Chapter 20 Acute heart failure
The pattern of heart failure seen in the community, outpatients clinics and specialist cardiac wards is dominated by the acute coronary syndromes and chronic heart failure, predominantly caused by ischaemic heart disease and hypertension.1,2 Heart failure is the commonest cause of hospital admission in people over 65 years of age and it has been estimated that in North America and Europe over 15 million patients have heart failure and 1.5 million new cases are diagnosed each year.3 Patients present with chest pain, shortness of breath, fatigue and oedema and will usually have single-organ failure. Management focuses on reducing cardiac work to relieve symptoms and prevent further myocardial damage.4,5
Patients either admitted with or who develop acute heart failure on the intensive care unit (ICU) frequently have overt or occult underlying coronary artery disease, but will usually have significant other organ dysfunction. Management in this setting focuses on both improving global and regional oxygen delivery and maintaining perfusion pressure, often with the use of drugs that stimulate rather than rest the myocardium.6–8 The resolution of this apparent paradox requires that for each patient management should attempt to achieve the frequently difficult balance between the best interests of the myocardium and the circulatory requirements of the other vital organs. The critical care physician should target the minimum necessary oxygen delivery and arterial pressure to maintain other organ function at maximum cardiac efficiency (e.g. ensuring adequate fluid resuscitation before starting beta-agonists) so that cardiac work and the risk of myocardial ischaemia and necrosis from exuberant beta-agonist use are minimised and the cardiologist should consider the wider circulation and other organ requirements when instituting strategies to protect the myocardium.
DIAGNOSIS OF ACUTE HEART FAILURE
The diagnosis of acute heart failure in critically ill patients can be more difficult than is commonly recognised. Although the underlying pathology in most patients with acute heart failure on intensive care will be coronary artery disease, other diagnoses must be considered (Table 20.1).
Table 20.1 Causes of acute heart failure on the intensive care unit
| Coronary artery disease |
| Infection – systemic sepsis,9 myocarditis |
| Mechanical – endocarditis, pulmonary emboli, valve problems, septal defects, tamponade, high intrathoracic pressure with inadequate preload |
| Drugs – beta-blockers, calcium antagonists, cytotoxic therapy |
| Hypoxaemia |
| Metabolic – acidaemia, thiamine deficiency, hypocalcaemia, hypophosphataemia |
| Myocardial contusion – blunt thoracic trauma |
| Myocardial infiltration – tumour, sarcoidosis, amyloidosis |
| Vasculitis – rare |
It is also important to reassess critically the patient referred with a diagnosis of acute heart failure to decide whether this is indeed the primary problem. The history, examination and initial investigations with routine blood tests, electrocardiogram (ECG) and chest X-ray may be compatible with this diagnosis but many such patients are elderly with multiple comorbidities and deciding whether the patient is suffering from a primary myocardial pathology as opposed to a pulmonary problem or indeed systemic sepsis9 can be difficult. Equally, patients believed to have a primary respiratory problem may fail to wean from ventilatory support because of a failure to realise that they have left ventricular failure with a high left atrial pressure and incipient pulmonary oedema, causing a reduction in pulmonary compliance, an increased work of breathing and respiratory distress when ventilatory support is withdrawn.
ECHOCARDIOGRAPHY (SEE CHAPTER 23)
Echocardiography is an extremely valuable investigation in the management of the critically ill patient with acute heart failure10 and the modern critical care physician should at least be able to perform a basic examination. It will frequently establish the underlying cardiac pathology and can be used to monitor the response to treatment. It will:
The images obtained with transthoracic echocardiography (TTE) may be poor in ventilated patients but the experienced operator can achieve considerable improvement using microbubble contrast techniques.11
MEASUREMENT OF TROPONIN AND BRAIN NATRIURETIC PEPTIDE
Myocardial injury and the development of acute heart failure are common but frequently unrecognised complications of critical illness occurring not only in patients with an overt acute coronary syndrome but also in other conditions such as sepsis and major pulmonary embolism (PE).12 Relying on blood tests alone to establish a diagnosis or to plan management is inadvisable but, when interpreted in conjunction with the wider clinical picture, brain-type natriuretic peptide (BNP) and cardiac troponin are two tests that are becoming routinely available and appear to be sensitive markers of myocardial stress and necrosis and to have significant prognostic significance.
BRAIN-TYPE NATRIURETIC PEPTIDE
BNP was first isolated from porcine brain but the major source is the ventricular myocardium. The main stimulus for synthesis and release is myocardial wall stress. As a triage tool in the emergency department it is able to discriminate patients with heart failure from those with pulmonary or other non-cardiac causes for acute dyspnoea and has been shown to reduce rates of ICU admission, length of hospital stay and cost.13 Several studies have demonstrated that, below a cut-off of 100 pg/ml, BNP has a sensitivity of almost 90% and a specificity approaching 80% as a test for excluding heart failure.14 It is now included in both the European and UK National Institute for Clinical Excellence guidelines for the management of heart failure15,16 and BNP has also been shown to be a marker of myocardial dysfunction and prognosis in severe sepsis.17
CARDIAC TROPONIN I AND T (cTnI, cTnT)
Troponin is part of the thin filament of the myocyte contractile apparatus and has three subunits: (1) I, which binds actin to inhibit actin-myosin contraction; (2) T, which binds tropomyosin to facilitate contraction; and (3) C, which binds calcium ions. The cardiac isoforms cTnI and cTnT are specific to the heart and can be measured in the blood after myocyte necrosis with 50% release by 4 hours, peaking at 12–24 hours and remaining elevated for up to 10 days. It is far more sensitive than the traditional cardiac enzyme tests such as creatine kinase and indeed it has substantially changed the diagnosis and management of acute myocardial infarction, as reflected in the new guidelines issued by the European Society of Cardiology15 and American College of Cardiologists. Troponin may be released in conditions other than acute coronary ischaemia18 such as sepsis and after chemotherapy and in the absence of evidence of myocardial necrosis as in acute heart failure or major PE, where it is believed that the acute ventricular dilatation causes increased membrane permeability. Raised troponin levels are also associated with increased morbidity and mortality in surgical ICU patients.19
In combination with BNP, cTnI is valuable in screening for massive PE: in massive and submassive PE both are raised and further investigation with computed tomography pulmonary angiogram (CTPA) is indicated but in minor PE both will be negative.
The remainder of this chapter addresses the assessment and principles of management of ventricular function in patients admitted to the ICU with acute heart failure. This inevitably involves reference to circulatory failure and the state of the peripheral circulation but the more detailed aspects of oxygen delivery and control of the regional and microcirculation are considered elsewhere20 (see Chapters 11 and 12), as are the acute coronary syndromes2 (see Chapter 16) and chronic heart failure.1
CIRCULATORY FAILURE OR ‘SHOCK’
Failure to maintain an adequate oxygen supply to the tissues with the consequent development of anaerobic cellular metabolism defines circulatory failure or ‘shock’, a term that benefits from brevity but little else since it implies neither cause nor prognosis, but its use is now widespread and inescapable. Table 20.2 classifies circulatory ‘shock’.
Table 20.2 Major categories of circulatory failure or ‘shock’
| Cardiogenic | Myocardial infarction, myocarditis, vasculitis, valve dysfunction (e.g. critical aortic stenosis, mitral regurgitation, acute endocarditis), post cardiac bypass surgery, drug overdose (β-blockers, calcium antagonists) |
| Hypovolaemic | Haemorrhage, burns, gastrointestinal fluid loss |
| Obstructive | Pulmonary embolus, cardiac tamponade, tension pneumothorax |
| Anaphylactic | Drugs, blood transfusion, insect sting |
| Septic | Bacterial infection, non-infective inflammatory conditions, e.g. pancreatitis, burns, trauma |
| Neurogenic | Intracranial haemorrhage, brainstem compression, spinal cord injury |
In considering these causes of circulatory failure, several points require emphasis:
ASSESSMENT OF VENTRICULAR FUNCTION
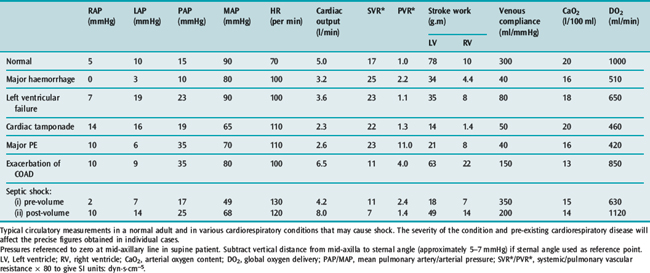
Table 20.3 illustrates typical values in normal subjects and in the common causes of circulatory failure with calculation of the associated vascular resistances and oxygen delivery. The values quoted are merely examples that indicate the pattern of circulatory derangement produced by these pathologies: pre-existing cardiopulmonary disease and the severity of the condition will affect the precise figures obtained in individual cases and the response to vasoactive therapy.
Table 20.3 Measurements in a normal 75-kg adult and in various conditions causing circulatory ‘shock’
Stroke volume (SV) is calculated from CO and heart rate:
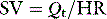
Three factors determine stroke volume: (1) preload; (2) afterload; and (3) myocardial contractility.
VENTRICULAR PRELOAD
Ventricular preload, traditionally assessed from the atrial filling pressures, determines the end-diastolic ventricular volume, which, according to Starling’s law of the heart and depending on ventricular contractility, dictates the stroke work generated by each ventricle at the next cardiac contraction. The resulting stroke volume depends on the resistance or afterload that confronts the ventricle.26
On the general ward the jugular venous pressure (JVP) is measured from the sternal angle but in ICU vascular pressures are measured from the mid-axillary line in the fifth intercostal space. From this reference point, in the supine position, the normal RAP is between 4 and 8 mmHg and the LAP, or wedge pressure, is between 8 and 12 mmHg. Relative changes in either the contractility of the two ventricles or the respective vascular resistances will change the relationship between the atrial pressures, which must then be independently assessed.27
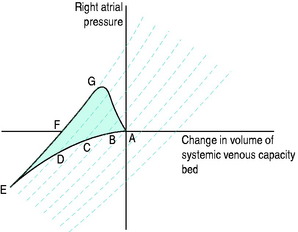
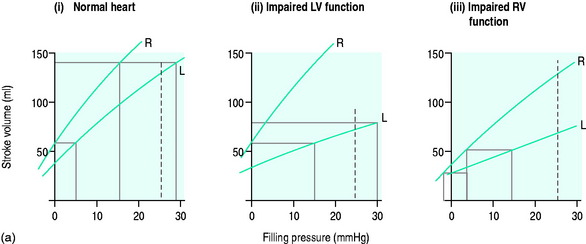
The systemic venous bed is the major intravascular capacitance or reservoir of the circulation with a compliance that can vary from 30 to over 300 ml/mmHg and which provides a buffer against the effects of intravascular volume loss. It also explains the response observed in major haemorrhage and subsequent transfusion. As volume is lost, venous tone increases, preventing the large falls in atrial filling pressures and CO that would otherwise occur. If the equivalent volume is returned over the subsequent few hours the RAP gradually returns to normal as the intravascular volume is restored and the reflex increase in sympathetic tone abates. However, rapid reinfusion of the same volume does not allow sufficient time for the venous and arteriolar tone to fall and may result in the LAP rising to a level that precipitates pulmonary oedema, although the intravascular volume has only been returned to the prehaemorrhage level and left ventricular function is normal (Figure 20.1).
In assessing preload, end-diastolic volume rather than pressure is relevant and when interpreting atrial pressures as measures of preload, two points must be considered:
Alternative methods of assessing ventricular preload are discussed later in this chapter under “Assessment of intravascular volume status” and in Chapter 12.
VENTRICULAR AFTERLOAD
The vascular resistance against which each ventricle works is calculated, by analogy with Ohm’s law, as the pressure gradient across the vascular bed divided by the CO (Table 20.4).
Table 20.4 Calculation of ventricular afterload and stroke work
| Systemic vascular resistance (SVR) = [(MAP − RAP)/Qt] × 80 dyn.s.cm−5 |
| = [(90 − 5)/5] × 80 = 1360 dyn.s.cm−5 |
| SVRI = SVR × BSA = 1360 × 1.65 = 2244 dyn.s.cm−9 |
| Pulmonary vascular resistance (PVR) = [(PAP − LAP)/Qt × 80 dyn.s.cm−5 |
| = [(15 − 5)/5] × 80 = 160 dyn.s.cm−5 |
| PVRI = PVR × BSA = 160 × 1.65 = 264 dyn.s.cm−9 |
| Stroke volume (SV) = Qt/HR = 72 ml |
| Stroke volume index (SVI) = 72/1.65 = 44 ml/m2 |
| Ventricular stroke work (VSV) = SV × (afterload − preload) |
| LVSW = SV × (MAP − LAP) × 0.0136 g.m |
| = 72 × (90 − 10) × 0.0136 = 78 g.m |
| LVSWI = 78/1.65 = 47 g.m |
| RVSW = SV × (PAP − RAP) × 0.0136 g.m |
| = 72 × (15 − 5) × 0.0136 = 10 g.m |
| RVSWI = 10/1.65 = 6 g.m |
MAP, mean arterial pressure; PAP, mean pulmonary artery pressure.
Pressures are measured in mmHg, cardiac output (Qt) in l/min.
Values for resistance, stroke work are frequently indexed by dividing by the patient’s body surface area (BSA) derived from height and weight.
In calculating ventricular stroke work, 0.0136 converts from ml.mmHg to SI units of g.m.
Example calculations assume a normal 75-kg individual with BSA 1.65 m2.
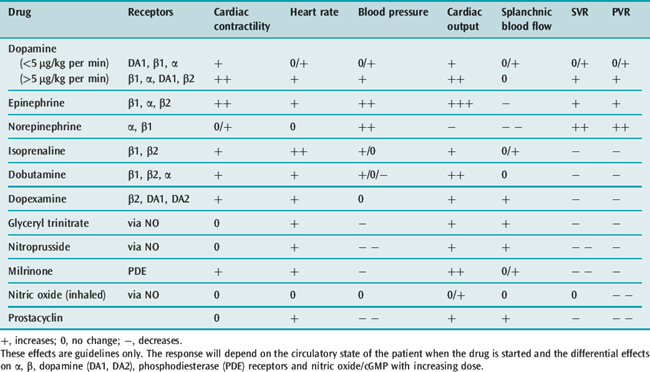
The effects of some of the commonly used vasoactive agents are shown in Table 20.5 and considered in more detail later (see Chapters 79 and 80).
VENTRICULAR CONTRACTILITY AND EFFICIENCY
The work that the ventricle performs under given loading conditions defines contractility.
For each ventricle it may be expressed mathematically as the gradient and intercept of the relationship between atrial filling pressure and stroke work (Figure 20.2). The resulting stroke volume varies with the resistance of the vascular bed into which the ventricle is ejecting. Although the right ventricle generates a much smaller stroke work, the afterload (pulmonary vascular resistance) against which it ejects is correspondingly lower since the right and left ventricular stroke volumes must necessarily be the same over time.
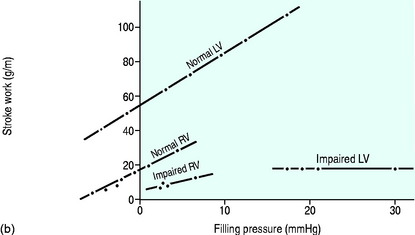
The work generated by each ventricle with each heart beat is the ventricular stroke work and is calculated as shown in Table 20.4.
Left ventricular efficiency is the ratio of work output to energy input and may be less than 20% in patients with acute heart failure, with over 80% of energy lost as heat. A technique, based on thermodynamic principles and requiring only measurement of temperature and oxygen content difference across the left ventricular capillary bed, is simpler, clinically applicable and more accurate than earlier methods.32 It will allow selection of therapies based on both global circulatory and myocardial metabolic considerations.
If circulatory failure is due to impaired myocardial contractility, as defined by a ‘flattened’ stroke work/filling pressure equation (see Figure 20.2), the atrial pressures will often already be raised. Provided such pressures reflect volume preload, further increases are not helpful since the ventricle becomes increasingly distended with high wall tension, as predicted by Laplace’s law:
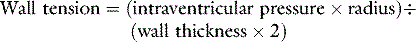
The remaining therapeutic options are:
HEART RATE AND RHYTHM
In cardiac failure the stroke volume is usually constant for rates up to 100/min and thereafter falls as restriction of diastolic filling time limits end-diastolic volume. Increasing the heart rate from 70 to 90/min will increase CO by almost 30%. Achieving this with a chronotrope such as the β1-agonist isoproterenol increases myocardial work and oxygen consumption and also ventricular irritability. In patients with ischaemic heart disease and particularly after a recent myocardial infarction, atrial or atrioventricular sequential pacing (which maintains coordinated atrial contraction in heart block) improves haemodynamics without stimulating myocardial metabolism and increasing myocardial irritability.35
Heart rates above 110 beats/min, particularly with an irregular rhythm, should be controlled by either drugs or DC cardioversion after ensuring that plasma potassium and magnesium levels have been corrected. If the rhythm is supraventricular and unstable with intermittent periods of sinus rhythm, pharmacological control is indicated using either digoxin or amiodarone. Digoxin is appropriate for atrial fibrillation and has a temporary positive inotropic effect.36 However amiodarone is suitable for all supraventricular rhythms and is more likely to restore sinus rhythm. A meta-analysis showed that, used prophylactically, it reduced the rate of arrhythmic episodes and sudden death in patients with recent myocardial infarction or congestive cardiac failure.37 It is however a negative inotrope and this can be significant in the patient with severe heart failure.
A fixed rate of 150/min suggests atrial flutter and should prompt careful inspection of the ECG and a trial of adenosine. A persistent sinus tachycardia unexplained by fever may be due to hypovolaemia, pain or anxiety.
ASSESSMENT OF MYOCARDIAL FUNCTION
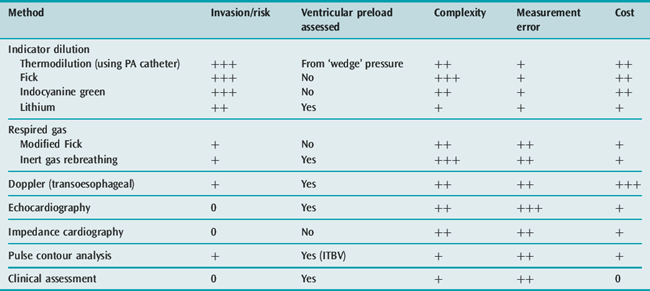
It is certainly not always necessary to use invasive monitoring.38 Initial management can be based on clinical assessment of intravascular volume and CO. The discipline of committing to an estimate of these key variables ensures that both the analysis of the circulation and the approach to treatment are logical. Further monitoring should be instituted if the initial management does not produce clinical improvement. Alternative, less invasive methods are available for assessing CO such as transoesophageal Doppler,39 lithium dilution,40 continuous CO by pulse contour analysis (PiCCO) and echocardiography10 – techniques which can also provide data on the volume rather than pressure preload of the left ventricle. Table 20.6 lists some features of the techniques available for measuring CO and whether they provide information on left ventricular preload. Further details of circulatory monitoring and these other techniques are described in the section on haemodynamic monitoring (see Chapter 12). A recent International Consensus Conference produced guidelines for the haemodynamic monitoring and management of patients with shock.41
KEY POINTS WHEN ASSESSING CARDIAC FUNCTION
PULMONARY ARTERY CATHETERISATION
This remains one of the most widely used methods for measurement of left atrial and PA pressures and assessment of CO using the thermodilution technique.42 Although generally viewed as the ‘gold standard’ for determining CO, the error is at least 10%, even with fastidious attention to technical detail.
Inflation of the balloon at the end of the catheter provides a PA occlusion or wedge pressure, which reflects left atrial pressure provided there are no significantpulmonary vascular bed abnormalities, as occur in chronic obstructive airways disease and long-standing mitral valve disease. Despite obtaining a good-quality wedge tracing, the measurement must be interpreted with caution since increased intrathoracic pressure and diastolic dysfunction make this pressure measurement an unreliable index of true left ventricular volume preload.
The focus on ‘goal-directed therapy’ led to the widespread use of PA catheters, but their indiscriminate use was challenged by a multicentre case-controlled study which suggested that patients managed with a PA catheter had a poorer outcome than those managed without such intervention.43 This study probably reflected the enthusiasm for inappropriate goal-directed therapy prevalent at that time, poor training in the use of the catheter and an inability of clinicians to respond appropriately to the data obtained.44
Table 20.7 lists the indications for PA catheterisation in heart failure. Other aspects of haemodynamic monitoring are discussed in Chapter 12.
Table 20.7 Indications for pulmonary arterial catheterisation in patients with heart failure
| Failure to improve with initial circulatory management and uncertainty about adequacy of cardiac output and relationship between atrial filling pressures |
| Assessment of left ventricular preload when the relationship between right and left atrial pressures is uncertain due to recent myocardial infarction, valvular abnormalities or high pulmonary vascular resistance. A low wedge pressure indicates that further volume is indicated but a high value does not necessarily exclude the need for further volume |
| Measurement of cardiac output by thermodilution to direct appropriate choice of vasoactive drug and to manipulate therapy, particularly when high doses are being used |
| Need to monitor pulmonary arterial pressures and assess right ventricular function |
ASSESSMENT OF INTRAVASCULAR VOLUME STATUS
CLINICAL
This is conventionally based on measurement of the right atrial filling pressure and the assumption that a normal relationship exists between the atrial filling pressures, which is not necessarily valid in the critically ill patient, particularly in cardiogenic shock.27 Although values < 12 mmHg suggest hypovolaemia, higher levels are more difficult to interpret, particularly in the ventilated patient. The RAP should therefore be interpreted carefully and in light of other clinical evidence. However such static tests for assessing the intravascular volume are less valuable than dynamic tests, such as the fluid challenge and the effect of positive-pressure ventilation, which assess the circulatory response to an intervention.
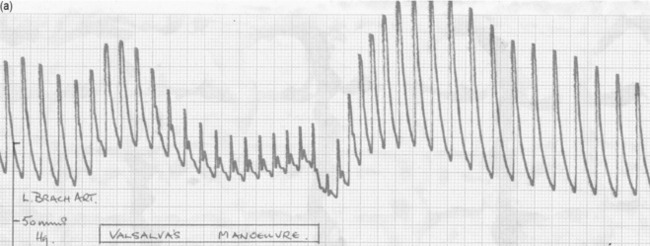
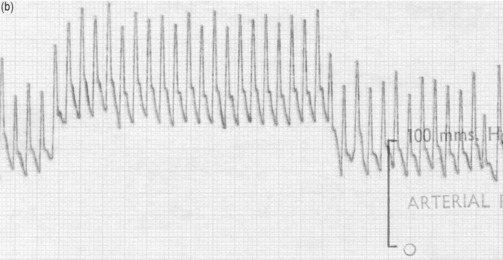
VALSALVA MANOEUVRE
The effect of changes in intrathoracic pressure can be used to assess intrathoracic blood volume and provide an estimate of true left ventricular preload. Figure 20.3 shows the classic Valsalva response in a normal subject and in a patient with a high intrathoracic blood volume. If a normal-type trace is observed on the monitor, further volume is indicated, whereas a square-wave response indicates an adequate left ventricular volume preload.45 This response can be quantified by calculating the ratio of the pulse pressure during phase 2 of the manoeuvre to the baseline value. This correlates with measurements of PA wedge pressure46 and can be applied at the bedside in sedated, ventilated patients.47 However, if the patient is breathing spontaneously, this test is difficult both to perform and to interpret.
ECHOCARDIOGRAPHY
Echocardiography is useful in identifying inadequate volume preload and the need for further fluid resuscitation, particularly when the preload pressures are high, as may occur with diastolic dysfunction. Performing serial studies to assess the response to therapy (fluid challenge, starting vasoactive therapy, changing ventilator settings) can be particularly valuable.
CORRECTION OF METABOLIC FACTORS
The following metabolic factors should be promptly corrected:
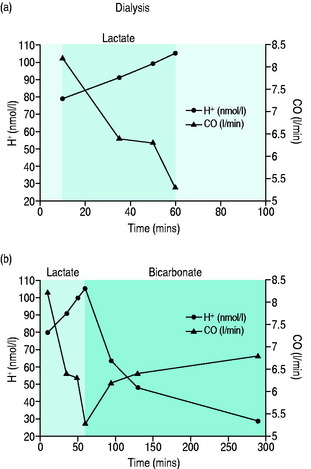
Metabolic acidaemia with pH < 7.20 or base deficit > 10 mmol/l should be corrected since myocardial contractility increases linearly with rising pH to values > 7.40. The suggestion that sodium bicarbonate should not be used as it produces a damaging paradoxical intracellular acidosis is misleading since the experiments demonstrating this effect were performed in vitro using non-physiological solutions, within a closed system that allowed no correction for any rise in carbon dioxide concentration and in which the sodium bicarbonate was given by bolus rather than by slow infusion.48 The case for using bicarbonate to correct a metabolic acidaemia in the clinical setting has been recognised49 and is supported by studies looking at the use of bicarbonate rather than lactate as the buffer solution in haemofiltration.50Figure 20.4 shows the effect of correcting a severe metabolic acidaemia on CO by changing from lactate to bicarbonate haemofiltration.
Although a prospective randomised study demonstrated an improved survival for critically patients if haemoglobin concentration was maintained at 7–9 g% rather than at 10–12 g%, this did not apply to the elderly and those with coronary artery disease, in whom the haemoglobin level should be maintained > 9 g%.51
Patients with poor dietary thiamine intake, chronic alcohol abuse and those on chronic furosemide or digoxin therapy are at risk of thiamine deficiency, resulting in impaired myocardial function. Oral thiamine (200 mg/day) improves left ventricular function in these patients.52
SELECTION OF APPROPRIATE VASOACTIVE AGENTS (SEE CHAPTER 82)
The choice of vasoactive agent when treating acute heart failure represents a balance between the global circulatory requirements and those of a stressed myocardium. The properties of commonly used agents are shown in Table 20.5.
Dopamine has been widely used in the erroneous belief that it selectively improves renal blood flow. However, if the patient has been fully volume-resuscitated and a modest inotropic effect with only a small increase in SVR and a natriuretic effect are required, then a low-dose dopamine infusion (< 4 μmol/kg per min) is appropriate.54
Dopexamine is used to improve splanchnic blood flow but, despite reported benefits when used with volume loading in perioperative patients,55 there is little evidence of outcome benefit in established shock.
Patients with chronic heart failure and those receiving long-term β-agonist infusion often develop tolerance with reduced catecholamine receptor responsiveness, resulting in less effect in raising intracellular cyclic adenosine monophosphate (cAMP) levels and increasing myocardial contractility. Phosphodiesterase inhibitors (enoximone, milrinone) offer an alternative strategy. Milrinone competitively inhibits the phosphodiesterase III isoenzyme, responsible for the breakdown of cAMP, thereby increasing intracellular cAMP levels and improving myocardial contractility independent of β-receptor stimulation. There is also improvement in ventricular diastolic relaxation. However, these agents are powerful vasodilators and hypotension frequently limits their use or requires a noradrenaline infusion. The dose should be reduced in renal failure.
Levosimendan is an intracellular calcium sensitiser and bypasses the receptors through which other inotropic agents act. Administered as an infusion over 3 days, it has a long-lasting metabolite which results in any improvement in myocardial contractility being sustained for several weeks.56 It has mainly been used in chronic heart failure and its role in severe acute heart failure and cardiogenic shock following acute myocardial infarction is uncertain but promising. As a potassium channel blocker, it dilates smooth muscle and may cause hypotension. In the patient with severe heart failure already on inotropic drugs, treatment should start with a low-dose infusion (0.05 μg/kg per min) and no loading dose should be given. If tachycardia develops or persists it is theoretically logical to consider addition of a beta-blocker but it is advisable to start with a small dose of a short-acting drug such as metoprolol or esmolol.
BETA-BLOCKADE
The use of β-blockers in patients with heart failure remains controversial. Large studies have demonstrated their benefit early after acute myocardial infarction4,57 and atenolol given intravenously perioperatively produces a survival benefit for up to 2 years after non-cardiac surgery in patients deemed to be at high risk of coronary artery disease.58 This evidence would appear to conflict with an increasing number of studies of perioperative optimisation that show benefit from increasing oxygen delivery with volume loading and the use of β-agonists.55 The explanation may be that in surgical patients the majority of the benefit derives from achieving adequate volume resuscitation and the additional use of a β-agonist is beneficial if the CO and oxygen delivery remain inadequate and the heart rate is less than 100/min, whereas the subset of patients with underlying coronary artery disease and a persisting tachycardia after fluid resuscitation may benefit from β-blockade rather than β-stimulation.
MECHANICAL SUPPORT FOR THE HEART
Continuous positive airway pressure (CPAP) by facemask59 and invasive positive-pressure ventilation are the most common forms of mechanical support provided in heart failure. The benefits result from improved oxygenation and reducing or eliminating the work of breathing, which may account for up to 30% of oxygen consumption.60 This reduction in oxygen consumption reduces left ventricular workload and alleviates myocardial ischaemia. When instituting mechanical ventilation the clinician must be prepared to give volume and even adrenaline as the sedation and other anaesthetic agents given for intubation will reduce endogenous levels of catecholamines, producing arteriolar and venular dilatation and potentially catastrophic hypotension.
The intra-aortic counterpulsation balloon pump (IABP) is physiologically attractive since it both improves coronary and peripheral circulatory perfusion and decreases cardiac work. This results in a more efficient cardiac performance and improved myocardial oxygenation.61
Left ventricular assist devices (LVAD) can temporarily take over myocardial function but are only indicated if all other treatment options have been explored and improvement in myocardial function can be anticipated.62 There are significant problems with bleeding, infection, thromboembolism and stroke. Both IABP and LVAD should be viewed as ‘bridges to recovery’ after cardiac surgery or recent myocardial infarction or when there is a realistic prospect of cardiac transplantation.
CARDIOGENIC SHOCK AFTER ACUTE MYOCARDIAL INFARCTION
1 Packer M. Pathophysiology and treatment of chronic heart failure. Lancet. 1992;340:88-95.
2 Simoons ML, Boersma E, van der Zwaan C, et al. The challenge of acute coronary syndromes. Lancet. 1999;353(suppl. II):1-4.
3 Redfield MM. Heart failure – an epidemic of uncertain proportions. N Engl J Med. 2002;347:1442-1444.
4 Yusuf S, Peto R, Lewis J, et alSleight P. Beta-blockade during and after myocardial infarction: an overview of the randomised trials. Prog Cardiovasc Dis. 1985;27:335-371.
5 MERIT-HF Study Group. Effect of metoprolol CR/XL in chronic heart failure: metoprolol CR/XL randomised intervention trial in congestive heart failure (MERIT-HF). Lancet. 1999;353:2001-2007.
6 Shoemaker WC, Appel PL, Kram HB, et al. Prospective trial of supranormal values of survivors as therapeutic goals in high-risk surgical patients. Chest. 1988;94:1176-1187.
7 Hayes MA, Timmins AC, Yau EH, et al. Elevation of systemic oxygen delivery in the treatment of critically ill patients. N Engl J Med. 1994;330:1717-1722.
8 Gattinoni L, Brazzi L, Pelosi P, et al. A trial of goal-orientated haemodynamic therapy in critically ill patients. N Engl J Med. 1995;333:1025-1032.
9 Turner A, Tsamitros M, Bellomo R. Myocardial cell injury in septic shock. Crit Care Med. 1999;27:1775-1780.
10 Price S, Nicol E, Gibson DG, et al. Echocardiography in the critically ill: current and potential roles. Intens Care Med. 2006;32:48-59.
11 Gmaurer G. Contrast echocardiography: clinical utility. Echocardiography. 2000;17:5-9.
12 Guest TM, Ramanathan AV, Tuteur AV, et al. Myocardial injury in critically ill patients: a frequently unrecognized complication. JAMA. 1995;273:1945-1949.
13 Mueller C, Scholer A, Laule-Kilian K, et al. Use of B-type natriuretic peptide in the evaluation and management of acute dyspnea. N Engl J Med. 2004;350:647-654.
14 McCullough PA, Nowak RM, McCord J, et al. B-type natriuretic peptide and clinical judgement in emergency diagnosis of heart failure: analysis from Breathing Not Properly (BNP) multinational study. Circulation. 2002;106:416-422.
15 Niemenen MS, Bohn M, Drexler H, et al. New practice guidelines of the European Society of Cardiology: management of acute heart failure. Eur Heart J. 2005;26:384-416.
16 NICE guidelines for the management of chronic heart failure in adults in primary and secondary care. Available online at: http://www.nice.org.uk, 2003.
17 Charpentier J, Luyt CE, Fulla Y, et al. Brain natriuretic peptide: a marker of myocardial dysfunction and prognosis during severe sepsis. Crit Care Med. 2004;32:660-665.
18 Arlati S, Brenna S, Prencipe L, et al. Myocardial necrosis in ICU patients with acute non-cardiac disease: a prospective study. Intens Care Med. 2000;26:31-37.
19 Relos RP, Hasinof IK, Beilman GJ. Moderately elevated serum troponin concentrations are associated with increased morbidity and mortality rates in surgical intensive care unit patients. Crit Care Med. 2003;31:2598-2603.
20 Leach RM, Treacher DF. Oxygen delivery and consumption in the critically ill. Thorax. 2002;57:170-177.
21 Shoemaker WC, Appel PL, Waxman K, et al. Clinical trial of survivors’ cardiorespiratory patterns as therapeutic goals in critically ill postoperative patients. Crit Care Med. 1982;10:398-403.
22 Corday E, Williams JH, DeVera LB, et al. Effect of systemic blood pressure and vasopressor drugs on coronary blood flow and the electrocardiogram. Am J Cardiol. 1959;3:626.
23 Parillo JE. Pathogenetic mechanisms in septic shock. N Engl J Med. 1993;328:1471-1477.
24 Kumar A, Thota V, Dee L, et al. Tumour necrosis factor-α and interleukin 1β are responsible for in vitro myocardial cell depression induced by human septic shock serum. J Exp Med. 1996;183:949-958.
25 Rivers E, Bguyen B, Havstad S, et al. Early goal directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med. 2001;345:1368.
26 Sarnov SJ, Bergland E. Ventricular function 1. Starling’s law of the heart studied by means of simultaneous right and left ventricular function curves. Circulation. 1954;9:706.
27 Bradley RD, Jenkins BS, Branthwaite MA. The influence of atrial pressure on cardiac performance following myocardial infarction complicated by shock. Circulation. 1970;42:827-837.
28 Vismara LA, Leamon DM, Zelis R. The effects of morphine on venous tone in patients with acute pulmonary oedema. Circulation. 1976;54:335-337.
29 Ross JJ. Afterload mismatch and preload reserve: a conceptual framework for the analysis of ventricular function. Prog Cardiovasc Dis. 1976;18:255-264.
30 Cournand A, Motley HL, Werko L, et al. Physiological studies of the effects of intermittent positive pressure breathing on cardiac output in man. Am J Physiol. 1948;152:162.
31 Buda AJ, Pinsky MR, Ingels NB, et al. Effect of intrathoracic pressure on left ventricular performance. N Engl J Med. 1979;301:453-459.
32 Stewart JT, Simpson IA. Left ventricular energetics: heat production by the human heart. Cardiovasc Res. 1993;27:1024-1032.
33 Francis GS. Vasodilators in the intensive care unit. Am Heart J. 1991;121:1875-1878.
34 Mueller H, Ayers SM, Gianelli S, et al. Effect of isoproterenol, L-norepinephrine and intra-aortic counterpulsation on haemodynamics and myocardial metabolism in shock following acute myocardial infarction. Circulation. 1972;45:335.
35 Chamberlain DA, Leinbach RC, Vassaux CE, et al. Sequential atrioventricular pacing in heart block complicating acute myocardial infarction. N Engl J Med. 1970;282:577-582.
36 Smith TW. Digoxin in heart failure. N Engl J Med. 1993;329:51-53.
37 Amiodarone Trials Meta-Analysis Investigators. Effect of prophylactic amiodarone on mortality after acute myocardial infarction and in congestive heart failure: meta-analysis of individual data from 6500 patients in randomised trials. Lancet. 1997;350:1417-1424.
38 Goodwin J. The importance of clinical skills. Br Med J. 1995;310:1281-1282.
39 Singer M, Clarke J, Bennett ED. Continuous haemodynamic monitoring by oesophageal Doppler. Crit Care Med. 1989;17:447-452.
40 Linton RAF, Band DM, O’Brien T, et al. Lithium dilution cardiac output measurement: a comparison with thermodilution. Crit Care Med. 1997;25:1796-1800.
41 Antonelli M, Levy M, Andrews PJD, et al. Haemodynamic monitoring in shock and implications for management. International Consensus Conference, Paris, France, April 2006. Intens Care Med. 2007;33:575-590.
42 Steingrub JS, Celori G, Vickers-Lahti M, et al. Therapeutic impact of pulmonary artery catheterisation in a medical/surgical ICU. Chest. 1991;99:1451-1455.
43 Connors AF, Speroff T, Dawson NV, et al. The effectiveness of right heart catheterisation in the initial care of critically ill patients. JAMA. 1996;276:889-897.
44 Iberti TJ, Fischer EP, Leibowitz AB, et al. A multi-centre study of physicians’ knowledge of the pulmonary artery catheter. JAMA. 1990;264:2928-2932.
45 Sharpey-Schafer EP. Effects of Valsalva’s manoeuvre on the normal and failing circulation. Br Med J. 1955;1:693-695.
46 McIntyre KM, Vita JA, Lambrew CT, et al. A non-invasive method of predicting pulmonary capillary wedge pressure. N Engl J Med. 1992;327:1715-1720.
47 Marik PE. The systolic blood pressure variation as an indicator of pulmonary capillary wedge pressure in ventilated patients. Anaesth Intens Care. 1993;21:405-408.
48 Ritter JM, Doctor H, Benjamin M. Paradoxical effect of bicarbonate on cytoplasmic pH. Lancet. 1990;335:1243-1246.
49 Narius RG, Cohen JJ. Bicarbonate therapy for organic acidosis: the case for its use. Ann Intern Med. 1987;106:615-618.
50 Hilton PJ, Taylor J, Formi LG, et al. Bicarbonate-based haemofiltration in the management of acute renal failure with lactic acidosis. Q J Med. 1998;91:279-283.
51 Hebert PC, Wells G, Blajchman MA, et al. A multicenter, randomized, controlled clinical trial of transfusion requirements in critical care. N Engl J Med. 1999;340:409-417.
52 Leslie D, Gheorghiade M. Is there a role for thiamine supplementation in the management of heart failure? Am Heart J. 1996;131:1248-1250.
53 Day NPJ, Phu NH, Bethell DP, et al. The effects of dopamine and adrenaline infusions on acid–base balance and systemic haemodynamics in severe infection. Lancet. 1996;348:219-223.
54 Australian and New Zealand Intensive Care Society (ANZICS) Clinical Trials Group. Low-dose dopamine in patients with early renal dysfunction: a placebo-controlled randomised trial. Lancet. 2000;356:2139-2143.
55 Wilson J, Woods I, Fawcett J, et al. Reducing the risk of major elective surgery: randomized, controlled trial of preoperative optimisation of oxygen delivery. Br Med J. 1999;318:1099-1103.
56 Hasenfuss G, Pieske B, Castell M, et al. Influence of the novel inotropic agent levosimendan on isometric tension and calcium coupling in failing human myocardium. Circulation. 1998;98:2141-2147.
57 Gottlieb SS, McCarter RJ, Vogel RA, et al. Effect of beta blockade on mortality among high risk and low risk patients after myocardial infarction. N Engl J Med. 1998;339:489-497.
58 Mangano DT, Layug EL, Wallace A, et al. Effect of atenolol on mortality and cardiovascular morbidity after noncardiac surgery. N Engl J Med. 1996;335:1713-1720.
59 Bersten AD, Holt AW, Vedig AE, et al. Treatment of severe cardiogenic pulmonary oedema with continuous positive airway pressure delivered by facemask. N Engl J Med. 1991;325:1825-1830.
60 Aubier M, Trippenbach T, Roussos C. Respiratory muscle fatigue during cardiogenic shock. J Appl Physiol Respir Environ Ex Physiol. 1981;51:499-508.
61 Nanas JN, Moulopouloss D. Counterpulsation: historical background, technical improvements, hemodynamic and metabolic effects. Cardiology. 1994;84:156-167.
62 Westaby S, Katsumata T, Houel R, et al. Jarvik 2000 heart: potential for bridge to myocyte recovery. Circulation. 1998;98:1568-1574.
63 Pfeffer MA, Braunwald E, Moye LA, et al. Effect of captopril on mortality and morbidity in patients with left ventricular dysfunction after myocardial infarction. Results of the survival and ventricular enlargement trial. N Engl J Med. 1992;327:669-677.
64 Pitt B, Remme W, Zannard F, et al. Epleronone, a selective aldosterone blocker in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med. 2003;348:1309-1321.
65 Mueller HS. Role of intra-aortic counterpulsation in cardiogenic shock and acute myocardial infarction. Cardiology. 1994;84:168-174.
66 Kohn JN, Guiha NH, Broder MI, et al. Right ventricular infarction: clinical and haemodynamic features. Am J Cardiol. 1974;33:209-214.
67 Hasdai D, Topol EJ, Califf RM, et al. Cardiogenic shock complicating acute coronary syndromes. Lancet. 2000;356:749-756.