Acute Respiratory Failure
Acute Respiratory Failure—Types 1 and 2
Acute respiratory failure is defined as the inability of the respiratory system to meet the oxygenation, ventilation, or metabolic requirements of the patient.1 Although the main function of the lungs appears to be related to gas exchange (i.e., oxygenation and ventilation), it should be remembered that the lung is a metabolically active organ as well.1,2 Respiratory failure has been divided into two main types. Type 1 is hypoxemic respiratory failure, and type 2 is hypercapnic failure with or without hypoxemic respiratory failure.2 More simply stated, type 1 respiratory failure is oxygenation failure and type 2 is ventilatory failure. Operationally, type 1 respiratory failure is defined by a partial pressure of oxygen in arterial blood (PaO2) less than 60 mm Hg and type 2 respiratory failure is defined by a partial pressure of carbon dioxide in arterial blood (PaCO2) greater than 50 mm Hg. The respiratory failure can be acute or chronic in nature, related to the onset and duration of the failure.2,3 Some patients may present with an acute deterioration or worsening of their chronic respiratory dysfunction termed acute-on-chronic respiratory failure.4
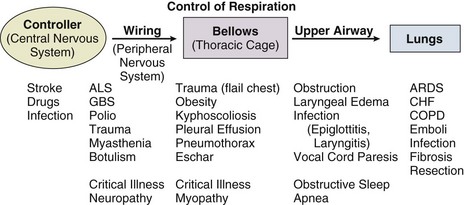
Acute respiratory failure is commonly encountered in the intensive care unit (ICU) setting and may be the primary reason for the admission or a complication of the patient’s medical condition(s) or treatment. Respiratory failure may be the result of a variety of causes, some of which may not directly involve the lungs or the respiratory muscles.2 Like a chain composed of individual links representing the brain, peripheral nervous system, upper airway, lower airway, respiratory muscles, cardiovascular system, and lungs (Fig. 37.1),2 respiratory failure may result when any of the links become sufficiently dysfunctional or weak. Like a chain, the body is only as strong as its weakest link, and respiratory failure may result when a component of the chain becomes sufficiently compromised. Common causes of hypoxemic and hypercapnic respiratory failure are listed in Box 37.1. This chapter reviews the basic mechanisms and clinical manifestations of type 1 and 2 respiratory failure and concludes with a more in-depth discussion of acute respiratory distress syndrome (ARDS), which includes clinical disorders in the type 1 category (ventilator management will be addressed in more depth in other chapters).
Hypoxemic Respiratory Failure
Basic Mechanisms
Hypoxemic respiratory failure refers to the inability of the respiratory system to maintain satisfactory levels of oxygen in the arterial blood.3 The five basic mechanisms of hypoxemia are listed in Box 37.2. Ventilation-perfusion abnormality is the most common.2,3 Multiple mechanisms may be instrumental in the development of respiratory failure. Most of the abnormalities will improve with the administration of supplemental oxygen, except for a shunt abnormality in which the PaO2 continues to be low despite the administration of high levels of supplemental oxygen. The shunt may be either intracardiac (such as a right-to-left shunt through a patent foramen ovale) or intrapulmonary (as seen with pneumonia or ARDS).5,6 A diffusion abnormality is infrequently the cause of hypoxemia in clinical practice and typically is only significant in the setting of tachycardia, high cardiac outputs, and when the diffusion capacity is below 25% of predicted.3
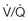 mismatch
mismatch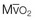
Assessment of Oxygenation
The hallmark of type 1 respiratory failure is hypoxemia, which is primarily assessed by the PaO2 of an arterial blood gas (ABG). An estimate of the patient’s oxygenation status can be obtained by measuring the oxygen saturation using a pulse oximeter. An appreciation of the sigmoid shape of the oxyhemoglobin dissociation curve allows for a rough estimation of the PaO2 (Fig. 37.2). The curve will shift rightward or leftward in relation to changes in the pH, PaCO2, temperature, and PO4−2 concentration.7 Determination of the ABG and knowledge of the exact FIO2 allows for the calculation of the alveolar-arterial oxygen gradient3 (A-a gradient) using the formula PAO2 − PaO2 = A-a gradient; thus,
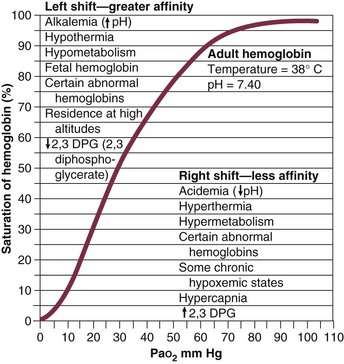
Figure 37.2 Oxyhemoglobin dissociation curve.
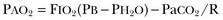
where
PAO2 Partial pressure of oxygen in the alveolus
PaO2 Partial pressure of oxygen in the arterial blood
FIO2 Fraction of inspired oxygen
PH2O Water vapor pressure at standard temperature and pressure
PaCO2 Partial pressure of carbon dioxide in the arterial blood
The A-a gradient is FIO2 dependent and will increase as the FIO2 increases. Because of this FIO2 dependency, some have preferred to evaluate oxygenation abnormalities by calculating the PaO2/PAO2 ratio, which is independent of the FIO2.3 For simplicity’s sake, many individuals use the PaO2/FIO2 ratio as a measure of oxygenation abnormality. This value is used in the definition of ARDS and has gained wide-scale acceptance as a measure of abnormal oxygenation.8 The shunt fraction or venous admixture can also be calculated while a patient breathes 100% oxygen and has reached steady-state oxygenation.
Hypercapnic Respiratory Failure
Hypercapnic respiratory failure can be the result of a variety of disorders as listed in Box 37.1.9 Acute hypercapnic respiratory failure, also termed acute ventilatory failure, occurs when a patient’s ABG reveals acute respiratory acidemia (pH <7.35) with a PaCO2 greater than 50 mm Hg. This definition is not generally applicable to patients with severe chronic obstructive lung disease or neuromuscular disorders who have developed a compensatory metabolic alkalemia in response to their chronic hypercapnia. However, these patients may have acute exacerbations or comorbid conditions that cause them to decompensate into acute-on-chronic respiratory failure.
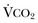 ) equals the rate of CO2 elimination. Carbon dioxide elimination (
) equals the rate of CO2 elimination. Carbon dioxide elimination (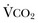 ) is equal to the alveolar ventilation (V
) is equal to the alveolar ventilation (V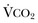 = V
= VNot all respired air is effective alveolar ventilation because some of the total tidal ventilation (VT) ventilates nonperfused areas or dead space (VD) (both anatomic and pathologic). This relationship between tidal volume, dead space volume, and alveolar volume can be expressed as3:
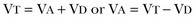
The minute ventilation (VE) is equal to the sum of the dead space ventilation (VD) and the alveolar ventilation (VA): VE = VD + VA. Combining and rearranging terms from these various equations yields the following relationship of carbon dioxide production and alveolar ventilation with the PaCO2.3
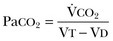
Therefore, elevated PaCO2 can result from a combination of any of three clinical alterations: increased CO2 production, decreased tidal ventilation, or increased dead space ventilation.9 Increased CO2 production arises from hypermetabolic states such as exercise, fever, sepsis, burns, trauma, excessive carbohydrate intake, and hyperthyroidism.9 In isolation, these relatively common conditions rarely induce hypercapnic respiratory failure because the normal physiologic response to an elevated PaCO2 is to increase minute (and therefore alveolar) ventilation to maintain eucapnia and a normal pH. Only those patients who are unable to increase their effective alveolar ventilation as a result of neuromuscular disorders affecting the muscles of respiration, the presence of excessive ventilation/perfusion (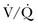 ) mismatching, or increased dead space ventilation develop hypercapnic respiratory failure caused by elevated CO2 production.
) mismatching, or increased dead space ventilation develop hypercapnic respiratory failure caused by elevated CO2 production.
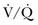 abnormalities, to account for the additional hypoxemia in these patients with hypercapnic respiratory failure.
abnormalities, to account for the additional hypoxemia in these patients with hypercapnic respiratory failure.Decreased tidal volume hypoventilation can be caused by disorders affecting any component of the “neuromuscular sequence” starting in the central nervous system (CNS) and ending at the muscles of respiration (see Fig. 37.1). Among the more common causes are CNS depressants such as narcotics and sedatives, which diminish respiratory drive; cerebral vascular disorders, especially those that involve the brainstem, which impair the efferent signals to breathe; and disorders of neuromuscular transmission such as myasthenia gravis and the paraneoplastic Eaton-Lambert myasthenic syndrome (Box 37.3).10,11 Abnormal respiratory mechanics can result from airflow obstruction, chest wall deformities, and loss of lung volume. Disorders that increase the respiratory load, such as a circumferential chest burn eschar or flail chest, or impair function of the respiratory muscles, such as kyphoscoliosis, will also lead to hypoventilation.
Assessment of Ventilation
The hallmark feature of inadequate ventilation is the presence of an elevated PaCO2 with or without the presence of hypoxemia.3,9 In the adult population the best method to detect an elevated PaCO2 is with an ABG measurement. Noninvasive evaluation of ventilation in the critically ill adult, using end-tidal CO2 (PetCO2) measurements or transcutaneous CO2 (PtcCO2) determinations, is not as reliable as it is in the healthy adult or the neonate, respectively.7
Clinical Manifestations
The risk factors for developing acute respiratory failure include the postoperative state, preexisting chronic illness, malnutrition, advanced age, morbid obesity, chronic bronchitis, and cigarette smoking. The clinical manifestations of respiratory distress may be subtle and nonspecific or may be obvious to even the untrained observer. The patient with respiratory distress may have an abnormal respiratory rate (too high or low) or display an irregular pattern of breathing. The patient with respiratory distress may evidence gasping ventilation, nasal flaring, or use of the accessory muscles of respiration. Intercostal retractions may be seen, and the patient may exhibit paradoxical respiratory movement. Typically, patients with acute respiratory failure, whether type 1 or type 2, will have altered heart rate and blood pressure. The majority will evidence a sympathetic response with tachycardia and hypertension, but patients may be hypotensive and bradycardic. Cardiac arrhythmias may be seen in both type 1 and type 2 respiratory failure. Patients with acute respiratory failure will usually be in distress and often appear apprehensive.2,9 Many will be diaphoretic and have altered mental status or level of consciousness. Hypercapnic patients may demonstrate signs of a respiratory encephalopathy including somnolence, coma, asterixis, seizures, tremors, or myoclonic jerks. Papilledema and congested conjunctivae may be present. Type 1 respiratory failure patients may appear cyanotic.9
Acute Respiratory Distress Syndrome
Since the initial 1967 description of acute catastrophic respiratory failure in 12 patients who were subsequently declared to have the acute respiratory distress syndrome, there has been a great deal of research and clinical study attempting to understand the mechanism of injury and improve the outcome of these critically ill patients.12–14 The hallmark of this disorder was the rapid onset of acute hypoxemic respiratory failure that was characterized by refractory hypoxemia despite the administration of high concentrations of supplemental oxygen; decreased pulmonary compliance; diffuse, bilateral pulmonary infiltrates; the absence of left-sided heart failure; and histologic evidence of alveolar damage with hyaline membrane formation, which was followed by fibrosis of the lung.8,12,13 The subsequent 10 to 20 years of ARDS management were associated with extremely high mortality rates, at times approaching 90%, despite the provision of extremely aggressive supportive care.15–20 Some of the initial clinical and basic investigations that attempted to improve the outcome of patients with ARDS were hampered by the lack of uniformly accepted definitions.21–25 In 1994 an American-European Consensus Conference (AECC) defined acute lung injury (ALI) and ARDS in an attempt to eliminate confusion related to terminology for these conditions.21,22 The definitions are clinically based and regard ALI as a continuum with the more severe oxygenation abnormalities reflective of ARDS. Among the main benefits of the uniform definition would be improved communication and enhanced clinical trial design.
The AECC definition of ALI and ARDS was challenged and there were noted discrepancies between the clinical and pathologic findings in individuals felt to have died with ARDS.25 Esteban and colleagues25 found that one third of the people who died with a clinical diagnosis of ARDS did not have histologic evidence of diffuse alveolar damage on postmortem examination. In addition, diffuse alveolar damage, the hallmark pathologic finding of ARDS, was evident in 10% of patients who died without a clinical diagnosis of ARDS. A subsequent study evaluated the sensitivity and specificity of the Murray Lung Injury Score, the AECC definition of ARDS, and a Delphi definition that incorporated oxygenation abnormality, X-ray findings, clinical onset, compatible clinical scenario, and absence of left-sided heart failure for ARDS.26 The combination of Lung Injury Score greater than 2.5 with either the AECC definition or the Delphi definition of ARDS resulted in the best combination of sensitivity and specificity. Since its introduction, the AECC definition has been used by the vast majority of clinical trials evaluating new treatment strategies for patients with ARDS, particularly those conducted by the ARDS Network investigators, supported by the U.S. National Heart, Lung, and Blood Institute of the National Institutes of Health.27–29 In 2011 experts from the European Society of Intensive Care Medicine, the American Thoracic Society, and the Society of Critical Care Medicine met in Berlin and proposed a new definition for staging ARDS as mild, moderate, or severe, based on the oxygenation impairment measured on a minimum of 5 cm H2O positive end-expiratory pressure (PEEP) (10 cm H2O for severe).30 The new Berlin definition did not drastically differ from the prior AECC definition, but it clarified several important concepts:
1. Timing—patients with ARDS should be identified within 72 hours of a recognized risk factor and nearly all patients should be identified within 7 days.
2. Chest imaging—bilateral opacities consistent with pulmonary edema are seen on chest radiograph or chest computed tomography (CT) scan.
3. Origin of pulmonary edema—ARDS may be present in the setting of coexisting cardiogenic edema or volume overload if in the opinion of the treating physicians the respiratory failure is not fully explained by these conditions and there is a recognized risk factor for ARDS. In the absence of a recognized risk factor for ARDS there should be some objective assessment of cardiac function such as echocardiography or pulmonary capillary wedge pressure measurement.
4. Oxygenation—because PEEP can affect the PaO2/FIO2 ratio there should be a minimum of 5 cm H2O PEEP and 10 cm H2O in those patients with severe ARDS (PaO2/FIO2 < 100).30
The Berlin definition for ARDS eliminates the use of the term “acute lung injury” to classify the severity of lung injury and defines mild ARDS as a PaO2/FIO2 300 or less but greater than 200, moderate ARDS as a PaO2/FIO2 200 or less but greater than 100, and severe ARDS as a PaO2/FIO2 100 or less, with morality rates associated with the different stages 27%, 32%, and 45%, respectively.30
To date there is no specific laboratory test that is pathognomonic for the diagnosis of ARDS.8,21,27 All of the current definitions for ARDS require the presence of bilateral pulmonary infiltrates on chest imaging, but interpretation is not always straightforward.21,31 When 21 experts were given 28 chest radiographs to evaluate for the presence of bilateral pulmonary infiltrates, they only agreed 43% of the time, and one third of the time five or more individuals differed in their interpretation.29 The range of ALI/ARDS diagnoses was from 36% to 71%.32
Risk Factors
It has been recognized that ARDS may arise in association with a number of clinical conditions or risk factors, with sepsis being the most common (Box 37.4).8,18,21,27,33–37 Various studies report that approximately 5% to 40% of septic patients will develop ARDS.8,18,36 Shock (prolonged hypotension) and the systemic inflammatory response syndrome (SIRS) are also common risk factors. Other frequently encountered clinical risk factors include multiple emergency transfusions; aspiration injury; near-drowning; pancreatitis; trauma (particularly lung contusion, fat emboli from long bone fractures); burns; cardiopulmonary bypass; and disseminated intravascular coagulation (DIC). These clinical risk factors are synergistic, and when more than one of the clinical risk factors is present, the likelihood of ARDS is greater than just the sum of the collective risk factors.8,17 The clinical risk factors associated with the development of ARDS appear to greatly influence the expected outcome. Whether ALI and ARDS arise from a direct pulmonary insult or from an extrapulmonary cause impacts the subsequent response to PEEP and ventilatory support (Box 37.5).19 Age also has an impact on the likelihood for ARDS development.38 Older individuals have a greater likelihood of developing ARDS from most of the risk factors noted earlier. In the setting of trauma the risk for ARDS seems to peak in the sixth and seventh decades and then declines.38
Negative pressure (postobstructive) pulmonary edema (NPPE or POPE) is a type of noncardiogenic pulmonary edema that can manifest as ARDS.39 It is rare (five cases reported from a large tertiary medical ICU in 4 years),40 can range in severity from mild hypoxemia treated with low-flow supplemental oxygen to profound hypoxemia requiring mechanical ventilation, and may even present as alveolar hemorrhage.41–45 It is believed to be caused by increased vascular permeability resulting in alveolar and capillary damage from large levels of negative intrathoracic pressure generated by attempting to inspire against an occluded airway or airway obstruction.46,47 This leads to a protein-rich alveolar exudate impairing gas exchange in mild-to-moderate cases and leaking of capillary blood into the alveoli in more severe circumstances. Most of the reported cases are related to surgical procedures: thyroidectomy,48,49 septorhinoplasty,50 tonsillectomy/adenoidectomy,51 mandibular open reduction and internal fixation,52 or cryosurgery for a tracheal obstruction.53 Nonoperative conditions leading to NPPE have included laryngospasm,54,55 occlusion of an endotracheal tube,56,57 excessive tube thoracostomy suction pressure,58 nonlethal hanging,59 and even hiccups.60,61 The common element is the rapid relief of a large airway occlusion. Patients often have no underlying cardiopulmonary disorders. NPPE may account for a small percentage of immediate postoperative extubation failures, particularly if an unrecognized mainstem intubation is present in an awake and spontaneously breathing patient. Clinical manifestations of hypoxemia and respiratory distress may be delayed up to 6 hours.47 Therapy is supportive with supplemental oxygen and, if needed, positive-pressure ventilation (invasive or noninvasive).
Elevated intracranial pressures, either from traumatic brain injury or intracerebral hemorrhage, or status epilepticus can induce an ARDS-like clinical syndrome termed neurogenic pulmonary edema (NPE).62,63 Although the exact mechanisms are not clear, an abrupt and massive release of catecholamines from injured neuronal tissues is thought to act directly on the lung parenchymal vasculature to promote a “capillary leak” of a protein-rich exudate as well as an increase in pulmonary vascular resistance64 in addition to inducing a neurohumoral (takotsubo) cardiomyopathy.65–67 The cardiomyopathy has the distinctive echocardiographic feature of ventricular apical ballooning and can persist for months.68
Incidence and Prevalence
The exact incidence and prevalence of ARDS has been variable and related to definitions used (Box 37.6).8,38 Early reports suggested that there were 150,000 patients with ARDS each year in the United States.8,33,36,69 This statistic led to an estimated incidence of 75 cases per 100,000 population. Proposed estimates have suggested that the incidence of ARDS ranges from 1.5 to 64 cases per 100,000 population.38,69,70 A recent study evaluated the incidence and outcome of ALI in mechanically ventilated patients aged 15 or older cared for at 21 hospitals in and around King County, Washington, over a 15-month observation period.38 The crude incidence of ALI was 78.9 per 100,000 person-years, and the incidence of ARDS was 58.7 per 100,000 person-years. On the basis of their results, the authors estimated the annual number of U.S. episodes was 332,100 cases of ARDS. The authors noted that increasing age was associated with an increased incidence and mortality rate in ARDS. A recent prospective observational study using the AECC definition and lung protective ventilatory support strategies in Spain (the ALIEN study) reported an incidence of 7.2 per 100,000 population per year for ARDS with an ICU and hospital mortality rate of 42.7% and 47.8%, respectively.71
Clinical Manifestations
When ARDS becomes clinically apparent, the patient is usually noted to be in significant distress manifesting dyspnea, tachypnea, visible signs of respiratory distress, and increased work of breathing.8,23,33,36 The typical presentation is manifest as an acute catastrophic complication in a patient who has one or more of the clinical risk factors for the development of this form of ARDS. The precipitating injury need not directly involve the pulmonary system.8,36,72 Past definitions have emphasized the need to exclude patients with previous or known chronic pulmonary or cardiovascular diseases.19–21,27 The AECC definition excluded patients with elevated left-sided heart filling pressures and chronic infiltrative lung disease as the cause of the radiographic or physiologic alterations, but the new Berlin definition will allow for the concomitant presence of ARDS and cardiac edema/volume overload as long as the predominant process is thought to be ARDS.21,30
The hallmark of ARDS is the presence of hypoxemia despite the administration of high concentrations of inspired oxygen, evidence of an increase in the shunt fraction, a decrease in pulmonary compliance, and an increase in the dead space ventilation.8,24,30 The chest radiographic manifestation of ARDS is the presence of diffuse, bilateral pulmonary infiltrates with a normal cardiac silhouette. Recent reports have cautioned that even among trained experts there is often disagreement concerning the interpretation of the chest radiograph.31,29,69,73 Chest CT has also demonstrated that the radiographic injury is not homogeneous and has a predominance in the dependent portions of the lung.8,43 It is important to remember that ARDS is a clinical syndrome and the diagnosis is made clinically, not on the basis of a single radiograph, ABG analysis, or laboratory test.
Pathologic Manifestations
Typically, type 1 alveolar cells compose the major gas exchange surface of the alveolus and are integral to the maintenance of the permeability barrier function of the alveolar membrane.8 Type 2 pneumocytes are the progenitors of type 1 cells and are responsible for surfactant production and homeostasis.8 During ARDS there is damage to the capillary endothelial and the alveolar epithelial cells.8 Cellular injury and alteration of the normal barrier function results in a permeability defect that gives way to flooding of the alveoli with protein-rich fluid and inflammatory cells.8,33,74 This results in the alteration of pulmonary mechanics, physiology, and gas exchange.8,74 There is alteration of surfactant that results from damage to the type 2 pneumocyte and from the inactivation and dilution of alveolar surfactant from the protein and fluid that have entered into the alveolar space, respectively.75,76 Surfactant dysfunction can lead to atelectasis and a further reduction in pulmonary compliance.8,75–78 In addition, dysfunction of the alveolar epithelial cells can impair the resorption of fluid from the alveolar space, which augments the parenchymal injury process and gas exchange abnormalities.8,79
The observed pathologic findings in ARDS depend on the timing of the tissue sampling. During the initial stages of clinically evident lung injury there is histologic evidence of diffuse alveolar damage.8 Histologic features of the injury include microthrombi composed of platelets and white blood cells within the capillary lumen, denudation of the alveolar epithelial lining cells, swelling of the capillary endothelial cells, interstitial and alveolar infiltration by polymorphonuclear leukocytes (PMNLs), and hyaline membrane formation within the alveoli.8 Grossly, the lungs appear heavy and wet. Later, areas of type 3 collagen deposition with fibrosis will be present.79 An intense inflammatory reaction involving PMNLs, activated monocytes, macrophages, and endothelial cells is present in the fibroproliferative phase of lung injury.76,80,81 Pro- and anti-inflammatory molecules produced by these activated cells may be found in the circulating blood and bronchoalveolar lavage (BAL) fluid.81–83 This phase may be followed by fibrosis, but this fibrosis does not appear to have the same permanence as typical fibrosis would have and may actually resolve over time in survivors of the injury.84
Pathophysiology
ARDS may develop as a result of epithelial or endothelial cell injury.8,13,85,86 Both sites of injury and cells are important for maintenance of normal barrier function and are capable of initiating an inflammatory response. In the majority of clinical settings the initial site of the ARDS involves the capillary endothelial cell, which may be the initial manifestation of a “panendothelial cell injury” resulting from SIRS.83 Endothelial cell injury compromises the integrity of the vascular barrier and results in transudation of fluid and inflammatory mediators into the interstitial tissues and ultimately into the alveoli.8,13 The frequent occurrence and early involvement of lung dysfunction as a component of multiple organ dysfunction/failure lends support to the hypothesis of a panendothelial cell injury as one of the target injuries in the setting of SIRS.87
The complex pathophysiologic processes that culminate in the production of ARDS involve a delicate balance between the body’s proinflammatory and anti-inflammatory responses to the inciting clinical event.83,87 The balance that exists between the various inflammatory molecules or mediators and the endogenous compensatory responses evoked by the inflammatory response will dictate whether or not lung injury and other forms of organ dysfunction will develop.81 The ensuing interaction between SIRS and the compensatory anti-inflammatory response syndrome (CARS) will thus determine whether a patient successfully deals with an injury or is predisposed to develop organ dysfunction (excessive SIRS response) or immunosuppression and infectious complications (excessive CARS response).83,87 This mixed antagonistic response syndrome (MARS) has tremendous impact on the fate of the critically ill patient.87
Management Strategies
Basic management strategies for patients with ARDS are listed in Box 37.7.8,18,21,23,33,36,88 Initial attention must be directed to providing lung protective ventilator support and identification of the predisposing underlying clinical risk factor(s) or condition(s), and there should be specific treatment directed at the underlying or predisposing disorder.8,21–23 The recognition of sepsis and infection as frequent causes of ARDS should prompt an aggressive search for undiagnosed foci of infection and the administration of appropriate antimicrobial treatment and use of surgical drainage procedures as indicated.15,17,89
The cornerstone of supportive management is the provision of mechanical ventilatory support.8,21–23 Recent experimental and clinical data report significant survival benefit from the use of lung protective ventilatory support strategies.90,91 The primary goal of this support is to improve oxygenation and ensure that the lung is allowed to heal and avoid further injury. Use of nonprotective ventilatory support strategies may have a role in the persistent proinflammatory response and the development of multiple organ dysfunction syndrome (MODS) and multiple organ failure (MOF).90–93
Mechanical Ventilation
See Chapter 11 for a more detailed discussion of mechanical ventilatory support in the management of patients with ARDS. Over the past 20 years there has been increasing recognition that the ventilatory support strategy used in the management of patients with ARDS may produce or augment lung injury or impair the healing process (Box 37.8).94–98 Results of experimental animal studies demonstrated clinical, physiologic, and histologic abnormalities similar to those observed in patients with ARDS when animals with normal lungs were ventilated with large tidal volumes or were given high inflation pressures.95–101 The alveolar overdistention produced by these ventilatory modes was felt to be the critical element in production of the lung injury.95–98 Lung injury could result from the administration of large tidal volumes or the administration of positive pressure or negative pressure breaths that were sufficient to produce alveolar overdistention (termed volutrauma).97–102 Other mechanisms that could potentially result in lung injury were the repetitive recruitment-derecruitment of distal airways (termed atelectotrauma) and alveoli or the disruption of alveoli resulting in translocation of organisms or air emboli.97,98 Alveolar overdistention can also give rise to systemic inflammatory molecules that may contribute to the SIRS response and drive the development of MODS/MOF.92,98 This has been termed biotrauma.98 Interestingly, the lung injury produced by high inflation pressures or large tidal volumes in these experimental models could be ameliorated by the addition of therapeutic amounts of PEEP.97,98 Subsequent human studies have evaluated the distribution of the radiographic lung injury in ARDS patients as determined by the use of CT scans of the chest and have noted the dependent nature of the injury.74,103 The injury to the lungs was not diffuse and homogeneous as once believed. These dependent areas of injury comprised regions of alveolar flooding from the gravitational accumulation of lung water, areas of lung injury, and normal lung regions. The relatively normal ventral (nondependent) lung has been referred to as the “baby lung,” and the injured (dependent) lung has been called the “sponge lung.”32,103 With the use of higher tidal volumes, PEEP, or high distending pressures, there was evidence of alveolar overdistention, particularly in the areas of normal lung. A multicenter trial of low versus traditional tidal volume ventilation in patients with ARDS demonstrated increased proinflammatory cytokines in the serum and BAL fluid of patients ventilated with the larger traditional tidal volumes, supporting the concept of biotrauma from alveolar overdistention.92 Pulmonary barotrauma (pneumomediastinum, pneumopericardium, pneumoperitoneum, pneumothorax, subcutaneous emphysema, pulmonary interstitial emphysema, and air embolism) is a well-recognized complication of ventilatory support that has been reported in 7% to 15% of patients with ARDS.104
The landmark study by NHLBI ARDS Network compared low tidal volumes (6 mL/kg of ideal body weight) against conventional tidal volumes (12 mL/kg of ideal body weight).94 A protocol governed the use of PEEP according to the FIO2 required to meet the oxygenation goals. A weaning protocol was used once the patient was on a reduced amount of ventilatory support. The trial was terminated after 861 patients were enrolled, following the determination of a significant (22% relative reduction) decrease in mortality rate associated with the use of low tidal volumes (39.8% vs. 31%; P = 0.007). A significant difference occurred between the low and high tidal volume groups in length of stay, ventilator days, and development of MODS.94 No difference occurred in the development of barotrauma between the two ventilator support strategies. On further analysis, this survival benefit was present irrespective of the patient’s body mass index (BMI).105
The use of lower tidal volumes typically results in a controlled hypoventilation or permissive hypercapnia and can lead to hypercapnic acidosis.106 Some experimental animal models of lung injury suggest that hypercapnic acidosis may be beneficial to the lung and produce less lung injury as measured by extravascular lung water (Box 37.9).105 Other reports have detailed potentially harmful effects of hypercapnia on epithelial and endothelial barrier function, lung edema, edema clearance, inate immunity, and host defenses.107
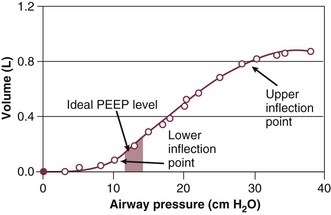
PEEP is a major component of the ventilatory support strategy and may actually have a direct therapeutic role in the prevention of ventilator-induced lung injury (VILI), as noted in some of the experimental models of VILI.8,96,108–117 PEEP is beneficial in the recruitment of atelectatic lung units and prevention of recruitment-derecruitment, and it increases the functional residual capacity (FRC), decreases the shunt fraction, and allows for a reduction to a less toxic FIO2 while still maintaining adequate oxygen saturation and tissue oxygen delivery.110,114–116,118 The application of the “right amount” of PEEP has recently taken on a more sophisticated approach. The ability to construct a pressure-volume curve to reflect the compliance of the patient’s lung has allowed for the identification of both the lower and upper inflection points (Fig. 37.3).117 Maintaining a PEEP level above the lower inflection point should potentially avoid the repetitive opening and closing of alveoli and the development of shear forces from the recruitment-derecruitment process, which may participate in the production or propagation of lung injury. If too much PEEP is applied, there may be overdistention of alveoli, which can potentiate volutrauma or barotrauma and have adverse consequences on pulmonary mechanics, hemodynamic function, and lung healing. Constructing pressure-volume curves to determine the “best PEEP” is a difficult undertaking and requires the patient to be heavily sedated or paralyzed and the use of a calibrated super syringe. Determining the precise lower inflection point on the inspiratory inflation curve is difficult, and the compliance curves may change (along with the inflection points) over time as the patient’s lung compliance changes. To simplify the determination of PEEP, the ARDS Network ventilatory support protocol used a monogram for PEEP based on the FIO2 requirements and the goal of maintaining a PAO2 between 55 and 80 mm Hg.94 For the protocol, end-inspiratory plateau pressure was kept lower than or equal to 30 cm H2O and the pH was maintained in the 7.30 to 7.45 range. Despite the demonstrated significant improvement in mortality rates associated with this protocol, some felt that additional benefit could be seen with the use of higher levels of PEEP. A subsequent large, prospective, randomized controlled trial conducted by the ARDS Network evaluated the PEEP protocol from the initial trial (PEEP levels from 5 to 24 cm H2O) versus higher levels of PEEP.119 During the study there was a change to a higher PEEP strategy to ensure a difference in the amount of applied PEEP between the two treatment arms. No significant difference in deaths before discharge home, breathing without assistance by day 28, ventilator-free days, ICU-free days, organ failure–free days, or barotrauma was found.119 Several studies have prospectively evaluated the benefits of higher versus lower PEEP strategies in the management of patients with ARDS, and a recent meta-analysis has failed to demonstrate a significant short-term mortality rate benefit associated with the use of higher PEEP.119–123
PEEP has the potential to have profound hemodynamic consequences in selected patients and clinical circumstances, and it is important to closely monitor patients with hemodynamic, echocardiographic, or other sophisticated monitors if there is a question of the adequacy of cardiac function when PEEP is applied.6,109–111 Right-to-left shunting through a patent foramen ovale can occur in up to a third of patients with severe ARDS, and increasing PEEP may not produce the expected oxygenation benefits in these individuals.124 The effect of PEEP may not be the same in the supine and prone positions. Apparently there is an enhanced ability to recruit alveoli when the patient is in the prone position as compared with the supine position when the patient is on the same amount of PEEP.125 There may be a different responsiveness to the use of PEEP depending on whether the ARDS results from a direct pulmonary injury or a nonpulmonary process that gives rise to ARDS.32
Despite the survival benefits demonstrated by the use of the lung protective ventilatory support strategy, there has not been universal adoption of this technique of ventilatory support for all patients with ARDS. Reasons for this slower than expected incorporation into daily practice are many and varied and certainly go beyond lack of awareness. Young and coworkers126 reported on the tidal volume used to ventilate ARDS patients at three large New England university hospitals. Prior to the publication and subsequent education of the ARDS Network results, the tidal volume averaged 12.3 mL/kg predicted body weight (9.8 mL/kg measured body weight). After the publication, the tidal volume dropped to 10.6 mL/kg predicted body weight (8 mL/kg measured body weight).
A multicenter French study has reported that the use of neuromuscular blockade with cisatracurium over the initial 48 hours of severe ARDS was associated with improved 90-day survival and longer time off ventilator support in contrast to conventional ventilator management.127 A prior study by these investigators noted decreased inflammatory response associated with the use of neuromuscular blockade in patients with ARDS.128 Although there is always a risk for prolonged neuromuscular weakness after use of neuromuscular blocking drugs, as well as in critical illness, there are many potential benefits in support of using neuromuscular blocking agents early in severe ARDS.129 Some of these potential benefits include better patient-ventilator synchrony and interaction, less VILI (barotraumas, atelectrauma, biotrauma), better recruitment effect from administered PEEP, and decreased oxygen consumption.129,130
Fluid Management and Vasoactive Support
There has been controversy regarding the approach to fluid management and oxygen delivery in the critically ill patient with ARDS.131–142 Patients with ARDS often require aggressive management to restore and maintain appropriate hemodynamic function.143–144 The SAFE trial demonstrated that resuscitation with saline is as beneficial as resuscitation with albumin in critically ill patients with shock.145 Correcting shock and hemodynamic derangements in patients with ARDS is important because these are potential causes for the lung injury and may result in organ dysfunction/failure.144 Several studies have reported increased survival associated with low pulmonary capillary occlusion pressure in the setting of ARDS.131,133 Concern for compromised organ perfusion and predisposition to the development of organ system dysfunction and the development of MODS/MOF led to additional studies from the ARDS Network to evaluate a liberal versus conservative fluid replacement strategy (Fluid and Catheter Treatment Trial [FACTT] study).146 In addition, this trial was designed to evaluate the utility and safety of using a pulmonary artery (PA) catheter to guide volume replacement as opposed to a central venous catheter.147 The safety and potential benefits of using PA catheters in the critically ill has been an area of controversy since the 1990s.148 In 1996, Connors and coworkers reported a lack of benefit and possible harm associated with the use of PA catheters in critically ill patients in the SUPPORT database.149
In a prospective, randomized controlled trial of PA catheters versus no PA catheters in the management of 676 adult patients with shock or ARDS, there was no difference in organ failure–free days, ventilator-free days, vasopressor-free days, or days in the ICU or hospital.150 This trial did not have an algorithm to direct management based on the hemodynamic data obtained from the use of the PA catheter. The ARDS Network trial evaluated liberal versus conservative fluid management on the basis of the central venous catheter (central venous pressure [CVP]) data versus PA catheter (PA occlusion pressure [PAOP]) data in 1000 patients with established ARDS.147 The use of a PA catheter did not significantly change 60-day survival or days of unassisted breathing in comparison with the use of a central venous catheter.147 There was no difference between the groups in lung or renal function, use of vasopressors, renal replacement therapy, or hypotension. The PA catheter group did have twice as many catheter-related complications, predominantly in the form of arrhythmias, compared with the central venous catheter group.
The liberal fluid management arm of the FACTT study had an average net gain of almost 7 L over the first 7 days, whereas the conservative fluid management group averaged a loss of 136 mL over 7 days of cumulative fluid balance.146 The use of the conservative fluid management strategy was associated with a significant improvement in oxygenation index and lung injury score and increased the number of ventilator-free days compared with the more liberal strategy. There was no difference in the development of shock or the need for renal replacement therapy between the two fluid management strategies.146
Prevention of Complications of Critical Illness
ARDS patients are also at risk for the complications that commonly complicate the course of the critically ill. Most of these complications are preventable, and prophylactic strategies should be employed whenever possible. Common complications include deep venous thrombosis and pulmonary embolism, stress-related gastrointestinal hemorrhage, ventilator-associated pneumonia (VAP) and nosocomial infections, metabolic abnormalities, critical illness polyneuropathy, and malnutrition.151–155 Anticipation and prevention of these complications are vitally important. Prophylactic strategies to prevent stress-related mucosal disease and gastrointestinal bleeding using H2 blockers, proton pump inhibitors, or possibly early enteral nutrition should occur in all patients unless otherwise contraindicated.155,156 Deep vein thrombosis and pulmonary embolism prophylaxis should also be administered unless there are contraindications.156 The use of enteral nutrition may be valuable in an attempt to prevent stress-related gastrointestinal bleeding and prevent translocation when normal barrier function of the gastrointestinal mucosa is compromised.155,157–160 Nutritional support may also be important to maintain the proper level of immune function and potentially to prevent the development of malnutrition in the catabolic critically ill patient. The use of enteral formulas designed to enhance the immune response with increased amounts of arginine, glutamine, or selected fatty acids remains controversial. The use of enteral formulas with increased amounts of eicosapentaenoic acid and γ-linolenic acid has been shown to improve organ dysfunction and improve oxygenation in clinical trials but has not been demonstrated to improve survival in patients with ARDS.161,162 The ARDS Network found that the use of twice daily enteral supplementation with ω-3 fatty acids, γ-linolenic acid, and antioxidants did not improve clinical outcome and was associated with a greater amount of diarrhea.163 A subsequent (EDEN) randomized trial by the same investigators found that ARDS patients who received trophic (minimal enteral) feeding did just as well and had less gastric residual volume, vomiting, and constipation and a lower blood sugar and insulin requirement than the patients who received full enteral feeding over the first 6 days.164 For those patients who do not tolerate enteral nutrition, waiting to provide parenteral nutritional support for up to 8 days was found to be well tolerated, was safer, had fewer complications, and was associated with a faster recovery in critically ill adult patients.165
A major goal of management should be the prevention of nosocomial or secondary infections/sepsis and multiple organ dysfunction/failure because these two conditions are currently responsible for the high mortality rate seen in patients with ARDS.18,89,151–153 VAP is a frequent complication in critically ill ventilated patients. As previously mentioned, prevention of VAP should be a primary management goal for patients with ARDS. Detection of a complicating VAP can be difficult in the setting of the pulmonary radiographic infiltrates seen in patients with ARDS. Diagnosis of VAP and identification of the offending pathogen often require the use of bronchoscopy with BAL or protected specimen brushes coupled with semiquantitative culture analysis.151,153 Recent data suggest that identifying the soluble triggering receptor expressed on myeloid cells (sTREM) may be beneficial in the detection of VAP but does not identify the specific etiologic organism.166 European investigators have been enthusiastic about the technique of selective digestive decontamination (SDD) as a method to decrease nosocomial lung infections in the critically ill patient.167 Keeping the head of the bed elevated above 30 degrees is also effective at preventing VAP.156 Additional measures designed to decrease the development of VAP include continuous subglottic suction, coated endotracheal tubes, closed suction systems, and kinetic therapy.156 Development of complications has been associated with increased morbidity rate, length of stay, cost of care, and possibly mortality rate.155,156
Pulmonary barotrauma has been reported to develop in approximately 7% to 15% of patients with ARDS.104 When a pneumothorax is detected in a mechanically ventilated patient, prompt recognition and chest tube insertion are required to prevent the development of tension physiology.104
Strategies to Improve Oxygenation
In patients with severe ARDS and persistent hypoxemia various strategies have been developed in an attempt to improve oxygenation and lessen FIO2 requirements. Included in these strategies are recruitment maneuvers, prone positioning, sighs, surfactant replacement therapy, partial liquid ventilation, inhaled nitric oxide, and enhanced edema clearance.125,168–194 These techniques remain investigational at this time and should be subjected to rigorous evaluation to adequately determine their ability to improve outcome for patients with ARDS. Importantly, the use of higher tidal volumes (12 mL/kg ideal body weight) in the ARDS Network trial was associated with an improvement in oxygenation but a significantly decreased survival in patients with ARDS.94
Recruitment maneuvers represent an attempt to open the atelectatic distal airways and alveoli on the border of the collapsed flooded alveoli that compose the dependent area of radiographic lung injury.98 Some experts believe that this maneuver should precede the provision of PEEP and ventilator support in the early phases of ARDS.195 The maneuver is accomplished by increasing the PEEP to 35 to 50 cm H2O and holding that level of pressure for 30 seconds. The ARDS Network attempted to define the value of a recruitment maneuver in 43 patients with ARDS by randomly assigning them to a recruitment maneuver with 35 to 40 cm H2O for 30 seconds versus a sham recruitment maneuver.179 The recruitment maneuver was assessed on the basis of a sustained improvement in oxygenation as judged by the ability to titrate PEEP/FIO2 on the basis of the network algorithm and changes in lung compliance. There was no significant difference in the magnitude or duration of the oxygenation effect, compliance, or change in PEEP/FIO2 titration. The group that received a recruitment maneuver did have a greater decrease in blood pressure during the maneuver. Other studies have reported that recruitment maneuvers are well tolerated from a hemodynamic standpoint and are not associated with increased cytokine release.180
Patients with ARDS may improve their oxygenation abnormalities when they are placed in the prone position.168–175 This position may be more physiologic for most mammals and result in improved secretion removal, better ventilation/perfusion matching, and better aeration of the dorsal lung units.172 The prone position may also prevent the heart from collapsing the left lower lobe and enhance the recruitment effects of PEEP by stabilizing the more flexible ventral chest wall.172 Prone positioning may also unload the right ventricle in the setting of severe ARDS.173 A number of trials have demonstrated an improvement in oxygenation during and after being placed in a prone position.170–174 One prospective randomized trial designed to demonstrate survival advantage failed to do so.175 A number of potential complications can result from the process of changing the patient from a supine to a prone position.183,184 Included in the list of potential complications are tube/catheter malposition/problems, pressure sores, blindness, and difficulty with patient assessment and resuscitation.183,184 Prone positioning should be considered in patients with low risk for such a position change who require high FIO2 despite optimization of ventilator strategy.
Inhaled Nitric Oxide
Inhaled nitric oxide is a bronchial and vascular smooth muscle dilator that also decreases platelet adherence and aggregation.173,196 It has been shown to improve oxygenation by improving ventilation/perfusion relationships in the lung.185,196,197 A reduction in PA pressure and pulmonary vascular resistance also occurs. These beneficial pulmonary effects are associated with minimal systemic effect from the inhaled nitric oxide because it is rapidly inactivated when the nitric oxide enters the circulation and is taken up by red blood cells. Multiple prospective, randomized, placebo-controlled clinical trials failed to demonstrate an improvement in survival despite the early improvement in oxygenation associated with the administration of inhaled nitric oxide.191,193,194
Surfactant Replacement Therapy
Surfactant abnormalities are present in patients with ARDS related to decreased production, inactivation by alveolar proteins and proteolytic enzymes, and dilution by the alveolar fluid.178 Theoretically, surfactant replacement should produce a survival benefit, just as it does in the infant respiratory distress syndrome.177,178 Anzueto and colleagues176 reported no difference in hemodynamic function, oxygenation, length of stay, duration of mechanical ventilation, and survival in 725 sepsis-induced ARDS patients who were prospectively randomized into a placebo-controlled trial of Exosurf (artificial surfactant) versus placebo. The researchers believe that the lack of associated surfactant proteins might account for the lack of efficacy. Trials are continuing to evaluate recombinant forms of surfactant replacement that include surfactant proteins.177 A meta-analysis from a group of small trials evaluating recombinant surfactant protein C replacement in patients with ARDS noted an improvement in oxygenation but no survival benefit.177 To date there has not been a survival benefit associated with the use of surfactant replacement therapy in adults with ARDS.77,78,173
Enhanced Edema Clearance
Accumulated fluid in the alveolus could potentially worsen the gas exchange, as well as produce adverse pulmonary mechanics with increased work of breathing. Recent strategies designed to improve edema clearance either using aquaporins or increasing the activity of the Na/K pump could potentially provide a benefit to patients with ARDS. The BALTI trial evaluated the use of intravenous β-agonist (salbutamol) in patients with ARDS and demonstrated a significant decrease in extravascular lung water at day 7.186 The salbutamol-treated group also had lower end-inspiratory plateau pressures, and there was a trend toward a lower Murray Lung Injury Score. This group also had more supraventricular arrhythmias. A subsequent trial by the same investigators (BALTI-2) demonstrated increased 28-day mortality rate (risk ratio 1.47) with the use of intravenous salbutamol (albuterol).187 The NHLBI ARDS Network investigators evaluated the use of nebulized salbutamol (albuterol 5 mg every 4 hours) and reported no change in ventilator-free days nor in in-hospital mortality rate.188 The amount of edema in the lung can also be increased when there is a low oncotic pressure. To evaluate the potential benefit of infusing albumin and furosemide as opposed to furosemide alone to patients with ARDS, Martin and colleagues190 conducted a randomized controlled trial in 40 hypoproteinemic patients with ALI. The albumin-infused group had an improvement in oxygenation, total protein, net fluid loss, increased number of shock-free days, and less hypotension. Although this is a small study, it does suggest a potential benefit of this maneuver in hypoproteinemic ARDS patients. Further investigation is necessary to determine the benefit of edema clearance strategies in the management of ARDS patients.
Experimental/Innovative Therapies
In an attempt to reduce the high mortality rate associated with ARDS, a number of experimental and innovative therapeutic approaches have been evaluated. A majority of these approaches target abnormalities that either produce or result from the systemic inflammatory response that is felt to be central to the pathogenesis of the injury. To date, none of these approaches has been demonstrated to offer significant benefit in well-conducted, prospective, randomized controlled, multicentered clinical trials. These approaches have included the early administration of high-dose corticosteroids, prostaglandin E1, nonsteroidal anti-inflammatory drugs, antiendotoxin and anticytokine therapy, inhaled nitric oxide, surfactant therapy, antioxidant therapy, positional changes, and partial liquid ventilation.169–174,177,185,191,192,198–234
Many investigations in experimental animal models of ARDS have demonstrated benefit from pretreatment and early treatment with high-dose corticosteroids.198–201 Similar benefit has been observed with a number of nonsteroidal anti-inflammatory agents in experimental animal models of lung injury.202,210,211,235 Unfortunately, the use of anti-inflammatory strategies in humans with sepsis or ARDS has repeatedly failed to demonstrate significant benefit.195,203,205,206,236,237 In fact, in subgroup analysis there was evidence of potential harm in patients with renal dysfunction who were administered high-dose methylprednisolone for treatment of severe sepsis and septic shock.206 To further complicate this clinical situation, it has been demonstrated that a significant proportion of patients with septic shock and other critical illnesses have relative adrenal insufficiency as defined by the inability to elevate the plasma cortisol level more than 9 µg/dL after adrenocorticotropic hormone (ACTH) stimulation.237 This relative adrenocortical deficiency has been implicated as a potential cause of the persistent shock state and impaired perfusion. However, these patients have not always been responsive to the administration of steroids, and the adrenergic hyporesponsiveness may be related to sepsis-induced nitric oxide production, desensitization, or downregulation of α- and β-adrenergic receptors.237 Lower-dose, more physiologic steroid replacement may restore the α- and β-adrenergic responsiveness and potentially turn off the inflammatory reaction to allow for better healing and less injury.237–239 Attempts to prevent the development of ARDS in high-risk patients with severe sepsis and septic shock with high-dose corticosteroids have not been shown to prevent the development of ARDS, improve the reversal of ARDS, or improve the outcome from ARDS.206
Steroid therapy has been shown to be beneficial in patients with severe Pneumocystis jiroveci (carinii) pneumonia and ARDS and possibly in patients with fat embolism.198 In the patient with adrenal insufficiency, stress dose steroids should be administered. The use of corticosteroids to treat patients with established ARDS is controversial and is discussed later. Ibuprofen, a nonsteroidal anti-inflammatory drug, failed to significantly improve the outcome of patients with severe sepsis or septic shock and failed to prevent the development of ARDS in a prospective, randomized, placebo-controlled, multicentered clinical trial.236
A plethora of clinical trials have evaluated “antimediators” that have targeted the potential proinflammatory compounds that can be identified in the blood or BAL of patients at risk for or diagnosed with ARDS.8,90,212,240,241 Despite encouraging results from preclinical experimental animal and early clinical studies, these innovative strategies have failed to demonstrate a significant survival benefit.8,206,213,216–218,221–229 Attempts to change the inflammatory response by changing the ratio of omega 3/omega 6 fatty acids have evaluated the potential benefit of an enteral nutritional formula rich in eicosapentaenoic acid and γ-linoleic acid.161 A large, multicentered, prospective, randomized trial demonstrated an improvement in lung injury score, oxygenation, and organ dysfunction as compared with an isocaloric, isonitrogenous enteral formulation.161 Unfortunately, a survival benefit was not seen.
Pentoxifylline and lisophylline are xanthine derivatives that were felt to have utility in the management of sepsis and ARDS. Pentoxifylline is a rheologic agent that has the ability to inhibit toxic oxygen radical release, decrease platelet aggregation, decrease phagocytosis, diminish the response to platelet-activating factor (PAF) stimulation, and inhibit the release of tumor necrosis factor (TNF) into the systemic circulation. Clinical evaluation of this treatment strategy by the National Institutes of Health (NIH) ARDS Network found no significant benefit.240
ARDS may be produced or worsened by the elaboration of toxic oxygen radicals from the activated inflammatory cells.8 The abundant production of toxic oxygen radicals may overwhelm the ability of the endogenous oxygen radical scavengers, superoxide dismutase (SOD), catalase, and the glutathione redux cycle. The administration of antioxidants such as N-acetylcysteine, procysteine, vitamin E, β-carotene, and vitamin C has been evaluated in the prevention and management of patients with ARDS.143,233,242,243 No survival benefit was seen associated with the administration of N-acetylcysteine or procysteine versus control in patients with ARDS.233
Prevention
The ability to prevent the development of ARDS would be a welcome achievement that to date has been elusive. Two older trials demonstrated a significant reduction in the development of ARDS in surgical patients when ketoconazole, an imidazole thromboxane A2 synthetase inhibitor, was administered to an at-risk population of patients.244–245 However, when ketoconazole was evaluated by the ARDS Network in the treatment of patients with ARDS there was no benefit on survival.246 The use of 3-hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors (statins) as a prevention for ARDS was evaluated in an observational study of a large cohort of patients at risk for developing ARDS and was not found to decrease the development of ARDS or improve mortality rates, organ failure–free days, or ventilator-free days.247
Fibroproliferative Phase
An improved understanding of the injury and repair phase of ARDS has resulted in the recognition of the late fibroproliferative phase.8,81–84,248 This stage of the injury/repair process is characterized by replacement of damaged epithelial cells and accumulation of mesenchymal cells and connective tissue products in the airspaces and the intra-acinar microvessels.79–81 Clinical manifestations include fever, leukocytosis, diffuse alveolar infiltrates on the chest radiograph, and persistent inflammatory mediators in the serum.79–81 Gallium scans demonstrate an increase in pulmonary uptake, and BAL typically contains markers of inflammation and type 3 procollagen peptide.79 Physiologic manifestations include the worsening of static pulmonary compliance, abnormal gas exchange, increased dead space ventilation, pulmonary hypertension, and lack of PEEP response.248 This picture of persistent inflammation requires a dedicated approach to ensure that there is not an ongoing uncontrolled infectious process that has not been adequately addressed.248 Once it is determined that this state is not the result of inadequately treated infection, therapy with corticosteroids is used by some experts.248 Several anecdotal reports have suggested that this therapy may have potential efficacy in patients with a persistent inflammatory response.198,248 A small, single-center, prospective, randomized, placebo-controlled, double-blind clinical trial in 24 patients with the fibroproliferative phase of ARDS demonstrated an improvement in survival, lung function, and organ system dysfunction.249 This study has been criticized because it included the crossover of patients at day 10 and had a smaller study population than a previously reported uncontrolled trial from the same center.249
The use of steroid rescue for the patient with persistent ARDS or the fibroproliferative phase of ARDS was evaluated by the NIH-sponsored ARDS Network.250 The study prospectively randomized 180 patients with ARDS for greater than or equal to 7 days into a placebo-controlled trial of methylprednisolone versus placebo. The primary efficacy outcome was alive at home at 60 days. Patients with septic shock, a defined need for corticosteroid therapy, disseminated fungal infection, or undrained abscess were excluded. The trial was conducted over 6 years and included a modification of the study protocol that included a reduction in the number of subjects from 400 to 180 and an increase in the inclusion PAO2/FIO2 ratio. The use of methylprednisolone was associated with an early improvement in mortality rate, PAO2/FIO2 ratio, blood pressure, ventilator, and ICU-free days. An increase in the white blood cell count and glucose level related to steroid administration occurred, along with a decrease in body temperature. No difference was measured in the primary efficacy end point and 60-day mortality rate, and no significant difference in 180-day outcome occurred between the two groups. The steroid treatment was not associated with an increase in serious infections. In fact, there was more pneumonia and septic shock seen in the placebo group than in the steroid-treated group. Unfortunately, the use of steroids was associated with more neuropathy and myopathy, but it is important to note that 30% of the steroid-treated patients were receiving neuromuscular blocking drugs.250 Meduri and colleagues evaluated a low-dose prolonged methylprednisolone infusion in patients with early severe ARDS and noted a significant decrease in lung injury score and C-reactive protein level along with a decrease in ICU mortality rates, duration of mechanical ventilation, and multiple organ dysfunction scores.251 The steroid infusion decreased levels of inflammatory biomarkers and coagulation.252 The prolonged infusion strategy appeared to be safe and well tolerated, but this was a small study of 91 patients and the topic of steroid treatment in ARDS remains controversial; we eagerly await more definitive studies.
Macrolide antibiotics have been said to have important immunomodulatory properties that could prove to be beneficial in patients with ARDS. The ARDS Network has reviewed initial antibiotic data on 235 patients enrolled in the LARMA trial and found that 20% received a macrolide antibiotic within the first 24 hours of the trial.253 These patients were more likely to have a pulmonary infection as a cause of ARDS. Of interest was the observation that the macrolide-treated patients had a 23% mortality rate compared to 36% mortality rate for nonmacrolide antibiotic regimens.253 This interesting observation may warrant additional study.
The use of extracorporal membrane oxygenation (ECMO) was evaluated in the 1970s with dismal outcomes, but with refinements in the ability to provide extracorporal support in the operative and pediatric settings there has been an increased utilization of the technique in adult patients with severe oxygenation issues related to ARDS.254–257 Improved catheters and understanding of the physiology of ECMO has brought the technique into more common use and was found to be associated with lower hospital mortality rate than non-ECMO support in ECMO referred patients with the severe 2009 influenza A (H1N1).257 Significant improvement in outcome was also seen in a report from England, which contrasted treatment at a tertiary care referral center capable of ECMO versus treatment at a center that had only conventional support in patients with severe ARDS.256 Because transfer to a center with ECMO capability did not necessarily lead to institution of ECMO, more stringent study is needed to clearly define the role for ECMO support in patients with significant oxygenation abnormalities associated with severe ARDS.
Multiple Organ Dysfunction/Failure
A frequent complication of an exaggerated proinflammatory state in the setting of sepsis, SIRS, and ARDS is the development of organ system dysfunction.8,18,36,89,258 This dysfunction may involve single or multiple organs.160,258 A recent consensus conference has suggested that the dysfunction of two or more organs such that normal homeostasis cannot be maintained in the setting of a systemic inflammatory response to a variety of insults is considered MODS.259 This dysfunction may be partial or complete, reversible or irreversible.160 A continuum of abnormalities ranging from dysfunction to failure for each organ is probable.160 Unfortunately, as of this time there has been no consensus on the threshold that separates these two phenomena or the threshold between reversible and irreversible organ system dysfunction.160
MODS and MOF are the most common causes of death in the noncoronary ICU.8,18,258,260,261 Many authorities consider ARDS as the earliest manifestation of an uncompensated systemic inflammatory process with excessive proinflammatory component.87,172 One hypothesis for this injury suggests that in the absence of a direct injury to a specific organ, there must be multiple inflammatory insults to produce the clinically apparent MODS/MOF.160 This hypothesis, called the “two-hit hypothesis,”160 suggests that an initial sensitizing insult is followed within a specific period of time by a second insult that is capable of initiating a more profound proinflammatory response because the target cells have been upregulated or primed by the initial insult.160 Multiple combinations of direct injury, ischemic injury, circulating humoral or inflammatory mediators, translocation of endotoxin or colonic bacteria, altered rheologic properties of the blood cells, or iatrogenic effects of the therapy administered may interact in the eventual production of MODS/MOF.160
Prognosis
Recent studies report a significant reduction in the mortality rate from ARDS using lung protective ventilatory support and proper levels of PEEP.8,83,241,243 Current trials report mortality rates in the range of 30% to 60% in comparison with 60% to 90% of the past.8,94,262,263 The 30-day mortality rate seen in patients enrolled in the NIH-sponsored ECMO trial of the 1970s was 91% with both conventional and ECMO treatment.262 Older reports from large tertiary referral centers documented mortality rates of 90% for patients with gram-negative septic shock and ARDS.263 A large multicenter trial of inhaled surfactant in 725 patients with septic-induced ARDS demonstrated survival rates of 60% at 28 days in both the treatment and the control groups.176 Today the mortality rate from ARDS appears to depend on the cause of the injury, the patient’s underlying disease status, patient age, and institutional factors.38,254 This improvement in mortality rate mandates the use of a concomitant control group as opposed to using historical control subjects in the assessment of new innovative therapeutic strategies.
The most common causes of death in patients with ARDS continues to be from MOF and recurrent sepsis.3,8,16,36,260,261 Fewer than 20% of patients die because of the inability to adequately oxygenate or ventilate them.260 The complexities of the balance between the proinflammatory and anti-inflammatory processes that encompass the pathophysiologic response of this injury direct the response from organ dysfunction secondary to an overzealous proinflammatory reaction to infectious complications. These complications result from the immune suppression of a predominant anti-inflammatory response.83,264 When infection is present, the lung is a frequent site for the process and may be extremely difficult to diagnose.35,89,151,152 Patients with pulmonary infections were found to typically have a septic clinical picture without definitive positive culture results.35 On the other hand, in patients who were found to have positive blood cultures without antemortem identification of a specific site, the site of occult infection was commonly found to be in the abdomen at postmortem examination.35 Predictors of high mortality rates from ARDS include the development of multiple organ dysfunction/failure, development of secondary sepsis, concomitant cancer, and the presence of cirrhosis or hepatic dysfunction.16 A study evaluating predictors of mortality rate in patients with ARDS receiving lung protective ventilatory support reported that the oxygenation index (mean airway pressure × FIO2 × 100 ÷ PaO2) was an independent predictor of mortality rate.265
After recovery from ARDS, the prognosis appears to be reasonably good. A recent report found that 80% of ARDS survivors discharged from the ICU were still alive 5 years later.266,267 Although most ARDS survivors have initial abnormalities in pulmonary function quality of life, the majority return to near their baseline pulmonary function status within 3 to 6 months.268,269 The major residual abnormality in pulmonary function is a restrictive pulmonary defect and a reduction in the carbon monoxide diffusion capacity.268,269 These alterations may result in exercise desaturation in some patients or, more commonly, a decrease in timed walked distance.268,269 When observed over the next 5 years, their pulmonary function did not improve a great deal after this initial improvement.266,270 However, most patients continue to experience exercise limitation at 5 years, despite the fact that 65% have returned to work.266
Survivors of ARDS have been found to have a decreased health-related quality of life, increased respiratory symptoms, insomnia, depression, anxiety, and posttraumatic stress disorder.271–274 ARDS survivors have been found to have a clinically significant reduction in their physical function and increased pulmonary symptoms in comparison with the matched survivors of critical illness.271–277 Survivors of ARDS are also found to have long-term cognitive impairment, problems coping with their disability, and relationship strains.274,275 Elderly patients, older than 70 years of age, seem to have worse outcomes with an increase in mortality rate compared with ARDS patients who are younger than 70 years of age.273
Future Considerations
The growing knowledge of molecular biology and the elaborate mechanisms that govern a person’s response to injury, repair, and cell death will likely have a major role in the management of patients with ARDS. Individuals with increased risk for ALI and ARDS development will no doubt be identified on the basis of their genetic profiles. This knowledge will likely affect future management. In years to come, scientists may potentially modify the genetic makeup or the biologic response of a susceptible individual by inserting selected genes or modifying the transcription or function of various regulatory proteins. Studies with mesenchymal stem cells have been conducted in rodents with lung injury and demonstrate potential promise for future therapy.278
References
1. Greene, KE, Peters, JI. Pathophysiology of acute respiratory failure. Clin Chest Med. 1994; 15:1–12.
2. Balk, RA, Bone, RC. Acute respiratory failure. Med Clin North Am. 1983; 67:351–356.
3. Tisi, GM. Pulmonary Physiology in Clinical Medicine, 2nd ed. Baltimore: Williams & Wilkins; 1983.
4. Derenne, JP, Fleury, B, Pariente, R. Acute respiratory failure of chronic obstructive pulmonary disease. Am Rev Respir Dis. 1988; 138:1006–1033.
5. Chen, WJ, Kuan, P, Lien, WP, Lin, FY. Detection of patent foramen ovale by contrast transesophageal echocardiography. Chest. 1992; 101:1515–1520.
6. Cujec, B, Polasek, P, Mayers, I, Johnson, D. Positive end-expiratory pressure increases the right-to-left shunt in mechanically ventilated patients with patent foramen ovale. Ann Intern Med. 1993; 119:887–894.
7. Tobin, MJ. Respiratory monitoring in the intensive care unit. Am Rev Respir Dis. 1988; 138:1625–1642.
8. Ware, LB, Matthay, MA. The acute respiratory distress syndrome. N Engl J Med. 2000; 342:1334–1349.
9. Weinberger, SE, Schwartzstein, RM, Weiss, JW. Hypercapnia. N Engl J Med. 1989; 321:1223–1231.
10. Parsons, PE. Respiratory failure as a result of drugs, overdoses, and poisonings. Clin Chest Med. 1994; 15:93–101.
11. Anzueto, A, Peters, JI, Tobin, MJ, et al. Effects of prolonged controlled mechanical ventilation on diaphragmatic function in healthy adult baboons. Crit Care Med. 1997; 25:1187–1190.
12. Ashbaugh, DG, Bigelow, DB, Petty, TL, Levine, BE. Acute respiratory distress in adults. Lancet. 1967; 2:319–323.
13. Matthay, MA, Zimmerman, GA. Acute lung injury and the acute respiratory distress syndrome: Four decades of inquiry into pathogenesis and rational management. Am J Respir Crit Cell Mol Biol. 2005; 33:319–327.
14. Jain, R, Dal Nogare, A. Pharmacological therapy for acute respiratory distress syndrome. Mayo Clin Proc. 2006; 81:205–212.
15. Petty, T. Indicators of risk, course, and prognosis in adult respiratory distress syndrome (ARDS). Am Rev Respir Dis. 1985; 132:471.
16. Doyle, RL, Szaflarski, N, Modin, GW, et al. Identification of patients with acute lung injury: Predictors of mortality. Am J Resp Crit Care Med. 1995; 152:1818–1824.
17. Fowler, AA, Hamman, RF, Good, JT, et al. Adult respiratory distress syndrome: Risk with common predispositions. Ann Intern Med. 1983; 98:593–597.
18. Balk, RA, Bone, RC. Adult respiratory distress syndrome. Med Clin North Am. 1983; 67:685–700.
19. Gattinoni, L, Pelosi, P, Suter, PM, et al. Acute respiratory distress syndrome caused by pulmonary and extrapulmonary disease. Different syndromes? Am J Respir Crit Care Med. 1998; 158:3–11.
20. Pepe, P, Potkin, R, Holtman Reus, D, et al. Clinical predictors of the adult respiratory distress syndrome. Am J Surg. 1982; 144:124–130.
21. Bernard, GR, Artigas, A, Brigham, KL, et al. The American-European Consensus Conference on ARDS: Definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am J Respir Crit Care Med. 1994; 149:818–824.
22. Artigas, A, Bernard, GR, Carlet, J, et al. The American-European Consensus Conference on ARDS, Part 2. Am J Respir Crit Care Med. 1998; 157:1332–1347.
23. American Thoracic Society. Round table conference: Acute lung injury. Am J Respir Crit Care Med. 1998; 158:675–679.
24. Abraham, E. Toward new definitions of acute respiratory distress syndrome. Crit Care Med. 1999; 27:237–238.
25. Esteban, A, Fernancez-Segoviano, P, Frutos-Villar, F, et al. Comparison of clinical criteria for the acute respiratory distress syndrome with autopsy findings. Ann Intern Med. 2004; 131:440–445.
26. Ferguson, ND, Frutos-Vivar, F, Esteban, A, et al. Acute respiratory distress syndrome: Underrecognition by clinician and diagnostic accuracy of three clinical definitions. Crit Care Med. 2005; 33:2228–2234.
27. Luce, JL. The imperfect diagnosis of acute respiratory distress syndrome. Crit Care Med. 2005; 33:2419–2420.
28. Diaz, JV, Brower, R, Calfee, CS, Matthay, MA. Therapeutic strategies for severe acute lung injury. Crit Care Med. 2010; 38:1644–1650.
29. Kallet, RH. What is the legacy of the National Institutes of Health acute respiratory distress syndrome network? Respir Care. 2009; 7:912–924.
30. The ARDS Definition Task Force. Acute respiratory distress syndrome: The Berlin definition. JAMA. 2012; 307:2526–2533.
31. Sheard, S, Rao, P, Devaraj, A. Imaging of acute respiratory distress syndrome. Resp Care. 2012; 57:607–612.
32. Rubenfeld, GD, Caldwell, E, Granton, J, et al. Interobserver variability in applying a radiographic definition for ARDS. Chest. 1999; 116:1347–1353.
33. Fulkerson, WJ, MacIntyre, N, Stamler, J, Crapo, JD. Pathogenesis and treatment of the adult respiratory distress syndrome. Arch Intern Med. 1996; 156:29–38.
34. Zilberberg, MD, Epstein, SK. Acute lung injury in the medical ICU. Am J Respir Crit Care Med. 1998; 157:1159–1164.
35. Seidenfeld, JJ, Pohl, DF, Bell, RC, et al. Incidence, site, and outcome of infections in patients with the adult respiratory distress syndrome. Am Rev Respir Dis. 1986; 134:12–16.
36. Kollef, MH, Schuster, DP. The acute respiratory distress syndrome. N Engl J Med. 1995; 332:27–37.
37. Brun-Buisson, C, for the ALIVE Study Group. Epidemiology and outcome of acute lung injury in European intensive care units: Results from the Alive Study. Intensive Care Med. 2004; 30:51–61.
38. Rubenfeld, GD, Caldwell, E, Peabody, E, et al. Incidence and outcomes of acute lung injury. N Engl J Med. 2005; 353:1685–1693.
39. Ackland, GL, Mythen, MG. Negative pressure pulmonary edema as an unsuspected imitator of acute lung injury/ARDS. Chest. 2005; 127:1867–1868.
40. Koh, MS, Hsu, AA, Eng, P. Negative pressure pulmonary edema in the medical intensive care unit. Intensive Care Med. 2003; 29:1601–1604.
41. Patel, AR, Bersten, AD. Pulmonary haemorrhage associated with negative-pressure pulmonary oedema: A case report. Crit Care Resuscitation. 2006; 8:115–116.
42. Sow Nam, Y, Garewal, D. Pulmonary hemorrhage in association with negative pressure edema in an intubated patient. Acta Anaesthesiol Scand. 2001; 45:911–913.
43. Broccard, AF, Liaudet, L, Aubert, JD, et al. Negative pressure post-tracheal extubation alveolar hemorrhage. Anesth Analg. 2001; 92:273–275.
44. Dolinski, SY, MacGregor, DA, Scuderi, PE. Pulmonary hemorrhage associated with negative-pressure pulmonary edema. Anesthesiology. 2000; 93:888–890.
45. Schwartz, DR, Maroo, A, Malhotra, A, Kesselman, H. Negative pressure pulmonary hemorrhage. Chest. 1999; 115:1194–1197.
46. Oswalt, CE, Gates, GA, Holmstrom, MG. Pulmonary edema as a complication of acute airway obstruction. JAMA. 1977; 238:1833–1835.
47. Tarrac, SE. Negative pressure pulmonary edema—A postanesthesia emergency. J Perianesth Nurs. 2003; 18:317–323.
48. Ikeda, H, Asato, R, Chin, K, et al. Negative-pressure pulmonary edema after resection of mediastinum thyroid goiter. Acta Otolaryngol. 2006; 126:886–888.
49. Sharma, ML, Beckett, N, Gormley, P. Negative pressure pulmonary edema following thyroidectomy. Can J Anaesth. 2002; 49:215.
50. Westreich, R, Sampson, I, Shaari, CM, Lawson, W. Negative-pressure pulmonary edema after routine septorhinoplasty: Discussion of pathophysiology, treatment, and prevention. Arch Facial Plast Surg. 2006; 8:8–15.
51. Thomas, CL, Palmer, TJ, Shipley, P. Negative pressure pulmonary edema after a tonsillectomy and adenoidectomy in a pediatric patient: Case report and review. AANA J. 1999; 67:425–430.
52. Lloyd, C, Kamisetty, A. Negative pressure pulmonary edema following open reduction and internal fixation of a fractured mandible. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2003; 95:2.
53. Gupta, S, Richardson, J, Pugh, M. Negative pressure pulmonary oedema after cryotherapy for tracheal obstruction. Eur J Anaesthesiol. 2001; 18:189–191.
54. Jouan, ZT, Jawan, B, Lee, JH. Pulmonary edema complicated by postextubation laryngospasm: A case report. Changgeng Yi Xue Za Zhi. 1997; 20:309–312.
55. Murray-Calderon, P, Connolly, MA. Laryngospasm and noncardiogenic pulmonary edema. J Perianesth Nurs. 1997; 12:89–94.
56. Dicpinigaitis, PV, Mehta, DC. Postobstructive pulmonary edema induced by endotracheal tube occlusion. Intensive Care Med. 1995; 21:1048–1050.
57. Liu, EH, Yih, PS. Negative pressure pulmonary oedema caused by biting and endotracheal tube occlusion—A case for oropharyngeal airways. Singapore Med J. 1999; 40:174–175.
58. Memtsoudis, SG, Rosenberger, P, Sadovnikoff, N. Chest tube suction-associated unilateral negative pressure pulmonary edema in a lung transplant patient. Anesth Analg. 2005; 101:38–40.
59. Kaki, A, Crosby, ET, Lui, AC. Airway and respiratory management following nonlethal hanging. Can J Anaesth. 1997; 44:445–450.
60. Stuth, EA, Stucke, AG, Berens, RJ. Negative-pressure pulmonary edema in a child with hiccups during induction. Anesthesiology. 2000; 93:282–284.
61. Mandal, NG. Negative-pressure pulmonary edema in a child with hiccups during induction. Anesthesiology. 2001; 94:378–379.
62. Baumann, A, Audibert, G, McConnell, J, Mertes, PM. Neurogenic pulmonary edema. Acta Anesth Scand. 2007; 51:447–455.
63. Davison, DL, Terek, M, Chawla, LS. Neurogenic pulmonary edema. Crit Care. 2012; 16:212–218.
64. Mascia, L. Acute lung injury in patients with severe brain injury: A double hit model. Neurocrit Care. 2009; 11:417–426.
65. Wittstein, IS, Thiemann, DR, Lima, JAC, et al. Neurohumoral features of myocardial stunning due to sudden emotional stress. N Engl J Med. 2005; 352:539–548.
66. Nguyen, H, Zaroff, JG. Neurogenic stunned myocardium. Curr Neurol Neurosci Rep. 2009; 9:486–491.
67. Sedy, J, Zicha, J, Kunes, J, et al. Mechanisms of neurogenic pulmonary edema development. Psysiol Res. 2008; 57:499–506.
68. Sharkey, SW, Windenburg, DC, Lesser, JR, et al. Natural history and expansive clinical profile of stress (tako-tsubo) cardiomyopathy. J Am Coll Cardiol. 2010; 55:333–341.
69. Moss, M, Goodman, PL, Heinig, M, et al. Establishing the relative accuracy of three new definitions of the adult respiratory distress syndrome. Crit Care Med. 1995; 23:1629–1637.
70. Goss, CH, for the ARDS Network. Incidence of acute lung injury in the United States. Crit Care Med. 2003; 31:1607–1611.
71. Villar, J, Blanco, J, Anon, JM, et al. The ALIEN study: Incidence and outcome of acute respiratory distress syndrome in the era of lung protective ventilation. Intensive Care Med. 2011; 37:1932–1941.
72. Hudson, L. New therapies for ARDS. Chest. 1995; 108:79S–91S.
73. Gattinoni, L, D’Andrea, L, Pelosi, P, et al. Regional effects and mechanism of positive-end expiratory pressure in early adult respiratory distress syndrome. JAMA. 1993; 269:2122–2127.
74. Weiland, JE, Davis, WB, Holter, JF, et al. Lung neutrophils in the adult respiratory distress syndrome. Am Rev Respir Dis. 1986; 133:218–225.
75. Shirley, HH, Jr., Wolfram, CG, Waserman, K, Mayerson, HS. Capillary permeability to macromolecules: Stretched pore phenomenon. Am J Physiol. 1957; 190:189–193.
76. Enhorning, G. Surfactant replacement in adult respiratory distress syndrome. Am Rev Respir Dis. 1989; 140:281–283.
77. Albert, RK. The role of ventilation-induced surfactant dysfunction and atelectasis in causing acute respiratory distress syndrome. Am J Respir Crit Care Med. 2012; 185:702–708.
78. Spragg, RG, Taut, FJH, Lewis, JF, et al. Recombinant surfactant protein C-based surfactant for patients with severe direct lung injury. Am J Respir Crit Care Med. 2011; 183:1055–1061.
79. Clark, JG, Milberg, JA, Steinberg, KP, Hudson, LD. Type III procollagen peptide in the adult respiratory distress syndrome. Ann Intern Med. 1994; 122:17–23.
80. Fowler, AA, Hamman, RF, Zerbe, GO, et al. Adult respiratory distress syndrome. Am Rev Respir Dis. 1985; 132:472–478.
81. Meduri, GU, Kohler, G, Headley, S, et al. Inflammatory cytokines in the BAL of patients with ARDS: Persistent elevation over time predicts poor outcome. Chest. 1995; 108:1303–1314.
82. Meduri, GU, Headley, S, Tolley, E, et al. Plasma and BAL cytokine response to corticosteroid rescue treatment in late ARDS. Chest. 1995; 108:1315–1325.
83. Meduri, GU, Headley, S, Kohler, G, et al. Persistent elevation of inflammatory cytokines predicts a poor outcome in ARDS: Plasma IL-1β and IL-6 levels are consistent and efficient predictors of outcome over time. Chest. 1995; 107:1062–1073.
84. Martin, C, Papazian, L, Payan, MJ, et al. Pulmonary fibrosis correlates with outcome in adult respiratory distress syndrome. Chest. 1995; 107:196–200.
85. Matthay, MA, Robriquet, L, Fang, X. Alveolar epithelium: Role in lung fluid balance and acute lung injury. Proc Am Thorac Soc. 2005; 2:206–213.
86. Martin, TR, Hagimoto, N, Nakamura, M, Matute-Bello, G. Apoptosis and epithelial injury in the lungs. Proc Am Thorac Soc. 2005; 2:214–220.
87. Bone, RC, Grodzin, CJ, Balk, RA. Sepsis: A new hypothesis for pathogenesis of the disease process. Chest. 1997; 117:235–243.
88. Fan, E, Needham, DM, Stewart, TE. Ventilatory management of acute lung injury and acute respiratory distress syndrome. JAMA. 2005; 294:2889–2896.
89. Bell, RC, Coalson, JJ, Smith, JD, Johanson, WG, Jr. Multiple organ system failure and infection in adult respiratory distress syndrome. Ann Intern Med. 1983; 99:293–298.
90. Suter, PM. Lung inflammation in ARDS—Friend or foe? N Engl J Med. 2006; 354:1739–1742.
91. Marini, JJ, Kelsen, SG. Retargeting ventilatory objectives in adult respiratory distress syndrome. Am Rev Respir Dis. 1992; 146:2–3.
92. Ranieri, VM, Suter, PM, Tortorella, C, et al. Effect of mechanical ventilation on inflammatory mediators in patients with acute respiratory distress syndrome. JAMA. 1999; 282:54–61.
93. Amato, MBP, Barbas, CSV, Medeiros, DM, et al. Effect of a protective ventilation strategy on mortality in the acute respiratory distress syndrome. N Engl J Med. 1998; 338:347–354.
94. The Acute Respiratory Distress Syndrome Network. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N Engl J Med. 2000; 342:1301–1308.
95. Slutsky, AS, Tremblay, LN. Multiple system organ failure: Is mechanical ventilation a contributing factor? Am J Respir Crit Care Med. 1998; 157:1721–1725.
96. Dreyfuss, D, Saumon, G. Ventilator induced lung injury: Lessons from experimental studies. Am J Respir Crit Care Med. 1998; 157:294–323.
97. Parker, JC, Hernandez, LA, Peevy, KJ. Mechanisms of ventilator induced lung injury. Crit Care Med. 1993; 21:131–143.
98. Pinhu, L, Whitehead, T, Evans, T, Griffiths, M. Ventilator-associated lung injury. Lancet. 2003; 361:332–340.
99. Esan, A, Hess, DR, Raoof, S, et al. Severe hypoxemic respiratory failure: Part 1. Ventilatory strategies. Chest. 2010; 137:1203–1216.
100. Marini, JJ. Point: Is pressure assist-control preferred over volume assist-control mode for lung protective ventilation in patients with ARDS? Yes. Chest. 2011; 140:286–289.
101. MacIntyre, N. Counterpoint: Is pressure assist-control preferred over volume assist-control mode for lung protective ventilation in patients with ARDS? No. Chest. 2011; 140:290–292.
102. Meade, MO, Cook, DJ, Kernerman, P, Bernard, G. How to use articles about harm: The relationship between high tidal volumes, ventilating pressures, and ventilator-induced lung injury. Crit Care Med. 1997; 25:1915–1922.
103. Vieira, ARR, Puybasset, L, Richecoeur, J, et al. A lung computed tomographic assessment of positive end-expiratory pressure-induced lung overdistention. Am J Respir Crit Care Med. 1998; 158:1571–1577.
104. Marini, JJ. Lung mechanics in the adult respiratory distress syndrome. Clin Chest Med. 1990; 11:673–690.
105. O’Brien, JM, Jr., Welsh, CH, Fish, RH, et al. Excess body weight is not independently associated with outcome in mechanically ventilated patients with acute lung injury. Ann Intern Med. 2004; 140:338–345.
106. Kacmarek, RM, Hickling, KG. Permissive hypercapnia. Respir Care. 1993; 38:373–387.
107. Vadasz, I, Hubmayr, RD, Nin, N, et al. Hypercapnia: A nonpermissive environment for the lung. Am J Respir Cell Mol Biol. 2012; 46:417–421.
108. Hall, JB. Respiratory system mechanics in adult respiratory distress syndrome: Stretching our understanding. Am J Respir Crit Care Med. 1998; 158:1–2.
109. Pesenti, A. PEEP: Blood gas cosmetics or a therapy for ARDS? Crit Care Med. 1999; 27:253–254.
110. Myers, J, Reilley, T, Cloutier, CT. Effect of positive end-expiratory pressure on extravascular lung water in porcine acute respiratory failure. Crit Care Med. 1988; 16:52.
111. Zwissler, B, Schosser, R, Schwickert, C, et al. Perfusion of the interventricular septum during ventilation with positive end-expiratory pressure. Crit Care Med. 1999; 19:1414.
112. Laffey, JG, Engelberts, D, Kavanagh, BP. Buffering hypercapnic acidosis worsens acute lung injury. Am J Resp Crit Care Med. 2000; 161:141–146.
113. Kregenow, DA, Rubenfeld, GD, Hudson, LD, Swenson, ER. Hypercapnic acidosis and mortality in acute lung injury. Crit Care Med. 2006; 34:1–7.
114. Walther, SM, Domino, KB, Glenny, RW, Hiastala, MP. Positive end-expiratory pressure redistributes perfusion to dependent lung regions in supine but not in prone lambs. Crit Care Med. 1999; 27:37–45.
115. Neumann, P, Berglund, JE, Mondejar, EF, et al. Effect of different pressure levels on the dynamics of lung collapse and recruitment of oleic-acid-induced lung injury. Am J Respir Crit Care Med. 1998; 158:1636–1643.
116. Ruiz-Bailen, M, Fernandez-Mondejar, E, Hurtado-Ruiz, B, et al. Immediate application of positive-end expiratory pressure is more effective than delayed positive-end expiratory pressure to reduce extravascular lung water. Crit Care Med. 1999; 27:380–384.
117. Jonson, B, Richard, JC, Straus, C, et al. Pressure-volume curves and compliance in acute lung injury. Am J Respir Crit Care Med. 1999; 159:1172–1178.
118. Carvalho, CRR, Barbas, CSV, Medeiros, DM, et al. Temporal hemodynamic effects of permissive hypercapnia associated with ideal PEEP in ARDS. Am J Respir Crit Care Med. 1997; 156:1458–1466.
119. The National Heart, Lung, and Blood Institute Acute Respiratory Distress Syndrome (ARDS) Clinical Trials Network. Higher versus lower positive end-expiratory pressures in patients with the acute respiratory distress syndrome. N Engl J Med. 2004; 351:327–336.
120. Meade, MO, Cook, DJ, Guyatt, GH, et al. Ventilation strategy using low tidal volumes, recruitment maneuvers, and high positive end-expiratory pressure for acute lung injury and acute respiratory distress syndrome: A randomized controlled trial. JAMA. 2008; 299:637–645.
121. Mercat, A, Richard, JC, Vielle, B, et al. Positive end-expiratory pressure setting in adults with acute lung injury and acute respiratory distress syndrome: A randomized controlled trial. JAMA. 2008; 299:646–655.
122. Talmor, D, Sarge, T, Malhotra, A, et al. Mechanical ventilation guided by esophageal pressure in acute lung injury. N Engl J Med. 2008; 359:295–2104.
123. Dasenbrook, EC, Needham, DM, Brower, RG, Fan, E. Higher PEEP in patients with acute lung injury: A systematic review and meta-analysis. Respir Care. 2011; 56:568–575.
124. Dessap, AM, Boissier, F, Leon, R, et al. Prevalence and prognosis of shunting across patient foramen ovale during acute respiratory distress syndrome. Crit Care Med. 2010; 38:1786–1792.
125. Mure, M, Glenny, RW, Domino, KB, Hlastala, MP. Pulmonary gas exchange improves in the prone position with abdominal distention. Am J Respir Crit Care Med. 1998; 157:1785–1790.
126. Young, MP, Manning, HL, Wilson, DL, et al. Ventilation of patients with acute lung injury and acute respiratory distress syndrome: Has new evidence changed clinical practice? Crit Care Med. 2004; 32:1260–1265.
127. Papazian, L, Forel, JM, Gacouin, A, et al. Neuromuscular blockers in early acute respiratory distress syndrome. N Engl J Med. 2010; 363:1107–1116.
128. Forel, JM, Roch, A, Marin, V, et al. Neuromuscular blocking agents decrease inflammatory response in patients presenting with acute respiratory distress syndrome. Crit Care Med. 2006; 34:2749–2757.
129. Slutsky, AS. Neuromuscular blocking agents in ARDS. N Engl J Med. 2010; 363:1176–1180.
130. Puthucheary, Z, Rawal, J, Ratnayake, G, et al. Neuromuscular blockade and skeletal muscle weakness in critically ill patients: Time to rethink the evidence? Am J Respir Crit Care Med. 2012; 185:911–917.
131. Humphrey, H, Hall, J, Sznajder, I, et al. Improved survival in ARDS patients associated with a reduction in pulmonary capillary wedge pressure. Chest. 1990; 97:1176–1180.
132. Hudson, LD. Fluid management strategy in acute lung injury. Am Rev Resp Dis. 1992; 145:988–989.
133. Mitchell, JP, Schuller, D, Calandrino, FS, Schuster, DP. Improved outcome based on fluid management in critically ill patients requiring pulmonary artery catheterization. Am Rev Respir Dis. 1992; 145:990–998.
134. Russell, JA, Phang, PT. The oxygen delivery/consumption controversy: Approaches to management of the critically ill. Am J Respir Crit Care Med. 1994; 149:533–537.
135. Tuchschmidt, J, Fried, J, Aziz, M, Rackow, E. Elevation of cardiac output and oxygen delivery improves outcome in septic shock. Chest. 1992; 102:216–220.
136. Hayes, MA, Timmins, AC, Yau, EHS, et al. Elevation of systemic oxygen delivery in the treatment of critically ill patients. N Engl J Med. 1994; 330:1717–1722.
137. Ronco, JJ, Fenwick, JC, Tweeddale, MG, et al. Identification of the critical oxygen delivery for anaerobic metabolism in critically ill septic and nonseptic humans. JAMA. 1993; 270:1724–1730.
138. Yu, M, Takanishi, D, Myers, SA, et al. Frequency of mortality and myocardial infarction during maximizing oxygen delivery: A prospective, randomized trial. Crit Care Med. 1995; 23:1025–1032.
139. Shoemaker, WC, Appel, PL, Kram, HB, et al. Prospective trial of supranormal values of survivors as therapeutic goals in high-risk surgical patients. Chest. 1988; 94:1176–1186.
140. Bishop, MH, Shoemaker, WC, Appel, WC, et al. Prospective, randomized trial of survivor values of cardiac index, oxygen delivery, and oxygen consumption as resuscitation endpoints in severe trauma. J Trauma Inj Infect Crit Care. 1995; 38:780–787.
141. Yu, M, Levy, MM, Smith, P, et al. Effect of maximizing oxygen delivery on morbidity and mortality rates in critically ill patients: A prospective, randomized, controlled study. Crit Care Med. 1993; 21:830–837.
142. Hayes, MA, Yau, EHS, Timmins, AC, et al. Response of critically ill patients to treatment aimed at achieving supranormal oxygen delivery and consumption. Chest. 1993; 103:886–895.
143. Goldstein, G, Luce, JM. Pharmacologic treatment of the adult respiratory distress syndrome. Clin Chest Med. 1990; 11:773–787.
144. Rivers, EP. Fluid-management strategies in acute lung injury-liberal, conservative, or both? N Engl J Med. 2006; 354:2598–2600.
145. The SAFE Study Investigators. A comparison of albumin and saline for fluid resuscitation in the intensive care unit. N Engl J Med. 2004; 350:2247–2256.
146. The National Heart, Lung, and Blood Institute Acute Respiratory Distress Syndrome (ARDS) Clinical Trials Network. Comparison of two fluid management strategies in acute lung injury. N Engl J Med. 2006; 354:2564–2575.
147. The National Heart, Lung, and Blood Institute Acute Respiratory Distress Syndrome (ARDS) Clinical Trials Network. Pulmonary-artery versus central venous catheter to guide treatment of acute lung injury. N Engl J Med. 2006; 354:2213–2224.
148. Shure, D. Pulmonary-artery catheters—Peace at last? N Engl J Med. 2006; 354:2273–2274.
149. Connors, AF, Jr., Speroff, T, Dawson, N, et al. The effectiveness of right heart catheterization in the initial care of critically ill patients. JAMA. 1996; 276:889–897.
150. Richard, C, Warszawski, J, Anguel, N, et al. Early use of the pulmonary artery catheter and outcomes in patients with shock and acute respiratory distress syndrome: A randomized controlled trial. JAMA. 2003; 290:2713–2720.
151. Chastre, J, Trouillet, JL, Vaugnat, A, et al. Nosocomial pneumonia in patients with acute respiratory distress syndrome. Am J Respir Crit Care Med. 1998; 157:1165–1172.
152. Winer-Muram, HT, Steiner, RM, Gurney, JW, et al. Ventilator-associated pneumonia in patients with adult respiratory distress syndrome: CT evaluation. Radiology. 1998; 208:193–199.
153. Meduri, GU, Reddy, RC, Stanley, T, El-Zeky, F. Pneumonia in acute respiratory distress syndrome. Am J Respir Crit Care Med. 1998; 158:870–875.
154. Delclaux, C, Roupie, E, Blot, F, et al. Lower respiratory tract colonization and infection during severe acute respiratory distress syndrome. Am J Respir Crit Care Med. 1997; 156:1092–1098.
155. Pingleton, SK. Complications of acute respiratory failure. Am Rev Respir Dis. 1988; 137:1463–1493.
156. Patel, G, Liberman, J, Gurka, D, et al. Complications of critical illness: Rationale for prophylactic strategies. Clin Pulm Med. 2005; 12:258–268.
157. Noseworthy, TW, Cook, DJ. Nosocomial pneumonia, prophylaxis against gastric erosive disease, and the clinically important gastrointestinal bleeding: Where do we stand? Crit Care Med. 1993; 21:1814–1816.
158. Bonten, MJM, Gaillard, CA, Van Der Gest, S, et al. The role of intragastric acidity and stress ulcer prophylaxis on colonization and infection in mechanically ventilated ICU patients. Am J Respir Crit Care Med. 1995; 152:1825–1834.
159. Prodhom, G, Leuenberger, P, Koerfer, J, et al. Nosocomial pneumonia in mechanically ventilated patients receiving antacid, ranitidine, or sucralfate as prophylaxis for stress ulcer. Ann Intern Med. 1994; 120:653–662.
160. Balk, RA. Pathogenesis and management of multiple organ dysfunction or failure in severe sepsis and septic shock. Crit Care Clin. 2000; 16:337–352.
161. Gadek, JE, DeMichele, SJ, Karlstad, MD, et al. Effect of enteral feeding with eicosapentaenoic acid, γ-linolenic acid, and antioxidants in patients with acute respiratory distress syndrome. Crit Care Med. 1999; 27:1409–1420.
162. Singer, P, Theilla, M, Fisher, H, et al. Benefit of an enteral diet enriched with eicosapentaenoic acid and gamma-linolenic acid in ventilated patients with acute lung injury. Crit Care Med. 2006; 34:1033–1036.
163. Rice, TW, Wheeler, AP, Thompson, BT, et al. Enteral omega-3 fatty acid, γ-linolenic acid, and antioxidant supplementation in acute lung injury. JAMA. 2011; 306:1574–1581.
164. The National Heart, Lung, and Blood Institute Acute Respiratory Distress Syndrome (ARDS) Clinical Trials Network. Initial trophic vs full enteral feeding in patients with acute lung injury: The EDEN randomized trial. JAMA. 2012; 307:795–803.
165. Casaer, MP, Mesotten, D, Hermans, G, et al. Early versus late parenteral nutrition in critically ill adults. N Engl J Med. 2011; 365:506–517.
166. Gibot, S, Cravoisy, A, Levy, B, et al. Soluble triggering receptor expressed on myeloid cells and the diagnosis of pneumonia. N Engl J Med. 2004; 350:451–458.
167. Jacobs, S, Foweraker, JE, Roberts, SE. Effectiveness of selective decontamination of digestive tract (SDD) in an ICU with a policy encouraging a low gastric pH. Clin Intensive Care. 1992; 3:52–58.
168. Chatte, G, Sab, JM, Dubois, JM, et al. Prone position in mechanically ventilated patients with severe acute respiratory failure. Am J Respir Crit Care Med. 1997; 155:473–478.
169. Broccard, AF, Shapiro, RS, Schmitz, LL, et al. Influence of prone position on the extent and distribution of lung injury in a high tidal volume oleic acid model of acute respiratory distress syndrome. Crit Care Med. 1997; 25:16–27.
170. Vollman, KM, Bander, JJ. Improved oxygenation utilizing a prone positioner in patients with acute respiratory distress syndrome. Intensive Care Med. 1996; 22:1105–1111.
171. Lamm, WJE, Graham, MM, Albert, RK. Mechanism by which the prone position improves oxygenation in acute lung injury. Am J Respir Crit Care Med. 1994; 150:184–193.
172. Pelosi, P, Tubiolo, D, Mascheroni, D, et al. Effects of the prone position on respiratory mechanics and gas exchange during acute lung injury. Am J Respir Crit Care Med. 1998; 157:387–393.
173. Raoof, S, Goulet, K, Esan, A, et al. Severe hypoxemic respiratory failure: Part 2. Nonventilatory strategies. Chest. 2010; 137:1437–1448.
174. Mure, M, Martling, C, Lindahl, S. Dramatic effect on oxygenation in patients with severe acute lung insufficiency treated in the prone position. Crit Care Med. 1997; 25:1539–1544.
175. Mancebo, J, Fernandez, R, Blanch, L, et al. A multicenter trial of prolonged prone ventilation in severe acute respiratory distress syndrome. Am J Respir Crit Care Med. 2006; 173:1233–1239.
176. Anzueto, A, Baughman, RP, Guntupalli, KK, et al. Aerosolized surfactant in adults with sepsis induced ARDS. N Engl J Med. 1996; 22:1417–1421.
177. Spragg, RG, Lewis, JF, Walmrath, HD, et al. Effect of recombinant surfactant protein C-based surfactant on the acute respiratory distress syndrome. N Engl J Med. 2004; 351:884–892.
178. Haas, CF, Weg, JG. Exogenous surfactant therapy: An update. Resp Care. 1996; 41:397–414.
179. The National Heart, Lung, and Blood Institute Acute Respiratory Distress Syndrome (ARDS) Clinical Trials Network. Effects of recruitment maneuvers in patients with acute lung injury and acute respiratory distress syndrome ventilated with high positive end-expiratory pressure. Crit Care Med. 2003; 31:2592–2597.
180. Talmor, D, Sarge, T, Legedza, A, et al. Cytokine release following recruitment maneuvers. Chest. 2007; 132:1434–1439.
181. Lim, C-M, Jung, H, Koh, Y, et al. Effect of alveolar recruitment maneuver in early acute respiratory distress syndrome according to antiderecruitment strategy, etiological category of diffuse lung injury, and body position of the patient. Crit Care Med. 2003; 31:411–418.
182. Pelosi, P, Bottino, N, Chiumello, D, et al. Sigh in supine and prone position during acute respiratory distress syndrome. Am J Respir Crit Care Med. 2003; 167:521–527.
183. Gattinoni, L, Vagginelli, F, Carlesso, E, et al. Decrease in PACO2 with prone position is predictive of improved outcome in acute respiratory distress syndrome. Crit Care Med. 2003; 31:2727–2733.
184. Guerin, C, Gaillard, S, Lemasson, S, et al. Effect of systematic prone positioning in hypoxemic acute respiratory failure: A randomized controlled trial. JAMA. 2004; 292:2379–2387.
185. Griffiths, MJD, Evans, TW. Inhaled nitric oxide therapy in adults. N Engl J Med. 2005; 353:2683–2695.
186. Perkins, GD, McAuley, DF, Thickett, DR, Gao, F. The β-agonist lung injury trial (BALTI). Am J Respir Crit Care Med. 2006; 173:281–287.
187. Smith, FG, Perkins, GD, Gates, S, et al. Effect of intravenous b-2 agonist treatment on clinical outcomes in acute respiratory distress syndrome (BALTI-2): A multicentre, randomized controlled trial. Lancet. 2012; 379:229–235.
188. The Acute Respiratory Distress Syndrome NetworkMatthay, MA, Brower, RG, Carson, S, et al. Randomized, placebo-controlled clinical trial of an aerosolized beta-2 agonist for treatment of acute lung injury. Am J Respir Crit Care Med. 2011; 342:1301e.
189. Kacmarek, RM, Wiedemann, HP, Lavin, PT, et al. Partial liquid ventilation in adult patients with acute respiratory distress syndrome. Am J Respir Crit Care Med. 2006; 173:882–889.
190. Martin, GS, Moss, M, Wheeler, AP, et al. A randomized, controlled trial of furosemide with or without albumin in hypoproteinemic patients with acute lung injury. Crit Care Med. 2005; 33:1681–1687.
191. Dellinger, RP, Zimmerman, JL, Taylor, RW, et al. Effects of inhaled nitric oxide in patients with acute respiratory distress syndrome: Results of a randomized phase II trial. Crit Care Med. 1998; 26:15–23.
192. Taylor, RW, Zimmerman, JL, Dellinger, RP, et al. Low-dose inhaled nitric oxide in patients with acute lung injury: A randomized controlled trial. JAMA. 2004; 291:1603–1609.
193. Lundin, S, Mang, H, Smithies, M, et al. Inhalation of nitric oxide in acute lung injury: Results of a European multicentre study. Intensive Care Med. 1999; 25:911–919.
194. Adhikari, NKJ, Burns, KEA, Friedrich, JO, et al. Effect of nitric oxide on oxygenation and mortality in acute lung injury: Systematic review and meta-analysis. BMJ. 2007; 334:1–8.
195. Marini, JJ. Are recruiting maneuvers necessary when ventilating acute respiratory distress syndrome? Crit Care Med. 2003; 31:2701–2703.
196. Rossaint, R, Falke, KJ, Lopez, F, et al. Inhaled nitric oxide for the adult respiratory distress syndrome. N Engl J Med. 1993; 328:399–405.
197. Luhr, O, Nathorst-Westfelt, U, Lundin, S, et al. A retrospective analysis of nitric oxide inhalation in patients with severe acute lung injury in Sweden and Norway. Acta Anaesthesiol Scand. 1997; 41:1238–1246.
198. Jantz, MA, Sahn, SA. Corticosteroids in acute respiratory failure. Am J Respir Crit Care Med. 1999; 160:1079–1100.
199. Hinshaw, LB, Archer, LT, Beller-Todd, BK, et al. Survival of primates in lethal septic shock following delayed treatment with steroid. Circ Shock. 1981; 8:291–300.
200. Brigham, KL, Bowers, RE, McKeen, CR. Methylprednisolone prevention of increased lung vascular permeability following endotoxemia in sheep. J Clin Invest. 1981; 67:1103–1110.
201. Hollenbach, SJ, DeGuzman, LR, Bellamy, RF. Early administration of methylprednisolone promotes survival in rats with intra-abdominal sepsis. Circ Shock. 1986; 20:161–168.
202. Rinaldo, JE, Dauber, JH. Effect of methylprednisolone and ibuprofen, a nonsteroidal antiinflammatory agent, on bronchoalveolar inflammation following endotoxemia. Circ Shock. 1985; 16:195–203.
203. Metz, CR, Sibbald, WK. Anti-inflammatory therapy for acute lung injury. A review of animal and clinical studies. Chest. 1991; 100:1110–1119.
204. Bernard, GR, Luce, JM, Sprung, CL, et al. High dose corticosteroids in patients with the adult respiratory distress syndrome. N Engl J Med. 1987; 317:1565–1570.
205. Luce, JM, Montgomery, AB, Marks, JD, et al. Ineffectiveness of high-dose methylprednisolone in preventing parenchymal lung injury and improving mortality in patients with septic shock. Am Rev Respir Dis. 1988; 138:62–68.
206. Bone, RC, Fisher, CJ, Jr., Clemmer, TP, et al. A controlled clinical trial of high-dose methylprednisolone in the treatment of severe sepsis and septic shock. N Engl J Med. 1987; 317:653–658.
207. Silverman, HJ, Slotman, G, Bone, RC, et al. Effects of prostaglandin E1 on oxygen delivery and consumption in patients with the adult respiratory distress syndrome. Chest. 1990; 98:405–410.
208. Holcroft, JW, Vassar, MJ, Weber, CJ. Prostaglandin E1 and survival in patients with the adult respiratory distress syndrome: A prospective trial. Ann Surg. 1986; 203:371–378.
209. Bone, RC, Slotman, G, Maunder, R, et al. Randomized, double-blind, multicenter study of prostaglandin E1 in patients with adult respiratory distress syndrome. Chest. 1989; 96:114–119.
210. Rinaldo, JE, Pennock, B. Effects of ibuprofen on endotoxin-induced alveolitis: Biphasic dose response and dissociation between inflammation and hypoxemia. Am J Med Sci. 1986; 291:29–38.
211. Balk, RA, Jacobs, RF, Tryka, AF, et al. Ibuprofen effects on neutrophil function and acute lung injury. Crit Care Med. 1988; 16:1121–1127.
212. Parsons, PE, Worthen, GS, Moore, EE, et al. The association of circulating endotoxin with the development of the adult respiratory distress syndrome. Am Rev Respir Dis. 1989; 140:294–301.
213. Fisher, CJ, Dhainaut, FA, Opal, SM, et al. Recombinant human interleukin 1 receptor antagonist in the treatment of patients with sepsis syndrome. JAMA. 1994; 271:1836–1848.
214. Fink, MP, O’Sullivan, BP, Menconi, MJ, et al. A novel leukotriene B4-receptor antagonist in endotoxin shock: A prospective, controlled trial in a porcine model. Crit Care Med. 1993; 21:1825–1837.
215. Marra, MN, Thornton, MB, Snable, JL, et al. Endotoxin-binding and neutralizing properties of recombinant bactericidal/permeability-increasing protein and monoclonal antibodies HA-1A and E5. Crit Care Med. 1994; 22:559–565.
216. Abraham, E, Wunderink, R, Silverman, H, et al. Efficacy and safety of monoclonal antibody to human tumor necrosis factor α in patients with sepsis syndrome. JAMA. 1995; 273:934–941.
217. Abraham, E, Baughman, R, Fletcher, E, et al. Liposomal prostaglandin E1 (TLC C-53) in acute respiratory distress syndrome: A controlled, randomized, double-blind, multicenter clinical trial. Crit Care Med. 1999; 27:1478–1485.
218. Opal, SM, Fisher, CJ, Dhainaut, J-FA, et al. Confirmatory interleukin-1 receptor antagonist trial in severe sepsis: A phase III, randomized, double-blind, placebo-controlled, multicenter trial. Crit Care Med. 1997; 25:1115–1124.
219. Reinhart, K, Wiegand-Lohnert, C, Grimminger, F, et al. Assessment of the safety and efficacy of the monoclonal anti-tumor necrosis factor antibody-fragment, MAK 195F, in patients with sepsis and septic shock: A multicenter, randomized, placebo-controlled, dose-ranging study. Crit Care Med. 1996; 24:733–742.
220. McCloskey, RV, Straube, RC, Sanders, C, for the CHESS Trial Study Group. Treatment of septic shock with human monoclonal antibody HA-1A. Ann Intern Med. 1994; 121:1–5.
221. Abraham, E, Anzueto, A, Gutierrez, G, et al. Double-blind randomized controlled trial of monoclonal antibody to human tumour necrosis factor in treatment of septic shock. Lancet. 1998; 351:929–933.
222. Abraham, E, Glauser, MP, Butler, T, et al. p55 tumor necrosis factor receptor fusion protein in the treatment of patients with severe sepsis and septic shock. JAMA. 1997; 277:1531–1538.
223. Sörensen, J, Kald, B, Tagesson, C, Lindahl, M. Platelet-activating factor and phospholipase A2 in patients with septic shock and trauma. Intensive Care Med. 1994; 20:555–561.
224. Bone, RC, Balk, RA, Fein, AM, for the E5 Sepsis Study Group. A second large controlled clinical study of E5, a monoclonal antibody to endotoxin: Results of a prospective, multicenter, randomized, controlled trial. Crit Care Med. 1995; 23:994–1005.
225. Greenman, RL, Schein, RM, Martin, MA, et al. A controlled clinical trial of E-5 murine monoclonal IgM antibody to endotoxin in the treatment of gram-negative sepsis. JAMA. 1991; 266:1097–1102.
226. Angus, DC, Birmingham, MC, Balk, RA, et al. E-5 murine monoclonal antiendotoxin antibody in gram-negative sepsis: A randomized controlled trial. JAMA. 2000; 283:1723–1730.
227. Ziegler, EJ, Fisher, CJ, Jr., Sprung, CL, et al. Treatment of gram negative bacteremia and septic shock with HA-1A human monoclonal antibody against endotoxin. A randomized, double-blind, placebo-controlled trial. N Engl J Med. 1991; 324:429–436.
228. Goldie, AS, Fearon, KCH, Ross, JA, et al. Natural cytokine antagonists and endogenous antiendotoxin core antibodies in sepsis syndrome. JAMA. 1995; 274:172–177.
229. Zwissler, B, Kemming, G, Habler, O, et al. Inhaled prostacyclin (PGI2) versus inhaled nitric oxide in adult respiratory distress syndrome. Am J Respir Crit Care Med. 1996; 154:1671–1677.
230. Dhainaut, JF, Tenaillon, A, Hemmer, M, et al. Confirmatory platelet-activating factor receptor antagonist trial in patients with severe gram negative bacterial sepsis: A phase III, randomized, double-blind, placebo-controlled, multicenter trial. Crit Care Med. 1998; 26:1963–1971.
231. Metzler, M, Balk, R, Schuster, D, et al. An evaluation of recombinant human platelet activating factor acetylhydrolase (rPAF-AH) in patients at risk for developing acute respiratory distress syndrome (ARDS). Crit Care Med. 1999; 27:A85.
232. Zeni, F, Freeman, B, Natanson, C. Anti-inflammatory therapies to treat sepsis and septic shock: A reassessment. Crit Care Med. 1997; 25:1095–1100.
233. Bernard, GR, Wheeler, AP, Arons, MM, et al. A trial of antioxidants N-acetylcysteine and procysteine in ARDS. Chest. 1997; 112:164–172.
234. Hirschl, RB, Pranikoff, T, Wise, C, et al. Initial experience with partial liquid ventilation in adult patients with the acute respiratory distress syndrome. JAMA. 1996; 275:383–389.
235. Bernard, GR, Reines, HD, Halushka, PV, et al. Prostacyclin and thromboxane A2 formation is increased in human sepsis syndrome. Effects of cyclooxygenase inhibition. Am Rev Respir Dis. 1991; 144:1095–1101.
236. Bernard, GR, Wheeler, AP, Russell, JA, et al. The effects of ibuprofen on the physiology and survival of patients with sepsis. N Engl J Med. 1997; 336:912–918.
237. Sprung, CL, Caralis, PV, Marcial, EH, et al. The effects of high-dose corticosteroids in patients with septic shock: A prospective, controlled study. N Engl J Med. 1984; 311:1137–1143.
238. Annane, D, Sebille, V, Troche, G, et al. A 3-level prognostic classification in septic shock based on cortisol levels and cortisol response to corticotropin. JAMA. 2000; 283:1038–1045.
239. Annane, D, Sebille, V, Bellissant, E, et al. Effect of low doses of corticosteroids in septic shock patients with or without early acute respiratory distress syndrome. Crit Care Med. 2006; 34:22–30.
240. The ARDS Clinical Trials Network. Randomized, placebo-controlled trial of lisofylline for early treatment of acute lung injury and acute respiratory distress syndrome. Crit Care Med. 2002; 30:1–6.
241. Fourrier, F, Chopin, C, Huart, JJ, et al. Double-blind, placebo-controlled trial of antithrombin III concentrates in septic shock with disseminated intravascular coagulation. Chest. 1993; 104:882–888.
242. Brochard, L. Clinical trials in acute respiratory distress syndrome: What is ARDS? Crit Care Med. 1999; 27:1657–1658.
243. Jepsen, S, Herlevson, P, Knudsen, P, et al. Antioxidant treatment with N-acetylcysteine during adult respiratory distress syndrome: A prospective, randomized, placebo-controlled study. Crit Care Med. 1992; 20:918–924.
244. Slotman, GJ, Burchard, KW, D’Arezzo, A, et al. Ketoconazole prevents acute respiratory failure in critically ill surgical patients. J Trauma. 1988; 28:648–654.
245. Yu, M, Thomas, G. A double-blind, prospective, randomized trial of ketoconazole, a thromboxane synthetase inhibitor, in the prophylaxis of the adult respiratory distress syndrome. Crit Care Med. 1993; 21:1635–1642.
246. Wiedemann, HP, Fisher, CH, Kormara, J, et al. Ketoconazole for early treatment of acute lung injury and acute respiratory distress syndrome. JAMA. 2000; 183:1995–2002.
247. Bajwa, EK, Malhotra, CK, Thompson, BT, et al. Statin therapy as prevention against development of acute respiratory distress syndrome: An observational study. Crit Care Med. 2012; 40:1470–1477.
248. Meduri, GU, Headley, AS, Golden, E, et al. Effect of prolonged methylprednisolone therapy in unresolving acute respiratory distress syndrome: A randomized controlled trial. JAMA. 1998; 280:159–165.
249. Meduri, GU, Chinn, AJ, Leeper, KV, et al. Corticosteroid rescue treatment of progressive fibroproliferation in late ARDS. Chest. 1994; 105:1516–1527.
250. The National Heart, Lung, and Blood Institute Acute Respiratory Distress Syndrome (ARDS) Clinical Trials Network. Efficacy and safety of corticosteroids for persistent acute respiratory distress syndrome. N Engl J Med. 2006; 354:1671–1684.
251. Meduri, GU, Golden, EM, Freire, AX, et al. Methylprednisolone infusion in early severe ARDS: Results of a randomized controlled trial. Chest. 2007; 131:954–963.
252. Seam, N, Meduri, GU, Wang, H, et al. Effects of methylprednisolone infusion on markers of inflammation, coagulation, and angiogenesis in early acute respiratory distress syndrome. Crit Care Med. 2012; 40:495–501.
253. Walkey, Aj, Wiener, RS. Macrolide antibiotics and survival in patients with acute lung injury. Chest. 2012; 141:1153–1159.
254. Bone, RC, Balk, R, Slotman, G, et al. Adult respiratory distress syndrome: Sequence and importance of development of multiple organ failure. Chest. 1992; 101:320–326.
255. Bone, RC, Balk, RA, Cerra, FB, et al. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. for the American College of Chest Physicians/Society of Critical Care Medicine Consensus Conference. Chest. 1992; 101:1644–1655.
256. Suchyta, MR, Clemmer, TP, Orne, JF, Jr., et al. Increased survival of ARDS patients with severe hypoxemia (ECMO criteria). Chest. 1991; 99:951–955.
257. Montgomery, AB, Stager, MA, Carrico, CJ, Hudson, LD. Causes of mortality in patients with the adult respiratory distress syndrome. Am Rev Respir Dis. 1985; 132:485–489.
258. Milberg, JA, Davis, DR, Steinberg, KP, Hudson, LD. Improved survival of patients with acute respiratory distress syndrome (ARDS) 983-1993. JAMA. 1995; 273:306–309.
259. Brodie, D, Bacchetta, M. Extracorporeal membrane oxygenation for ARDS in adults. N Engl J Med. 2011; 365:1905–1914.
260. Peek, GJ, Mugford, M, Tiruvoipati, R, et al. Efficacy and economic assessment of conventional ventilator support versus extracorporeal membrane oxygenation for severe adult respiratory failure (CESAR): A multicentre randomized controlled trial. Lancet. 2009; 374:1351–1363.
261. Noah, MA, Peek, GJ, Finney, SJ, et al. Referral to an extracorporeal membrane oxygenation center and mortality among patients with severe 2009 influenza A (H1N1). JAMA. 2011; 306:1659–1668.
262. Zapol, WM, Snider, MT, Hill, JD, et al. Extracorporeal membrane oxygenation in severe acute respiratory failure. A randomized prospective study. JAMA. 1979; 242:2193–2196.
263. Fein, AM, Lippman, M, Holtzman, H, et al. The risk factors, incidence, and prognosis or ARDS following septicemia. Chest. 1983; 83:40–42.
264. Faist, E, Wichmann, M, Kim, C. Immunosuppression and immunomodulation in surgical patients. Curr Opin Crit Care. 1997; 3:293–298.
265. Seeley, E, McAuley, DF, Eisner, M, et al. Predictors of mortality in acute lung injury during the era of lung protective ventilation. Thorax. 2008; 63:994–998.
266. Cheung, AM, Tansey, CM, Tomlinson, G, et al. Two-year outcomes, health care use, and costs of survivors of acute respiratory distress syndrome. Am J Respir Crit Care Med. 2006; 174:538–544.
267. Herridge, MS, Tansey, CM, Matte, A, et al. Functional disability 5 years after acute respiratory distress syndrome. N Engl J Med. 2011; 364:1293–1304.
268. Elliott, CG. Pulmonary sequelae in survivors of the adult respiratory distress syndrome. Clin Chest Med. 1990; 11:789–800.
269. Peters, JI, Bell, RC, Prihoda, TJ, et al. Clinical determinants of abnormalities in pulmonary functions in survivors of the adult respiratory distress syndrome. Am Rev Respir Dis. 1989; 139:1163–1168.
270. Herridge, MS, Am, Cheung, Tansey, CM, et al. One year outcomes in survivors of the acute respiratory distress syndrome. N Engl J Med. 2003; 348:683–693.
271. Heyland, DK, Groll, D, Caeser, M. Survivors of acute respiratory distress syndrome: Relationship between pulmonary dysfunction and long-term health-related quality of life. Crit Care Med. 2005; 33:1549–1556.
272. Angus, DC, Musthafa, AA, Clermont, G, et al. Quality-adjusted survival in the first year after acute respiratory distress syndrome. Am J Respir Crit Care Med. 2001; 163:1389–1394.
273. Ely, EW, Wheeler, AP, Thompson, BT, et al. Recovery rate and prognosis in older persons who develop acute lung injury and the acute respiratory distress syndrome. Ann Intern Med. 2002; 136:25–36.
274. Mikkelsen, ME, Christie, JD, Lanken, PN, et al. The adult respiratory distress syndrome cognitive outcomes study: Long-term neuropsychological function in survivors of acute lung injury. Am J Respir Crit Care Med. 2012; 185:1307–1315.
275. Cox, CE, Docherty, SL, Brandon, DH, et al. Surviving critical illness: Acute respiratory distress syndrome as experienced by patients and their caregivers. Crit Care Med. 2009; 37:2702–2708.
276. Davidson, TA, Caldwell, ES, Curtis, JR, et al. Reduced quality of life in survivors of acute respiratory distress syndrome compared with critically ill control patients. JAMA. 1999; 281:354–360.
277. Weinert, CR, Gross, CR, Kangas, JR, et al. Health related quality of life after acute lung injury. Am J Respir Crit Care Med. 1997; 156:1120–1128.
278. Curley, GF, Hayes, M, Ansari, B, et al. Mesenchymal stem cells enhance recovery and repair following ventilator-induced lung injury in the rat. Thorax. 2012; 67(6):496–501.