[level-membership-for-emergency-medicine-category]
55 Acute Coronary Syndrome
 Key Points
Key Points• Acute coronary syndrome (ACS) occurs as a spectrum of diseases that includes unstable angina pectoris, non–ST-segment elevation myocardial infarction, and ST-segment elevation myocardial infarction.
• ACS is classically manifested as chest tightness or pressure with associated dyspnea, nausea, and diaphoresis.
• ACS is diagnosed through a careful history and analysis of the 12-lead electrocardiogram.
• Treatment of the spectrum of ACS involves oxygen, aspirin, beta-blockers, nitrates, and anticoagulants.
• Patients with evidence of myocardial infarction also benefit from clopidogrel and glycoprotein IIb/IIIa receptor inhibitors.
• Patients with ST-segment elevation myocardial infarction require early revascularization therapy with either fibrinolysis or primary percutaneous coronary intervention.
• Immediate complications of ACS include congestive heart failure, cardiogenic shock, and rhythm disturbances, both tachyarrhythmias and bradyarrhythmias.
Epidemiology
Nonetheless, the burden of ACS remains significant both from a health care perspective and from an economic perspective. More than 1 million acute MIs occur in the United States annually, and 20% of affected patients die before reaching the hospital, primarily from arrhythmias during the first hours of symptoms.1 Many survivors of acute MI are left with impaired cardiac function, which adversely affects their ability to perform activities of daily living and their quality of life. Approximately 6 million emergency department (ED) visits in the United States are made annually for the evaluation of chest pain, and as many as one in three of these patients are ultimately found to have ACS.2 The annual cost of providing care for patients with ACS, both immediately and then later for those who survive, is more than $100 billion.3 Finally, despite advances in diagnostic techniques, 2% to 5% of patients with acute MI are discharged from the ED because their disease is not identified.4 These “missed MI” patients represent the highest mean payments for emergency medicine–related medical malpractice claims.
Definitions
Myocardial infarction is defined as myocardial necrosis. Clinical criteria for the presence of an acute, evolving, or recent MI, which have been laid out jointly by the American College of Cardiology and the European Society of Cardiology, focus on any evidence of myocardial cell death. The exact definition of an acute or evolving MI is a rise above the upper limit of normal and subsequent fall in levels of cardiac biomarkers specific for myocardial necrosis (troponin or the MB fraction of creatine kinase MB [CK-MB]) along with at least one of the following5:
• Symptoms consistent with ACS
• ECG evidence of myocardial ischemia, specifically, ST-segment elevation or depression or T-wave inversions
Myocardial infarction is further classified as STEMI and non–ST-elevation MI (NSTEMI). STEMI is present when the patient has (1) cardiac biomarkers for necrosis as previously defined and (2) new or presumed new ST-segment elevation in two or more contiguous ECG leads. The cutoff point for ST-segment elevation is 0.1 mV.6 Contiguous leads are defined in the chest leads as V1 through V6 and in the frontal plane as the sequence aVL, I, inverted aVR, II, aVF, and III. Patients who meet the clinical criteria for STEMI and left bundle branch block (LBBB) and are not old or who have ECG evidence of an isolated true posterior MI are also considered, for treatment algorithm purposes, to have STEMI. NSTEMI is present when the patient meets the criteria for MI as previously defined but exhibits no evidence of ST-segment elevation, new LBBB, or ECG evidence of an isolated posterior wall MI.
Differential Diagnosis and Medical Decision Making
The differential diagnosis in patients with ACS includes a host of other diseases that can be manifested as chest pain or dyspnea: stable angina, pericarditis, myocarditis, pulmonary embolism (PE), aortic dissection, pneumonia, pleurisy, pneumothorax, Boerhaave syndrome, esophageal reflux, esophageal spasm, gastritis, biliary colic, pancreatitis, peptic ulcer disease, musculoskeletal pain, and herpes zoster. One of the historical features that tends to favor a diagnosis of ACS is chest pressure or tightness rather than a sharp pain, which is more commonly associated with pericarditis, pleurisy, pneumothorax, PE, and aortic dissection. In addition, the chest discomfort in patients with ACS tends to gradually worsen, unlike the pain associated with PE or aortic dissection, which is generally worst at the onset and then persistently severe. Pain of a pleuritic nature tends to favor PE, pleurisy, or pneumothorax, whereas pain that is worse on palpation tends to suggest a chest wall musculoskeletal cause. Discomfort that is positional in nature tends to favor pericarditis or gastrointestinal causes rather than ACS. However, it is very important to remember that a significant percentage of patients with ACS have pleuritic, positional, or palpable chest pain and that these historical features cannot be used to exclude the diagnosis.7
Diagnostic Testing
Electrocardiogram
Electrocardiographic Findings in ST-Segment Elevation Myocardial Infarction
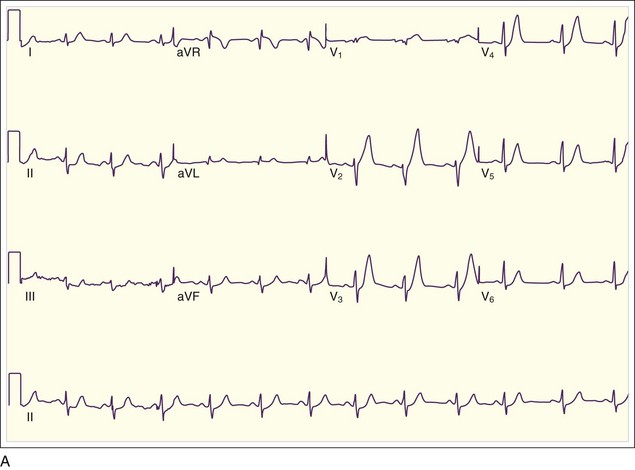
The initial ECG abnormality that occurs in patients with epicardial coronary artery occlusion is peaked hyperacute T waves in the distribution supplied by the IRA. T waves become tall and sharply peaked within minutes of occlusion of the IRA (Fig. 55.1, A). Peaked T waves may also be seen in patients with hyperkalemia, pericarditis, early repolarization, and LBBB. In the next several minutes, ST-segment elevation becomes evident on the ECG (see Fig. 55.1, B). To be diagnostic, the ST-segment elevation must be at least 1 mm above the baseline; this is generally considered the TP segment. Most typically, this ST elevation is convex or domed, though less commonly it may be straight or, rarely, concave. Concave ST-segment elevations are more characteristic of other conditions associated with ST-segment elevation (Box 55.1).
Box 55.1 Differential Diagnosis of ST-Segment Elevation on Electrocardiography
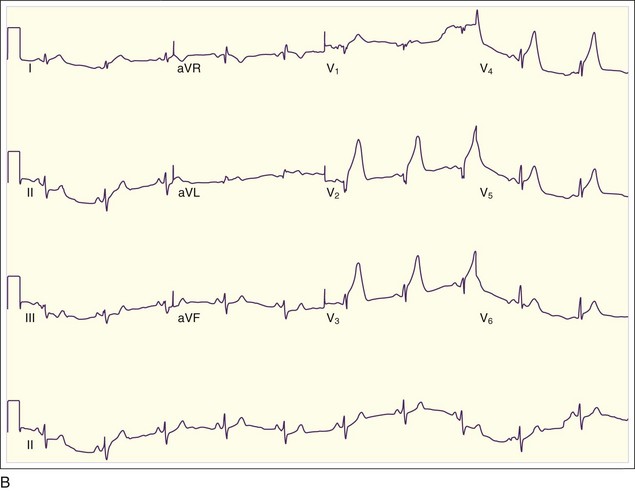
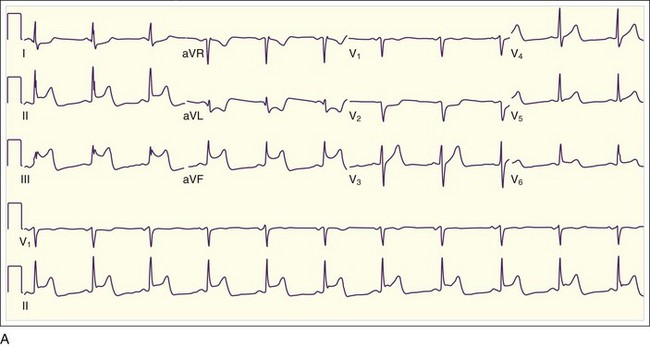
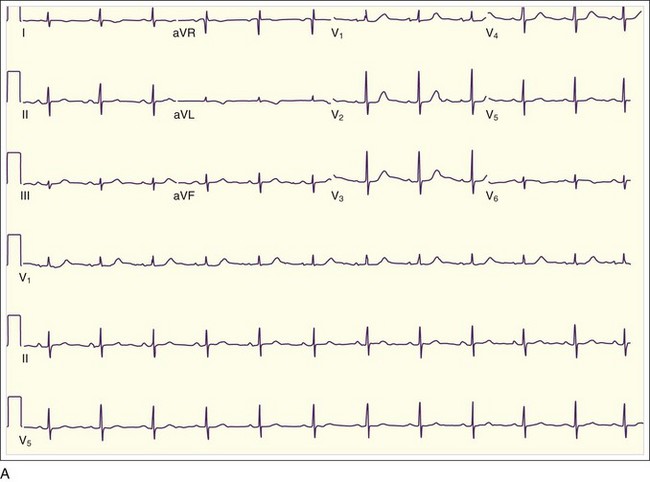
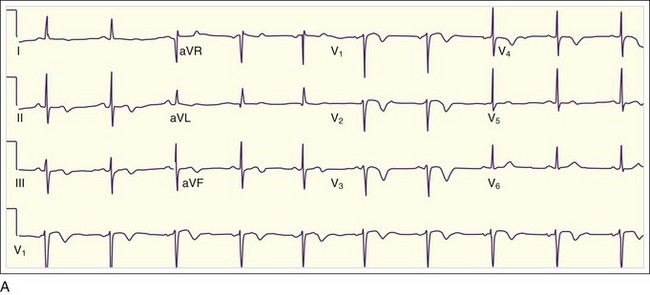
In addition to the clinical situation, a factor distinguishing STEMI from other conditions is the dynamic nature of the ST-segment changes with STEMI; serial ECGs commonly show waxing and waning ST-segment elevation. Hours to days later, the ST segments return toward baseline, the T waves invert, and pathologic Q waves develop in areas of the ECG that correspond to the IRA. The location of the ST elevations and other findings on the ECG generally correspond to the anatomic location of the myocardium and the associated IRA. Anterior infarctions exhibit ST elevation in leads V1 through V4 (Fig. 55.2). Findings in leads V1 and V2 indicate involvement of the septum. MIs with these findings are caused by occlusion of the left anterior descending (LAD) coronary artery. When additional ST elevations are seen in leads V5, V6, I, and aVL, the location of the LAD occlusion is probably proximal to the first diagonal branch, which causes an anterolateral infarction (see Fig. 55.1, B). Inferior infarctions are characterized by ST elevations in leads II, III, and aVF (Fig. 55.3, A) and are due most commonly to right coronary artery (RCA) occlusion. Reciprocal ST depressions may be present in leads I and aVL.
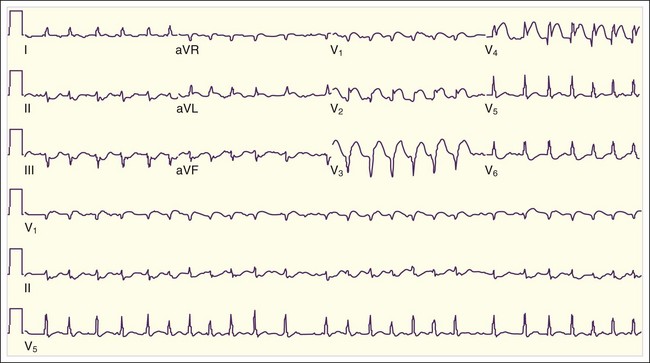
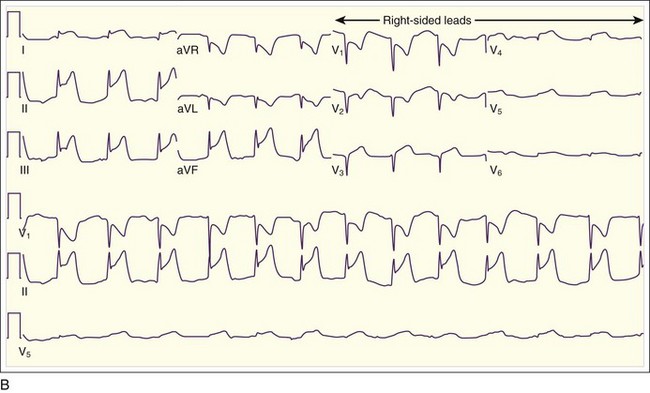
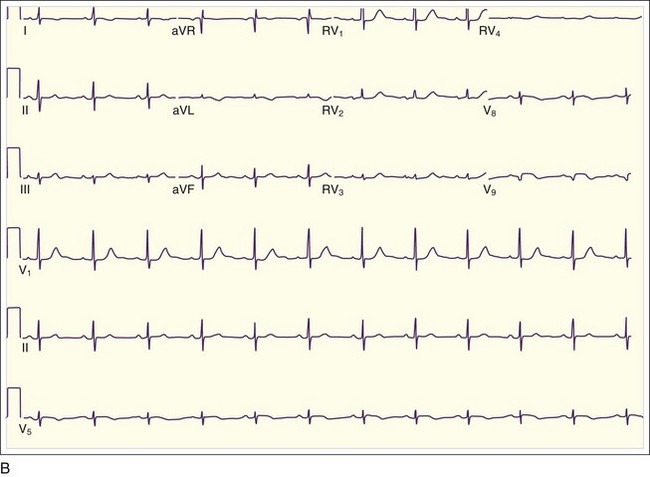
Inferior MIs are associated with concomitant right ventricular infarction, which can be evident on right-sided ECG leads, particularly in RV4 and RV5 (see Fig. 55.3, B). Inferior MIs are also frequently associated with posterior wall involvement, which is seen on the ECG as ST depressions in leads V1 through V3 and, on occasion, early R-wave progression with tall R waves in leads V1 through V3 (Fig. 55.4).
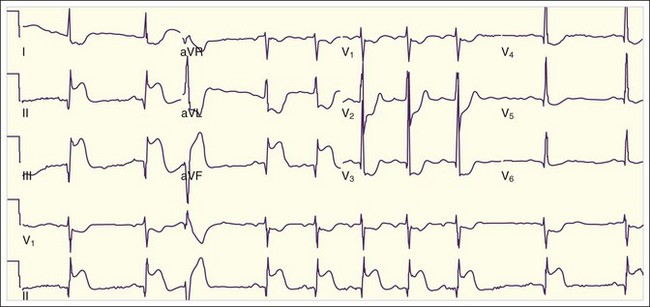
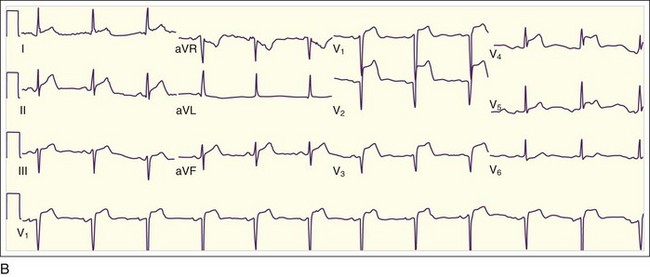
Isolated posterior MIs, the rarest of transmural MIs, are the most easily misdiagnosed because the 12-lead ECG may show ST depressions in V1 through V3 and sometimes V4 and V5, often with tall R waves in V1 through V3 but without evidence of ST elevations (Fig. 55.5). This situation can be confused with anterior wall ischemia. Clues on the ECG to the diagnosis of isolated posterior wall MI include horizontal (rather than sloping) ST depressions with prominent R waves and tall upright T waves in leads V1 through V3. Occasionally, isolated posterior wall MIs are manifested as a nondiagnostic 12-lead ECG (Fig. 55.6, A), with small pathognomonic ST-segment elevations evident only when extended ECG leads are placed inferior to the tip of the left scapula (V8) and in the left paraspinal line at the same level (V9) (see Fig. 55.6, B). Posterior wall MIs are the result of occlusion of the posterior descending coronary artery or the posterior left ventricular branch, either of which can arise from the RCA (more commonly) or the left circumflex coronary artery.
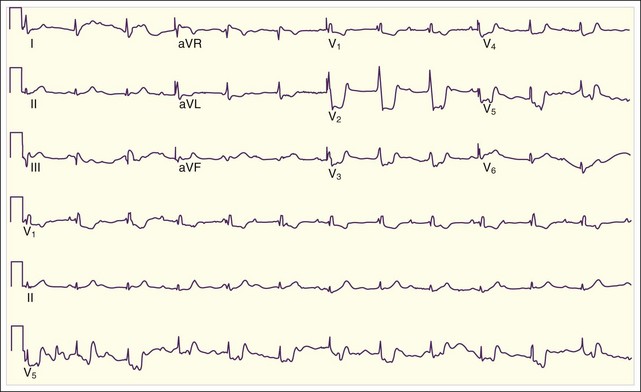
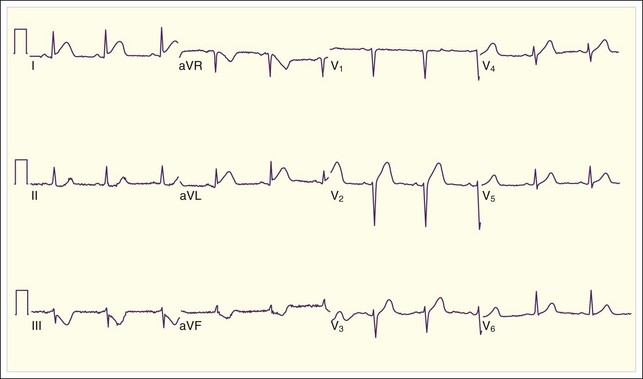
Lateral wall MIs, characterized by ST-segment elevation in some or all of leads I, aVL, V5, and V6, may be associated with anterior MI as previously described or with inferior or posterior MI or may occur in isolation. This is because the lateral wall of the heart is variably supplied with blood by the LAD, the RCA, and the left circumflex artery. Isolated lateral wall MIs are most commonly associated with left circumflex artery occlusion; the ECG may show reciprocal ST depressions in leads II, III, and aVF (Fig. 55.7).
Electrocardiographic Findings in Non–ST-Segment Elevation Acute Coronary Syndrome
In the clinical setting of NSTEMI, the ECG may be normal or unchanged from baseline, although more commonly it will show ST-segment depressions, T-wave abnormalities, or both in the area of the ECG representing the area of ischemia or infarction in the heart (Fig. 55.8). As mentioned, ST-segment depressions in the precordial leads may also represent true posterior wall transmural infarction. In addition, ST depressions may also represent reciprocal changes, with STEMI occurring in another location; this is most commonly seen in the lateral leads in patients with an inferior STEMI or in the inferior leads in patients with a lateral STEMI (see Fig. 55.7). T-wave inversions are nonspecific findings, particularly when seen in isolation (without ST-segment depressions), but they do suggest ACS in the right clinical setting, especially when comparison with previous tracings shows that the findings are new. Note that T waves are normally inverted in leads aVR and V1 and are variably inverted in leads III, aVF, aVL, and V2.
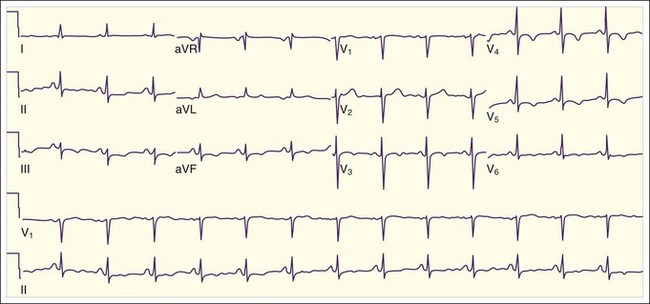
One important subgroup of T-wave inversions occurs in the precordial leads. The changes may be symmetric deep T-wave inversions (Fig. 55.9, A) or more subtle biphasic T-wave changes. This pattern, referred to as Wellen syndrome, represents an unstable lesion in the LAD. Without prompt appropriate treatment, this lesion may lead to an anterior STEMI (see Fig. 55.9, B). The differential diagnosis of inverted T waves includes not only ACS but also left ventricular hypertrophy, LBBB, pericarditis, myocarditis, pulmonary embolism, Wolfe-Parkinson-White syndrome, ischemic or hemorrhagic stroke, hypokalemia, and a persistent juvenile pattern. These findings may also be normal variants. Occasionally, patients with chronically inverted T waves are found to have new upright T waves in the setting of chest pain or anginal equivalent. This finding, referred to as pseudonormalization of the T waves, is highly suggestive of ACS.
Cardiac Biomarkers
Numerous cardiac biomarkers become elevated in the setting of myocardial cell death and are thus indicators of MI. The most sensitive and specific of these biomarkers at present are troponins, which are detectable in serum 4 to 10 hours after the onset of MI. Consequently, a single “negative” troponin value cannot be used to exclude MI. In addition to troponins, CK-MB and myoglobins are also useful and widely used. However, none of the cardiac biomarker measurements currently available represent an adequate test for unstable angina (without MI). For a complete discussion of cardiac biomarkers, see Chapter 54.
Other Tests
Cardiac ultrasonography, nuclear imaging, and stress testing can be very important in confirming the diagnosis of ACS or in suggesting an alternative cause of the symptoms. Most recently, contrast-enhanced multidetector computed tomography of the coronary arteries has been shown to have a role in the evaluation of patients with chest pain. These tests are discussed in detail in Chapter 56.
Treatment
Prehospital Management
Hospital Management
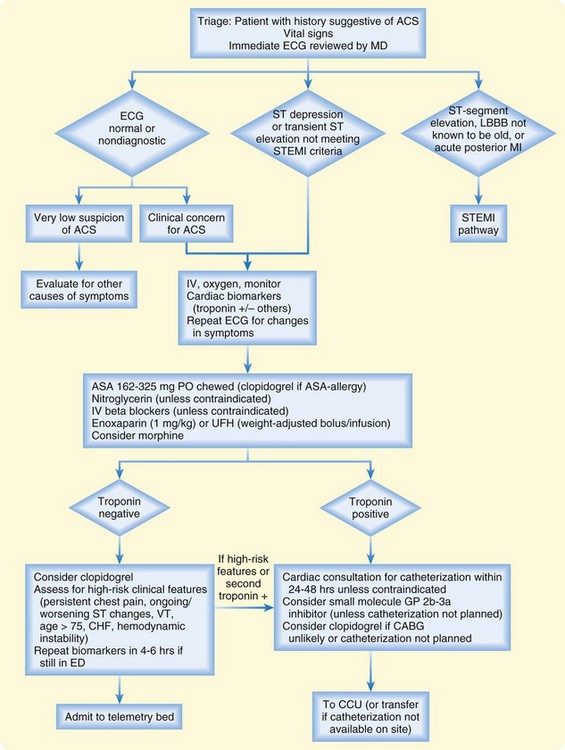
Figures 55.10 and 55.11 present treatment algorithms for STEMI and non–ST-segment acute coronary symptoms.
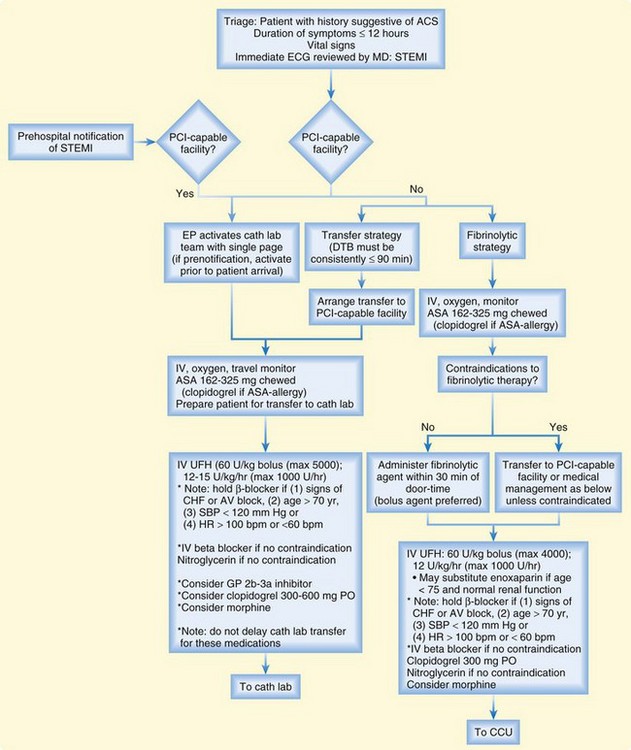
Fig. 55.10 Assessment and treatment algorithm for ST-segment elevation myocardial infarction (STEMI).
Platelet Inhibitors
Aspirin remains the cornerstone of therapy across the spectrum of ACS. It is highly cost-effective and remains one of a few drugs with a mortality benefit in patients with ACS. In patients with STEMI, aspirin independently reduces mortality by approximately 23%.8 Aspirin is an antiplatelet agent that irreversibly inactivates platelet cyclooxygenase and also reduces the formation of prostacyclin by endothelial cells. It should be administered in the ED orally (chewed and swallowed) or rectally; the standard dose is 162 to 325 mg. The EP should take care to avoid using enteric-coated preparations in the acute treatment of ACS. The only true contraindication to aspirin in patients with ACS is a history of severe allergic reaction.
Clopidogrel is one of several currently available thienopyridines, a class of drugs that inhibits adenosine diphosphate–mediated platelet aggregation. These drugs are more potent platelet inhibitors than aspirin is. Ticlopidine, another drug in this class, is not generally used because of its slow onset of action and concerns about its adverse effects, which include neutropenia and, rarely, agranulocytosis. Prasugrel, a more potent thienopyridine recently approved for use in patients with ACS, may play an increasing important role in the future, but at present clopidogrel remains the most commonly used drug in this class. Clopidogrel has been shown to improve clinical outcomes in patients with non–ST-segment elevation ACS, particularly in those who undergo PCI.9,10 Clinical benefit is demonstrable within 24 hours of dosing, but it has been associated with a small increase in bleeding in those who undergo coronary artery bypass grafting (CABG) within 5 days of discontinuation of clopidogrel.9 The traditional loading dose of clopidogrel has been 300 mg orally, but 600 mg appears to be equally safe and may be more efficacious. Clopidogrel (300 to 600 mg orally) should be administered to all patients with ACS and documented aspirin allergy, as well as to those in whom a noninterventional approach is planned.11 Clopidogrel should also be administered to patients with non–ST-segment elevation ACS in whom emergency CABG is deemed unlikely11; this may best be determined in consultation with a cardiologist. For patients with STEMI, clopidogrel, 300 mg, is indicated as an adjunct to fibrinolytic therapy.12 In patients with STEMI who will undergo PCI, it is reasonable to administer clopidogrel, 300 to 600 mg, in the ED, provided that transfer to the catheterization laboratory is not delayed as a result.
GP IIb/IIIa inhibitors have been shown to provide benefit to patients with ACS treated by PCI. Some data also show benefit in troponin-positive patients who test positive for troponins, but not in patients who test negative or who are managed medically. Consequently, current recommendations for the use of this class of drugs are as follows11: GP IIb/IIIa inhibitors should be administered, along with aspirin and a heparin preparation, to patients with ACS in whom PCI is planned, even when clopidogrel is also given. However, the best available evidence suggests that no benefit is seen when GP IIb/IIIa inhibitors are administered in the ED versus delayed provisional use in the cardiac catheterization laboratory.13,14 Furthermore, delaying this decision to the time of catheterization may reduce the likelihood of medication administration error, as well as the incidence of adverse effects and overall cost. With this in mind, the decision to use these drugs in the ED setting should be made with caution and limited to patients who have a compelling history of ACS, test positive for troponins, have no contraindication to cardiac catheterization, and are expected to have a delay in prompt interventional treatment.
Beta-Blocking Agents
Beta-blockers are an important first-line therapy for patients with ACS. These agents act by reducing the effects of catecholamines on the heart—they slow the heart rate, reduce myocardial contractility, and thereby lower myocardial demand for oxygen. They are also potent antiarrhythmic agents and lessen the likelihood of ventricular and atrial tachyarrhythmias. However, they have also been shown, when given intravenously to patients with STEMI, to increase risk for the development of cardiogenic shock, which counterbalances the salutary effects of beta-blockers and results in no overall mortality benefit.15 Thus beta-blockers should be administered intravenously with caution in the ED setting to patients with STEMI and specifically withheld, per the most recent American College of Cardiology and American Heart Association guidelines for STEMI, in the following settings16:
Anticoagulants
UFH has a synergistic salutary effect on ischemic outcomes when combined with aspirin in patients with ACS, particularly those with MI. Several LMWH preparations have shown efficacy in patients with ACS, but only enoxaparin has demonstrated an improvement over UFH. Therefore, enoxaparin is the LMWH of choice for the treatment of ACS. Current guidelines recommend the administration of UFH or enoxaparin to patients with ACS in conjunction with antiplatelet therapy.11 For non–ST-segment elevation ACS, enoxaparin (1 mg/kg given subcutaneously twice daily) is the preferred agent unless urgent CABG is planned.11 UFH is dosed as an intravenous bolus of 60 U/kg (maximum, 4000 units), followed by an infusion at 12 U/kg/hr (maximum, 1000 U/hr).6,11 UFH use must be monitored by serial prothrombin time determinations; such monitoring is not necessary in patients treated with enoxaparin. Either drug should be discontinued immediately in patients with evidence of bleeding or if thrombocytopenia develops.
Fondaparinux is a relative newcomer to the class of anticoagulant drugs. It is a pentasaccharide molecule that represents the terminal five saccharide moieties of heparin. Principally a factor Xa inhibitor, this agent is administered as a subcutaneous injection, with dose reductions required in patients with renal insufficiency. Fondaparinux already has indications for the treatment of venous thromboembolic disease, and data suggest that it is similar to enoxaparin in terms of safety and efficacy for the treatment of non–ST-segment elevation ACS.17 Current published guidelines recommend fondaparinux as an acceptable alternative to UFH or enoxaparin in patients with non–ST-segment elevation ACS.11
Revascularization Therapy
Patients with STEMI who arrive at the ED within 12 to 24 hours of the onset of symptoms require urgent revascularization therapy. It can be accomplished mechanically with primary PCI or pharmacologically with fibrinolytic therapy. Although fibrinolytic therapy remains the most common strategy worldwide, use of primary PCI for STEMI has been growing rapidly in the United States and has been deemed a preferable approach in terms of safety and efficacy. If a primary PCI strategy is chosen, the IRA must be opened within 90 minutes of patient arrival at the health care system to achieve maximal efficacy.5 This interval includes time spent at the initial hospital if transfer to a PCI-capable facility is necessary. If the “door-to-balloon” target time of 90 minutes cannot be routinely achieved, a fibrinolytic strategy is preferable, particularly for patients who are seen early (within 3 hours of symptom onset).5
For patients in cardiogenic shock or those with contraindications to fibrinolytic therapy, primary PCI should be performed as soon as possible. In addition, patients in whom fibrinolytic therapy fails, as evidenced by ongoing anginal symptoms and ST-segment elevations continuing an hour or more after therapy, should be referred for rescue PCI, which should be performed as soon as possible. Patients with STEMI who undergo primary PCI should also receive aspirin, clopidogrel, and UFH. It is reasonable to administer a GP IIb/IIIa inhibitor to these patients as well, and abciximab is the preferred agent in this setting. However, current evidence suggests that this decision can be safely deferred to the time of catheterization and PCI,13 which may allow the drug to be given more safely.
It is important to emphasize that none of these adjunctive therapies should delay transfer of the patient from the ED to the cardiac catheterization laboratory, which is the first priority. A number of validated strategies should be used to decrease door-to-balloon time.18 Those that specifically affect the ED are (1) empowering the EP to activate the entire cardiac catheterization laboratory team with a single phone call; (2) increasing, when possible, the capacity to obtain prehospital ECG tracings in patients with chest pain and activation of the catheterization laboratory team while the patient is still en route to the hospital; and (3) providing prompt feedback from a multidisciplinary quality improvement team to all clinical providers involved in care of the patient.18
Fibrinolytic therapy remains an important treatment option for patients with STEMI, particularly those who go to community hospitals that do not have the capability of performing PCI. Rapid initiation of treatment is the standard of care, with a target goal “door-to-needle” time of less than 30 minutes. Fibrinolytic therapy is indicated for patients who have symptoms consistent with ACS within 12 hours of symptom onset, meet ECG criteria (Box 55.2), and do not have an absolute contraindication to the therapy (Box 55.3). The presence of relative contraindications (Box 55.4) must be weighed against the risk of treatment delay if primary PCI is not readily available. Advanced age alone is not a contraindication, and although elderly patients treated with fibrinolytic therapy do have a higher incidence of hemorrhage, they also have a significantly higher mortality rate, which can be mitigated with therapy.
Box 55.3 Absolute Contraindications to Fibrinolytic Therapy
Previous history of intracranial hemorrhage
Known malignant intracranial neoplasm
Known cerebrovascular lesion (e.g., arteriovenous malformation)
Active bleeding (excluding menses) or known bleeding diathesis
Significant closed-head or facial trauma within the previous 3 months
Ischemic stroke within the previous 3 months (except if within 3 hours)
Box 55.4 Relative Contraindications to Fibrinolytic Therapy
History of chronic, severe, poorly controlled hypertension
Uncontrolled hypertension at initial evaluation (systolic blood pressure higher than 180 mm Hg or diastolic blood pressure higher than 110 mm Hg)
Previous ischemic stroke more than 3 months earlier
Traumatic or prolonged (>10 minutes) cardiopulmonary resuscitation or major surgery within less than 3 weeks
Recent (within 2 to 4 weeks) internal bleeding
All patients with STEMI treated with fibrinolytic therapy should also receive aspirin and clopidogrel. Beta-blockers should be given with caution and perhaps should be limited in the ED setting to oral use in appropriate patients as discussed earlier. If a fibrin-specific agent is administered, UFH should be given in a bolus dose of 60 U/kg (maximum, 4000 units) and as an infusion of 12 U/kg/hr (maximum, 1000 U/hr). Enoxaparin can be safely and effectively substituted for UFH in patients with normal renal function who are younger than 75 years.19 If streptokinase is administered, either heparin preparation should be withheld.
Combination pharmacologic treatment of STEMI has attracted considerable interest. The most promising combination has been half-dose fibrinolytic therapy coupled with a GP IIb/IIIa inhibitor, which has been demonstrated to provide better angiographic outcomes in the IRA at 90 minutes. However, large-scale clinical trials have failed to show a mortality benefit of this combination, although they have suggested that it achieves reductions in the risk for recurrent MI and the need for rescue angioplasty.20 At present, combination therapy, because it is more expensive and cumbersome to administer, should be considered for use only in facilities whose remote locations make transfer of a patient for rescue PCI very difficult.
 Priority Actions
Priority Actions
1. Immediate 12-lead electrocardiogram for all patients with chest pain or anginal equivalent
2. Administration of aspirin to patients with acute coronary syndrome, the only contraindication being a true aspirin allergy
3. Prompt revascularization for patients with ST-segment elevation myocardial infarction (“door-to-needle” time of less than 30 minutes or “door-to-balloon” time of less than 90 minutes)
4. Early recognition and treatment of electrical and mechanical complications of acute coronary syndrome
5. Admission to a telemetry unit or coronary care unit for all patients with acute coronary syndrome
Follow-Up and Next Steps in Care
 Patient Teaching Tips
Patient Teaching Tips
Recognize chest pain and anginal equivalents as an indicator of potential acute coronary syndrome.
Call 911 for new-onset symptoms or those that do not resolve promptly with cessation of exertion or the use of sublingual nitroglycerin.
Do not ascribe symptoms to gastrointestinal or other noncardiac causes.
Diet, exercise, and smoking cessation should be encouraged for all patients.
For patients with a history of acute coronary syndrome, carrying an accurate medication list and a copy of the baseline electrocardiogram can be useful.
 Red Flags
Red Flags
Many patients with acute coronary syndrome, particularly the elderly, have atypical manifestations.
Normal electrocardiographic findings do not exclude acute coronary syndrome.
A single set of “negative” cardiac biomarkers does not exclude acute myocardial infarction.
Recognize that patients with ST-segment elevation myocardial infarction who are transferred for primary percutaneous coronary intervention may not be revascularized within the recommended treatment window; consider fibrinolysis for these patients instead.
Avoid nitrates and intravenous beta-blockers in situations in which their use is contraindicated.
Anderson JL, Adams CD, Antman EM, et al. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non ST-elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines for the Management of Patients With Unstable Angina/Non ST-Elevation Myocardial Infarction). Circulation. 2007;116:e148–e304.
Antman EM, Hand M, Armstrong PW, et al. 2007 Focused update of the ACC/AHA 2004 Guidelines for the Management of Patients With ST-Elevation Myocardial Infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines: developed in collaboration with the Canadian Cardiovascular Society endorsed by the American Academy of Family Physicians: 2007 Writing Group to Review New Evidence and Update the ACC/AHA 2004 Guidelines for the Management of Patients With ST-Elevation Myocardial Infarction, Writing on Behalf of the 2004 Writing Committee. Circulation.. 2008;117:296–329.
Bradley E, Herrin H, Wang Y, et al. Strategies to reduce the door-to-balloon time in acute myocardial infarction. N Engl J Med. 2006;355:2308–2320.
Chen ZM, Pan HC, Chen YP, et al. Early intravenous then oral metoprolol in 45,852 patients with acute myocardial infarction: randomised placebo-controlled trial. Lancet. 2005;366:1622–1632.
1 Spertus JA, Radford MJ, Every NR, et al. Challenges and opportunities in quantifying the quality of care for acute myocardial infarction: summary from the Acute Myocardial Infarction Working Group of the American Heart Association/American College of Cardiology First Scientific Forum on Quality of Care and Outcomes Research in Cardiovascular Disease and Stroke. Circulation. 2003;107:1681–1691.
2 McCaig LF, Nawar EW. National hospital ambulatory medical care survey: 2004 emergency department summary. Adv Data. 2006;372:1–29.
3 Weinstein MD, Stason WB. Cost-effectiveness of interventions to prevent or treat coronary heart disease. Annu Rev Public Health. 1985;6:41–63.
4 Storrow AB, Gibler WB. Chest pain centers: diagnosis of acute coronary syndrome. Ann Emerg Med. 2000;35:449–461.
5 Alpert JS, Thygesen K, Antman E, et al. Myocardial infarction redefined—a consensus document of the joint European Society of Cardiology/American College of Cardiology Committee for the redefinition of myocardial infarction. J Am Coll Cardiol. 2000;36:959–969.
6 Antman EM, Anbe DT, Armstrong PW, et al. for the American College of Cardiology; American Heart Association; Canadian Cardiovascular Society. ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction—executive summary. A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 1999 Guidelines for the Management of Patients With Acute Myocardial Infarction). J Am Coll Cardiol. 2004;44:671–719.
7 Lee TH, Cook EF, Weisberg M, et al. Acute chest pain in the emergency room: identification and examination of low-risk patients. Ann Intern Med. 1985;145:65–69.
8 Randomised trial of intravenous streptokinase, oral aspirin, both or neither among 17,187 cases of suspected acute myocardial infarction: ISIS-2. ISIS-2 (Second International Study of Infarct Survival) Collaborative Group. Lancet. 1988;2:349–360.
9 Yusuf S, Zhao F, Mehta SR, et al. Clopidogrel in Unstable Angina to Prevent Recurrent Events Trial Investigators: effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med. 2001;345:494–502.
10 Mehta SR, Yusuf S, Peters RJ, et al. Clopidogrel in Unstable Angina to Prevent Recurrent Events Trial (CURE) Investigators: effects of pretreatment with clopidogrel and aspirin followed by long-term therapy in patients undergoing percutaneous coronary intervention: the PCI-CURE study. Lancet. 2001;358:527–533.
11 Anderson JL, Adams CD, Antman EM, et al. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non ST-elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines for the Management of Patients With Unstable Angina/Non ST-Elevation Myocardial Infarction). Circulation. 2007;116:e148–e304.
12 Sabatine MS, Cannon CP, Gibson CM, et al. Clopidogrel as Adjunctive Reperfusion Therapy (CLARITY)–Thrombolysis in Myocardial Infarction (TIMI) 28 Investigators: addition of clopidogrel to aspirin and fibrinolytic therapy for myocardial infarction with ST-segment elevation. N Engl J Med. 2005;352:1179–1189.
13 Ellis SG, Tendera M, de Belder MA, et al. for the FINESSE Investigators. Facilitated PCI in patients with ST-elevation myocardial infarction. N Engl J Med. 2008;358:2205–2217.
14 Giugliano RP, White JA, Bode C, et al. for the EARLY ACS Investigators. Early versus delayed, provisional eptifibatide in acute coronary syndromes. N Engl J Med. 2009;360:2176–2190.
15 Chen ZM, Pan HC, Chen YP, et al. Early intravenous then oral metoprolol in 45,852 patients with acute myocardial infarction: randomised placebo-controlled trial. Lancet. 2005;366:1622–1632.
16 Antman EM, Hand M, Armstrong PW, et al. 2007 Focused update of the ACC/AHA 2004 Guidelines for the Management of Patients With ST-Elevation Myocardial Infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines: developed in collaboration With the Canadian Cardiovascular Society endorsed by the American Academy of Family Physicians: 2007 Writing Group to Review New Evidence and Update the ACC/AHA 2004 Guidelines for the Management of Patients With ST-Elevation Myocardial Infarction, Writing on Behalf of the 2004 Writing Committee. Circulation. 2008;117:296–329.
17 Yusuf S, Mehta SR, Chrolavicius S, et al. Fifth Organization to Assess Strategies in Acute Ischemic Syndromes Investigators: comparison of fondaparinux and enoxaparin in acute coronary syndromes. N Engl J Med. 2006;354:1464–1476.
18 Bradley E, Herrin H, Wang Y, et al. Strategies to reduce the door-to-balloon time in acute myocardial infarction. N Engl J Med. 2006;355:2308–2320.
19 Antman EM, Morrow DA, McCabe CH, et al. Enoxaparin versus unfractionated heparin with fibrinolysis for ST-elevation myocardial infarction. N Engl J Med. 2006;354:1477–1488.
20 Topol E. J, for the GUSTO V Investigators. Reperfusion therapy for acute myocardial infarction with fibrinolytic therapy or combination reduced fibrinolytic therapy and platelet glycoprotein IIb/IIIa inhibition: the GUSTO V randomised trial. Lancet. 2001;357:1905–1914.
[/level-membership-for-emergency-medicine-category][not-level-membership-for-emergency-medicine-category]
55 Acute Coronary Syndrome
• Acute coronary syndrome (ACS) occurs as a spectrum of diseases that includes unstable angina pectoris, non–ST-segment elevation myocardial infarction, and ST-segment elevation myocardial infarction.
• ACS is classically manifested as chest tightness or pressure with associated dyspnea, nausea, and diaphoresis.
• ACS is diagnosed through a careful history and analysis of the 12-lead electrocardiogram.
• Treatment of the spectrum of ACS involves oxygen, aspirin, beta-blockers, nitrates, and anticoagulants.
• Patients with evidence of myocardial infarction also benefit from clopidogrel and glycoprotein IIb/IIIa receptor inhibitors.
• Patients with ST-segment elevation myocardial infarction require early revascularization therapy with either fibrinolysis or primary percutaneous coronary intervention.
• Immediate complications of ACS include congestive heart failure, cardiogenic shock, and rhythm disturbances, both tachyarrhythmias and bradyarrhythmias.
Epidemiology
Nonetheless, the burden of ACS remains significant both from a health care perspective and from an economic perspective. More than 1 million acute MIs occur in the United States annually, and 20% of affected patients die before reaching the hospital, primarily from arrhythmias during the first hours of symptoms.1 Many survivors of acute MI are left with impaired cardiac function, which adversely affects their ability to perform activities of daily living and their quality of life. Approximately 6 million emergency department (ED) visits in the United States are made annually for the evaluation of chest pain, and as many as one in three of these patients are ultimately found to have ACS.2 The annual cost of providing care for patients with ACS, both immediately and then later for those who survive, is more than $100 billion.3 Finally, despite advances in diagnostic techniques, 2% to 5% of patients with acute MI are discharged from the ED because their disease is not identified.4 These “missed MI” patients represent the highest mean payments for emergency medicine–related medical malpractice claims.
Definitions
Myocardial infarction is defined as myocardial necrosis. Clinical criteria for the presence of an acute, evolving, or recent MI, which have been laid out jointly by the American College of Cardiology and the European Society of Cardiology, focus on any evidence of myocardial cell death. The exact definition of an acute or evolving MI is a rise above the upper limit of normal and subsequent fall in levels of cardiac biomarkers specific for myocardial necrosis (troponin or the MB fraction of creatine kinase MB [CK-MB]) along with at least one of the following5:
• Symptoms consistent with ACS
• ECG evidence of myocardial ischemia, specifically, ST-segment elevation or depression or T-wave inversions
Myocardial infarction is further classified as STEMI and non–ST-elevation MI (NSTEMI). STEMI is present when the patient has (1) cardiac biomarkers for necrosis as previously defined and (2) new or presumed new ST-segment elevation in two or more contiguous ECG leads. The cutoff point for ST-segment elevation is 0.1 mV.6 Contiguous leads are defined in the chest leads as V1 through V6 and in the frontal plane as the sequence aVL, I, inverted aVR, II, aVF, and III. Patients who meet the clinical criteria for STEMI and left bundle branch block (LBBB) and are not old or who have ECG evidence of an isolated true posterior MI are also considered, for treatment algorithm purposes, to have STEMI. NSTEMI is present when the patient meets the criteria for MI as previously defined but exhibits no evidence of ST-segment elevation, new LBBB, or ECG evidence of an isolated posterior wall MI.
Differential Diagnosis and Medical Decision Making
The differential diagnosis in patients with ACS includes a host of other diseases that can be manifested as chest pain or dyspnea: stable angina, pericarditis, myocarditis, pulmonary embolism (PE), aortic dissection, pneumonia, pleurisy, pneumothorax, Boerhaave syndrome, esophageal reflux, esophageal spasm, gastritis, biliary colic, pancreatitis, peptic ulcer disease, musculoskeletal pain, and herpes zoster. One of the historical features that tends to favor a diagnosis of ACS is chest pressure or tightness rather than a sharp pain, which is more commonly associated with pericarditis, pleurisy, pneumothorax, PE, and aortic dissection. In addition, the chest discomfort in patients with ACS tends to gradually worsen, unlike the pain associated with PE or aortic dissection, which is generally worst at the onset and then persistently severe. Pain of a pleuritic nature tends to favor PE, pleurisy, or pneumothorax, whereas pain that is worse on palpation tends to suggest a chest wall musculoskeletal cause. Discomfort that is positional in nature tends to favor pericarditis or gastrointestinal causes rather than ACS. However, it is very important to remember that a significant percentage of patients with ACS have pleuritic, positional, or palpable chest pain and that these historical features cannot be used to exclude the diagnosis.7
Diagnostic Testing
Electrocardiogram
Electrocardiographic Findings in ST-Segment Elevation Myocardial Infarction
The initial ECG abnormality that occurs in patients with epicardial coronary artery occlusion is peaked hyperacute T waves in the distribution supplied by the IRA. T waves become tall and sharply peaked within minutes of occlusion of the IRA (Fig. 55.1, A). Peaked T waves may also be seen in patients with hyperkalemia, pericarditis, early repolarization, and LBBB. In the next several minutes, ST-segment elevation becomes evident on the ECG (see Fig. 55.1, B). To be diagnostic, the ST-segment elevation must be at least 1 mm above the baseline; this is generally considered the TP segment. Most typically, this ST elevation is convex or domed, though less commonly it may be straight or, rarely, concave. Concave ST-segment elevations are more characteristic of other conditions associated with ST-segment elevation (Box 55.1).
Box 55.1 Differential Diagnosis of ST-Segment Elevation on Electrocardiography
In addition to the clinical situation, a factor distinguishing STEMI from other conditions is the dynamic nature of the ST-segment changes with STEMI; serial ECGs commonly show waxing and waning ST-segment elevation. Hours to days later, the ST segments return toward baseline, the T waves invert, and pathologic Q waves develop in areas of the ECG that correspond to the IRA. The location of the ST elevations and other findings on the ECG generally correspond to the anatomic location of the myocardium and the associated IRA. Anterior infarctions exhibit ST elevation in leads V1 through V4 (Fig. 55.2). Findings in leads V1 and V2 indicate involvement of the septum. MIs with these findings are caused by occlusion of the left anterior descending (LAD) coronary artery. When additional ST elevations are seen in leads V5, V6, I, and aVL, the location of the LAD occlusion is probably proximal to the first diagonal branch, which causes an anterolateral infarction (see Fig. 55.1, B). Inferior infarctions are characterized by ST elevations in leads II, III, and aVF (Fig. 55.3, A) and are due most commonly to right coronary artery (RCA) occlusion. Reciprocal ST depressions may be present in leads I and aVL.
Inferior MIs are associated with concomitant right ventricular infarction, which can be evident on right-sided ECG leads, particularly in RV4 and RV5 (see Fig. 55.3, B). Inferior MIs are also frequently associated with posterior wall involvement, which is seen on the ECG as ST depressions in leads V1 through V3
[/not-level-membership-for-emergency-medicine-category]
