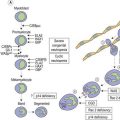
CHAPTER 18 Acute myeloid leukemias
Introduction
The acute myeloid leukemias (AML) are malignancies of immature precursors of the non-lymphoid hematopoietic lineages, involving the bone marrow (BM), peripheral blood (PB), and possibly other tissues. Taken together, this heterogeneous group of neoplasms has an estimated incidence between 1.6 and 3.7 per 100 000 population per year in the US and Western Europe.1–3 Yearly incidence rates appear to increase with patient age, and the median age at diagnosis is 65.2 AML is the predominant form of acute leukemia in adults, but accounts for only 15–20% of acute leukemias in pediatric patients.4 Environmental exposures or therapies that damage DNA, and a number of clinical syndromes affecting DNA repair pathways or tumor suppressor genes lead to increased risk of AML. Intensive study has revealed many somatic genetic lesions that appear to contribute to the development of AML. Approximately 20% of cases show recurrent cytogenetic abnormalities, while fewer than 10% of cases have a normal karyotype and fail to show mutations in genes such as FMS-like tyrosine kinase 3 (FLT3), nucleophosmin (NPM1), CCAAT/enhancer-binding protein alpha (CEBPA) and myeloid/lymphoid or mixed lineage leukemia (MLL) that are commonly altered in AML.5,6 High-resolution studies of genomic copy number alterations in AML with comparison to normal somatic cell DNA from the affected patients, as well as initial full-genome sequencing efforts have given indications that there may be additional recurrent genetic lesions in this malignancy, as well as many other mutations that may be very rare, pathogenic only in concert with numerous other mutations, or entirely unrelated to pathogenesis.7–9 Prognosis of patients with AML shows a striking relationship with the cytogenetic and other mutational features of the malignant blasts, with the implication that a complete diagnostic report for AML should contain this information, despite the logistical challenges of combining data from several laboratory disciplines into a unified synopsis.
Classification systems
The rapid evolution of classification schemes for AML has resulted from several successive waves of new technology entering the clinical hematology laboratory and being validated in clinical trials. The French-American-British (FAB) classification of 1976 and its later modifications primarily relied upon morphologic examination, cytochemical tests, and limited immunophenotyping to establish lineage-related classification of myeloid blasts. In the years that followed, additional data including recognition of common cytogenetic abnormalities, clinical history and correlation with clinical outcomes led to the World Health Organization (WHO) classification scheme in 2001, with distinct AML categories defined by: 1) recurrent cytogenetic abnormalities; 2) presence of myelodysplastic changes; 3) history of methylating agent or topoisomerase 2 inhibitor chemotherapy drug exposure, or 4) a ‘not otherwise specified’ category encapsulating the FAB morphologic/cytochemical classification.10 The defining BM blast percentage for AML was decreased from 30% to 20% at this time, largely because myelodysplastic syndrome (MDS) patients with over 20% blasts had been found to have a poor prognosis very similar to those with AML.11 The transition from FAB to WHO 2001 was a pragmatic effort to generate a clinically useful classification system providing prognostic information relevant to the therapeutic options available, while incorporating cytogenetic findings likely related to disease pathogenesis, and still retaining familiar entities defined by their morphology and antigen expression. Incorporating the response to therapy as part of the basis for the classification scheme may necessitate future revisions if new therapies with distinctive properties come into wide use. For the practicing hematopathologist, knowledge of the preceding classification systems is essential, given that long-running clinical trials often refer to the older systems, and reports with the correct current classification and parallel classification by the older terminology are appreciated by clinicians and trial researchers.
A revised WHO classification was published in 2008 (Table 18.1), and builds on the insights of the 2001 system with an increased number of recurrent cytogenetic abnormalities, and the creation of provisional categories for AML cases with mutations in the NPM1 or CEBPA genes.6 The key role of cytogenetic data is further increased by the stipulation that cases with the t(8;21)(q22;q22), inv(16)(p13.1q22), t(16;16)(p13.1;q22), or t(15;17)(q22;q12) need not have >20% blasts to be diagnosed as AML. Additional weight is given to cytogenetic abnormalities in the AML with myelodysplasia-related changes category, where specific MDS-associated cytogenetic findings can take the place of morphologic evidence of myelodysplasia. Two entities found in patients with Down syndrome are now classified separately, and the therapy-related AML category has been simplified to a single entity for patients with history of exposure to any of a broad range of cytotoxic drugs or ionizing radiation therapy. Adjustments to the criteria for AML with myelodysplasia-related changes, and the addition of blastic plasmacytoid dendritic cell neoplasm as an AML-related precursor neoplasm are the other significant changes from the WHO 2001 scheme.6 In the sections that follow, we will use the WHO 2008 framework to organize discussion of these malignancies, after a brief review of the clinical features and laboratory methods required for their analysis.
Table 18.1 World Health Organization classification of acute myeloid leukemia (AML) and related precursor neoplasms
Methods for diagnosis and classification
Peripheral blood and bone marrow cell count and morphology
Complete blood counts and morphologic review of PB smears usually reveal multiple abnormalities in AML patients. Reduced counts of red cells, platelets and the normal leukocytes are commonly seen, but the white blood cell count (WBC) is most often elevated due to circulating blasts. Patients usually present before the WBC count rises above 50 × 109 cells per liter, but a minority of patients have WBC counts over 100 × 109 cells per liter, and occasional cases show WBC counts as low as 1 × 109 cells per liter with few circulating blasts. BM evaluation should be performed from aspirate smears of adequate quality reviewed in conjunction with bone marrow trephine biopsy (BMTB) sections. With the exception of a few cytogenetically-defined subcategories, blasts in the PB or BM must be ≥20% of total nucleated cells for a diagnosis of AML, based on a 200-cell count in the blood or a 500-cell count in the BM. The presence of Auer rods (linear filaments of primary granules) (Fig. 18.1) is the only definitive morphologic finding sufficient to distinguish myeloid blasts from lymphoid ones, but other features typical of myeloid blasts include larger blast size, fine chromatin, multiple often easily visualized nucleoli and cytoplasmic granulation. Blasts of some cases of AML (corresponding to those formerly classified in the FAB M0 and M1 categories) may be morphologically similar to lymphoblasts, small to medium in size with few nucleoli and minimal amounts of agranular cytoplasm, while others (corresponding to monoblasts and promonocytes in the FAB M4 and M5 categories) show monocytic differentiation, being large in size with round to delicately folded or convoluted nuclei, fine lacy chromatin, visible nucleoli and basophilic cytoplasm with absent to minimal granulation. A background of dysplasia in other BM myeloid lineages may also provide evidence that blasts are more likely to be of myeloid type. In some subcategories of AML, modifications of the blast count are part of the WHO 2008 classification: promonocytes are considered as blasts in AML cases with myelomonocytic, monoblastic or monocytic differentiation; the abnormal promyelocytes in APL are considered blasts; and in AML not otherwise specified erythroid/myeloid erythroleukemia cases, the blast percentage is calculated from non-erythroid BM nucleated cells.6 Patients with myeloid sarcomas are considered to have AML regardless of PB or BM blast counts.
Immunophenotyping
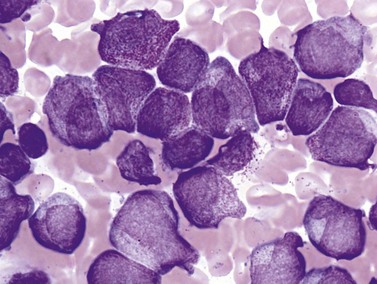
Flow cytometry (FCM) is the preferred method of characterizing surface and internal antigen expression in leukemia cases, as modern flow cytometers and fluorescent antibodies enable four or more antigens to be measured simultaneously on each cell assayed, together with forward and side light scatter properties that correlate with cell size and internal complexity or granularity. Given the large number of different antigens that may be expressed by AML or other leukemic blasts, panels of antibodies used for diagnosis can be extensive, and are usually designed with one common antibody (typically binding the leukocyte marker CD45) present in all tubes used for staining cells, so that cell populations defined by CD45 expression and forward or side light scatter can be uniformly compared across all antibody stains in the panel. Myeloid surface antigens include CD13, CD33, CD117 (for more immature precursors), and CD15 (for more mature cells), while monocytic markers include CD14 and CD64. Megakaryocytic lineage is suggested by the platelet antigens CD41a and CD61, while erythroid markers include CD71, glycophorin and hemoglobin A. Surface expression of CD34 is a broad marker of immaturity seen in myeloid and lymphoid lineage blasts. Internal antigens helpful for characterizing myeloid blasts include myeloperoxidase (MPO) and TdT, which, as its role in lymphoid biology would suggest, is usually negative in AML but can be expressed aberrantly in a subset of cases. Many AML cases can be demonstrated to express antigens of other hematopoietic lineages: for example, t(8;21)-containing cases often show expression of the B cell antigens CD19, Pax5 or cytoplasmic CD79a; and T cell markers CD2 or CD7 are not uncommon in several subtypes of AML.12 The aberrant expression of such markers typically does not warrant a diagnosis of biphenotypic or mixed phenotype acute leukemia (see Chapter 19), but may increase the sensitivity and specificity of detecting residual or recurrent disease following therapy.
Cytochemistry
A key part of lineage determination in the FAB classification scheme, cytochemical testing on air-dried blood or BM aspirate smear slides is performed to a more limited degree in most modern laboratories, with the result that many leukemias may not be fully evaluated by FAB criteria under current protocols.13 Cytochemical stains for myeloperoxidase (MPO) and the somewhat less specific Sudan Black B (SBB) are used to identify myeloid lineage, although these stains are negative in very early myeloblasts and monoblasts, as well as in erythroid and megakaryocytic blasts. SBB has the advantage of being usable even when the air-dried smears have not been recently prepared. The nonspecific esterase (NSE) activity that characterizes monocytes and monoblasts can be detected by reactivity with alpha naphthyl butyrate esterase, or sodium fluoride-inhibited reactivity with alpha naphthyl acetate esterase. Naphthol-ASD-chloroacetate esterase (CAE) activity is specific for neutrophil and mast cell lineages. Using these stains, AML with minimal differentiation cases are defined by having less than 3% of blasts positive for MPO, SBB or CAE, and lacking NSE activity; the designation of these cases as acute myeloid leukemias relies on immunophenotypic detection of expression of some combination of CD13, CD117 or CD33 and lack of lymphoid differentiation markers. AML without maturation is distinguished from AML with minimal differentiation by having >3% MPO or SBB-positive blasts, or the presence or Auer rods (which should also stain for both MPO and SBB). Monoblasts and promonocytes seen in the acute myelomonocytic or acute monoblastic or monocytic leukemia categories show blast-equivalent cells with NSE reactivity but usually no MPO or SBB reactivity. Acute megakaryoblastic leukemias may show NaF-resistant NSE activity for alpha naphthyl acetate, but are MPO and SBB negative.
Cytogenetics and molecular testing
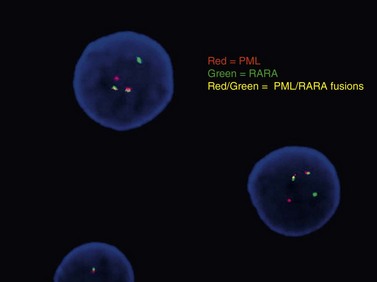
Conventional karyotype analysis, fluorescence in situ hybridization (FISH) assays for detecting translocations, and molecular biology tests for point mutations or other small genetic lesions have become an integral part of evaluating and sub-classifying acute myeloid leukemias, and can guide therapy, most notably for APL carrying t(15;17) that respond to all-trans retinoic acid (ATRA) treatment (Fig. 18.2). Cytogenetic and molecular testing additionally impacts therapeutic decision-making for patients with favorable cytogenetic or gene mutation-containing leukemias such as cases with low WBC counts and t(8;21), inv(16), t(16;16), or NPM1 mutation without cytogenetic abnormalities and without FLT3-internal tandem duplication, in which allogeneic stem cell transplant does not appear to confer increased survival.14,15 The key cytogenetic and mutational lesions in AML will be discussed in greater detail in the following sections dealing with each classification category.
Acute myeloid leukemia with recurrent genetic abnormalities
These leukemias feature balanced translocations or inversions, or belong to new provisional categories defined by mutation of the NPM1 or CEPBA genes. These cytogenetic or mutationally-defined features have been associated with prognostic significance in clinical trials (Table 18.2). Most cases with recurrent translocations or inversions have some characteristic morphologic, immunophenotypic or clinical features.16–19 If the classic t(8;21), inv(16), t(16;16), or t(15;17) translocations are present, 20% blood or BM blasts are not required for diagnosis. It should be noted that AML in patients with a history of chemotherapy or radiation therapy should be classified in the therapy-related AML category regardless of other findings. Multilineage dysplasia is commonly seen in the recurrent translocation/inversion cases with inv(3), t(3;3), or t(6;9), but the cytogenetic findings take priority for classification of these entities. Since the latter chromosomal changes were found also in patients clinically presenting as MDS, cases with inv(3), t(3;3), or t(6;9) can be classified as AML only when ≥20% blasts are present.
Table 18.2 AML with recurrent genetic abnormalities: prognostic correlations
| AML with t(8;21)(q22;q22); (RUNX1-RUNX1T1) | Good prognosis |
| AML with inv(16)(p13.1q22) or t(16;16)(p13.1;q22); (CBFB-MYH11) | Good prognosis |
| Acute promyelocytic leukemia (APL) with t(15;17)(q22;q12); (PML-RARA) | Good prognosis |
| AML with t(9;11)(p22;q23); (MLLT3-MLL) | Intermediate prognosis; better than AML with other 11q23 translocations |
| AML with t(6;9)(p23;q34); (DEK-NUP214) | Poor prognosis, similar to AML with myelodysplasia-related changes |
| AML with inv(3)(q21q26.2) or t(3;3)(q21;q26.1); (RPN1-EVI1) | Poor prognosis |
| AML (megakaryoblastic) with t(1;22)(p13;q13); (RBM15-MKL1) | Probably good prognosis with intensive AML chemotherapy |
| AML with mutated NPM1 (provisional) | Good prognosis |
| AML with mutated CEBPA (provisional) | Good prognosis |
AML with t(8;21)(q22;q22); (RUNX1-RUNX1T1)
Cases with the t(8;21) (q22;q22) translocation most commonly present in younger patients, and comprise 5–10% of AML diagnoses. Morphologic features are typically consistent with the former FAB M2 category (‘with maturation’) with blasts displaying ample cytoplasm with abundant granules and some blasts containing Auer rods (Fig. 18.3). Telltale features suggesting this diagnosis include prominent perinuclear hofs, the presence of larger pink granules among the more typical darker cytoplasmic granules, and frequent aberrant expression of B-cell antigens such as CD19, Pax5, or cytoplasmic CD79a. Other morphologic findings that can be seen are Auer rods in more mature granulocytes, abnormal very large granules in blasts, and varying degrees of dyspoietic morphology in granulocytes, including Pelger–Huetoid bilobed neutrophils. CD34 (often overexpressed) and myeloid antigens CD13, CD33 and MPO are usually positive by FCM, although CD33 may be weak. The RUNX1 gene (synonyms: AML1, CBFA) encodes the core binding factor alpha protein (CBFA), one component of a heterodimeric hematopoietic transcription factor; the abnormal fusion to the RUNX1T1 gene (synonym: ETO, for eight-twenty-one) likely impairs or misdirects gene-regulatory activity.20 Prognosis for these cases is good with modern therapies, although high presenting WBC counts (>20 × 109 cells per liter) and KIT mutations (see below) lead to an intermediate prognosis.
AML with inv(16)(p13.1q22) or t(16;16)(p13.1;q22); (CBFB-MYH11)
The inv(16) or t(16;16) cases are also predominantly diagnosed in younger patients and account for 5–10% of all AML. Lesions affecting 16p13.1 involve the gene for the second half of the core binding factor (CBF) transcription factor (CBFB core-binding factor beta) fused to a smooth muscle myosin heavy chain gene (MYH11), possibly indicating an underlying biological basis for the good prognosis and younger demographic that these cases share with t(8;21) AML.21 Clinically, these leukemias show a high rate of extramedullary disease, approaching 50% of cases. The morphologic features of inv(16) or t(16;16) AML are highly distinctive, with most but not all cases showing granulocytic or monocytic blasts (often consistent with FAB M4 criteria) accompanied by abnormal eosinophil precursors in the BM (Fig. 18.4). The eosinophil precursors at the promyelocyte and myelocyte-equivalent stages show occasional to numerous large purple or basophilic granules of variable shape, intermixed with eosinophilic granules. FCM often documents several blast populations with myeloid or monocytic marker expression, and frequent (but nonspecific) aberrant expression of the T-cell marker CD2. FISH or real-time PCR (RT-PCR) testing is more sensitive than conventional karyotyping for detection of the inv(16)(p13.1q22) abnormality.
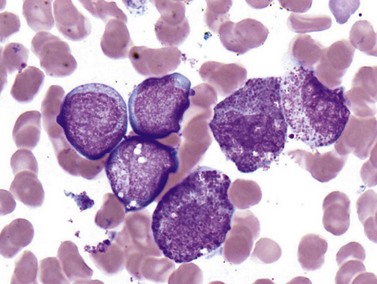
Acute promyelocytic leukemia (APL) with t(15;17)(q22;q12); (PML-RARA)
The dangerous prospect of DIC in patients with APL, the responsiveness of cases with the t(15;17)(q22;q12) translocation to all-trans retinoic acid (ATRA), and the good overall prognosis if acute life-threatening bleeding or clotting events are avoided make it imperative for the pathologist to recognize the morphologic features of these leukemias, and ensure that prompt confirmatory molecular testing is performed. Frequently, treatment with ATRA is initiated even before cytogenetic confirmation of the diagnosis is obtained. The disease may appear throughout adult life but is most common in the middle-aged. Roughly two-thirds of cases present with low WBC counts and show classic ‘hypergranular’ morphology, with abnormal promyelocytes displaying bilobed nuclei, dense granulation with large granules ranging in color from pink to violet, and frequent Auer rods (Fig. 18.5). Some cells may contain numerous and overlapping Auer rods. The ‘microgranular’ morphologic variant often has higher WBC counts but relatively sparse granulation of the blasts, with fewer cells containing Auer rods. The presence of bilobed nuclei is consistent and may be the best morphologic clue to the diagnosis in the microgranular variant.22–24 FCM of hypergranular cases usually highlights blasts and promyelocytes with a broad range of side-scatter of light, staining negative for CD34 and HLA-DR, with bright CD33, variable CD13, and strong MPO. Microgranular cases are more often positive for CD34 (at least in a fraction of blasts), more likely to show aberrant expression of CD2, and unfortunately can occasionally express the monocytic marker CD64, necessitating careful review of morphology and cytogenetic correlation to distinguish these cases from monocytic AML with folded nuclei and monocytic FCM immunophenotype. APL cases are usually negative for adhesion molecules CD11b and CD11c that are most often strongly expressed in AML with monocytic differentiation. Cytochemical stains for MPO or SBB are strongly positive in both variant and classic cases. The aberrant RARA (retinoic acid receptor alpha) fusion protein generated by the t(15;17)(q22;q12) translocation appears to prevent normal myeloid maturation, and is targeted by ATRA therapy, leading to progressive granulocytic differentiation and eventual cell death. Several less common variant translocations with the 17q12 RARA gene have been reported in AML cases with APL-like morphology and immunophenotype, including the ATRA-resistant t(11;17)(q23;q12) ZBTB16-RARA and t(17;17)(q11.2;q12) STAT5B-RARA cases, as well as likely ATRA-responsive t(11;17)(q13;q12) NUMA1-RARA and t(5;17)(q35;q12) NPM1-RARA.25 These variant translocations appear to correlate with morphologic differences, such as in t(11;17)(q23;q12) ZBTB16-RARA cases featuring non-bilobed nuclei, no Auer rods and Pelgeroid neutrophils. The WHO 2008 classification indicates that cases with RARA translocations other than the classic t(15;17)(q22;q12) should be diagnosed as ‘AML with a variant RARA translocation’.
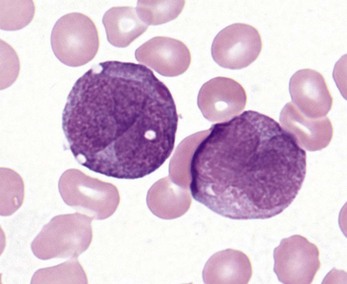
AML with t(9;11)(p22;q23); (MLLT3-MLL)
The WHO 2008 classification splits t(9;11)(p22;q23); (MLLT3-MLL) cases out from the former WHO 2001 category featuring a variety of 11q23 translocations of the MLL gene, to reflect the intermediate prognosis of MLLT3-MLL cases compared to the poor prognosis of the other 11q23 entities.26,27 The t(9;11) translocation is usually seen in pediatric patients, and blasts show monocytic or myelomonocytic morphology (FAB M5 or M4). As with other monocytic AML cases, there can be extramedullary disease including involvement of gums, skin and other tissues. The product of the MLL gene has been shown to have histone methyltransferase activity and likely plays a role in gene regulation by acting with other chromatin remodeling proteins. The numerous variant translocation partners reported for this gene in AML suggest that there are various ways of preventing normal MLL function, or misdirecting its activity, that can contribute to leukemogenesis.
AML with t(6;9)(p23;q34); (DEK-NUP214)
t(6;9) AML is an uncommon variety that can affect a wide age range of patients, and is most distinctive for the presence of BM or blood basophilia (>2% basophils), seen in approximately half of cases (Fig. 18.6).18,28 WBC counts are typically low in the peripheral blood in adult patients (median value 12 × 109 cells per liter), anemia and thrombocytopenia are common, and myelodysplasia can be seen in all lineages. Patients occasionally present before the blast count has risen to 20%, in which case careful monitoring is recommended. No distinctive morphologic features have been reported for the blasts, which can be myeloid or monocytic, with unremarkable myeloid or monocytoid FCM immunophenotype and cytochemistry.18 The t(6;9) is most commonly the sole detectable cytogenetic abnormality in these cases, and is given priority for classification over any findings of myelodysplasia. The effects of the fusion of the chromatin-associated protein DEK and the nuclear pore protein NUP214 on the cell have not been elucidated, but could involve derangement of gene expression or nuclear transport, among other possibilities. The prognosis of these cases is generally poor.
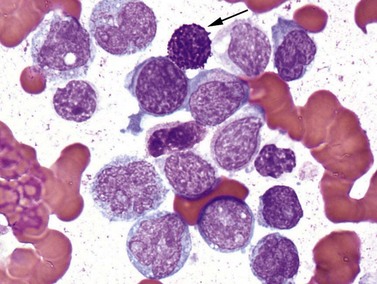
AML with inv(3)(q21q26.2) or t(3;3)(q21;q26.1); (RPN1-EVI1)
AML with inv(3) or t(3;3) is relatively rare (1–2% of AML cases), and is primarily a disease of adults. It is commonly found in association with myelodysplastic findings in granulocytes and platelets, with lesser dyspoiesis of the erythroid lineage. PB platelet counts are often within the normal range, or increased. Patients may present before the blast count exceeds 20%. The most striking morphologic feature in the BM is the presence of monolobated or hypolobated megakaryocytes that may be increased in number (Fig. 18.7).16,17,29 Blasts are myeloid, monocytic or megakaryoblastic with an unremarkable myeloid blast immunophenotype, including possible expression of CD41 and CD61 in megakaryoblastic cases. The EVI1 (ecotropic virus integration-1) gene is an oncogene with zinc finger protein homology, proposed to have transcriptional repression activity via recruitment of histone deacetylases to chromatin.30 Fusion to the gene for proteasome component RPN1 may serve primarily to drive high expression of EVI1. Some patients with chronic myelogenous leukemia (CML) develop inv(3) or t(3;3) chromosomal abnormalities, often as their disease accelerates or enters blast crisis, but such cases with documented t(9;22) translocations should be treated as CML rather than AML with inv(3) or t(3;3). Prognosis is usually poor in all AML cases with these chromosome 3 abnormalities.
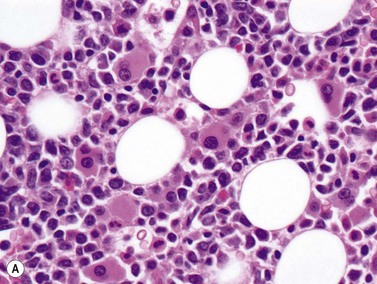
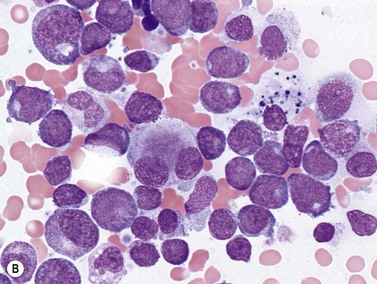
AML (megakaryoblastic) with t(1;22)(p13;q13); (RBM15-MKL1)
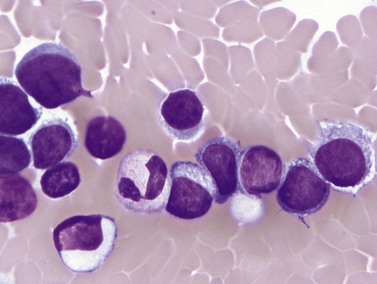
This is very rare form of infant leukemia usually seen in patients without Down syndrome. It is commonly associated with hepatosplenomegaly or other organomegaly. The blasts show megakaryocytic differentiation similar to those of the FAB M7 type, and are medium-sized or larger, with rounded nuclei containing fine reticular chromatin, visible nucleoli, agranular basophilic cytoplasm, and occasional cytoplasmic blebbing (Fig. 18.8).31–33 The FCM immunophenotype commonly shows blasts lacking CD45, CD34 and HLA-DR but expressing myeloid markers CD13 and CD33 as well as megakaryocyte antigens CD41, CD61 and possibly CD42. Overt multilineage dysplastic features are not usually present in the BM, although micromegakaryocytes are common. As with other megakaryoblastic leukemias, BM collagen fibrosis is typically present and may result in aspirates with falsely low blast counts, necessitating correlation with BMTB. The fusion partners in the translocation are RBM15, a gene encoding a protein with RNA-binding motifs, and MKL1, a transcriptional coactivator of serum response factor (SRF).34 Prognosis for these patients appears to be relatively good, although the number of patients reported in the literature is low.35
Acute myeloid leukemia with gene mutations
Many gene mutations have been detected in AML blasts, but a handful of specific mutations appear to occur most frequently or be most potently selected in the process of leukemogenesis.5 Mutations in the tyrosine kinase receptor genes FLT3 (FMS-like tyrosine kinase 3) and KIT are relatively common. FLT3 mutations are found in up to one-third of the 40–50% of adult AML cases that have a normal karyotype, and take the form of internal tandem duplications (ITD) and point mutations of the kinase domain (TKD) that correlate with a poor prognosis.36 FLT3 mutation is also seen in AML with recurrent cytogenetic abnormalities and worsens prognosis, although the mutations are uncommon in the context of t(8;21), inv(16) and 11q23 translocations, and may not worsen the already bad prognosis of t(6;9) and inv(3) cases. KIT mutations are most common in the core binding factor translocation categories t(8;21) and inv(16), and herald a poorer prognosis.37
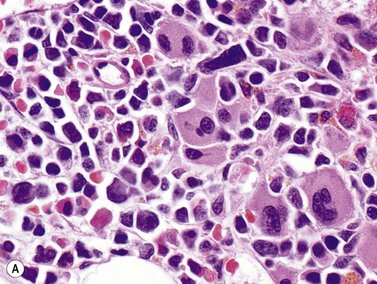
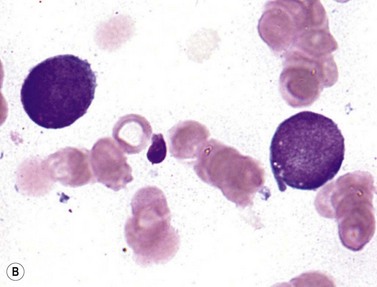
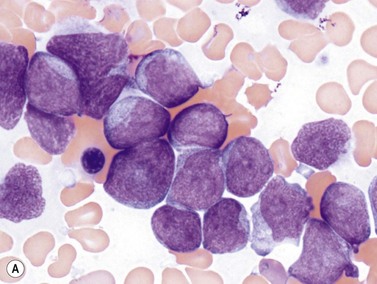
AML with mutated NPM1
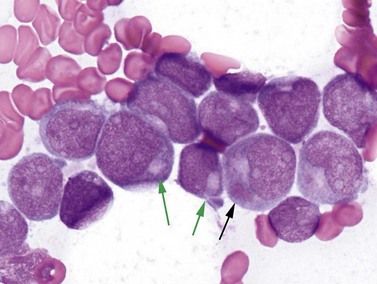
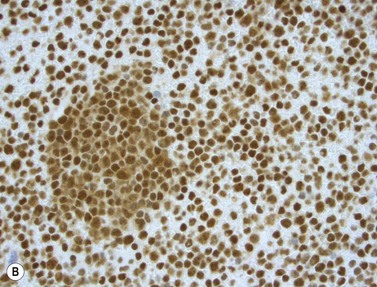
The NPM1 gene at 5q35 encodes a nuclear shuttling protein reported to have roles in ribosome and centrosome biology as well as regulation of other cellular systems such as the ARF-TP53 tumor suppressor pathway. NPM1-mutated AML cases are more common in adults than children (approximately 30% of adult cases and 5% of pediatric ones) and in women compared to men.38 Most have a normal karyotype, and there is little overlap with cases having recurrent translocations, partial tandem duplication of MLL, or mutations in CEPBA. About half of adult normal karyotype AML cases have mutated NPM1; about 40% of these (20% of normal karyotype cases) have both NPM1 and FLT3-ITD mutations. Morphologically, most NPM1-mutant cases are myelomonocytic or monocytic without myelodysplastic findings, but other morphologies including AML without maturation (Fig. 18.9A), AML with maturation and erythroleukemia, as well as small numbers of cases with myelodysplasia have also been reported. The blast immunophenotype is typical for myelomonocytic or monocytic AML, but consistent lack of expression of CD34 is a hallmark regardless of blast morphology. A useful immunohistochemical surrogate for mutation in NPM1 is detection of aberrant cytoplasmic localization of the protein (Fig. 18.9B). Mutations of the gene are typically detected by PCR-based methods, with the most common being tetranucleotide insertions in exon 12 that change the reading frame, ablate a nuclear localization signal and create a spurious nuclear export signal, contributing to the cytoplasmic accumulation of the protein.39 Isolated NPM1 mutation in normal karyotype AML confers a good prognosis, and even cases with coexisting FLT3-ITD mutation appear to benefit, compared to those with FLT3-ITD alone; however, NPM1 mutations should always be studied in conjunction with FLT3 mutations. The significance of NPM1 mutation in the context of myelodysplasia-related findings, or additional chromosomal abnormalities is less clear.
AML with mutated CEBPA
The CEBPA gene at 19q31.1 is a tumor suppressor and transcription factor implicated in the differentiation of many disparate cell lineages, including granulocytes.40 There is evidence that the t(8;21) RUNX1-RUNX1T1 fusion product may act by repressing expression of CEBPA. CEBPA mutations are detected in approximately 10% of all AML cases, and 15–18% of cases with normal karyotype, with no apparent association with patient age or sex. There is little overlap with cases having NPM1 mutation, while FLT3-ITD mutations occur in approximately 25% of CEBPA-mutant cases. Taken together, the clinical features of CEBPA-mutant AML suggest relative preservation of erythropoiesis, worse thrombocytopenia, higher blast count in the blood, and less frequent extramedullary disease than AML without CEBPA mutation. Blast morphology is most often consistent with FAB M1 (without maturation) and M2 (with maturation), with rarer examples having myelomonocytic or monocytic morphology. Immunophenotype is typical for myeloid blasts, and CD34 and HLA-DR are most often positive. A high percentage of cases show aberrant expression of the T-cell marker CD7. Gene sequencing is needed for full evaluation, given that over 100 distinct mutations have been reported throughout the gene, with 5′ exons often showing frame-shifting insertions or deletions producing truncated dominant-negative protein products, while 3′ exons more commonly have frame-preserving small insertions or deletions.41,42 The prognosis of isolated CEBPA-mutant AML with normal karyotype is favorable; consensus about the effect of this mutation in the context of FLT3-ITD or other cytogenetic abnormalities has not yet been reached.
Acute myeloid leukemia with myelodysplasia-related changes (AML-MRC)
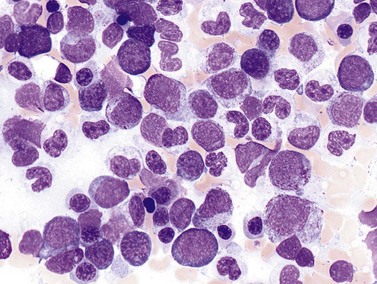
AML-MRC is characterized by a myeloid blast count ≥20% in the blood or bone marrow of a patient with any or all of the following: 1) prior history of MDS or myelodysplastic/myeloproliferative neoplasm; 2) characteristic myelodysplasia-associated cytogenetic abnormalities; 3) multilineage dysplasia in at least 50% of cells in two cell lineages. The kind of data supporting the AML-MRC diagnosis should be included in the bottom line of the pathology report. There should be no history of cytotoxic chemotherapy or radiation treatment (which would warrant classification as therapy-related AML) and no evidence for ‘AML with recurrent genetic abnormality’ cytogenetic abnormalities, which take diagnostic precedence. This diagnosis is more common in older patients and very rare in the pediatric population, but with the new cytogenetic criteria introduced in the WHO 2008 classification, the rate of diagnosis in children will likely increase. The clinical features of the case may be dominated by severe cytopenias. For establishing morphologic multilineage dysplasia, the following findings should be documented: neutrophils with hypogranular cytoplasm, hyposegmented nuclei, or bizarre nuclear segmentation patterns; erythroid precursors with megaloblastoid changes (with delayed nuclear maturation relative to cytoplasmic hemoglobinization), multiple nuclei, irregular nuclear contours, nuclear fragments, or the presence of ring sideroblasts, cytoplasmic inclusions, or periodic acid-Schiff (PAS) staining vacuoles; and megakaryocytes with micromegakaryocyte morphology, hyposegmented nuclei, or widely-separated nuclear lobes (Fig. 18.10).43 Blast morphology is variable, but blast with features of AML with maturation and acute myelomonocytic leukemia are most common. Some cases are designated as hypocellular AML, if BM cellularity is less than 30%, or less than 20% in patients over 60 years of age; these cases often represent progression of hypocellular MDS. Immunophenotyping is of limited utility for diagnosis, as the blasts show typical myeloid marker expression and common non-specific aberrancies such as expression of CD7 or TdT. The defining cytogenetic abnormalities for AML-MRC are similar to those defining MDS and listed in Table 20.5. Many of these are monosomies or chromosome arm losses, but a number of balanced translocations can also be used to support the diagnosis once a prior history of chemotherapy or radiation exposure is ruled out. Strikingly, up to half of cases with these defining cytogenetic abnormalities do not show sufficient morphologic evidence to meet the criteria for multilineage dysplasia. It appears from clinical trials that the AML-MRC-defining cytogenetic abnormalities portend a worse prognosis than do isolated morphologic findings of multilineage dysplasia. The presence of mutations in other significant AML-associated genes such as FLT3, NPM1 and CEBPA should be evaluated and noted in the report in cases with normal cytogenetics but multilineage dysplasia. Prognosis in AML-MRC is poor, and appears worse still for patients with characteristic monosomies or expression of the EVI1 gene.43–45
Therapy-related myeloid neoplasms
The previous, 2001 WHO classification included categories for AML arising following alkylating agent therapy (usually 5–7 years after treatment, with a period of MDS seen) or topoisomerase II inhibitor therapy (usually 1–3 years after treatment). In the 2008 WHO classification, a single category of therapy-related AML (t-AML) is used to encompass these entities as well as AML arising following other cytotoxic drug treatments including fludarabine and anti-tubulin drugs, as well as ionizing radiation if used on large fields containing active bone marrow. These cases together comprise approximately 15% of AML diagnoses, and most show multilineage dysplasia, often with myelodysplasia-associated chromosome abnormalities such as loss of part or all of chromosomes 5 or 7, a complex karyotype, or, in patients with a history of topoisomerase II inhibitor treatment, translocations involving 11q23.46,47 A variety of myeloid blast morphologies can be seen, with monoblastic and myelomonocytic cytology being most common. Immunophenotype is not specific for t-AML and is usually consistent with the blast morphology. Prognosis is poor, but appears to be influenced by the underlying cytogenetic abnormalities as well as the initial malignancy for which the patient was treated.
Acute myeloid leukemia, not otherwise specified (AML, NOS)
AML without maturation
AML without maturation corresponds in part with FAB M1 AML and is defined by cases where blasts are ≥90% of non-erythroid cells, and there is minimal differentiation to more mature granulocyte forms. Older epidemiologic data indicate that 5–10% of all AML cases meet these criteria. Blast morphology can be similar to that of AML with minimal differentiation, or can show basophilic cytoplasmic granulation or even Auer rods, but by definition at least 3% of blasts must show cytochemical staining for MPO or SBB. Immunophenotype shows cytoplasmic staining for MPO, as well as surface expression of any or all of the myeloid markers CD13, CD33, CD117, or more rarely, CD11b. CD34 and HLA-DR are most often positive, and there is usually no staining for mature granulocyte markers CD15 or CD65 or monocyte markers CD14 and CD64. Aberrant expression of less-specific lymphoid markers such as CD7 is not infrequent, but more specific cytoplasmic lymphoid markers such as cCD3, cCD79a and cCD22 are not seen. Many cases of FAB M1 AML are now classified as AML with NPM1 mutation (with or without concurrent FLT3 mutation); these are associated with some specific features, including a high absolute blast count and cup-like nuclear invaginations in the blasts, lack of CD34 or HLA-DR expression, and normal karyotype (Fig. 18.9A). A second group of reclassified cases comprises those with myelodysplasia-related changes. The less common AML with CEBPA mutation category has a predominance of cases with FAB M1 morphology immunophenotype and cytochemistry.
Acute monoblastic and monocytic leukemia
In these cases, which correspond in part with FAB M5a and M5b AML, monoblasts, promonocytes and monocytes comprise at least 80% of non-erythroid nucleated BM cells, while maturing neutrophil lineage cells must be <20%. In monoblastic cases, monoblasts make up at least 80% of the monocytic cells, whereas acute monocytic cases have a majority of promonocytes. Clinically, there is a tendency towards extramedullary disease, particularly involving the skin, gums or CNS. Monoblasts are typically relatively large blasts with rounded nuclear contours, fine lacy chromatin with visible nucleoli, and ample variably basophilic cytoplasm that may be agranular or contain MPO-negative fine basophilic granules. Auer rods are not seen in monoblasts. MPO cytochemistry is usually negative, and NSE is positive. Promonocytes are considered blast-equivalents, and often have less basophilic, finely granulated cytoplasm and show characteristic delicate nuclear folds not seen in monoblasts. Cytoplasmic vacuoles can be seen in both monoblasts and promonocytes. The blast immunophenotype in these cases often lacks CD34, but includes HLA-DR and myeloid antigens such as CD13, CD15, CD65, or CD33, which is often bright. Several or many monocytic markers including CD4, CD11b, CD11c, CD14, CD36, CD64, CD163 or lysozyme may be expressed. Many AML cases with features of the FAB M5 category contain translocations involving MLL at 11q23. If a case shows the t(9;11)(p22;q23); (MLLT3-MLL), it should be classified as AML with recurring genetic abnormality; other 11q23 cases, if not in a patient with a history of cytotoxic therapy, can be classified as AML, NOS, with the chromosomal abnormality noted in the report. If hemophagocytosis is seen in cases with maturation or with monocytic or myelomonocytic features, it may indicate the presence of the relatively uncommon t(8;16)(p11.2;p13.3) translocation, which does not define an AML with recurrent genetic abnormalities category but is frequently associated with hemophagocytosis.48
Acute erythroid leukemia
Erythroid precursors and/or blasts are the predominant cell type of this leukemia, which partially corresponds to FAB M6, is relatively rare (<5% of AML) and is found mainly in adults. In the more common erythroid/myeloid variant, erythroid precursors represent at least 50% of total BM nucleated cells, and myeloblasts are at least 20% of the remaining non-erythroid nucleated cells. The less common pure erythroid leukemia is diagnosed when at least 80% of BM nucleated cells are undifferentiated or proerythroblastic cells, without evidence for a significant myeloblast population among non-erythroid nucleated cells (Fig. 18.11).49 The erythroid/myeloid variant usually presents with pancytopenia and circulating nucleated red cells, while the BM erythroid precursors can be strikingly abnormal, with megaloblastoid changes, fragmented or multiple nuclei, and blocky cytoplasmic PAS staining. Dysplastic features are common in megakaryocytes as well, and many cases meet the morphologic criteria of AML with myelodysplasia-related changes and should be classified as such. The myeloid blasts typically show a lack of maturation by morphology and immunophenotype. FCM of erythroblasts in these cases often shows a population lacking CD34 and HLA-DR, without myeloid antigens, and expressing aberrant dim CD71 (transferrin receptor), glycophorin, and possibly hemoglobin A. The proerythroblasts and early basophilic erythroblasts of pure erythroid leukemia show a similar immunophenotype. Expression of megakaryocytic markers such as CD41 or CD61 may complicate distinguishing erythroleukemia cases from acute megakaryoblastic leukemias. A variety of non-neoplastic causes of dyserythropoiesis and erythroid hyperplasia must be ruled out, including vitamin B12 or folate deficiency, drug effects, heavy metal poisoning, and congenital dyserythropoietic disorders.
Acute megakaryoblastic leukemia
Most AML cases with megakaryoblasts, corresponding to AML FAB M7, now meet the 2008 WHO criteria for AML with t(1;22), AML with myelodysplasia-related changes, or the Down syndrome-associated disorders discussed below. The small number of remaining cases may, like other megakaryoblastic leukemias, demonstrate extensive marrow fibrosis limiting marrow aspiration. The blasts are relatively large with dense smooth chromatin, variable nucleoli, and scant to moderate amounts of cytoplasm. The cytoplasmic membrane may show irregular contours, and some examples show membrane protrusions or blebs. A spectrum of morphologies toward micromegakaryocytes is sometimes seen. FCM often reveals blasts lacking CD34, HLA-DR and CD45, variable expression of myeloid markers such as CD13 or CD33, and frequent expression of megakaryocyte antigens CD41 or CD61. At least 50% of blasts must show evidence of megakaryocytic differentiation. Ultrastructural analysis and ultracytochemistry can be diagnostic if demarcation membranes or bulls-eye granules are visualized, or if peroxidase activity is visualized in the nuclear membrane and endoplasmic reticulum but not the Golgi body or granules.50,51 Prognosis in both adults and children is poor.
Acute basophilic leukemia
This is a vanishingly rare leukemia showing blasts with aberrant basophilic differentiation and occasional involvement of skin, bones or other tissues, sometimes with symptoms of hyperhistaminemia. Reported immunophenotypes show lack of CD117, with variable CD34 and HLA-DR, and frequent expression of CD13 or CD33 with CD123, CD22 and CD11b. Cytochemistry is negative for MPO or SBB reactivity. One report using electron microscopy methods suggests that this may be an under-recognized entity.52,53 Acute basophilic leukemia may be difficult to distinguish from another rare entity, acute mast cell leukemia, which is described in Chapter 26.
Acute panmyelosis with myelofibrosis (APMF)
Another very rare disease, acute panmyelosis, must, by definition, present de novo rather than evolving from another condition. Acute panmyelosis with myelofibrosis (APMF) features pancytopenia, myelofibrosis, a proliferation of all myeloid lineages (panmyelosis) and increased blasts.54,55 There is usually prominent reticulin fibrosis, with fewer cases showing collagen fibrosis. Megakaryocytic dysplasia is often seen, but there must be insufficient evidence to diagnosis AML with myelodysplasia-related changes in order to consider this diagnosis. Myelodysplastic syndrome with fibrosis must also be ruled out, based on the blast count. The presence of dysplastic features and a lack of splenomegaly distinguish this entity from most myeloproliferative neoplasms. APMF typically differs from acute megakaryoblastic leukemia by showing expression of CD34 on blasts and by the panmyeloid proliferation seen amid the marrow fibrosis. The prognosis is typically poor.
Myeloid sarcoma
Mass involvement of a tissue site by myeloid blasts defines myeloid sarcoma. These lesions can present in isolation, or may accompany blood or BM myeloid diseases including MDS, myeloproliferative neoplasms, or myelodysplastic/myeloproliferative varieties. In any case, myeloid sarcoma is equivalent to a diagnosis of AML regardless of blast counts in blood or BM. The subcategories of myeloid sarcoma in the WHO 2001 classification have now been consolidated into a single category. Pediatric cases are less likely than adult cases to present without blood or BM leukemia, but are more likely to show 11q23 or (8;21) translocations, although the source of tissue for karyotypic analysis may be a possible confounder of these data, given that most adult samples are biopsied tissues, whereas pediatric samples are typically from BM blast populations. Inclusion of this entity in the differential diagnosis for tissue masses is critical, as many of the most helpful immunohistochemical stains may not be part of routine panels used to evaluate suspected lymphomas or small round blue cell tumors. Immunostains for myeloperoxidase, CD33, CD68, CD4, CD163, CD34, CD117, CD43, LAT and CD61 can help to evaluate suspected cases, although CD34 is commonly negative on the monocytic leukemic blasts that are a frequent cause of these lesions.56 CD68 is the most commonly positive marker in myeloid sarcomas overall, but is not particularly specific. In the unlikely circumstance that a portion of the specimen has not already been formalin-fixed at the time of morphologic review, FCM and cytochemical stains can greatly assist diagnosis.
Myeloid proliferations related to Down syndrome
Transient abnormal myelopoiesis (TAM)
Roughly one-tenth of patients with Down syndrome are born with clinical findings of acute megakaryoblastic leukemia, but in approximately 75% of cases the disease undergoes a mysterious spontaneous remission within a few months.57,58 Despite this favorable behavior, some cases cause life-threatening complications such as cardiac or respiratory compromise, hyperviscosity, hepatic or renal dysfunction (particularly hepatic fibrosis), splenic necrosis or disseminated intravascular coagulopathy. The blasts have morphology consistent with the former FAB M7 megakaryoblastic category, and most often express CD34, CD4, typical myeloid markers such as CD13, CD33 and CD117, megakaryocytic markers CD41, CD42, CD61, or CD71, and aberrant CD7 and CD56, without expression of myeloperoxidase, glycophorin, HLA-DR or monocytic markers CD14 or CD64. An unusual genetic feature of these cases beyond the patient’s trisomy 21 is the frequent mutation of the GATA1 transcription factor gene.59 There is little consensus about optimal therapeutic strategies for this usually transient condition.
Myeloid leukemia associated with Down syndrome
In 20–30% of Down syndrome patients who develop transient abnormal myelopoiesis, the transient disease is followed within 1–3 years by a recurrent and non-remitting AML. PB cytopenias and BM myelodysplastic features often precede the development of ≥20% blasts in the blood or BM, but no distinction is made between MDS and AML in this context. The BM may show extensive fibrosis. Extramedullary disease, particularly in the liver and spleen, is common. The blast immunophenotype is similar to that seen in TAM, except that about half of cases have CD34-negative blasts, and a third of cases fail to express CD41 and CD56. In addition to trisomy 21, these leukemias share the GATA1 mutations seen in TAM, and have a relatively high rate of complete or partial trisomy 8 and trisomy 1.59 The prognosis is very good compared to other childhood cases of AML. If an older Down syndrome child develops AML without GATA1 mutation, the leukemia should be classified as a conventional, non-Down syndrome case would be.
Blastic plasmacytoid dendritic cell (BPDC) neoplasm
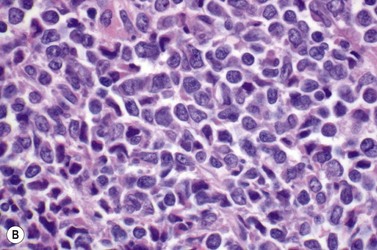
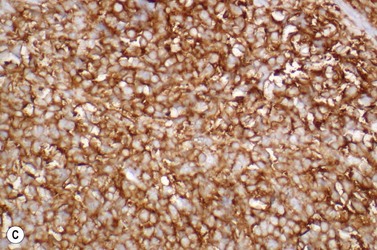
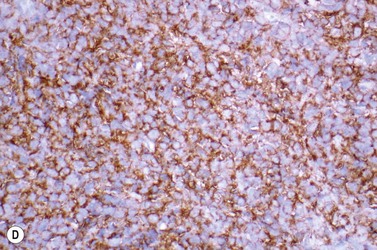
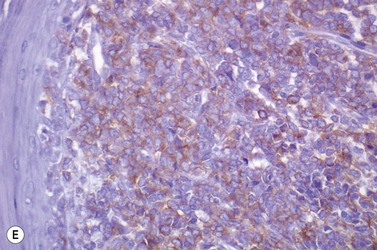
This rare entity, formerly referred to as blastic NK lymphoma or agranular CD4+ CD56+ hepatodermic neoplasm, is an aggressive malignancy often presenting in the skin with frequent BM and systemic leukemic involvement.60–62 The closeness of its relationship to other acute myeloid leukemias in the 2008 WHO classification is debatable; indeed, the underlying developmental pathways and physiology of plasmacytoid dendritic cells are subjects of current research.63 It appears that the plasmacytoid dendritic cells seen in the T-cell zones of lymphoid tissues are the predominant source of type I interferon in response to viral infections. Apparently reactive proliferations of plasmacytoid dendritic cells can be seen in association with CMML, hyaline vascular Castleman’s disease, viral infections and granulomatous lymphadenitis, and can usually be distinguished from BPDC neoplasm by their normal morphology and lack of CD56 expression. The violaceous nodules formed by the neoplasm in the skin typically show sparing of the epidermis and infiltration of the dermis and subcutaneous fat. Most patients also show BM involvement ranging from sparse to full infiltration. In approximately 65% of cases, circulating tumor cells in the blood are seen. Roughly half of cases have disease in the lymph nodes and a third have disease in the spleen. The tumor cells are medium-sized blasts with irregular nuclear contours, smooth chromatin, one to several visible nucleoli, and scant agranular blue-gray cytoplasm sometimes containing microvacuoles (Fig. 18.12A, B). The blastic cells are negative for CD34 and CD117, but express CD4 (Fig.18.12C), CD56 (Fig.18.12D), TCL1, CD43, CD45RA, CD68 (as small cytoplasmic dots), the plasmacytoid dendritic cell marker CD123 (the IL-3 receptor alpha chain, Fig.18.12E), BDCA-2 (CD303), CLA (cutaneous lymphocyte-associated antigen), the interferon-alpha induced MxA (myxovirus-resistance protein A), CD2-AP (CD2 adapter protein) and, in about one-third of cases, TdT.64,65 Expression of CD34, CD117, CD14, CD13 or lysozyme argues against BPDC, and would favor a diagnosis of AML. Cytogenetic analysis usually shows a complex karyotype with loss of 5q, 12p, 6q, and deletions of chromosomes 13, 9 and 15 being relatively common abnormalities. If one of the recurrent genetic abnormalities of AML in the 2008 WHO classification is detected, then the case should be classified as AML. Similarly, cytochemical stains for MPO or NSE should be negative, and prompt classification as AML if positive. The prognosis of the disease is poor, with median survival of approximately 1 year.
1 Bhayat F, Das-Gupta E, Smith C, et al. The incidence of and mortality from leukaemias in the UK: a general population-based study. BMC Cancer. 2009;9:252.
2 Deschler B, Lubbert M. Acute myeloid leukemia: epidemiology and etiology. Cancer. 2006 Nov 1;107(9):2099-2107.
3 Yamamoto JF, Goodman MT. Patterns of leukemia incidence in the United States by subtype and demographic characteristics, 1997–2002. Cancer Causes Control. 2008 May;19(4):379-390.
4 Aquino VM. Acute myelogenous leukemia. Curr Probl Pediatr Adolesc Health Care. 2002 Feb;32(2):50-58.
5 Mrozek K, Marcucci G, Paschka P, et al. Clinical relevance of mutations and gene-expression changes in adult acute myeloid leukemia with normal cytogenetics: are we ready for a prognostically prioritized molecular classification? Blood. 2007 Jan 15;109(2):431-448.
6 Swerdlow SH, Campo E, Harris NL, et al, editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed, Lyon: IARC, 2008.
7 Ley TJ, Mardis ER, Ding L, et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature. 2008 Nov 6;456(7218):66-72.
8 Mardis ER, Ding L, Dooling DJ, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009 Sep 10;361(11):1058-1066.
9 Walter MJ, Payton JE, Ries RE, et al. Acquired copy number alterations in adult acute myeloid leukemia genomes. Proc Natl Acad Sci USA. 2009 Aug 4;106(31):12950-12955.
10 Jaffe ES, Harris NL, Stein H, Vardiman JW. World Health Organization Classification of Tumours: Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC Press; 2001.
11 Greenberg P, Cox C, LeBeau MM, et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood. 1997 Mar 15;89(6):2079-2088.
12 Khalidi HS, Medeiros LJ, Chang KL, et al. The immunophenotype of adult acute myeloid leukemia: High frequency of lymphoid antigen expression and comparison of immunophenotype, French-American-British classification, and karyotypic abnormalities. American Journal of Clinical Pathology. 1998;109:211-220.
13 Bennett JM, Catovsky D, Daniel MT, et al. Proposals for the classification of the acute leukaemias. French-American-British (FAB) co-operative group. Br J Haematol. 1976 Aug;33(4):451-458.
14 de Labarthe A, Pautas C, Thomas X, et al. Allogeneic stem cell transplantation in second rather than first complete remission in selected patients with good-risk acute myeloid leukemia. Bone Marrow Transplant. 2005 Apr;35(8):767-773.
15 Schlenk RF, Dohner K, Krauter J, et al. Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. New England Journal of Medicine. 2008;358(18):1909-1918.
16 Bitter MA, Neilly ME, Le Beau MM, et al. Rearrangements of chromosome 3 involving bands 3q21 and 3q26 are associated with normal or elevated platelet counts in acute nonlymphocytic leukemia. Blood. 1985 Dec;66(6):1362-1370.
17 Secker-Walker LM, Mehta A, Bain B. Abnormalities of 3q21 and 3q26 in myeloid malignancy: a United Kingdom Cancer Cytogenetic Group study. Br J Haematol. 1995 Oct;91(2):490-501.
18 Slovak ML, Gundacker H, Bloomfield CD, et al. A retrospective study of 69 patients with t(6;9)(p23;q34) AML emphasizes the need for a prospective, multicenter initiative for rare ‘poor prognosis’ myeloid malignancies. Leukemia. 2006 Jul;20(7):1295-1297.
19 Arber DA, Carter NH, Ikle D, Slovak ML. Value of combined morphologic, cytochemical, and immunophenotypic features in predicting recurrent cytogenetic abnormalities in acute myeloid leukemia. Hum Pathol. 2003 May;34(5):479-483.
20 Downing JR. The AML1-ETO chimaeric transcription factor in acute myeloid leukaemia: biology and clinical significance. British Journal of Haematology. 1999;106:296-308.
21 Byrd JC, Mrozek K, Dodge RK, et al. Pretreatment cytogenetic abnormalities are predictive of induction success, cumulative incidence of relapse, and overall survival in adult patients with de novo acute myeloid leukemia: results from Cancer and Leukemia Group B (CALGB 8461). Blood. 2002 Dec 15;100(13):4325-4336.
22 Golomb HM, Rowley JD, Vardiman JW, et al. ‘Microgranular’ acute promyelocytic leukemia: a distinct clinical, ultrastructural, and cytogenetic entity. Blood. 1980;55(2):253-259.
23 McKenna RW, Parkin J, Bloomfield CD, et al. Acute promyelocytic leukaemia: a study of 39 cases with identification of a hyperbasophilic microgranular variant. British Journal of Haematology. 1982;50:201-214.
24 Neame PB, Soamboonsrup P, Leber B, et al. Morphology of acute promyelocytic leukemia with cytogenetic or molecular evidence for the diagnosis: charaterization of additional microgranular variants. American Journal of Hematology. 1997;6:131-142.
25 Redner RL. Variations on a theme: the alternate translocations in APL. Leukemia. 2002 Oct;16(10):1927-1932.
26 Rubnitz JE, Raimondi SC, Tong X, et al. Favorable impact of the t(9;11) in childhood acute myeloid leukemia. Journal of Clinical Oncology. 2002;20(9):2302-2309.
27 Mrozek K, Heinonen K, Lawrence D, et al. Adult patients with de novo acute myeloid leukemia and t(9;11)(p22;q23) have a superior outcome to patients with other translocations involving band 11q23: a Cancer and Leukemia Group B study. Blood. 1997;90(11):4532-4538.
28 Pearson MG, Vardiman JW, Le Beau MM, et al. Increased numbers of marrow basophils may be associated with a t(6;9) in ANLL. Am J Hematol. 1985 Apr;18(4):393-403.
29 Sweet DL, Golomb HM, Rowley JD, Vardiman JW. Acute myelogenous leukemia and thrombocythemia associated with an abnormality of chromosome no. 3. Cancer Genetics and Cytogenetics. 1979;1(1):33-37.
30 Cattaneo F, Nucifora G. EVI1 recruits the histone methyltransferase SUV39H1 for transcription repression. J Cell Biochem. 2008 Oct 1;105(2):344-352.
31 Carroll A, Civin C, Schneider N, et al. The t(1;22) (p13;q13) is nonrandom and restricted to infants with acute megakaryoblastic leukemia: a Pediatric Oncology Group Study. Blood. 1991;78(3):748-752.
32 Chan WC, Carroll A, Alvarado CS, et al. Acute megakaryoblastic leukemia in infants with t(1;22)(p13;q13) abnormality. American Journal of Clinical Pathology. 1992;98(2):214-221.
33 Lion T, Haas OA, Harbott J, et al. The translocation t(1;22)(p13;q13) is a nonrandom marker specifically associated with acute megakaryocytic leukemia in young children. Blood. 1992;79(12):3325-3330.
34 Cen B, Selvaraj A, Prywes R. Myocardin/MKL family of SRF coactivators: key regulators of immediate early and muscle specific gene expression. J Cell Biochem. 2004 Sep 1;93(1):74-82.
35 Duchayne E, Fenneteau O, Pages MP, et al. Acute megakaryoblastic leukaemia: a national clinical and biological study of 53 adult and childhood cases by the Groupe Francais d’Hematologie Cellulaire (GFHC). Leuk Lymphoma. 2003 Jan;44(1):49-58.
36 Kottaridis PD, Gale RE, Linch DC. Flt3 mutations and leukaemia. Br J Haematol. 2003 Aug;122(4):523-538.
37 Paschka P, Marcucci G, Ruppert AS, et al. Adverse prognostic significance of KIT mutations in adult acute myeloid leukemia with inv(16) and t(8;21): a Cancer and Leukemia Group B Study. J Clin Oncol. 2006;24(24):3904-3911.
38 Falini B, Nicoletti I, Martelli MF, Mecucci C. Acute myeloid leukemia carrying cytoplasmic/mutated nucleophosmin (NPMc+ AML): biologic and clinical features. Blood. 2007;109(3):874-885.
39 Bolli N, Nicoletti I, De Marco MF, et al. Born to be exported: COOH-terminal nuclear export signals of different strength ensure cytoplasmic accumulation of nucleophosmin leukemic mutants. Cancer Research. 2007;67(13):6230-6237.
40 Koschmieder S, Halmos B, Levantini E, Tenen DG. Dysregulation of the C/EBPalpha differentiation pathway in human cancer. Journal of Clinical Oncology. 2009;27(4):619-628.
41 Ahn JY, Seo K, Weinberg O, et al. A comparison of two methods for screening CEBPA mutations in patients with acute myeloid leukemia. JMolDiagn. 2009;11(4):319-323.
42 Ho PA, Alonzo TA, Gerbing RB, et al. Prevalence and prognostic implications of CEBPA mutations in pediatric acute myeloid leukemia (AML): a report from the Children’s Oncology Group. Blood. 2009;113(26):6558-6566.
43 Arber DA, Stein AS, Carter NH, et al. Prognostic impact of acute myeloid leukemia classification. Importance of detection of recurring cytogenetic abnormalities and multilineage dysplasia on survival. Am J Clin Pathol. 2003 May;119(5):672-680.
44 Lugthart S, van Drunen E, van Norden Y, et al. High EVI1 levels predict adverse outcome in acute myeloid leukemia: prevalence of EVI1 overexpression and chromosome 3q26 abnormalities underestimated. Blood. 2008;111(8):4329-4337.
45 Breems DA, van Putten WL, De Greef GE, et al. Monosomal karyotype in acute myeloid leukemia: a better indicator of poor prognosis than a complex karyotype. Journal of Clinical Oncology. 2008;26(29):4791-4797.
46 Rund D, Ben-Yehuda D. Therapy-related leukemia and myelodysplasia: evolving concepts of pathogenesis and treatment. Hematology. 2004 Jun;9(3):179-187.
47 Rund D, Krichevsky S, Bar-Cohen S, et al. Therapy-related leukemia: clinical characteristics and analysis of new molecular risk factors in 96 adult patients. Leukemia. 2005 Nov;19(11):1919-1928.
48 Haferlach T, Kohlmann A, Klein HU, et al. AML with translocation t(8;16)(p11;p13) demonstrates unique cytomorphological, cytogenetic, molecular and prognostic features. Leukemia. 2009;23(5):934-943.
49 Garand R, Duchayne E, Blanchard D, et al. Minimally differentiated erythroleukaemia (AML M6 ‘variant’): a rare subset of AML distinct from AML M6. British Journal of Haematology. 1995;90:868-875.
50 Eguchi M, Ozawa T, Sakakibara H, et al. Ultrastructural and ultracytochemical differences between megakaryoblastic leukemia in children and adults. Analysis of 49 patients. Cancer. 1992 Jul 15;70(2):451-458.
51 Zipursky A, Christensen H, De Harven E. Ultrastructural studies of the megakaryoblastic leukemias of Down syndrome. Leuk Lymphoma. 1995 Jul;18(3–4):341-347.
52 Peterson LC, Parkin JL, Arthur DC, Brunning RD. Acute basophilic leukemia. A clinical, morphologic, and cytogenetic study of eight cases. American Journal of Clinical Pathology. 1991;96(2):160-170.
53 Shvidel L, Shaft D, Stark B, et al. Acute basophilic leukaemia: eight unsuspected new cases diagnosed by electron microscopy. Br J Haematol. 2003;120(5):774-781.
54 Bearman RM, Pangalis GA, Rappaport H. Acute (‘malignant’) myelosclerosis. Cancer. 1979;43(1):279-293.
55 Sultan C, Sigaux F, Imbert M, Reyes F. Acute myelodysplasia with myelofibrosis: a report of eight cases. BrJ Haematol. 1981;49(1):11-16.
56 Pileri SA, Ascani S, Cox MC, et al. Myeloid sarcoma: clinico-pathologic, phenotypic and cytogenetic analysis of 92 adult patients. Leukemia. 2007;21(2):340-350.
57 Massey GV, Zipursky A, Chang MN, et al. A prospective study of the natural history of transient leukemia (TL) in neonates with Down syndrome (DS): Children’s Oncology Group (COG) study POG–9481. Blood. 2006;107(12):4606-4613.
58 Ganick DJ. Hematological changes in Down’s syndrome. Crit Rev Oncol Hematol. 1986;6(1):55-69.
59 Pine SR, Guo Q, Yin C, et al. Incidence and clinical implications of GATA1 mutations in newborns with Down syndrome. Blood. 2007;110(6):2128-2131.
60 Brody JP, Allen S, Schulman P, et al. Acute agranular CD4-positive natural killer cell leukemia. Comprehensive clinicopathologic studies including virologic and in vitro culture with inducing agents. Cancer. 1995 May 15;75(10):2474-2483.
61 Feuillard J, Jacob MC, Valensi F, et al. Clinical and biologic features of CD4(+)CD56(+) malignancies. Blood. 2002 Mar 1;99(5):1556-1563.
62 DiGiuseppe JA, Louie DC, Williams JE, et al. Blastic natural killer cell leukemia/lymphoma: a clinicopathologic study. American Journal of Surgical Pathology. 1997;21(10):1223-1230.
63 Herling M, Jones D. CD4+/CD56+ hematodermic tumor: the features of an evolving entity and its relationship to dendritic cells. Am J Clin Pathol. 2007 May;127(5):687-700.
64 Pilichowska ME, Fleming MD, Pinkus JL, Pinkus GS. CD4+/CD56+ hematodermic neoplasm (‘blastic natural killer cell lymphoma’): neoplastic cells express the immature dendritic cell marker BDCA-2 and produce interferon. Am J Clin Pathol. 2007 Sep;128(3):445-453.
65 Marafioti T, Paterson JC, Ballabio E, et al. Novel markers of normal and neoplastic human plasmacytoid dendritic cells. Blood. 2008 Apr 1;111(7):3778-3792.