[level-membership-for-anesthesiology-category]
Chapter 26 Acute respiratory failure in chronic obstructive pulmonary disease
The terms ‘chronic obstructive pulmonary or airways disease’ (COPD or COAD) are applied to patients with chronic bronchitis and/or emphysema. COPD affects 5% of the adult population, is the fifth most common cause of death worldwide and is the only major cause of death that is increasing in prevalence.1 Despite this, when an acute deterioration occurs, most precipitating factors are reversible and the outcome is usually good.2 This justifies aggressive management in the majority of patients.
AETIOLOGY
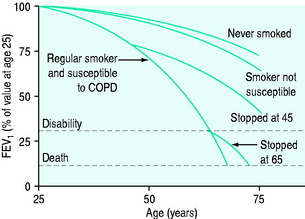
The causes of COPD can be divided into environmental and host factors. Environmental factors include tobacco smoke, air pollution, indoor fumes (e.g. indoor cooking with solid biomass fuel) and poor socioeconomic status. The biggest single factor in over 95% of patients with COPD is tobacco smoking (Figure 26.1). However, only approximately 15% of smokers develop COPD. Marijuana smoking may cause premature and quite advanced bullous emphysema compared with tobacco smokers due to extremely hot and toxic inhaled smoke held at peak inspiration for prolonged periods of time.3 Host factors are the balance between circulating proteases and antiproteases (e.g. alpha-1 antitrypsin deficiency) and the intake of antioxidant vitamins (A, C, E).4
PATHOPHYSIOLOGY
Pulmonary hyperinflation has both static and dynamic components. The static component remains at the end of an expiratory period long enough for all expiratory airflow to cease (30–120 s), enabling the lungs and chest wall to reach their static functional residual capacity (FRC). This component of hyperinflation is due to loss of parenchymal elastic recoil, chest wall adaptation5 and airway closure that occurs throughout expiration. Dynamic pulmonary hyperinflation is the further increase in hyperinflation due to slow expiratory airflow not allowing completion of expiration before the arrival of the next breath. The extent of dynamic hyperinflation depends on the severity of airflow obstruction, the amount inspired (tidal volume) and the expiratory time.6 Thus, the degree of hyperinflation may vary in a patient with changes in minute ventilation due to changes in CO2 production (depending on exercise, diet or the metabolic response to illness) or dead space, as well as with changes in airflow obstruction during an exacerbation.
Central respiratory drive may also be impaired, or poorly responsive to physiological triggers – hypoxaemia or hypercapnia – and lead to chronic hypercapnia. This may occur in the setting of sleep (i.e. obstructive sleep apnoea), obesity or drugs (sedatives, antiepileptics, alcohol).
CHRONIC BRONCHITIS OR EMPHYSEMA?
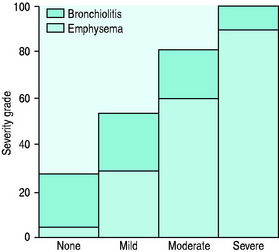
The value of labelling patients as chronic bronchitis or emphysema is uncertain as the two disease processes usually coexist and the principles of management are similar. Five pathophysiologic processes may be present to varying degrees in each patient with COPD: (1) inflammatory airway narrowing (bronchiolitis); (2) loss of connective tissues tethering airways; (3) loss of alveoli and capillaries; (4) hyperinflation; and (5) increased pulmonary vascular resistance. Early/mild COPD tends to be dominated by bronchiolitis with a minimal component of emphysema (Figure 26.2), whereas when COPD becomes severe, the reverse is true. However, recognition that COPD is dominated by one of these patterns is helpful with regard to clinical pattern and prognosis.
CLINICAL FEATURES OF ACUTE RESPIRATORY FAILURE IN COPD
Acute respiratory failure (ARF) in COPD can present with two distinct clinical patterns7 (Table 26.1).
Table 26.1 Clinical differences between normocapnic and hypercapnic chronic obstructive pulmonary disease
| Normocapnic (PaCO2 35–45 mmHg) | Hypercapnic (PaCO2 > 45 mmHg) |
|---|---|
| Emphysema > chronic bronchitis | Chronic bronchitis > emphysema |
| Thin | Obese |
| Pursed-lip breathing | Central nervous system depression: consider the role of oxygen therapy |
| Accessory muscle use | Alcohol, sedatives, analgesics |
| Hyperinflated | Sleep-related hypoventilation |
| Right heart failure late | Right heart failure early |
PRECIPITANTS OF ACUTE RESPIRATORY FAILURE
In approximately 50% of patients, there is an infective cause, in 25% heart failure and in the remaining 25% retained secretions, air pollution, coexistent medical problems (e.g. pulmonary embolus, medication compliance or side-effects) or no cause can be identified8 (Table 26.2).
Table 26.2 Precipitants of acute respiratory failure in chronic obstructive pulmonary disease
| Infective (including aspiration) |
| Left ventricular failure (systolic and diastolic failure) |
| Sputum retention (postoperative, traumatic) |
| Pulmonary embolism |
| Pneumothoraces and bullae |
| Uncontrolled oxygen |
| Sedation |
| Medication – non-compliance or side-effects |
| Nutritional (K, PO4, Mg deficiency, CHO excess) |
| Sleep apnoea |
The most common bacterial isolates are Streptococcus pneumoniae and Haemophilus influenzae in 80% of exacerbations.9S. viridans,10Moraxella (previously Branhamella) catarrhalis,11Mycoplasma pneumoniae12 and Pseudomonas aeruginosa may also be found. Viruses can be isolated in 20–30% of exacerbations13 and include rhinovirus, influenza and parainfluenza viruses, coronaviruses and occasionally adenovirus, and respiratory syncytial virus. Whether these organisms are pathogens or colonisers is often unclear.
PNEUMONIA
Pneumonia has been estimated to account for 20% of presentations requiring mechanical ventilation.13 It is most commonly caused by S. pneumoniae and H. influenzae but Mycoplasma, Legionella, enteric Gram-negatives and viruses are occasional causes.
LEFT VENTRICULAR FAILURE
Left ventricular (LV) systolic failure may result from coexisting ischaemic heart disease, fluid overload, tachyarrhythmias or biventricular failure secondary to cor pulmonale. LV diastolic failure occurs commonly and is precipitated by hypoxaemia, tachycardia,14 pericardial constraint due to intrinsic positive end-expiratory pressure (PEEPi) or right ventricular (RV) dilation. Increased work of breathing related to COPD will also increase by up to 10-fold the amount of blood flow to the respiratory pump muscles,15 thereby causing an increased demand upon the overall cardiac output. In patients with borderline cardiac status, this may precipitate heart failure. The components of right and LV failure can be accurately distinguished by Doppler echocardiography. Pulmonary congestion can be difficult to diagnose because of the abnormal breath sounds and chest X-ray appearance which are commonly present in COPD. In a recent publication, 51% of patients with acute exacerbation of COPD had echocardiographic evidence of left heart failure (systolic 11%, diastolic 32%, systolic and diastolic 7%).16
DIAGNOSIS AND ASSESSMENT
DIAGNOSIS
The clinical examination findings of COPD depend upon severity.
A chest X-ray will commonly show hyperinflated lung fields, as suggested by 10 ribs visible posteriorly, six ribs visible anteriorly or large airspace anterior to heart (> 1/3 of the length of the sternum), flattened diaphragms (best seen on lateral chest X-ray) and a paucity of lung markings. Pulmonary hypertension is manifest by enlarged proximal and attenuated distal vascular markings and by RV and atrial enlargement. Lung bullae may be evident.
A high-resolution computed tomographic (CT) scan of the chest (1–2-mm slices) can demonstrate characteristic appearance and regional distribution of emphysema. It can also assess for coexistent bronchiectasis, LV failure17 and pulmonary fibrosis. Such scans are less sensitive than standard chest CT scans (1-cm slice) for detecting pulmonary lesions (e.g. neoplasms). Nuclear ventilation perfusion scans can also provide a characteristic appearance of COPD.
DIFFERENTIAL DIAGNOSIS
Chronic heart failure (CHF) may be a differential diagnosis of COPD, or simply coexist, as both disorders are common in smokers.14,16 Orthopnoea and paroxysmal nocturnal dyspnoea are features which correlate with heart failure severity. A past history of myocardial ischaemia or atrial fibrillation should alert one to the possibility of heart failure. An echocardiogram and high-resolution CT (looking for shift in interstitial oedema with changes in posture from supine to prone)17 are sensitive markers of CHF.
NON-VENTILATORY MANAGEMENT
OXYGEN THERAPY
Oxygen given by low-flow intranasal cannulae or 24–35% Venturi mask should be titrated to achieve a saturation (SpO2) of 90 ± 2% as these levels will avoid significant increases in PaCO2 in the majority of COPD patients with ARF. Increases in PaCO2 are most common in patients with initial PaCO2 > 50 mmHg and pH < 7.35.18 If the rise in PaCO2 is excessive (> 10 mmHg or 1.33 kPa), then FiO2 should be reduced, titrating SpO2 to 2–3% below the previous value, and arterial blood gases should be repeated. If no PaCO2 rise occurs with oxygen therapy, then a higher SpO2 may be targeted with repeat blood gases.
Inadequate reversal of hypoxia (e.g. SpO2 < 85%) is suggestive of an additional problem such as pneumonia, pulmonary oedema or embolus, or a pneumothorax. Investigation of this should commence and a higher O2 delivery system should be used (see Chapter 24). Although high levels of O2 should be avoided, reversal of hypoxia is important and O2 should not be withheld in the presence of hypercapnia, or withdrawn if it worsens.
BRONCHODILATORS
Bronchodilators are routinely given in all acute exacerbations of COPD because a small reversible component of airflow obstruction is common, and bronchodilators improve mucociliary clearance of secretions.19 However, a large meta-analysis of 22 large randomised controlled long-term trials of ambulatory COPD patients involving either anticholinergics and/or β2 agonists (short- and long-acting) over 3–60 months indicates that anticholinergics are more favourable than placebo in terms of acute exacerbations, hospitalisations and respiratory deaths.20 There were no favourable advantages with β2-agonists compared with placebo for acute exacerbations or hospitalisations, and placebo was better than β2-agonists in terms of respiratory death.20
ANTICHOLINERGIC AGENTS
Anticholinergic agents, such as ipratropium bromide, have been shown to have a similar or greater bronchodilator action than β-agonists in COPD,1,21,22 and also to have fewer side-effects and no tachyphylaxis. Anticholinergic agents should be used routinely in COPD with ARF and many now believe them to be the agent of first choice.1 An ipratropium bromide nebule of 0.5 mg in 2 ml should be nebulised initially 2-hourly, then every 4–6 hours. Long-term use of ipratropium bromide has been shown to reduce the incidence of exacerbations23 and is therefore recommended for chronic use in ambulatory COPD. Long-acting anticholinergics (e.g. tiotropium) offer potential of once-daily dosing.
NEBULISED β2-AGONISTS
Nebulised β-agonists are also effective bronchodilators in COPD,21,22 although they may cause tachycardia, tremor, mild reductions in potassium and PaCO2 (due to pulmonary vasodilatation) and tachyphylaxis. Nebulised β-agonists (e.g. salbutamol, terbutaline or fenoterol) given 2–4-hourly should be used routinely in combination with ipratropium. This combination has been shown to be more effective than either agent alone.1 Parenteral sympathomimetic agents are rarely indicated and not recommended for routine use. In stable patients, long-term use of β-agonists may improve symptoms of dyspnoea, particularly in the subgroup of COPD with an objective bronchodilator response. Long-acting β-agonists may also have a beneficial effect on symptoms, quality of life and exercise capacity in COPD.24
AMINOPHYLLINE
Aminophylline is a weak bronchodilator in COPD. It improves diaphragm contractility,25 stimulates respiratory drive,26 improves mucociliary transport27 and right heart function,28 is anti-inflammatory29 and is a weak diuretic. Some studies have shown no benefit and significant side-effects,30 whereas others have shown small benefit31 instable COPD. Theophylline has a number of additional effects that may also be of benefit in COPD, although the clinical importance of these is not yet established. For an exacerbation, aminophylline (loading dose 5–6 mg/kg IV over 30 min, followed by an infusion of 0.5 mg/kg per h) is commonly also given, despite doubt about its overall benefit. Serum theophylline levels must be monitored regularly to reduce risk of toxicity. The low therapeutic range should be targeted (55–85 mmol/l) for the best ratio of effect to side-effect.32 The high therapeutic range (85–110 mmol/l) has little additional benefit and a significant increase in side-effects,32 but is thought necessary for diaphragm contractility and respiratory stimulation effects. There is some evidence in favour of theophylline use in the long-term management33 of COPD but, due to its narrow therapeutic window, it should be used with caution.
STEROIDS
In acute exacerbations of COPD, short-term steroids have been shown to improve airflow obstruction,34 including those patients requiring mechanical ventilation for COPD.35 Doses similar to those for acute asthma should be used. Methylprednisolone 0.5 mg/kg, given 6-hourly for 72 h, was used in the study by Albert et al.,34 demonstrating benefit in patients with an exacerbation of COPD. Current American Thoracic Society guidelines recommend the equivalent to oral prednisolone at 0.5 mg/kg body weight for up to 10 days, then ceasing; however, this will depend upon the response to treatment, and their premorbid use.1 Steroids should be avoided if the deterioration is clearly due to bacterial pneumonia without bronchospasm.
Longer-term oral steroids in COPD are associated with a substantial increased risk of side-effects (osteoporosis, diabetes, peptic ulcer, myopathy, systemic hypertension, fluid retention, weight gain), which are likely to impair quality of life and precipitate readmission and are therefore not recommended.1 A small group of patients (15%) may demonstrate a significant bronchodilator response; coexistent asthma is likely in these patients and longer-term high-dose (oral or inhaled) steroids may be necessary. In the majority of patients, long-term inhaled steroids do not improve lung function or survival; however, they do improve quality of life and reduce admissions and are therefore recommended as long-term treatment at low to medium dose.1
ANTIBIOTICS
Antibiotics have an accepted role in the treatment of infection-induced exacerbations of COPD. Amoxicillin is a suitable first-line agent against Haemophilus influenzae and Streptococcus pneumoniae for outpatient exacerbations.36 Serious exacerbations requiring hospital admission require newer agents such as ciprofloxacin or a third-generation cephalosporin.37 Antibiotics for pneumonia are discussed elsewhere in this volume.
SECRETION CLEARANCE TECHNIQUES
Clearance of lower respiratory secretions is of crucial importance.
NEBULISED MUCOLYTIC AGENTS
Nebulised mucolytic agents, such as acetylcysteine, continue to be proposed, although their benefit has never been established in acute exacerbations of COPD.38 Oral mucolytics have been shown to reduce cough frequency and severity in stable COPD.39
OTHER MEASURES
Other adjunctive measures are applicable to some patients.
HYDRATION, DIURETICS, DIGOXIN AND VASODILATORS
COPD patients are sensitive to changes in fluid status and intravenous hydration should be undertaken with care and minimised. Diuretics and digoxin are of benefit if LV failure is present. Even if evidence of LV failure is minimal, a trial of diuresis is worthwhile in patients refractory to usual treatment. Digoxin has been shown to improve LV function in patients with cor pulmonale.40
Diuretics will reduce fluid overload in cor pulmonale; however, care should be taken with severe pulmonary hypertension where a decrease in RV filling pressure may result in a low-output state. Digoxin is of no established benefit to RV function in cor pulmonale, where the primary problem is increased afterload.41 Pulmonary hypertension is commonly present in severe COPD and is associated with a poor prognosis. In stable COPD pulmonary vasodilators may have acutely beneficial effects42 but have not yet been shown to improve outcome.43 In ARF, pulmonary hypertension invariably increases and may precipitate acute cor pulmonale. Although not clinically proven, pulmonary vasodilators have a rational basis in this setting. Numerous agents have been or are undergoing trials to reduce pulmonary hypertension in COPD. These agents include calcium channel blockers (e.g. nifedipine), endothelin receptor antagonists (e.g. bosentan), phosphodiesterase inhibitors (e.g. sildenafil) and prostacycline analogues (e.g. epoprostenil, iloprost and trepostinil).42 Side-effects include systemic hypotension, tachycardia, worsening of hypoxia and failure to improve haemodynamics. A trial of vasodilators appears reasonable if cor pulmonale is present with ARF, provided that response to therapy is carefully monitored. The diagnosis and response to treatment of pulmonary hypertension may require a right heart catheter.
ELECTROLYTE CORRECTION
Electrolyte correction is important. Hypophosphataemia is common and may cause respiratory muscle weakness.44 Hypomagnesaemia,45 hypocalcaemia46 and hypokalaemia may also be present and may impair respiratory muscle function. Hyponatraemia may occur with inappropriate antidiuretic hormone release or with excess use of diuretics and inappropriate intravenous fluids.
RESPIRATORY STIMULANTS
Many drugs have been shown to increase respiratory drive and lower PaCO2. These include acetazolamide, medroxyprogesterone, naloxone, doxapram and almitrine. Use of such agents presupposes that the limiting factor is reduced respiratory drive, whereas the majority of ARF is limited by a reduced capacity to deal with an increased load to breathing. Side-effects include increased dyspnoea and fatigue, impaired sleeping, with almitrine, and increased pulmonary hypertension. Respiratory stimulants have not been shown to improve either short-term or long-term outcome. As a result respiratory stimulants are not recommended or used in ARF. Narcotic- or benzodiazepine-induced respiratory depression is best managed with the appropriate antagonist – naloxone or flumazenil.
NON-INVASIVE VENTILATION (NIV)
NIV is a technique in which ventilatory support is provided via a nasal or facial mask without endotracheal intubation. There have been several randomised controlled trials of NIV in COPD patients with acute hypercapnic respiratory failure which have demonstrated improved respiratory physiology, reduced mortality (up to 12 months), reduced iatrogenic complications, reduced need for intubation and mechanical ventilation and reduced length of stay in hospital.47–50 All studies have shown good tolerance of the technique (∼80% of patients), with few side-effects, improvements in both oxygenation and PaCO2 compared with medically treated control patients.51,52
Indications for NIV are a deterioration of COPD with: (1) acute dyspnoea; (2) respiratory rate > 28 breaths/min; (3) PaCO2 > 45 mmHg with a pH < 7.35, despite optimal medical treatment and not related to excessive supplemental oxygen. Although these indications are for mild exacerbations, most randomised studies have used these as entry guidelines.47–50 Initial guidelines recommended NIV use to be limited to patients with pH in the range 7.25–7.35; however, recent evidence suggests that NIV is useful even in those patients with lower pH values (to as low as 7.0) and associated more severe hypercapnia (as high as 140 mmHg).53 Included in the indications are recently extubated patients in whom NIV has been shown to reduce reintubation rates significantly.54 Recently NIV has been advocated for use in patients with hypoxic respiratory failure,55 but success is significantly less in the setting of hypoxaemia and either normocapnia or hypocapnia.56 NIV may also have a role in some patients where mechanical ventilation is considered inappropriate.
Side-effects of NIV have included discomfort, intolerance, skin necrosis, gastric distension and aspiration. Pressure support has been reported as better tolerated than assist/control.51
INVASIVE MECHANICAL VENTILATION
OUTCOME OF ARF AND THE DECISION TO VENTILATE INVASIVELY
Patients with COPD have a reduced life expectancy compared with an age-matched general population group and this life expectancy decreases in proportion with severity of COPD as assessed by FEV1 (Figure 26.3).
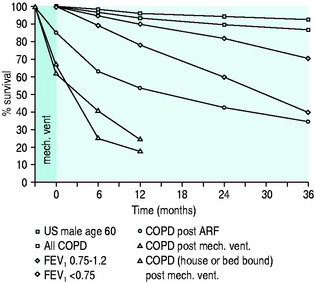
Figure 26.3 Survival curves for various patient groups with chronic obstructive pulmonary disease (COPD). Note: these are approximations only, based on other studies13,57 with different patient numbers, criteria and study periods. FEV1, forced expiratory volume in 1 second; ARF, acute respiratory failure.
An episode of ARF further decreases survival (see Figure 26.3). ARF precipitated only by bronchitis has a better outcome,2 whereas ARF due to more serious causes, such as pneumonia, LV failure and pulmonary embolus, has a worse outcome57 and studies including all such outcomes have lower survival rates (see Figure 26.3).
If ARF requires invasive mechanical ventilation, survival decreases further still (see Figure 26.3). Although the majority of patients do not require mechanical ventilation, the short-term survival in this more severe subset is still good, with a hospital survival rate in some series as high as 80%,58 but 2- and 3-year survival is significantly lower (see Figure 26.3).59,60 The severity of ARF and the severity of underlying COPD based on FEV1, lifestyle score and dyspnoea score are also predictors of outcome.13,57 Lifestyle13 and dyspnoea61 categories may be the most useful factors in the decision to withhold mechanical ventilation. Lifestyle categories 3 (house-bound and at least partly dependent) and 4 (bed- or chair-bound) indicate both a poor outcome (see Figure 26.3)13 and quality of life that may not justify aggressive treatment.
INDICATIONS FOR INVASIVE MECHANICAL SUPPORT
MECHANICAL VENTILATION TECHNIQUE
Excessive dynamic hyperinflation must be avoided by using a low minute ventilation – 115/ml per kg is a guideline6 – and allowing adequate time for expiration. This should be achieved by the use of a small tidal volume (8 ml/kg) and a ventilator rate < 14 breaths/min.6,62 Dynamic hyperinflation can be assessed clinically, by visualising the expiratory flow–time curve, and by measuring plateau airway pressure (Pplat) or PEEPi. Pplat should be measured by applying an end-inspiratory pause of 0.5 s. This should only be applied following a single breath as it shortens expiratory time and, if it is applied to a series of breaths, it increases dynamic hyperinflation, resulting in an increased Pplat level and increased risk to the patient. If Pplat is > 25 cmH2O, there is likely to be dynamic hyperinflation, and the ventilator rate should be reduced. However, Pplat may be high without dynamic hyperinflation if chest wall compliance is low. Intrinsic PEEP measured as a prolonged end-expiratory pause more directly assesses dynamic hyperinflation. Provided PEEPi is accurately measured, it is a useful tool to follow dynamic hyperinflation. In severe airflow limitation it may be necessary to accept low levels of PEEPi, but as PEEPi rises above 8–10 cmH2O, further prolongation of expiratory time must be considered. Although still controversial, the use of a high inspiratory flow rate is recommended,6,63 as it results in a shorter inspiratory time and hence a longer expiratory time for a given ventilatory rate. It has been shown to reduce dynamic hyperinflation and alveolar pressure6 further and to improve gas exchange.63
If dynamic hyperinflation is excessive and causing circulatory compromise or risk of barotrauma, then minute ventilation should be decreased, hypercapnic acidosis accepted and spontaneous ventilation, which will only increase dynamic hyperinflation, should be discouraged by sedation. Muscle relaxants should be avoided unless essential. When dynamic hyperinflation is critical during controlled mechanical ventilation, PEEPi increases pulmonary hyperinflation and should not be applied.64
If dynamic hyperinflation is not excessive then spontaneous ventilation should be encouraged to promote ongoing respiratory muscle activity and to minimise wasting. Flow-by, pressure support and low-level CPAP may all reduce the work of spontaneous breathing and promote a better ventilatory pattern. CPAP approximately equal to the level of PEEPi is most commonly recommended.65 Care must be taken with all of these supports as each can increase dynamic hyperinflation by a different mechanism, leading to circulatory compromise or risk of barotrauma. Flow-by increases resistance through the expiratory valve, pressure support increases tidal volume and may increase inspiratory time and CPAP increases FRC.
WEANING FROM INVASIVE MECHANICAL VENTILATION
Weaning a patient with severe COPD from ventilatory support can be difficult and prolonged. Numerous criteria have been proposed to assess the capacity of the patient to wean;66 however, the predictive value of any of these individual criteria is limited. The simple criterion of patient respiration rate/tidal volume < 100 breaths/min per litre had the best predictive value for weaning success, but the advantage of this overly simple clinical assessment during weaning is uncertain.66 Weaning or withdrawal of ventilation is further discussed elsewhere in this volume.
TRACHEOSTOMY
Consider removing the tracheostomy when:
POST INTENSIVE UNIT CARE
REHABILITATION
Rehabilitation should be considered for all patients with COPD, particularly those following ARF. There are numerous randomised controlled trials showing improvements in exercise physiology, lung function, quality of life and reduced hospitalisation rates.67,68 Such programmes include extensive education and aerobic upper and lower-limb and respiratory muscle exercise, usually three times per week over a 6-week period. Patients are recommended to continue with exercises regularly and independently thereafter.
VACCINATION
Vaccination should be considered in all patients with COPD when stable. Annual influenza and 5-yearly pneumococcal vaccination is recommended.1
LUNG VOLUME REDUCTION SURGERY
Lung volume reduction surgery is a palliative surgical procedure for patients aged < 75 years, with advanced disabling emphysema. Poorly perfused and poorly ventilated areas of one or both lungs, thought to compress relatively preserved areas of well-functioning lung tissue, are excised by either midline sternotomy or endoscopically, with video assistance.69 Such areas in the lung, usually apically placed, are determined by V/Q scans and high-resolution CT scans. Suitable patients must have undergone a pulmonary rehabilitation programme, be on optimal medical treatment and have disabling dyspnoea and anatomically suitable (apical bullous) emphysema. Patients with profoundly severe emphysema (e.g. FEV1 or TLCO < 20% predicted) are unlikely to benefit.69 Group mean data suggest that the mean FEV1 increases from 0.5 to 0.91 litres, and the mean 6-min walk distance increases from 205 to 290 metres, both associated with significant improvement in quality of life. The peak improvements in physiology occur at 1–2 years and thereafter decline to presurgical levels. Improvements in survival are yet to be determined.69
LUNG TRANSPLANTATION
Lung transplantation is another palliative surgical procedure for patients with advanced disabling COPD who are aged < 65 years, are not ventilator-dependent, are on less than 10 mg prednisolone/day and are free of significant coexistent disease. It may be used following lung volume reduction surgery. The current 1-, 2- and 5-year international survival figures are 75, 66 and 50% respectively.70 Common complications are systemic hypertension, bronchiolitis obliterans, acute rejection, viral infection with cytomegalovirus and neoplasms.66 Its widespread application in COPD is in doubt because of the very limited supply of donor lungs.71
PROGNOSIS
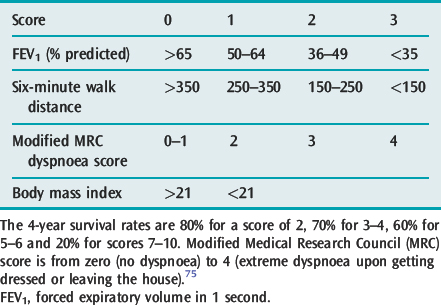
Patients with sufficiently severe COPD to warrant hospital admission incur an inpatient mortality of 7%, and 90-day mortality of 15% in the UK.72 Predictors of mortality were performance status, age and admission urea, albumin, pH and SpO2 plus the presence of respiratory physicians involved in the care.72 Although in-hospital mortality for hypercapnic COPD patients may reach 62%,73 in an Australian report, in-hospital mortality for hypercapnic COPD patients treated with NIV was 11% and all deaths were with palliative intent, which followed time to allow for patient and family discussions. In Hong Kong, acute hypercapnic COPD patients have a 12-month readmission rate of 80% and a 49% 1-year mortality.74 The body mass index, airflow obstruction, dyspnoea and exercise capacity (BODE) index75 is a 10-point scale made up from the following four variables: (1) body mass index; (2) airflow obstruction; (3) severity of dyspnoea; and (4) exercise capacity, and has been found to be very useful in predicting survival in ambulatory patients with COPD (Table 26.3).
1 Pauwels RA, Buist AS, Ma P, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. NHLBI/WHO global initiative for chronic obstructive lung disease (GOLD) workshop summary. Am J Respir Crit Care Med. 2001;163:1256-1276.
2 Martin T, Lewis S, Albert R. The prognosis of patients with chronic obstructive pulmonary disease after hospitalization for acute respiratory failure. Chest. 1982;82:310-314.
3 Hii S, Lam J, Thompson BR, et al. Bullous lung disease due to marijuana. Respirology. 2008;13:122-127.
4 Britton JR, Pavord ID, Richards KA, et al. Dietary antioxidant vitamin intake and lung function in the general population. Am J Respir Crit Care Med. 1995;151:1383-1387.
5 Burrows B, Fletcher C, Heard B. The emphysematous and bronchial types of chronic airways obstruction. A clinico-pathological study of patients in London and Chicago. Lancet. 1966;7442:1830-1835.
6 Tuxen D, Lane S. The effects of ventilatory pattern on hyperinflation, airway pressures, and circulation in mechanical ventilation of patients with severe airflow obstruction. Am Rev Respir Dis. 1987;136:872-879.
7 Fahey P, Hyde R. ‘Won’t breathe’ versus ‘can’t breathe’. Detection of depressed ventilatory drive in patients with obstructive pulmonary disease. Chest. 1983;84:19-25.
8 Connors AFJr., Dawson NV, Thomas C, et al. Outcomes following acute exacerbations of severe chronic obstructive lung disease. Am J Respir Crit Care Med. 1996;154:959-967.
9 Schreiner A, Bjerkestrand G, Digrannes A. Bacteriologic findings in the trans-tracheal aspirate from patients with acute exacerbations of chronic bronchitis. Infection. 1978;6:54-56.
10 Irwin R, Erickson A, Pratter M. Prediction of tracheobronchial colonization in current cigarette smokers with chronic obstructive bronchitis. J Infect Dis. 1982;145:234-241.
11 Christensen J, Gadeberg O, Bruvn B. Branhamella catarrhalis: significance in pulmonary infections and bacteriological features. Acta Pathol Microbiol Immunol Scand. 1986;94:89-95.
12 Smith CB, Golden CA, Kanner RE, et al. Association of viral and Mycoplasma pneumoniae infections with acute respiratory illness in patients with chronic obstructive pulmonary disease. Am Rev Respir Dis. 1980;121:225-232.
13 Menzies R, Gibbons W, Goldberg P. Determinants of weaning and survival among patients with COPD who require mechanical ventilation for acute respiratory failure. Chest. 1989;95:398-405.
14 Baum GL, Schwartz A, Llamas R, et al. Left ventricular function in chronic obstructive lung disease. N Engl J Med. 1971;285:361-365.
15 Robertson C, Foster G, Johnson R. The relationship of respiratory failure to the oxygen consumption of, lactate production by, and distribution of blood flow among respiratory muscles during increasing inspiratory resistance. J Clin Invest. 1977;59:31-42.
16 Abroug F, Ouanes-Besbes L, Nciri N, et al. Association of left-heart dysfunction with severe exacerbation of chronic obstructive pulmonary disease: diagnostic performance of cardiac biomarkers. Am J Respir Crit Care Med. 2006;174:990-996.
17 Kato S, Nakamoto TK, Iizuka M. Early diagnosis and estimation of pulmonary congestion and edema in patients with left-sided heart disease from histogram of pulmonary CT number. Chest. 1996;109:1439-1445.
18 Bone R, Pierce A, Johnson R. Controlled oxygen administration in acute respiratory failure in chronic obstructive pulmonary disease. Am J Med. 1978;65:896-902.
19 Mossberg B, Strandberg K, Philipson K, et al. Tracheobronchial clearance and beta-adrenoceptor stimulation in patients with chronic bronchitis. Scand Respir Dis. 1976;57:281-289.
20 Salpeter SE, Buckley NS, Salpeter EE. Anticholinergics but not beta agonists reduce severe exacerbations and respiratory mortality in COPD. J Gen Int Med. 2006;21:1011-1019.
21 Braun SR, McKenzie WN, Copeland C, et al. A comparison of effect of ipratropium and albuterol (salbutamol) in chronic obstructive airway disease. Arch Intern Med. 1989;149:544-547.
22 Karpel JP, Schacter EN, Fanta C, et al. A comparison of ipratropium and albuterol vs albuterol alone for the treatment of acute asthma. Chest. 1996;110:611-616.
23 Friedman M, Serby CW, Menjoge SS, et al. Pharmacoeconomic evaluation of a combination of ipratropium plus albuterol compared with ipratropium alone and albuterol alone in COPD. Chest. 1999;115:635-641.
24 Boyd G, Morice A, Pounsford J. An evaluation of salmeterol in the treatment of chronic obstructive pulmonary disease. Eur Respir J. 1997;10:815-821.
25 Aubier M, De Troyer A, Sampson M, et al. Aminophylline improves diaphragmatic contractility. N Engl J Med. 1981;305:249-252.
26 Berry RB, Desa MM, Branum JP, et al. Effect of theophylline on sleep and sleep-disordered breathing in patients with chronic obstructive pulmonary disease. Am Rev Respir Dis. 1991;143:245-250.
27 Wanner A. Effects of methylxanthines on airway mucociliary function. Am J Med. 1985;79:16-21.
28 Matthay RA, Berger HJ, Davies R. Improvement in cardiac performance by oral long-acting theophylline in chronic obstructive pulmonary disease. Am Heart J. 1982;104:1022-1026.
29 Pauwels R. New aspects of the therapeutic potential of theophylline in asthma. J Allergy Clin Immunol. 1989;83:548-553.
30 Rice KL, Leatherman JW, Duane PG, et al. Aminophylline for acute exacerbations of chronic obstructive pulmonary disease. Ann Intern Med. 1987;107:305-309.
31 Guyatt GH, Townsend M, Pugsley SO, et al. Bronchodilators in chronic airflow limitation. Effects on airway function, exercise capacity, and quality of life. Am Rev Respir Dis. 1987;135:1069-1074.
32 Rogers R, Owens G, Pennock B. The pendulum swings again: toward a rational use of theophylline. Chest. 1985;87:280-282.
33 Chrystyn H, Mulley B, Peake M. Dose response relation to oral theophylline in severe chronic obstructive airways disease. Br Med J. 1988;297:1506-1510.
34 Albert R, Martin T, Lewis S. Controlled clinical trial of methylprednisolone in patients with chronic bronchitis and acute respiratory insufficiency. Ann Intern Med. 1980;92:753-758.
35 Rubini S, Rampullua C, Nava S. Acute effect of corticosteroids on respiratory mechanics in mechanically ventilated patients with chronic airflow obstruction and acute respiratory failure. Am J Respir Crit Care Med. 1994;149:306-310.
36 Hosker H, Cooke N, Hawkey P. Antibiotics in chronic obstructive pulmonary disease. Br Med J. 1994;308:871-872.
37 Basran GS, Joseph J, Abbas AM, et al. Treatment of acute exacerbations of chronic obstructive airways disease – a comparison of amoxycillin and ciprofloxacin. J Antimicrob Chemother. 1990;26(Suppl. F):19-24.
38 Siafakas NM, Vermeire P, Pride NB, et al. Optimal assessment and management of chronic obstructive pulmonary disease (COPD). Eur Respir J. 1995;8:1398-1420.
39 Petty T. The National Mucolytic study. Results of a randomized, double-blind, placebo-controlled study of iodinated glycerol in chronic obstructive bronchitis. Chest. 1990;97:75-83.
40 Mathur P, Pugsley S, Powles A. Effect of digitalis on left ventricular function in chronic cor pulmonale. Am Rev Respir Dis. 1980;121:163.
41 Green L, Smith T. The use of digitalis in patients with pulmonary disease. Ann Intern Med. 1977;87:459-465.
42 Sajkov D, McEvoy R, Cowie R. Felodipine improves pulmonary hemodynamics in chronic obstructive pulmonary disease. Chest. 1993;103:1354-1361.
43 Salvaterra C, Rubin L. Investigation and management of pulmonary hypertension in chronic obstructive pulmonary disease. Am Rev Respir Dis. 1994;148:1414-1417.
44 Aubier M, Murciano D, Lecocguic Y. Effect of hypophosphatemia on diaphragmatic contractility in patients with acute respiratory failure. N Engl J Med. 1985;313:420-424.
45 Dhingra S, Solven F, Wilson A, et al. Hypomagnesemia and respiratory muscle power. Am Rev Respir Dis. 1984;129:497-498.
46 Aubier M, Viires N, Piquet J. Effects of hypocalcemia on diaphragmatic strength generation. J Appl Physiol. 1985;58:2054-2061.
47 Brochard L, Mancebo J, Wysocki M, et al. Noninvasive ventilation for acute exacerbations of chronic obstructive pulmonary disease. N Engl J Med. 1995;333:817-822.
48 Bott J, Carroll MP, Conway JH, et al. Randomised controlled trial of nasal ventilation in acute ventilatory failure due to chronic obstructive airways disease. Lancet. 1993;341:1555-1557.
49 Kramer N, Meyer TJ, Meharg J, et al. Randomised prospective trial of noninvasive positive pressure ventilation in acute respiratory failure. Am J Respir Crit Care Med. 1995;151:1799-1806.
50 Plant P, Owen J, Elliott M. Early use of noninvasive ventilation for acute exacerbations of chronic obstructive pulmonary disease on general respiratory wards: a multicentre randomised controlled trial. Lancet. 2000;355:1931-1935.
51 Mehta S, Hill NS. Noninvasive ventilation. Am J Respir Crit Care Med. 2001;163:540-577.
52 Hillberg RE, Johnson DC. Noninvasive ventilation. N Engl J Med. 1997;337:1746-1752.
53 Crummy F, Buchan C, Miller B, et al. The use of non-invasive ventilation in COPD with severe hypercapnic respiratory acidosis. Respir Med. 2007;101:53-61.
54 Nava S, Ambrosino N, Clini E, et al. Noninvasive mechanical ventilation in the weaning of patients with respiratory failure due to chronic obstructive pulmonary disease. A randomized controlled trial. Ann Intern Med. 1998;128:721-728.
55 Antonelli M, Conti G, Rocco M, et al. A comparison of noninvasive positive-pressure ventilation and conventional mechanical ventilation in patients with acute respiratory failure. N Engl J Med. 1998;339:429-435.
56 Wysocki M, Tric L, Wolff MA, et al. Noninvasive pressure support ventilation in patients with acute respiratory failure. Chest. 1993;103:907-913.
57 Hudson L. Survival data in patients with acute and chronic lung disease requiring mechanical ventilation. Am Rev Respir Dis. 1989;140:S19-24.
58 Petty T. Acute respiratory failure in chronic obstructive pulmonary disease. In: Sheomaker W, Thompson W, Holbrook P, editors. Textbook of Critical Care. Sydney: WB Saunders; 1984:264-272.
59 Burk R, George R. Acute respiratory failure in chronic obstructive pulmonary disease (immediate and long term prognosis). Arch Intern Med. 1972;132:865-868.
60 Admandsson T, Kilburn K. Survival after respiratory failure (145 patients observed 5 to 8.5 years). Ann Intern Med. 1974;80:54-59.
61 Ferris B. Epidemiology standardization project. Am Rev Respir Dis. 1978;118(Suppl):1-120.
62 Curtis J, Hudson L. Emergent assessment and management of acute respiratory failure in COPD. Clin Chest Med (Respir Emerg II). 1994;15:481-500.
63 Connors A, McCaffree D, Gray B. Effect of inspiratory flow rate on gas exchange during mechanical ventilation. Am Rev Respir Dis. 1981;124:537-543.
64 Tuxen D. Detrimental effects of positive end-expiratory pressure during controlled mechanical ventilation of patients with severe airflow obstruction. Am Rev Respir Dis. 1989;140:5-9.
65 Biagorri F, De Monte A, Blanch L. Hemodynamic response to external counterbalancing of auto-positive end expiratory pressure in mechanically ventilated patients with chronic obstructive pulmonary disease. Crit Care Med. 1994;22:1782-1791.
66 Esteban A, Frutos F, Tobin MJ, et al. A comparison of four methods of weaning patients from mechanical ventilation. N Engl J Med. 1995;332:345-350.
67 Lacasse Y, Ferreira I, Brooks D, et al. Critical appraisal of clinical practice guidelines targeting chronic obstructive pulmonary disease. Arch Intern Med. 2001;161:69-74.
68 Goldstein RS, Gort EH, Stubbing D, et al. Randomised controlled trial of respiratory rehabilitation. Lancet. 1994;344:362-368.
69 Stirling GR, Babidge WJ, Peacock MJ, et al. Lung volume reduction surgery in emphysema: a systematic review. Ann Thoracic Surg. 2001;72:641-648.
70 Hosenpud JD, Bennett LE, Keck BM, et al. The registry of the International Society for Heart and Lung Transplantation: seventeenth official report – 2000. J Heart Lung Transplant. 2000;19:909-931.
71 Snell GI, Griffiths A, Macfarlane L, et al. Maximising thoracic organ transplant opportunities: the importance of efficient co-ordination. J Heart Lung Transplant. 2000;19:410-417.
72 Price LC, Lowe D, Hosker HSR, et al. UK national COPD audit 2003: impact of hospital resources and organisation of care on patient outcome following admission for acute COPD exacerbation. Thorax. 2006;61:827-842.
73 Squadrone E, Frigerio P, Fogliati C, et al. Noninvasive vs invasive ventilation in COPD patients with severe acute respiratory failure deemed to require ventilatory assistance. Intens Care Med. 2004;30:1303-1310.
74 Chu CM, Chan VL, Lin AWN, et al. Readmission rates and life threatening events in COPD survivors treated with non-invasive ventilation for acute hypercapnic respiratory failure. Thorax. 2004;59:1020-1025.
75 Celli BR, Cote CG, Marin JM, et al. The body-mass index, airflow obstruction, dyspnea and exercise capacity index in chronic obstructive pulmonary disease. N Engl J Med. 2005;350:1005-1012.
[/level-membership-for-anesthesiology-category][not-level-membership-for-anesthesiology-category]
Chapter 26 Acute respiratory failure in chronic obstructive pulmonary disease
The terms ‘chronic obstructive pulmonary or airways disease’ (COPD or COAD) are applied to patients with chronic bronchitis and/or emphysema. COPD affects 5% of the adult population, is the fifth most common cause of death worldwide and is the only major cause of death that is increasing in prevalence.1 Despite this, when an acute deterioration occurs, most precipitating factors are reversible and the outcome is usually good.2 This justifies aggressive management in the majority of patients.
AETIOLOGY
The causes of COPD can be divided into environmental and host factors. Environmental factors include tobacco smoke, air pollution, indoor fumes (e.g. indoor cooking with solid biomass fuel) and poor socioeconomic status. The biggest single factor in over 95% of patients with COPD is tobacco smoking (Figure 26.1). However, only approximately 15% of smokers develop COPD. Marijuana smoking may cause premature and quite advanced bullous emphysema compared with tobacco smokers due to extremely hot and toxic inhaled smoke held at peak inspiration for prolonged periods of time.3 Host factors are the balance between circulating proteases and antiproteases (e.g. alpha-1 antitrypsin deficiency) and the intake of antioxidant vitamins (A, C, E).4
PATHOPHYSIOLOGY
Pulmonary hyperinflation has both static and dynamic components. The static component remains at the end of an expiratory period long enough for all expiratory airflow to cease (30–120 s), enabling the lungs and chest wall to reach their static functional residual capacity (FRC). This component of hyperinflation is due to loss of parenchymal elastic recoil, chest wall adaptation5 and airway closure that occurs throughout expiration. Dynamic pulmonary hyperinflation is the further increase in hyperinflation due to slow expiratory airflow not allowing completion of expiration before the arrival of the next breath. The extent of dynamic hyperinflation depends on the severity of airflow obstruction, the amount inspired (tidal volume) and the expiratory time.6 Thus, the degree of hyperinflation may vary in a patient with changes in minute ventilation due to changes in CO2 production (depending on exercise, diet or the metabolic response to illness) or dead space, as well as with changes in airflow obstruction during an exacerbation.
Central respiratory drive may also be impaired, or poorly responsive to physiological triggers – hypoxaemia or hypercapnia – and lead to chronic hypercapnia. This may occur in the setting of sleep (i.e. obstructive sleep apnoea), obesity or drugs (sedatives, antiepileptics, alcohol).
CHRONIC BRONCHITIS OR EMPHYSEMA?
The value of labelling patients as chronic bronchitis or emphysema is uncertain as the two disease processes usually coexist and the principles of management are similar. Five pathophysiologic processes may be present to varying degrees in each patient with COPD: (1) inflammatory airway narrowing (bronchiolitis); (2) loss of connective tissues tethering airways; (3) loss of alveoli and capillaries; (4) hyperinflation; and (5) increased pulmonary vascular resistance. Early/mild COPD tends to be dominated by bronchiolitis with a minimal component of emphysema (Figure 26.2), whereas when COPD becomes severe, the reverse is true. However, recognition that COPD is dominated by one of these patterns is helpful with regard to clinical pattern and prognosis.
CLINICAL FEATURES OF ACUTE RESPIRATORY FAILURE IN COPD
Acute respiratory failure (ARF) in COPD can present with two distinct clinical patterns7 (Table 26.1).
Table 26.1 Clinical differences between normocapnic and hypercapnic chronic obstructive pulmonary disease
| Normocapnic (PaCO2 35–45 mmHg) | Hypercapnic (PaCO2 > 45 mmHg) |
|---|---|
| Emphysema > chronic bronchitis | Chronic bronchitis > emphysema |
| Thin | Obese |
| Pursed-lip breathing | Central nervous system depression: consider the role of oxygen therapy |
| Accessory muscle use | Alcohol, sedatives, analgesics |
| Hyperinflated | Sleep-related hypoventilation |
| Right heart failure late | Right heart failure early |
PRECIPITANTS OF ACUTE RESPIRATORY FAILURE
In approximately 50% of patients, there is an infective cause, in 25% heart failure and in the remaining 25% retained secretions, air pollution, coexistent medical problems (e.g. pulmonary embolus, medication compliance or side-effects) or no cause can be identified8 (Table 26.2).
Table 26.2 Precipitants of acute respiratory failure in chronic obstructive pulmonary disease
| Infective (including aspiration) |
| Left ventricular failure (systolic and diastolic failure) |
| Sputum retention (postoperative, traumatic) |
| Pulmonary embolism |
| Pneumothoraces and bullae |
| Uncontrolled oxygen |
| Sedation |
| Medication – non-compliance or side-effects |
| Nutritional (K, PO4, Mg deficiency, CHO excess) |
| Sleep apnoea |
The most common bacterial isolates are Streptococcus pneumoniae and Haemophilus influenzae in 80% of exacerbations.9S. viridans,10Moraxella (previously Branhamella) catarrhalis,11Mycoplasma pneumoniae12 and Pseudomonas aeruginosa may also be found. Viruses can be isolated in 20–30% of exacerbations13 and include rhinovirus, influenza and parainfluenza viruses, coronaviruses and occasionally adenovirus, and respiratory syncytial virus. Whether these organisms are pathogens or colonisers is often unclear.
PNEUMONIA
Pneumonia has been estimated to account for 20% of presentations requiring mechanical ventilation.13 It is most commonly caused by S. pneumoniae and H. influenzae but Mycoplasma, Legionella, enteric Gram-negatives and viruses are occasional causes.
LEFT VENTRICULAR FAILURE
Left ventricular (LV) systolic failure may result from coexisting ischaemic heart disease, fluid overload, tachyarrhythmias or biventricular failure secondary to cor pulmonale. LV diastolic failure occurs commonly and is precipitated by hypoxaemia, tachycardia,14 pericardial constraint due to intrinsic positive end-expiratory pressure (PEEPi) or right ventricular (RV) dilation. Increased work of breathing related to COPD will also increase by up to 10-fold the amount of blood flow to the respiratory pump muscles,15 thereby causing an increased demand upon the overall cardiac output. In patients with borderline cardiac status, this may precipitate heart failure. The components of right and LV failure can be accurately distinguished by Doppler echocardiography. Pulmonary congestion can be difficult to diagnose because of the abnormal breath sounds and chest X-ray appearance which are commonly present in COPD. In a recent publication, 51% of patients with acute exacerbation of COPD had echocardiographic evidence of left heart failure (systolic 11%, diastolic 32%, systolic and diastolic 7%).16
DIAGNOSIS AND ASSESSMENT
DIAGNOSIS
The clinical examination findings of COPD depend upon severity.
A chest X-ray will commonly show hyperinflated lung fields, as suggested by 10 ribs visible posteriorly, six ribs visible anteriorly or large airspace anterior to heart (> 1/3 of the length of the sternum), flattened diaphragms (best seen on lateral chest X-ray) and a paucity of lung markings. Pulmonary hypertension is manifest by enlarged proximal and attenuated distal vascular markings and by RV and atrial enlargement. Lung bullae may be evident.
A high-resolution computed tomographic (CT) scan of the chest (1–2-mm slices) can demonstrate characteristic appearance and regional distribution of emphysema. It can also assess for coexistent bronchiectasis, LV failure17 and pulmonary fibrosis. Such scans are less sensitive than standard chest CT scans (1-cm slice) for detecting pulmonary lesions (e.g. neoplasms). Nuclear ventilation perfusion scans can also provide a characteristic appearance of COPD.
DIFFERENTIAL DIAGNOSIS
Chronic heart failure (CHF) may be a differential diagnosis of COPD, or simply coexist, as both disorders are common in smokers.14,16 Orthopnoea and paroxysmal nocturnal dyspnoea are features which correlate with heart failure severity. A past history of myocardial ischaemia or atrial fibrillation should alert one to the possibility of heart failure. An echocardiogram and high-resolution CT (looking for shift in interstitial oedema with changes in posture from supine to prone)17 are sensitive markers of CHF.
NON-VENTILATORY MANAGEMENT
OXYGEN THERAPY
Oxygen given by low-flow intranasal cannulae or 24–35% Venturi mask should be titrated to achieve a saturation (SpO2) of 90 ± 2% as these levels will avoid significant increases in PaCO2 in the majority of COPD patients with ARF. Increases in PaCO2 are most common in patients with initial PaCO2 > 50 mmHg and pH < 7.35.18 If the rise in PaCO2 is excessive (> 10 mmHg or 1.33 kPa), then FiO2 should be reduced, titrating SpO2 to 2–3% below the previous value, and arterial blood gases should be repeated. If no PaCO2 rise occurs with oxygen therapy, then a higher SpO2 may be targeted with repeat blood gases.
Inadequate reversal of hypoxia (e.g. SpO2 < 85%) is suggestive of an additional problem such as pneumonia, pulmonary oedema or embolus, or a pneumothorax. Investigation of this should commence and a higher O2 delivery system should be used (see Chapter 24). Although high levels of O2 should be avoided, reversal of hypoxia is important and O2 should not be withheld in the presence of hypercapnia, or withdrawn if it worsens.
BRONCHODILATORS
Bronchodilators are routinely given in all acute exacerbations of COPD because a small reversible component of airflow obstruction is common, and bronchodilators improve mucociliary clearance of secretions.19 However, a large meta-analysis of 22 large randomised controlled long-term trials of ambulatory COPD patients involving either anticholinergics and/or β2 agonists (short- and long-acting) over 3–60 months indicates that anticholinergics are more favourable than placebo in terms of acute exacerbations, hospitalisations and respiratory deaths.20 There were no favourable advantages with β2-agonists compared with placebo for acute exacerbations or hospitalisations, and placebo was better than β2-agonists in terms of respiratory death.20
ANTICHOLINERGIC AGENTS
Anticholinergic agents, such as ipratropium bromide, have been shown to have a similar or greater bronchodilator action than β-agonists in COPD,1,21,22 and also to have fewer side-effects and no tachyphylaxis. Anticholinergic agents should be used routinely in COPD with ARF and many now believe them to be the agent of first choice.1
[/not-level-membership-for-anesthesiology-category]
