Chapter 16 Acute cardiac syndromes, investigations and interventions
Cardiovascular disease (CVD) accounts for 35–40%1 of deaths in western industrialised society, with coronary artery disease (CAD) being responsible for about half of these. In patients over age 40, acute myocardial infarction (MI) is the cause of approximately 20% of all deaths. Up to 80–90% of these deaths occur outside hospital. Of patients admitted to hospital, mortality is 8–9%,1 much higher in at-risk groups. Lowering in-hospital mortality from CAD requires us to identify rapidly patients who are at risk and to implement evidence-based treatment regimens.
MYOCARDIAL INFARCTION
Acute MI can occur in association with or result from a number of different pathological and epidemiological mechanisms:2
Each mechanism may have a different long-term prognosis despite similar biomarker and ECG changes. Such classifications may be clinically important when interpreting the results of clinical trials.2
ACUTE CORONARY SYNDROMES (ACS)
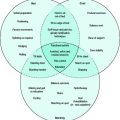
ACS represent the largest group of patients developing MI. They describe the spectrum of patients who present with chest discomfort or other symptoms caused by acute myocardial ischaemia (Figure 16.1). ACS can be further divided into acute MI and unstable angina (USA). Both are invariably caused by recent thrombus formation on pre-existing coronary artery plaque leading to impaired myocardial oxygen supply. In this sense they differ from stable angina, which is usually precipitated by increased myocardial oxygen demand with severe background coronary artery narrowing. Both represent medical emergencies and are one of the most frequent causes of hospital and coronary care unit (CCU) admission.

AETIOLOGY AND RISK FACTORS
Atheroma deposits in the walls of coronary arteries provide the substrate for the development of ACS. Major risk factors for the development of coronary artery atheroma are seen in Figure 16.2. Up to 90% of the adult population possess at least one risk factor for the development of atheroma and CAD.
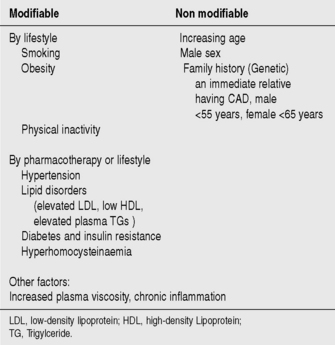
Cessation of smoking, lowering plasma cholesterol (diet and medications) and adoption of a more active lifestyle can all help prevent the development of CAD. Treatment of hypertensive patients produces a large and early reduction in stroke incidence (35–40%) and mortality, and a significant fall in MI (20–25%).3 Modification of risk factors is one of the most important means of decreasing the prevalence and mortality from CVD.
PATHOPHYSIOLOGY
Formation of thrombus upon disrupted, fissured or eroded atheromatous plaque is the usual precipitant of an ACS.4 Atherosclerotic plaque formation is probably initiated by injury to the vessel wall that may commence even as early as childhood. Highly activated macrophages are attracted to the site of injury and differentiate into tissue macrophages. Macrophages incorporate blood stream lipids into the connective tissue fibres of the plaque, forming a thrombogenic soft lipid core. Plaque development is slow, but is rapidly accelerated in people with risk factors.
‘Vulnerable plaque’ is often rich in lipid and covered by a thin fibrin cap. The cause of plaque rupture or fissuring is unknown but exposes thrombogenic lipid and collagen, which are potent activators of platelets. Development of thrombus upon this eroded plaque results from: (1) platelet adherence and activation; and (2) coagulation pathway activation.
Although many pathways initiate platelet activation, the final common pathway of thrombus formation is via activation of the glycoprotein (GP) IIb/IIIa receptor, the platelet surface membrane receptor for fibrinogen. Activated GP IIb/IIIa receptors cross-link fibrinogen between activated platelets, promoting the formation of platelet thrombi. Platelets aggregate to form ‘white thrombus’; however this thrombus is seldom totally occluding. Activation of coagulation pathways by exposed lipid and fibrin, as well as by the now activated platelets, leads ultimately to thrombin activation and the laying-down of fibrin clot. Red cells are enmeshed in this so-called ‘red thrombus’ complex, which surrounds the ‘white thrombus’. Sudden artery occlusion by thrombus may thus complicate even only moderate-sized plaque; 70% of ACS patients may have a < 50% stenotic lesion and in only 14% is the underlying stenosis > 70% of the lumen diameter (Figure 16.3).
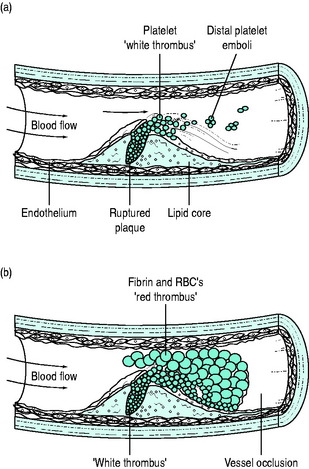
These processes have immediate relevance to treatment:
Totally occluding thrombus causes myocardial necrosis unless there is good collateral flow or the thrombus is rapidly cleared. Occlusion is often accompanied by ST-segment elevation on the ECG. If thrombus is largely ‘white thrombus’, with minimal or non-occlusive red thrombus, ST-segment elevation is far less likely. Non-occlusive thrombus may be asymptomatic, may cause USA or may cause MI, especially if spasm or distal embolisation of thrombus occurs. Although non-occlusive thrombus is less likely to be associated with early or sudden death, it is indicative of unstable plaque and is strongly associated with reinfarction and death in following months.
CLINICAL PRESENTATION
The diagnosis of myocardial ischaemia is usually made (suspected) on the basis of clinical history and ECG.2
HISTORY
Typically, the pain of acute MI:
Sweating, nausea, pallor, dyspnoea and anxiety are common.
The pain may sometimes be atypical:
These features do not necessarily exclude infarction.5 Differential diagnosis includes:
Atypical or silent presentations are common: 20–60% of non-fatal infarctions are unrecognised at onset.4 This presentation is more common in patients who are elderly, diabetic, have hypertension, who smoke or take non-steroidal anti-inflammatory agents.
PHYSICAL EXAMINATION
Examination of patients with USA is often unremarkable. With more severe infarction and extensive myocardial injury, signs of autonomic activation (pallor, sweating, agitation, clamminess) as well as heart failure and even shock may be apparent. Pericardial friction rubs occur frequently after MI but are usually transient.
Cardiogenic shock, hypotension, oliguria and other features of low cardiac output are associated with particularly poor outcome. Shock may be present without hypotension6 and may develop many hours after onset of symptoms. Right ventricle (RV) infarction results in hypotension and marked elevation of the jugular venous pressure (also seen with major LV dysfunction, usually in association with marked pulmonary venous congestion).
Conditions with similar presentations that do not benefit from thrombolysis are:
INVESTIGATIONS
The presence of MI should also be qualified by:2
Technological advances allow accurate detection of very small infarcts (myocardial necrosis < 1.0 g)7 that would not have been detected in earlier eras.
ELECTROCARDIOGRAPHY
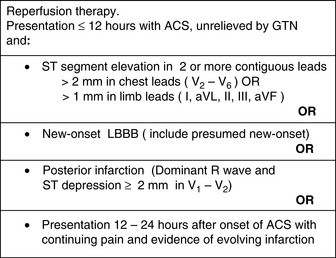
Identification of classical acute and early changes where ST-segment elevation MI (STEMI) is present identifies patients in whom reperfusion therapy may interrupt, prevent or minimise myocardial necrosis (Figure 16.4). These are:
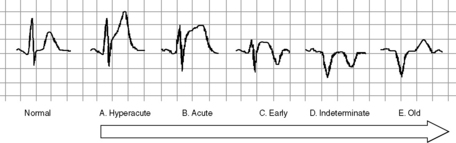
The Takotsubo syndrome is characterised by precordial ST-segment elevation, apical ballooning on echocardiography but normal vessels on angiography. It may follow the onset of recent severe stress and may cause up to 1–2% of STEMI.9 It has been recognised in critical illness.10
Patients with ACS but without significant ST-segment elevation (generically, non-ST-segment elevation ACS (NSTEACS), until further subdivided by biomarker studies) may still be at high risk of infarction and death. They likely have active, non-occluding thrombus or, if it is occluding, then some collateral flow is present. ECGs in these patients may be normal or display:
LOCALISATION OF INFARCTION
The left anterior descending (LAD) coronary artery supplies the anterior two-thirds of the interventricular septum (septal perforators), the anterior and lateral wall of the LV (diagonal branches) and sometimes part of the RV. The left circumflex artery supplies the LV lateral (anterolateral marginal branches) and posterior walls, and occasionally its inferior aspect (posterior LV arteries: 15% of patients) and the posterior septum. The right coronary artery (RCA) supplies the RV wall, and usually the posterior septum and inferior (diaphragmatic) wall of the left ventricle (posterior LV arteries; 85% of people). The RCA is ‘dominant’ (as opposed to the circumflex) if it gives rise to the posterior descending coronary artery (PDA) (Figure 16.5).
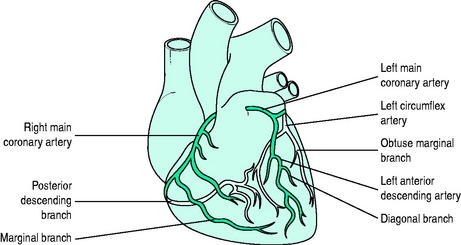
It is usual to use the ECG in initial clinical assessments to localise the area of myocardial ischaemia. The pattern of lead involvement may thus assist with localisation of the MI (Figure 16.6).8,11,12 There is a reasonable correlation between the site of infarction as defined by the ECG and the occluded coronary artery and the infarcted region of myocardium (Figure 16.7). However, ECG localisation may differ from angiographic, echocardiographic and autopsy findings, especially where there is collateral circulation or previous CABG. Anterior wall infarctions usually result from occlusion of the LAD; inferior, true posterior and RV infarctions result from occlusion of the RCA or circumflex arteries.
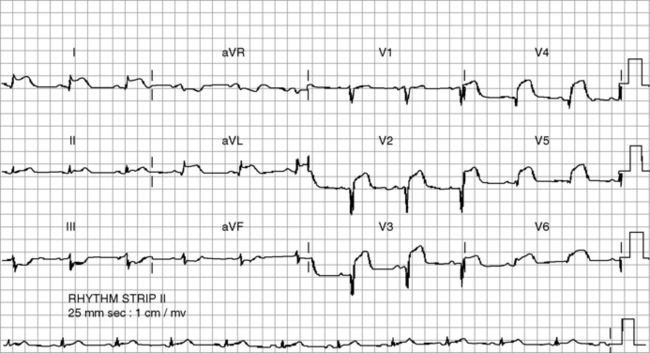
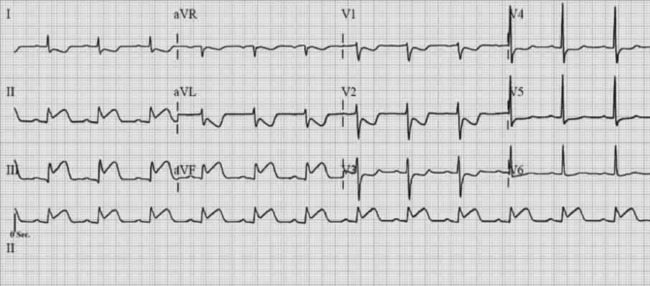
Common ECG patterns of infarction are shown in Figure 16.6. Approximately 40–50% of patients present with anterior infarction and 50% with inferior infarction. Anterior wall infarctions may be extensive or localised (septal, anterior, lateral) whereas inferior infarctions may similarly involve extension to the lateral, posterior or RV myocardium. Of clinical importance:
The resting ECG does not have sufficient predictive value to stratify patients with NSTEACS reliably into those with infarction (NSTEMI) and those without (USA). Up to 18% of patients with MI show no changes on the initial ECG and up to 20% of patients with NSTEACS have normal or minimal CAD.11,12 Cardiac biomarkers are necessary to confirm myocardial cellular injury and meet diagnostic criteria for MI.
CARDIAC BIOCHEMICAL MARKERS
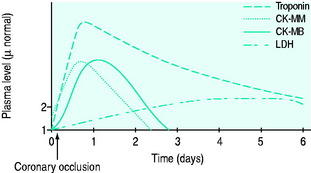
The typical rise and fall of cardiac markers after infarction are shown in Figure 16.8.
TROPONINS
The increased sensitivity of troponins compared to CK means that a third of patients previously considered to have USA are now recognised or redefined as having evidence of myocardial necrosis. Troponins also have better specificity than CK, similar to that of its isomer CK-MB. They do not differentiate the cause of the myocardial injury (e.g. ischaemia, myocarditis, trauma), however, and thus clinical context must always be considered.2 Troponins are more persistent in the serum (up to 7–10 days) and thus may be useful in diagnosis when presentation is delayed. Whilst CK-MB has traditionally been thought to be a better predictor of reinfarction, a rise in troponin of > 20% some 3–6 hours after onset of suspected reinfarction is also significant.2
Troponins should be checked in all patients with ACS and aggressive therapies should be targeted at patients with elevated levels.5
TROPONINS IN CRITICAL ILLNESS
Cardiac-specific troponins are frequently elevated in critical illness, including but not confined to sepsis, pulmonary embolus and renal insufficiency.13 It is likely that they are of cardiac origin and are indicative of myocardial injury. Their elevation does correspond with adverse outcomes. Most will not be due to ‘unstable plaque’, however, and the role of standard NSTEMI therapies is uncertain. Clinical context is necessary to decide those likely to have significant underlying CAD that may require acute or subsequent investigation.2
Please also see the latest information at the end of the chapter.
ECHOCARDIOGRAPHY
Echocardiography detects regional wall motion abnormalities, which can help confirm or exclude the diagnosis of MI in the small percentage of cases where diagnosis is uncertain (e.g. left bundle branch block (LBBB) or old infarction with atypical presentations). Regional wall motion abnormality and loss of wall thickening with contraction are often present in these cases if due to ischaemia, while their absence suggests that ischaemia is not acute. It is useful for excluding differential diagnoses (e.g. aortic dissection or pericardial effusions), again in a small percentage of patients.2
STRESS TESTING
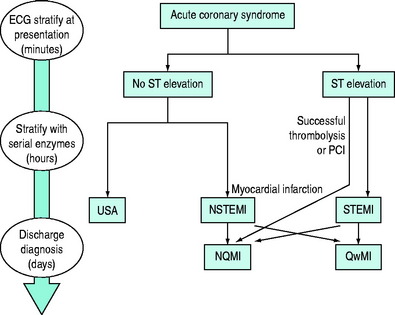
STEMI
STEMI (including new, presumed new LBBB) with persistent pain is the most lethal form of ACS and is usually due to complete occlusion of a coronary artery (> 90% of patients).5 It is an indication for reperfusion therapy. Such therapy may be successful and significantly reduce the size of the potential infarction, although some rise in troponin is usually inevitable. Patients who develop ST-segment elevation after admission should also then be stratified to this group.
NSTEACS
The therapy of NSTEMI aligns more clinically with that of USA than with that of STEMI. These two conditions, both forms of NSTEACS, represent a spectrum of disease and require common treatments directed at platelet inactivation and ‘plaque stabilisation’. The term ‘NSTEACS’ recognises that they are clinically indistinguishable at presentation. The more severe the ischaemia, the higher the need for more aggressive anticoagulation and invasive procedures. Early diagnostic classification using both ECG and troponins allows early risk stratification and evidence-based therapy. Only 35–75% of patients have evidence of coronary thrombus formation and thrombolytic therapy is not beneficial in this group. Indeed, it is associated with worse outcomes.5,14
Figure 16.10 displays the incidence of major coronary events over the following 12 months in patients presenting with and without ST-segment elevation. ST-segment depression has lesser early mortality but a similar or higher mortality at 6 months and at 10 years than those presenting with ST-segment elevation.15,16 Features that correlate with risk are:
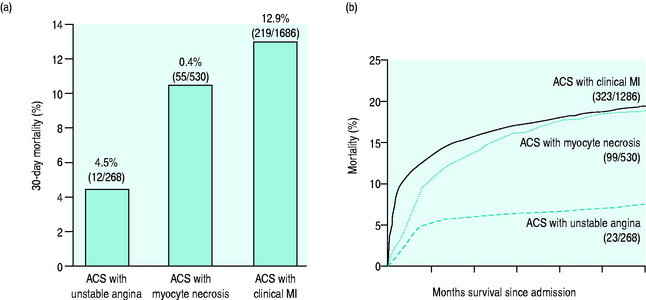
Q-wave MI (QwMI) or non-Q-wave MI (NqwMI) are older terms used to describe MI. The 10-year mortality of NqwMI (70%) is 10% higher than that of QwMI.16
IMMEDIATE MANAGEMENT OF ACUTE CORONARY SYNDROMES
PREHOSPITAL CARE
Review of trials comparing prehospital to in-hospital thrombolysis suggests a 17% decrease (95% confidence interval, 2–29%) in 30-day mortality.17
IMMEDIATE HOSPITAL CARE
ACUTE MANAGEMENT OF STEMI (Figure 16.12)
REPERFUSION THERAPY
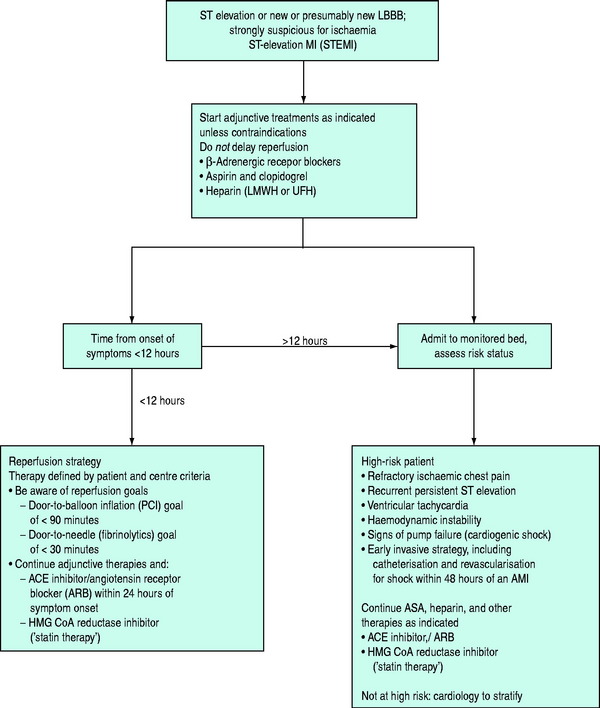
Reperfusion therapy should be considered for all patients presenting within 12 h of onset of STEMI.5,20
Indications and contraindications are given in Figures 16.11 and 16.13. Restoration of patency reduces infarct size, preserves LV function, reduces mortality and prolongs survival.

PERCUTANEOUS TRANSLUMINAL CORONARY INTERVENTION
The survival benefit of primary PCI compared with thrombolytic therapy is of the order of 20 lives per 1000 patients treated. These benefits are not uniform and are significantly influenced by its better outcomes in high-risk patients.
Urgent PCI in STEMI should be strongly considered in patients with:
Very few patients have contraindications to PCI,17 although caution is needed in those at risk of contrast-associated renal failure.
THROMBOLYTIC THERAPY
Thrombolysis within 6 h of symptom onset saves 30 lives per 1000 patients treated; within 7–12 h, the rate is 20 lives per 1000 patients treated, but there is no overall benefit for therapy beyond this time. Up to 40 lives per 1000 could be saved (relative risk reduction 50%) if treatment was given in the first hour.22 Benefit is still present at 20-year follow-up.23 Largest benefits are seen in anterior infarction (3.7% absolute mortality reduction), less in inferior (0.8% reduction).5,14,24
Age has a major impact upon survival following STEMI. Compared with controls thrombolysis reduces mortality at age < 55 years (3.4% versus 4.6%,), age 55–64 years (7.2% versus 8.9%, 18 lives per 1000 patients treated), age 65–75 years (11.1% versus 12.7%, 27 lives per 1000 patients treated) and age > 75 years (24.3% versus 25.3%, 10 lives per 1000). Thus, although the relative reduction in elderly patients is small (4%), the absolute reduction in mortality is still significant.14
THROMBOLYTIC AGENTS (FIBRINOLYTIC AGENTS)
Major and minor contraindications are shown in Figure 16.13.
SIDE-EFFECTS AND CHOICE OF AGENT
More efficient (fibrin-specific) thrombolytic agents improved upon the initial results of streptokinase. In Global Utilization of Streptokinase and Tissue Plasminogen Activator to Treat Occluded Arteries (GUSTO-1), accelerated rt-PA regimens were shown to reduce mortality compared to streptokinase (6.3% versus 7.3%, relative rate 14% reduction), but at the price of a small excess of haemorrhagic strokes.26,27 This represented an extra 10 lives saved per 1000 patients.
The tPA congeners reteplase and tenecteplase have very short initial half-lives, allowing convenient bolus dose administration. Trials of these agents against ‘accelerated rt-PA’ demonstrated equivalence.28 Accordingly these agents have come into frequent use because of their ease of administration. Tenecteplase can be given as a single agent, making it useful for prehospital and emergency hospital use.
Aggressive thrombolysis regimens are associated with lower all-cause mortality rates despite higher rates of cerebral haemorrhage5 that are fatal in 40–60% of cases.25 Of concern with these regimens is that the number of non-fatal strokes (severe disability occurs in 50%) can be problematic in high-risk groups. Accelerated t-PA regimens may result in 5 disabled stroke survivors per 1000 patients treated; streptokinase with subcutaneous heparin may result in 3 per 1000 patients treated.25 Age, recent stroke and hypertension on arrival significantly increase the risk of both lethal and non-lethal stroke. Since these patients were often excluded from trials, specialist input is required on a case-by-case basis to ascertain whether a less aggressive fibrinolytic agent (streptokinase) may be of use.26 Alternatively, strong consideration is given to PCI when thrombolysis is likely to be associated with a high incidence of lethal or non-lethal stroke.
Failure to adjust the dose of fibrinolytic correctly to body weight may be associated with increased mortality and intracerebral haemorrhage.17
PRIMARY PCI VERSUS THROMBOLYTIC THERAPY
Despite the advantages of PCI over thrombolysis, many patients present to centres that do not offer primary PCI. Given the advantage of PCI over thrombolysis, the possible benefit of transfer to such a centre needs to be considered. Transfer, however, involves an inherent delay and, if this is prolonged, then the advantage of more reliable revascularisation may be rapidly removed by the death of myocardium occurring as a result of the delay.17,29 Studies of groups of patients suggest that this advantage for groups may be lost if the transfer adds an additional 60–110 min to the process. This may represent an erosion of PCI’s mortality advantage of 0.29–0.40% for every 10-min delay. Well-developed systems need to be in place to minimise transport delays.
INVASIVE STRATEGY (PCI) IS GENERALLY PREFERRED (CONSIDER TRANSFER) IF
FIBRINOLYSIS GENERALLY PREFERRED IF:17
There is increasing evidence that, in the presence of failed thrombolysis where predicted mortality is high, rescue PCI confers survival benefit.18 Trials of ‘facilitated PCI’ where pharmacological therapy is routinely followed immediately by PCI are controversial and current evidence favours worse outcomes with this routine approach.17,18
ADJUNCTIVE THERAPY USED WITH THROMBOLYSIS AND REPERFUSION (Figure 16.14)
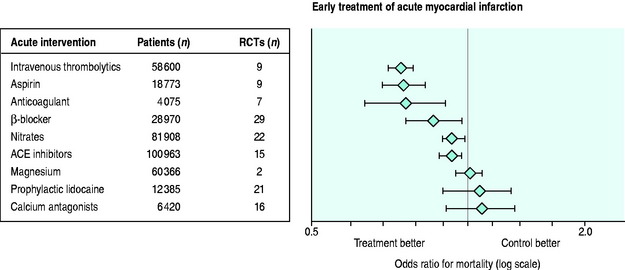
Adjunctive therapy is necessary after thrombolysis due to the following:
The major complication of adjunctive therapy is generally an increase in bleeding.
ASPIRIN AND CLOPIDOGREL
All patients undergoing reperfusion therapy for STEMI (PCI or fibrinolysis) should be given aspirin and clopidogrel unless contraindicated.18
Aspirin is one of the most significant and cost-effective treatments for STEMI. Given acutely in the second International Study of Infarct Survival (ISIS-2) it reduced mortality by 23% and gave a nearly 50% reduction in reinfarction and stroke. Streptokinase also reduced mortality (by 25%), while the combination had an additive effect and reduced mortality by 42%.24,30 Treatment for a mean duration of 1 month resulted in approximately 25 fewer deaths and 10–15 fewer episodes of non-fatal reinfarction and non-fatal stroke for every 1000 patients treated.30 It does not appear to increase bleeding and benefit is still present after 10 years.
Clopidogrel confers significant additional benefit. Given in conjunction with thrombolytics and aspirin, it reduces the risk of early vessel reocclusion without a significant rise in bleeding.29,31 Patients who undergo PCI and have a stent inserted also benefit if they have received clopidogrel. Benefit is less evident in patients who undergo PCI but do not have a stent placed, and bleeding is problematic.
GLYCOPROTEIN IIB/IIIA INHIBITORS
In theory, GPIs override the platelet activation caused by plasminogen activators, improving patency rates and allowing better reperfusion of small vessels beyond the occluding thrombus. In the setting of PCI, they are proven to be a most important adjunct to the procedure if a stent is placed or lesion dilated. Trials of GPIs in combination with full-dose thrombolytics found increased bleeding. These trials were done with half-dose thrombolytics. Results were again disappointing, with increased bleeding (especially in the elderly) but no survival benefit. In STEMI patients who have not received reperfusion therapy, GPIs do not appear to confer benefit.29,31,32
UNFRACTIONATED HEPARIN (UFH, STANDARD) AND LOW-MOLECULAR-WEIGHT HEPARIN
Prior to the use of fibrinolytics and aspirin, a limited number of small trials suggested that UFH reduced mortality (from 13.1% to 9.2%, 20–25% relative rate reduction). With the introduction of fibrinolytics and aspirin, UFH administration was continued. It was usually administered for 24–48 h after the fibrin-specific agents alteplase, reteplase or tenecteplase, reflecting the basis on which the drugs were trialled and licensed. Heparins do not lyse clot, but may decrease rethrombosis.
Despite UFH use, few trials (1239 patients) examined whether it added survival benefit to patients already treated with both thrombolytics and aspirin. At most the effect of UFH seems to be small (perhaps 5 lives saved per 1000 patients treated) and increased bleeding can be problematic.24,33
Comparison of UFH and LMWH has often proven difficult because of large variations in duration of therapy. LMWH is probably superior17,33–35 to UFH, resulting in lower rates of reinfarction, although the bleeding can be problematic in high-risk patients and caution should be used in this group.17
Meta-analysis34 of LMWH versus placebo has suggested that, across the whole ACS spectrum, adjunctive antithrombin therapy with enoxaparin is associated with significantly superior efficacy. Among STEMI34 patients, approximately 21 death or MI events were prevented for every 1000 patients treated with enoxaparin, at the cost of an increase of 4 non-fatal major bleeds.
The pentasaccharide factor Xa inhibitor fondaparinux has recently demonstrated a moderate reduction in mortality and reinfarction over ‘usual care’ (placebo or UFH). These benefits were seen without an increase in bleeding.36 Like LMWH it has the advantage of ease of administration and absence of need for monitoring but is associated with fewer bleeding complications. Benefits were present in patients at higher risk and not undergoing PCI. End-catheter thrombus was problematic in patients undergoing PCI, suggesting the need for UFH if this therapy is subsequently required. Some benefit was seen in patients who did not receive reperfusion therapy.36
β-BLOCKERS
Significant benefit from IV β-blockers was seen in the prethrombolysis era, but benefits appear small or absent in the postthrombolysis era.18 More recent studies have suggested that the use of IV β-blockers is associated with a decrease in reinfarction and VF but with an increase in cardiogenic shock, producing no overall benefit. IV therapy may be useful in patients with hypertension or tachycardia (although it is wise to assess LV function, perhaps with echocardiography). IV β-blockers given to patients receiving PCI produce an early absolute mortality reduction of 1.7%: the benefit is confined to patients not on β-blockers at time of admission.19
OTHER THERAPIES
Although nitrates may have had a small benefit in prethrombolysis days, there is little evidence of outcome benefit in the postthrombolysis era and indeed complicating hypotension may be harmful. Glucose–insulin–potassium therapies have not been shown to provide benefit under trial conditions.37
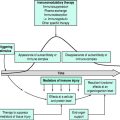
COMPLICATIONS OF MYOCARDIAL INFARCTION (Figure 16.15)
ARRHYTHMIAS
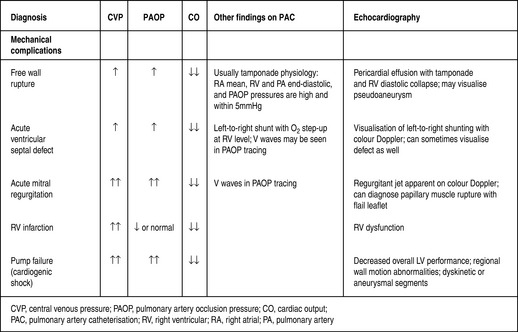
Prophylactic lidocaine tends to increase mortality and is thus reserved for the treatment of life-threatening ventricular arrhythmias. Prophylactic or routine IV magnesium (< 4 h) was of benefit in the Second Leicester Intravenous Magnesium Intervention Trial (LIMIT II) study but not in the larger ISIS-4 study, and is not of proven benefit.5
FAILED THROMBOLYSIS (RESCUE PCI)
A number of patients with STEMI will fail to show resolution of ST-segment elevation with fibrinolysis, suggesting failed reperfusion. Whether this should be treated with further fibrinolysis or PCI, or whether no intervention should be undertaken, has been uncertain. Meta-analysis of a small number of trials suggests that further thrombolysis is not associated with survival or reinfarction benefit and indeed may be harmful. Rescue PCI, compared to conservative management, conferred no survival benefit but was associated with significant reductions in heart failure, requiring 9 patients to be treated to achieve this benefit. Treatment was associated with an increased risk of bleeding and possibly stroke. Consideration for rescue PCI should probably be considered on a case-by-case basis; most benefit is likely to be achieved in shocked or haemodynamically unstable patients.
CARDIAC FAILURE
RV failure secondary to RV infarction38 should be distinguished from RV failure secondary to LV failure and should be considered in any patient with inferior MI. It is associated with significantly increased mortality. These patients have a markedly elevated jugular venous pressure with little or no pulmonary congestion. Treatment principles are different to those for LV dysfunction. Patients with RV infarction often respond to volume loading and it is important to maintain RV preload. The latter is guided by clinical response and echocardiography. Pulmonary flotation catheters, seeking to maintain an optimal LV filling pressure at around 16–18 mmHg (2.1–2.4 kPa), are now used less often. Diuretic therapy, afterload reduction and unrecognised hypovolaemia may aggravate hypotension and renal insufficiency in these patients.
CARDIOGENIC SHOCK (SEE Figure 16.15)
Mortality from cardiogenic shock remains very high (55–70%)39 and is the major cause of hospital mortality from STEMI. Patients with cardiogenic shock may have ‘stunned’ myocardium that is capable of improvement with revascularisation and initial intensive medical support. Patients presenting with cardiogenic shock are treated with acute interventional revascularisation (PCI or CABG) in preference to thrombolysis and medical management: the benefits are still present beyond 5 years (survival 32.8% versus 19.6%).21
POSTINFARCTION ANGINA AND REINFARCTION
Postinfarction angina unresponsive to medical therapy or with significant ECG changes is an indication for aggressive anti-ischaemic therapy and early PCI. Reinfarction in the 10 days following MI occurs in up to 5–10%; it can be treated with further thrombolysis or angioplasty. Where streptokinase has previously been given, t-PA is preferred because of the possible presence of antibodies that may neutralise activity or cause anaphylaxis.
CARDIAC RUPTURE20
SYSTEMIC EMBOLI
Embolic (ischaemic) stroke occurs in approximately 1% of patients during their hospital admission and in 2% by 12 months.40 Most of these cases occur following extensive anterior MI.41
MANAGEMENT
POST-MI INJURY SYNDROME (DRESSLER’S SYNDROME) AND PERICARDITIS
Pericarditis is a common early complication of extensive anterior and inferior infarction.
MANAGEMENT OF UNSTABLE ANGINA AND NSTEMI (NSTEACS) (Figure 16.16)
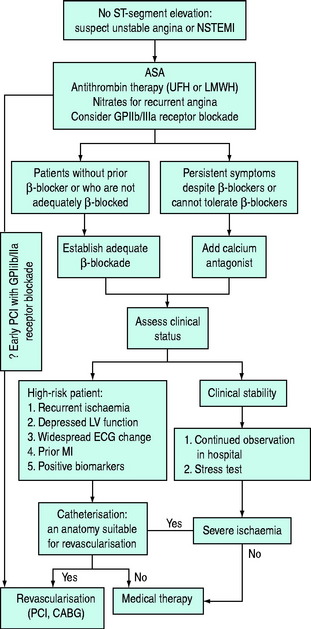
General principles of treatment are:
Thrombolytic agents are generally contraindicated in the treatment of NSTEACS; their routine administration is associated with poorer outcome.14,42
EARLY INVASIVE VERSUS MEDICAL THERAPY IN NSTEACS
While medical therapy is introduced to all patients, keen observation should be continued for signs of instability which suggest that an invasive approach is appropriate. Prior to the introduction of potent antiplatelet inhibitors as ‘upstream therapy’ and coronary stents, early invasive therapy was associated with minimal benefit or with adverse outcome despite its theoretical attractiveness.43 The development of better adjunctive agents (principally GPI blockers) has led to more recent studies suggesting that early invasive strategies are associated with reduced rates of MI and death.44 The benefits of PCI are dependent upon the baseline risk to the patient. Emergent PCI is demonstrated to offer clinical benefit to patients with high-risk features such as elevated troponins, recurrent chest pain and recurrent ECG changes.43
ANTI-ISCHAEMIC AGENTS
ANTIPLATELET THERAPIES
ANTITHROMBINS AND ANTICOAGULANTS
Meta-analyses that compared regimens of LMWHs to regimens of UFH found a minimal or less significant reduction in the composite of ‘death, MI and need for urgent revascularisation’ in favour of LMWHs than found when compared in STEMI patients.33–3551 However, despite higher acquisition costs, their simplicity, lack of need for monitoring and acceptable safety profile perhaps favour their administration.
Fondaparinux compared to enoxaparin in NSTEACS had similar early efficacy but appeared to do so with fewer bleeding complications. At later follow-up, fondaparinux was associated with lower mortality and cerebrovascular accidents; most of the benefit seemed to accrue from its lesser bleeding rate.52 Substitution of fondaparinux for enoxaparin might save 6 lives per 1000 patients treated and might result in 19 less bleeds. There is concern that fondaparinux is not sufficient to prevent stent occlusion and concomitant UFH use is recommended in patients undergoing PCI.
ONGOING AND DISCHARGE CARE OF ACS (SECONDARY PREVENTION) (Figure 16.17)
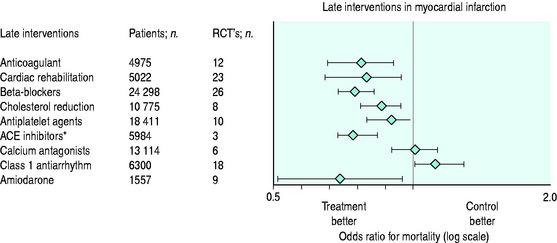
The aldosterone antagonist eplerenone reduces mortality in asymptomatic patients with low EF post-MI and already receiving β-blockers and ACE inhibitors (see Chapter 20).
MYOCARDIAL INFARCTION IN THE INTENSIVE CARE UNIT
Myocardial ischaemia is a common problem in the ICU. It also commonly complicates perioperative care of major surgery, with mortality of up to 15–25%. Diagnostic criteria are uncertain but a system has been proposed by Devereaux et al.60 There are few randomised controlled trials to guide therapy of postoperative infarction, or infarction complicating the care of the critically ill. Many patients with such presentations were excluded from trials of ACS therapy.
The pathophysiology of postoperative infarction and infarction in ICU patients is probably different to that of ACS.61 Studies suggest that, in the presence of severe ischaemia, left main disease and triple-vessel disease are common and that ischaemia is secondary to oxygen supply-and-demand problems rather than thrombosis. However, data on this are conflicting.61 The absence of thrombosis as an underlying pathological mechanism in many suggests that standard aggressive ‘antithrombus’ therapies will have different risk–benefit profiles, and that harm is exacerbated by the often high bleeding risk of these patients.
Echocardiography may be useful in confirming regional wall motion abnormality and in confirming the amount of myocardium at risk. Of interest is the Takotsubo syndrome, where anterior ST-segment elevation and apical ballooning on echocardiography, often in association with elevated troponins, may occur in the presence of normal coronary arteries.9,10
BLEEDING COMPLICATIONS POSTREPERFUSION THERAPY
The increased use of aggressive fibrinolytic regimens and of adjunctive reperfusion agents has led to troublesome bleeding in some patients. Some knowledge of reversal of these agents is necessary.62
OUTCOME OF MYOCARDIAL INFARCTION
The in-hospital mortality from acute MI has been steadily decreasing over the past three decades from 15% to 30% in the 1970s to approximately 10% in 1980 and now to around 8–9% in the new millennium.1 Despite improved mortality, 60% of all deaths occur within the first hour (usually from VF), and usually before reaching a medical facility.1 Modern management of acute MI has undoubtedly contributed to decreased mortality. Further significant reductions in mortality must come from management strategies within the first hours of the onset of symptoms.
The major in-hospital changes are likely to come from:63
The latter cannot be underestimated. In one study, the in-hospital mortality was 5.7% for those who received thrombolysis but 14.8% for those who were eligible for but did not receive such therapy (9.3% versus 18% in eligible women who did not receive therapy, 10.5% versus 19% in eligible elderly). Up to 24% of eligible patients do not receive reperfusion therapy.63
NEW INFORMATION
1 Rosamond W, Flegal K, Furie K, et al. Heart disease and stroke statistics – 2008 update. A report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2008;117:e25-146.
2 Thygesen K, Alpert JS, White HD. Universal definition of myocardial infarction. J Am Coll Cardiol. 2007;50:2173-2195.
3 Chobanian AV, Bakris GL, Black HR, et al. The seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA. 2003;289:2560-2572.
4 Anderson JL, Adams CD, Antman EM, et al. ACC/AHA guidelines for the management of patients with unstable angina/non-ST-elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee to Revise the 2002 Guidelines for the Management of Patients with unstable angina/Non-ST-Elevation Myocardial Infarction). J Am Coll Cardiol. 2007;50:e1-157.
5 Antman EM, Anbe DT, Armstrong PW, et al. ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee to Revise the 1999 Guidelines for the Management of Patients with Acute Myocardial Infarction). Circulation. 2004;110:e82-292.
6 Menon V, Slater JN, White HD, et al. Acute myocardial infarction complicated by systemic hypoperfusion without hypotension: report of the SHOCK trial registry. Am J Med. 2000;108:374-380.
7 Alpert JS, Thygesen K, Antman E, et al. Myocardial infarction redefined – a consensus document of the Joint European Society of Cardiology/American College of Cardiology Committee for the redefinition of myocardial infarction. J Am Coll Cardiol. 2000;36:959-969.
8 Zimetbaum PJ, Josephson ME. Use of the electrocardiogram in acute myocardial infarction. N Engl J Med. 2003;348:933-940.
9 Prasad A. Apical ballooning syndrome: an important differential diagnosis of acute myocardial infarction. Circulation. 2007;115:e56-59.
10 Tsuchihashi K, Ueshima K, Uchida T, et al. Transient left ventricular apical ballooning without coronary artery stenosis: a novel heart syndrome mimicking acute myocardial infarction. J Am Coll Cardiol. 2001;38:11-18.
11 Sgarbossa EB, Birnbaum Y, Parrillo JE. Electrocardiographic diagnosis of acute myocardial infarction: current concepts for the clinician. Am Heart J. 2001;141:507-517.
12 Owens CG, Adgey AA. Electrocardiographic diagnosis of non-ST-segment elevation acute coronary syndromes: current concepts for the physician. J Electrocardiol. 2006;39:271.
13 Fromm REJr. Cardiac troponins in the intensive care unit: common causes of increased levels and interpretation. Crit Care Med. 2007;35:1-5.
14 Appleby P, Baigent C, Collins R, et al. Indications for fibrinolytic therapy in suspected acute myocardial infarction: collaborative overview of early mortality and major morbidity results from all randomised trials of more than 1000 patients. Lancet. 1994;343:311.
15 Das R, Kilcullen N, Morrell C, et al. The British Cardiac Society Working Group definition of myocardial infarction: implications for practice. Heart. 2006;92:21-26.
16 Herlitz J, Karlson BW, Sjolin M, et al. Ten year mortality in subsets of patients with an acute coronary syndrome. Heart. 2001;86:391-396.
17 Boden EB, Eagle K, Granger CB. Reperfusion strategies in acute ST-segment elevation myocardial infarction. J Am Coll Cardiol. 2007;50:917-929.
18 Antman EM, Hand M, Armstrong PW, et al. 2007 focused update on the ACC/AHA 2004 guidelines for the management of patients with ST-elevation myocardial infarction (a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines). J Am Coll Cardiol. 2008;51:210-247.
19 Halkin A, Grines CL, Cox DA, et al. Impact of intravenous beta-blockade before primary angioplasty on survival in patients undergoing mechanical reperfusion therapy for acute myocardial infarction. J Am Coll Cardiol. 2004;43:1780-1787.
20 Van de Werf F, Ardissino D, Betriu A, et al. Management of acute myocardial infarction in patients presenting with ST-segment elevation. The Task Force on the Management of Acute Myocardial Infarction of the European Society of Cardiology. Eur Heart J. 2003;24:28-66.
21 Hochman JS, Sleeper LA, Webb JG, et al. Early revascularization and long-term survival in cardiogenic shock complicating acute myocardial infarction. JAMA. 2006;295:2511-2515.
22 Boersma E, Maas ACP, Decker JW, et al. Early thrombolytic treatment in acute myocardial infarction: reappraisal of the golden hour. Lancet. 1996;348:771.
23 van Domburg RT, Sonnenschein K, Nieuwlaat R, et al. Sustained benefit 20 years after reperfusion therapy in acute myocardial infarction. J Am Coll Cardiol. 2005;46:15-20.
24 Collins R, Peto R, Baigent C, et al. Aspirin, heparin, and fibrinolytic therapy in suspected acute myocardial infarction. N Engl J Med. 1997;336:847-860.
25 Gore JM, Granger CB, Simoons ML, et al. Stroke after thrombolysis. Mortality and functional outcomes in the GUSTO-I trial. Global use of strategies to open occluded coronary arteries. Circulation. 1995;92:2811-2818.
26 Walley T, Dundar Y, Hill R, et al. Superiority and equivalence in thrombolytic drugs: an interpretation. Q J Med. 2003;96:155-160.
27 The GUSTO Investigators. An international randomized trial comparing four thrombolytic strategies for acute myocardial infarction. N Engl J Med. 1993;329:673-682.
28 Sinnaeve P, Alexander J, Belmans A, et al. One-year follow-up of the ASSENT-2 trial: a double-blind, randomized comparison of single-bolus tenecteplase and front-loaded alteplase in 16,949 patients with ST-elevation acute myocardial infarction. Am Heart J. 2003;146:27-32.
29 Ting HH, Yang EH, Rihal CS. Narrative review: reperfusion strategies for ST-segment elevation myocardial infarction. Ann Intern Med. 2006;145:610-617.
30 Awtry EH, Loscalzo J. Aspirin. Circulation. 2000;101:1206-1218.
31 Clappers N, Brouwer MA, Verheugt FW. Antiplatelet treatment for coronary heart disease. Heart. 2007;93:258-265.
32 De Luca G, Suryapranata H, Stone GW, et al. Abciximab as adjunctive therapy to reperfusion in acute ST-segment elevation myocardial infarction: a meta-analysis of randomized trials. JAMA. 2005;293:1759-1765.
33 Eikelboom JW, Quinlan DJ, Mehta SR, et al. Unfractionated and low-molecular-weight heparin as adjuncts to thrombolysis in aspirin-treated patients with ST-elevation acute myocardial infarction: a meta-analysis of the randomized trials. Circulation. 2005;112:3855-3867.
34 Murphy SA, Gibson CM, Morrow DA, et al. Efficacy and safety of the low-molecular weight heparin enoxaparin compared with unfractionated heparin across the acute coronary syndrome spectrum: a meta-analysis. Eur Heart J. 2007;28:2077-2086.
35 Yusuf S, Mehta SR, Xie C, et al. Effects of reviparin, a low-molecular-weight heparin, on mortality, reinfarction, and strokes in patients with acute myocardial infarction presenting with ST-segment elevation. JAMA. 2005;293:427-435.
36 Yusuf S, Mehta SR, Chrolavicius S, et al. Effects of fondaparinux on mortality and reinfarction in patients with acute ST-segment elevation myocardial infarction: the OASIS-6 randomized trial. JAMA. 2006;295:1519-1530.
37 Mehta SR, Yusuf S, Diaz R, et al. Effect of glucose-insulin-potassium infusion on mortality in patients with acute ST-segment elevation myocardial infarction: the CREATE-ECLA randomized controlled trial. JAMA. 2005;293:437-446.
38 O’Rourke RA, Dell’italia LJ. Diagnosis and management of right ventricular myocardial infarction. Curr Probl Cardiol. 2004;29:6-47.
39 Dauerman HL, Goldberg RJ, Malinski M, et al. Outcomes and early revascularization for patients > or = 65 years of age with cardiogenic shock. Am J Cardiol. 2001;87:844-848.
40 Witt BJ, Ballman KV, Brown RDJr, et al. The incidence of stroke after myocardial infarction: a meta-analysis. Am J Med. 2006;119:e1-19.
41 Cairns JA, Theroux P, Lewis HDJr, et al. Antithrombotic agents in coronary artery disease. Chest. 2001;119(Suppl.):228S-252S.
42 Antman EM, Anbe DT, Armstrong RW, et al. ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction – executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 1999 Guidelines for the Management of Patients with Acute Myocardial Infarction). Circulation. 2004;110:588-636.
43 Cannon CP, Weintraub WS, Demopoulos LA, et al. Comparison of early invasive and conservative strategies in patients with unstable coronary syndromes treated with the glycoprotein IIb/IIIa inhibitor tirofiban. N Engl J Med. 2001;344:1879-1887.
44 Bavry AA, Kumbhani DJ, Rassi AN, et al. Benefit of early invasive therapy in acute coronary syndromes: a meta-analysis of contemporary randomized clinical trials. J Am Coll Cardiol. 2006;48:1319-1325.
45 Antiplatelet Trialists Collaboration. Collaborative overview of randomised trials of antiplatelet therapy – I: Prevention of death, myocardial infarction, and stroke by prolonged antiplatelet therapy in various categories of patients. Br Med J. 1994;308:81-106.
46 The Clopidogrel in Unstable angina to prevent Recurrent Events (CURE). Investigators trial. I. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med. 2001;345:494-502.
47 Topol EJ, Moliterno DJ, Herrmann HC, et al. Comparison of two platelet glycoprotein IIb/IIIa inhibitors, tirofiban and abciximab, for the prevention of ischemic events with percutaneous coronary revascularization. N Engl J Med. 2001;344:1888-1894.
48 Topol EJ. A contemporary assessment of low-molecular-weight heparin for the treatment of acute coronary syndromes: factoring in new trials and meta-analysis data. Am Heart J. 2005;149(Suppl. 1):S100-106.
49 Kastrati A, Mehilli J, Neumann FJ, et al. Abciximab in patients with acute coronary syndromes undergoing percutaneous coronary intervention after clopidogrel pretreatment: the ISAR-REACT 2 randomized trial. JAMA. 2006;295:1531-1538.
50 Simoons ML. Effect of glycoprotein IIb/IIIa receptor blocker abciximab on outcome in patients with acute coronary syndromes without early coronary revascularisation: the GUSTO IV-ACS randomised trial. Lancet. 2001;357:1915-1924.
51 Eikelboom JW, Anand SS, Malmberg K, et al. Unfractionated heparin and low-molecular-weight heparin in acute coronary syndrome without ST elevation: a meta-analysis. Lancet. 2000;355:1936.
52 Yusuf S, Mehta SR, Chrolevicius S, et al. Comparison of fondaparinux and enoxaparin in acute coronary syndromes. N Engl J Med. 2006;354:1464-1476.
53 Hennekens CH, Albert CM, Godfried SL, et al. Adjunctive drug therapy of acute myocardial infarction – evidence from clinical trials. N Engl J Med. 1996;335:1660-1668.
54 Gheorghiade M, Goldstein S. Beta-blockers in the post-myocardial infarction patient. Circulation. 2002;106:394-398.
55 Hunt SA. ACC/AHA 2005 Guideline update for the diagnosis and management of chronic heart failure in the adult: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Update the 2001 Guidelines for the Evaluation and Management of Heart Failure). J Am Coll Cardiol. 2005;46:e1-82.
56 Flather MD, Yusuf S, Kober L, et al. Long-term ACE-inhibitor therapy in patients with heart failure or left-ventricular dysfunction: a systematic overview of data from individual patients. Lancet. 2000;355:1575.
57 Latini R, Tognoni G, Maggioni AP, et al. Clinical effects of early angiotensin-converting enzyme inhibitor treatment for acute myocardial infarction are similar in the presence and absence of aspirin: systematic overview of individual data from 96 712 randomized patients. J Am Coll Cardiol. 2000;35:1801-1807.
58 Cannon CP, Braunwald E, McCabe CH, et al. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med. 2004;350:1495-1504.
59 Pratt CM, Moye LA. The Cardiac Arrhythmia Suppression Trial: background, interim results and implications. Am J Cardiol. 1990;65:20B-29B.
60 Devereaux PJ, Goldman L, Yusuf S, et al. Surveillance and prevention of major perioperative ischemic cardiac events in patients undergoing noncardiac surgery: a review. CMAJ. 2005;173:779-788.
61 Devereaux PJ, Goldman L, Cook DJ, et al. Perioperative cardiac events in patients undergoing noncardiac surgery: a review of the magnitude of the problem, the pathophysiology of the events and methods to estimate and communicate risk. CMAJ. 2005;173:627-634.
62 Schroeder WS, Gandhi PJ. Emergency management of hemorrhagic complications in the era of glycoprotein IIb/IIIa receptor antagonists, clopidogrel, low molecular weight heparin, and third-generation fibrinolytic agents. Curr Cardiol Rep. 2003;5:310-317.
63 Gibson CM. NRMI and current treatment patterns for ST-elevation myocardial infarction. Am Heart J. 2004;148(Suppl. 1):29-33.