.
117 Tajimi K, Kasai T, Nakatani T, et al. Extracorporeal lung assist (ECLA) for a patient with hypercapnia due to status asthmaticus. Intens Care Med. 1988;14:588-589.
118 Leatherman J, Fluegel W, David W, et al. Muscle weakness in mechanically ventilated patients with severe asthma. Am J Respir Crit Care Med. 1996;153:1686-1690.
119 Behbehani NA, Al-Mane F, D’Yachkova Y, et al. Myopathy following mechanical ventilation for acute severe asthma: the role of muscle relaxants and corticosteroids. Chest. 1999;115:1627-1631.
120 Hirano M, Ott B, Raps E, et al. Acute quadriplegic myopathy: a complication of treatment with steroids, nondepolarizing blocking agents, or both. Neurology. 1992;42:2082-2087.
121 Marquette C, Saulnier F, Leroy O, et al. Long-term prognosis of near-fatal asthma. Am Rev Respir Dis. 1992;146:76-81.
[/level-membership-for-anesthesiology-category][not-level-membership-for-anesthesiology-category]
Chapter 31 Acute severe asthma
Acute severe asthma is a medical emergency associated with significant morbidity and mortality. The majority of adverse outcomes are attributed to underestimation of severity with delayed and/or inadequate treatment1–3 and are potentially preventable.
The worldwide prevalence of asthma varies widely (2–37% in children),4 but it is now clear that the overall prevalence has increased worldwide,5,6 with life-threatening episodes affecting an estimated 0.5% of asthmatics per year.7 Australia, New Zealand and the UK have amongst the highest incidences.7 In Australia, 9% have asthma as a long-term condition8 and up to 40% of children have asthma symptoms at some time.5
Although the prevalence of asthma is increasing, many countries have achieved reductions in hospital presentations and admissions,9,10 reduced intensive care admissions and reduced overall asthma mortality.9,10 Improved community management of asthma,1,2 more widespread use of inhaled corticosteroids3 and other preventive measures have been given the credit for these improvements.
Despite reducing admissions, significant and potentially preventable mortality continues to occur in those patients who do require intensive care or mechanical ventilation.11,12
CLINICAL DEFINITION
Asthma has been defined as a lung disease with the following characteristics:13
Exacerbations of asthma are characterised by increasing dyspnoea, cough, wheeze, chest tightness and decreased expiratory airflow. Status asthmaticus has had varying definitions. However, for practical purposes, any patient not responding to initial doses of nebulised bronchodilators should be considered to have status asthmaticus.14
AETIOLOGY
The pathogenesis of asthma is complex, with both genetic and environmental influences. The increase in asthma prevalence has been attributed to the ‘hygiene hypothesis’15 which suggests that reduced exposure to childhood infections as a result of antibiotics and hygienic lifestyle promotes an imbalance in T-cell phenotype leading to inflammatory cytokine overproduction. Immunoglobulin (Ig) E-dependent mechanisms appear to be particularly important in generating the characteristic state of airway inflammation and bronchial hyperreactivity with the allergens in the local environment dictating the specificity of the antibody response.16 Triggers of acute asthma can be non-specific (cold air, exercise, atmospheric pollutants), specific allergens (housemite, pollen, animal danders), modifiers of airway control (aspirin, β-blockers) or stress and emotion. No precipitant can be identified in over 30% of patients.
PATHOPHYSIOLOGY
The postmortem airway pathology of patients who die from acute asthma includes bronchial wall thickening from oedema and inflammatory cell infiltrate, hypertrophy and hyperplasia of bronchial smooth muscle and submucosal glands, deposition of collagen beneath the epithelial basement membrane and prominent intraluminal secretions. These secretions may narrow or occlude the small airways and postmortem studies frequently report extensive plugging and atelectasis.17,18 When interpreting the latter findings it is important to remember that during life in very severe asthma, the lungs are hyperinflated close to total lung capacity (TLC) by maximal inspiratory effort and to beyond TLC during mechanical ventilation. At this time radiological atelectasis is rare, suggesting that most airways are communicating at these high lung volumes, even if airways are very narrowed. Only at death does the prolonged apnoea allow lung deflation, widespread airway closure and alveolar gas absorption to give the postmortem appearance of extensive complete occlusion that was not present to the same degree during life.
In some deaths bronchial mucus is absent; in these cases airway obstruction may be mainly due to intense smooth bronchoconstriction.
This observation may account for two patterns of progression of asthma:
CLINICAL FEATURES AND ASSESSMENT OF SEVERITY
Triage and assessment of severity of the acute asthma attack are crucial. Underestimation or non-measurement of asthma severity is associated with increased mortality.26,27 Assessment has two key features: assessment of initial severity and ongoing assessment of response to treatment.
HISTORY
Any history of prior intubation and mechanical ventilation for asthma is a predictor for life-threatening asthma.26,27 A history of poor asthma control and multiple recent medical presentations for asthma are recognised risk factors. Other risk factors include a poor response to prior treatments and poor psychosocial circumstances.
PHYSICAL EXAMINATION
The general appearance and level of distress can be an important indicator of severity (Table 31.1). Use of accessory muscles, suprasternal retraction, markedly diminished breath sounds or a silent chest, central cyanosis, inability to speak (sentences, phrases, single words), a disturbance in the level of consciousness, upright posture and diaphoresis all suggest a severe attack.28 A respiratory rate > 30 breaths/min, pulse rate > 120 beats/min and pulsus paradoxus of > 15 mmHg are associated with severe asthma, though their absence does not preclude life-threatening asthma.
VENTILATORY FUNCTION TESTS
The patient may not be able to perform these due to breathlessness. However, forced expiratory volume in 1 second (FEV1) and peak expiratory flow rate (PEFR) are useful indicators of severity and response to treatment when done serially. An FEV1 < 1.0 litre or PEFR < 100 l/min indicates very severe asthma at significant risk of requiring mechanical ventilation. In some patients forced expiration may worsen symptoms; these measurements should cease if this occurs.29
PULSE OXIMETRY
Pulse oximetry (SpO2) is usually readily available and provides a rapid assessment of oxygenation. It is also very valuable in regulation oxygen therapy, both for avoiding hypoxia (SpO2 > 90%)30 and avoiding potential side-effects of hyperoxia (SpO2 < 95%).31 Of course, pulse oximetry cannot assess arterial PCO2 or acid–base state or lactate and hence does not replace the need for blood gases
ARTERIAL BLOOD GASES
Arterial hypoxaemia is almost invariably present in a patient with severe asthma breathing room air, though it usually responds well to low-level oxygen supplementation (28–35%).30 Blood gases should not delay initiation of treatment, and are not required in mild asthma or moderate asthma that is responding well to treatment. They are very important in severe asthma, or moderate asthma with inadequate treatment response. The arterial PaCO2 is an important measure of severity and, if hypercapnia is present, an important guide to treatment response.
Ventilation is initially increased in an acute attack, leading to hypocapnia and a respiratory alkalosis.30 As the asthmatic attack worsens, the work of breathing, V/Q mismatch and adverse cardiopulmonary interactions all increase, and the minute ventilation required to maintain the same alveolar ventilation and PaCO2 increases. Eventually the patient is incapable of meeting this demand and the PaCO2 rises. The presence of hypercapnic acidosis is associated with an FEV1 of < 20% predicted and reliably indicates that asthma is severe. A metabolic acidosis may also be present, and this is most commonly due to lactic acidosis32 associated with intravenous or continuous nebulised β-agonists.33,34
CHEST X-RAY
Although not generally helpful in assessing severity,35 a chest X-ray should be performed when asthma is severe or refractory to treatment, when barotrauma or lower respiratory tract infection is suspected or when the diagnosis is in doubt. It is not required in milder attacks that respond well to treatment.
ASSESSMENT OF TREATMENT RESPONSE
Repeated evaluation of the patient’s response to treatment is a valuable tool in assessing severity of acute asthma. The response to the first 2 hours of treatment is an important predictor of outcome.36 Adverse events are associated with underestimation of severity, inadequate observation after initial assessment and under treatment. Ongoing evaluation of response to therapy is critical. This evaluation should include repeated assessments of:
MANAGEMENT
ESTABLISHED TREATMENTS
Initial therapy of acute severe asthma should include the following.
OXYGEN
Hypoxaemia contributes to life-threatening events that complicate acute severe asthma.37 Humidified supplemental oxygen should be titrated to achieve a SpO2 > 90%. The risk of oxygen-induced increasing hypercapnia with coexisting chronic obstructive pulmonary disease or pre-existing chronic hypercapnia is a well-known reason to maintain a lower SpO2 (e.g. 90–92%). This phenomenon was not believed to be relevant to the majority of patients with acute asthma; however, there is now emerging evidence that hyperoxia may be harmful to a more widespread group38 by releasing pulmonary hypoxic vasoconstriction, worsening V/Q matching and increasing hypercapnia. A recent randomised controlled trial31 showed a decrease in PaCO2 in a group receiving 28% O2 and an increase in PaCO2 in a group receiving 100% O2.
β-AGONISTS
Short-acting β-agonists remain the first-line bronchodilator therapy of choice.6,39–41 Agents include salbutamol (albuterol), terbutaline, isoprenaline and epinephrine. Salbutamol is generally the agent of first choice as it has relative β2-selectivity, with decreased β1-mediated cardiac toxicity. Long-acting β-agonists such as salmeterol have no role in status asthmaticus due to slow onset of action and association with fatalities in this setting.14 Eformotarol is a combined long- and short-acting β-agonist which could theoretically be used for acute asthma; however it is in dry powder form and unlikely to be helpful in intubated and mechanically ventilated patients. β-agonists cause bronchodilatation by stimulation of β2-receptors on airway smooth muscle and may reduce bronchial mucosal oedema.42
β-agonists are best given by metered-dose inhaler (MDI) and a spacer device. There are data to suggest that, in non-intubated patients, MDIs combined with a spacer device are more effective than nebulisers and are cheaper to use.43,44 In intubated patients both nebulisers and MDIs have been used effectively.45 The dose per MDI inhalation is 100 μg and this can be given, in severe asthma, in repeated doses at 1–5-minute intervals.
Alternatively, β-agonists can be given by nebuliser in high and repeated doses.46 The typical adult dose of salbutamol is 5–10 mg (in 2.5–5.0 ml diluent volume) every 2–4 hours but more frequent doses with a higher total dose are often required in severe asthma. It should be noted that less than 10% of the nebulised drug reaches the lung even under ideal conditions.47 Continuous nebulisation appears to be superior to intermittent doses and is commonly used at the beginning of treatment in severe asthma.14,48 The nebuliser should be driven by oxygen with the flow at 10–12 l/min and a reservoir volume of 2–4 ml so as to produce particles in the desired 1–3 μm range.49 The total dose should be modulated by response to treatment and the level of toxic side-effects.
Two-thirds of patients presenting acutely will respond well to inhaled β-agonists,50 irrespective of the method of administration. The remaining one-third are refractory even to high doses and usually require longer periods of intense treatment, including multiple other agents.
Intravenous β-agonists remain controversial. There is no clear evidence of benefit51 and significant side-effects. Despite this, intravenous β-agonists have a theoretical benefit of additional access to lung units with severe airflow obstruction and poor nebulised drug delivery, and some studies have demonstrated improved response when intravenous β-agonist is used.52 Intravenous β-agonists continue to be considered if the patient is not responding to continuous nebulisation.53 The typical dose is 5–20 μg/min but doses > 10 μg/min should be used with caution because of side-effects, which should be monitored closely. Salbutamol 100–300 μg may also be given intravenously to non-intubated patients in extremis or delivered down an endotracheal tube if there is not time to gain intravenous access.
Side-effects of β-agonists include tachycardia, arrhythmias, hypertension, hypotension, tremor, hypokalaemia, worsening of ventilation–perfusion mismatch and hyperglycaemia,54 but the most common side-effect of parenteral β-agonist – also occasionally seen with continuous nebulised β-agonists – is lactic acidosis. This occurs in over 70% of patients, has an onset within 2–4 hours of commencing an infusion or following an intravenous statim dose, levels may reach 4–12 mmol/l and may significantly add to respiratory acidosis and respiratory distress.34,55,56 Parenteral infusions should be initially limited to 10 μg/min and statim doses should not exceed 250 μg. Serum bicarbonate and lactate should be regularly monitored. If lactic acidosis becomes significant, the salbutamol infusion should be reduced or ceased. Lactic acidosis will generally resolve within 4–6 hours of infusion cessation and is seldom a problem with infusions in place for more than 24 hours.
Long-term high-dose β-agonist use has been associated with increased mortality,57 but whether high-dose β-agonists are a marker of disease severity, an indicator of suboptimal inhaled steroids or a direct cause of death is unclear. These concerns do not apply in the treatment of the acute asthma attack.
ANTICHOLINERGICS
Anticholinergics cause bronchodilatation by decreasing parasympathetic-mediated cholinergic bronchomotor tone.58 Ipratropium bromide is the most commonly used anticholinergic for asthma and is a quaternary derivative of atropine. A number of studies and meta-analyses now suggest clear additional benefit and few side-effects when ipratropium bromide is added to the β-agonist regimen59,60 and ipratropium is now considered accepted first-line therapy for acute severe asthma in conjunction with β-agonist therapy. Preservative-induced bronchoconstriction has been reported in a few patients and can be prevented by using preservative-free solutions.61 The bronchodilatation effect of ipratropium bromide appeared to be maximal with a dose of 250 μg when studied in children between 9 and 17 years of age. The optimal dose is not known in adults; a reasonable regimen would be to add 500 μg ipratropium bromide to the salbutamol nebuliser every 2–6 hours; however, initial dose intervals as low as 10–20 minutes have been recommended.62
CORTICOSTEROIDS
The role of corticosteroids in the acute asthma attack has been well established. Systemic steroids should be considered in all but mild exacerbations of asthma.41 Their benefits include increased β-responsiveness of airway smooth muscle, decreased inflammatory cell response and decreased mucus secretion. Early treatment with corticosteroids has been shown to decrease the likelihood of hospitalisation and decrease the mortality rate from acute asthma. Systematic reviews41,63 suggest that effects commence within 6–12 hours, that oral administration is as effective as intravenous and that there is little evidence of benefit for initial daily doses exceeding 800 mg/day hydrocortisone (160 mg/day methylprednisolone) given in four divided doses.
Inhaled steroids have established long-term benefit and are believed to be a major factor in asthma mortality reduction.1–3 There is now emerging evidence that inhaled steroids may also have a role during an acute attack64,65 and it appears reasonable to use them routinely from day 1 as they may also enable more rapid dose reduction of parenteral steroids, potentially reducing side-effects.
Side-effects of corticosteroids include hyperglycaemia, hypokalaemia, hypertension, acute psychosis and myopathy,66,67 though they are usually well tolerated acutely. The immunosuppressive effects can increase the risk of infections, including Legionella, Pneumocystis carinii and varicella,68,69 especially when the patient is on long-term corticosteroids. Allergic reactions including anaphylaxis have been reported with the use of most corticosteroid preparations.
AMINOPHYLLINE
There have been conflicting reports regarding the efficacy of aminophylline in acute asthma ranging from no benefit70 to improved lung function and improved outcome.71 However it is accepted that aminophylline is an inferior bronchodilator, with a narrow therapeutic range and frequent side-effects,72 including headache, nausea, vomiting and restlessness; cardiac arrhythmias and convulsions can occur at serum levels above 200 μmol/l (40 mg/l).
As a result, aminophylline is not a first-line treatment.40,41,53 Aminophylline may be given to patients with acute asthma who are not showing a favourable response to full treatment with first-line agents. Careful administration and monitoring are required with an initial loading dose of 3 mg/kg (maximum 6 mg/kg, omitted if the patient is already taking oral theophylline) and an infusion of 0.5 mg/kg per hour. This should be reduced in patients with cirrhosis, cardiac failure or chronic obstructive pulmonary disease and in patients taking cimetidine, erythromycin or antiviral vaccines. Drug levels should be taken after a loading dose (if given), and then 24 hours later, aiming for a level of 30–80 μmol/l (5–12 mg/l). Levels should be repeated daily thereafter until stability has been achieved. The duration should be based on the response to treatment.
