[level-membership-for-neurology-category]
76 Acquired Myopathies
Clinical Vignette
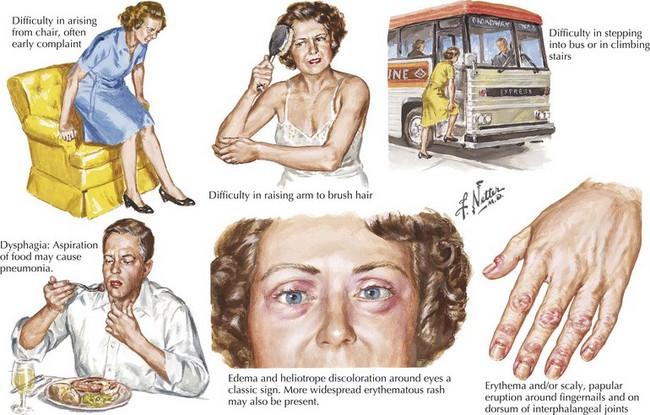
The common nongenetically determined myopathies are classified into those having a primary inflammatory process, an underlying endocrinopathy, a toxic pathophysiology, or an underlying associated systemic disorder. Much less commonly, a few infectious agents, such as trichinosis, may lead to a primary myopathy. Myopathies typically present with symmetric symptoms and signs of muscle weakness affecting the proximal limbs and paraspinal musculature (Fig. 76-1). Asymmetric, distal, generalized, or regional patterns of weakness also occur in certain distinct myopathies such as inclusion body myositis (IBM). Less commonly, ventilatory muscles or cardiac muscles are primarily affected. Myopathies occasionally present with periodic weakness, exercise-induced muscle pain, or stiffness.
Diagnostic Approach
Laboratory Evaluation
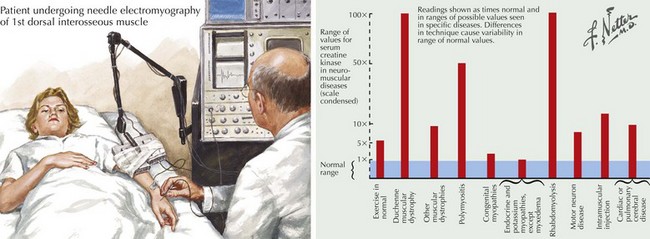
The serum CK is characteristically increased in many myopathies; this may vary from a 2- to 50-fold increase, although in most myopathies CKs are usually in the 500–5000 IU/mL range (Fig. 76-2). When this enzyme is abnormally elevated, its serum levels do not closely parallel disease severity or activity. Serum aldolase levels are also frequently elevated in myopathies; its increase generally parallels the increase in CK, although many clinical neuromuscular specialists do not routinely order an aldolase level. However on occasion it may be elevated with a normal CK as illustrated in the Cushing syndrome vignette reported later in this chapter.
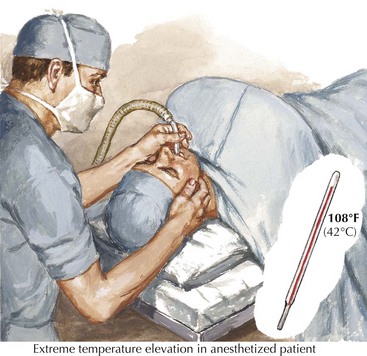
Patients with persistently increased CK levels sometimes associated with muscle pain but without clinically demonstrable weakness, family history, or exposure to potentially myotoxic substances are classified as having hyperCKemia. Despite thorough clinical and laboratory examination, it is often difficult to assign a specific pathophysiologic mechanism to this finding. HyperCKemia is often an elusive clinical challenge. However, it is important to emphasize that although no diagnosis per se is defined, the finding of hyperCKemia deserves serious consideration. Such individuals are at an increased risk of developing malignant hyperthermia (MH) if they require surgery under general anesthesia. Certain induction agents, particularly the halogenated ones, namely halothane, are particularly prone to inducing this life-threatening complication in patients with hyperCKemia. Therefore, we suggest that our hyperCKemia patients wear a MedAlert bracelet to always call the attention of anesthesiologists to this finding and thus potentially prevent an episode of MH (Fig. 76-3).
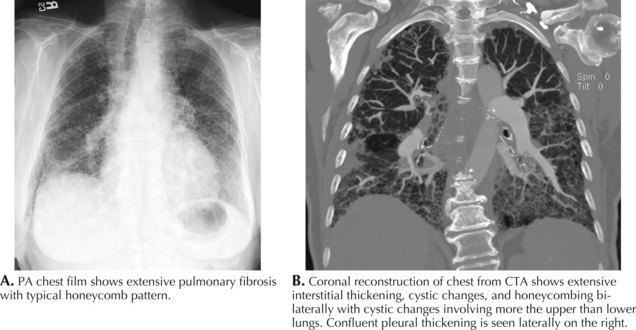
The serum myositis-specific and myositis-associated antibodies are other testing parameters that are useful in the evaluation of some patients with a myopathy. However, as these are present in fewer than half of all patients with polymyositis and dermatomyositis, routine serologic testing for these antibodies is of limited use. The presence of anti-Jo-1 (antibody to histidyl t-RNA synthetase) antibodies suggests potential end organ comorbidity, for example, interstitial lung disease (Fig. 76-4). Signal recognition particle antibodies are most often associated with necrotizing myopathies and may suggest a poor treatment response.
Electromyography
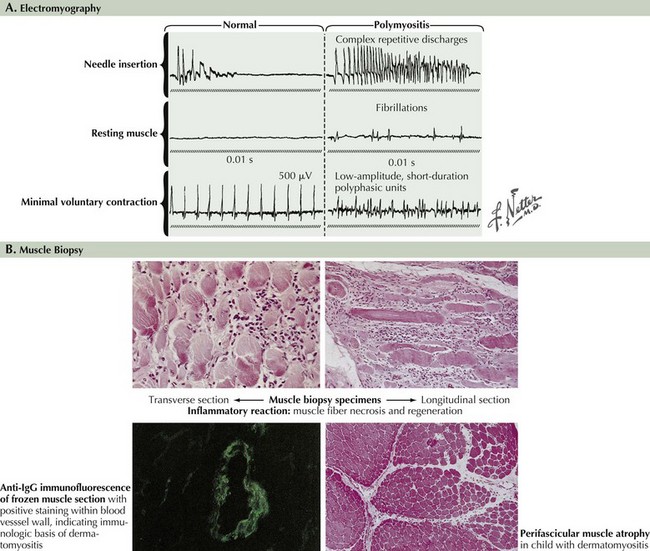
EMG evaluation of patients with suspected myopathies is important (Fig. 76-5, and see Fig. 76-2). Results of routine nerve conduction studies are normal in myopathies, with the exception of diminished compound muscle action potential amplitudes in more severe disorders. The primary EMG abnormalities in the myopathies are classically found at the time of the needle examination. Classic findings of a myopathy include the presence of abnormally low amplitude, short duration, and polyphasic motor unit potentials (MUPs). It is typical for these patients to have both an early recruitment and increased numbers of MUPs early on in the muscle activation for a given effort. Destruction of myofibrils or muscle membrane results in abnormal insertional activity, particularly fibrillation potentials and complex repetitive discharges. Inflammatory myopathies, several dystrophies, and various myotonic muscle disorders may be distinguished by the presence of myotonic potentials on needle EMG.
Muscle Biopsy
Muscle biopsy is the definitive diagnostic tool for many myopathies (Fig. 76-6). The selection of the biopsy site is important; muscles that are unaffected, that are severely affected (are at end stage), or have been recently subjected to EMG evaluation should be avoided. Muscles commonly biopsied include the vastus lateralis, deltoid, and biceps brachii. The gastrocnemius muscle is often avoided due to the possibility of incidentally discovered neurogenic atrophy. The upper lumbosacral muscles, thoracic paraspinal muscles, such as the multifidus, and much less commonly the cervical paraspinal muscles provide an alternative site for biopsy. On reflection one recognizes that these muscles are indeed the most proximal ones and thus more prone to show early changes of an active myopathic process.
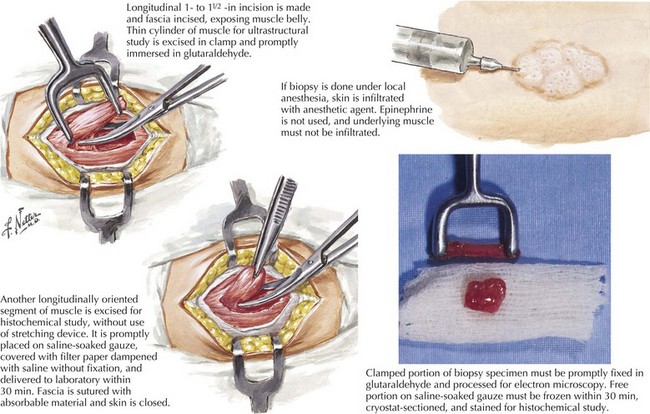
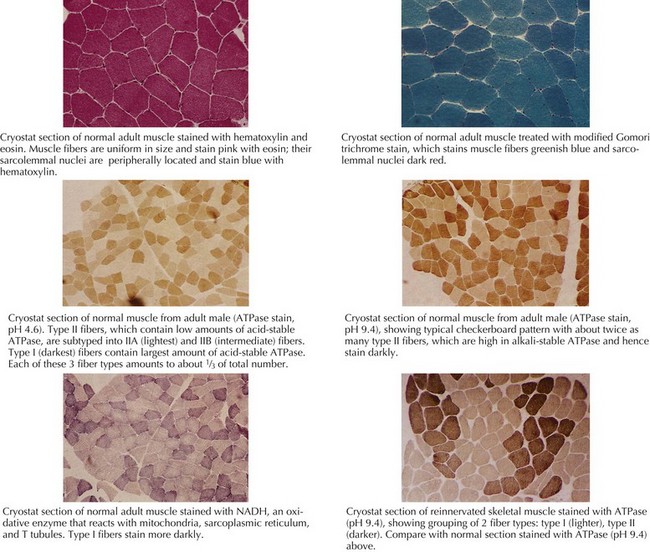
The muscle biopsy specimen per se is divided into separate aliquots for formalin fixation, paraffin embedding, and immediate freezing. The formalin-fixed piece is stained with hematoxylin and eosin (H and E) because this permits a rapid means for initial evaluation. This is especially useful for identifying inflammatory myopathies where such a diagnosis offers the potential for successful therapeutic intervention. Frozen specimens are best for other stains, including nicotinamide adenosine dinucleotide dehydrogenase (NADH), modified Gomori trichrome, adenosine triphosphatase, and lipid and glycogen stains (Fig. 76-7 and see Fig. 76-5).
Specific Inflammatory Myopathic Disorders
Polymyositis
EMG is often abnormal and may show characteristic findings, myopathic motor units, and increased insertional activity, with fibrillation potentials and complex repetitive discharges. Laboratory studies reveal an elevated CK level. Muscle biopsy demonstrates perimysial and endomysial inflammatory infiltrate with CD8+ T cells invading nonnecrotic muscle fibers (see Fig. 76-5). Interstitial lung disease can be seen in 10–20% of patients with PM and may be associated with positive anti-Jo1 antibody. Cardiac involvement (cardiomyopathy and congestive heart failure) is common, although the incidence of these associated conditions is unknown.
Dermatomyositis
Dermatomyositis (DM) is also seen in both children and adults. Proximal weakness develops insidiously over weeks. The characteristic rash may accompany or precede the myopathy (see Fig. 76-1). The rash is present over the exposed areas of the face, neck, and arms. Other dermatologic manifestations include heliotrope rash over the eyelids and erythematous rash over the knuckles, known as Gottron papules (Fig. 76-1, bottom). Nail bed examination will often demonstrate capillary telangiectasia. Occasional DM patients never develop this classic rash; here the differential diagnosis from PM is made on the classic pathologic findings of perifascicular atrophies in the muscle biopsy. In contrast, some patients present with the classic DM rash but paradoxically have no signs of a clinical myopathy (amyopathic DM). Other systemic manifestations include calcinosis, dysphagia, cardiomyopathy, and interstitial lung disease (ILD) (see Fig. 76-4). As in PM, ILD may be associated with positive anti-Jo 1 antibodies in some patients.
Laboratory tests typically demonstrate the elevated CK. ANA and anti-Jo1 may be elevated. MRI usually shows inflammation in affected muscles. EMG will reveal characteristic myopathic changes in established disease. The characteristic histopathology in DM is perifascicular atrophy although this may not be seen in early disease (see Fig. 76-5). Inflammation is not prominent; when present it is seen in the perimysial and perivascular regions.
Other Acquired Myopathies
Toxic Myopathies
Many pharmacologic agents may cause myopathies as rare adverse effects of their use (Box 76-1). The almost ubiquitously utilized HMG–CoA reductase inhibitor (statin) class of lipid-lowering agents may cause a necrotizing myopathy in a small percentage of these patients. Muscle biopsies in severely affected patients demonstrate necrosis and mitochondrial changes.
Hypokalemic Myopathies
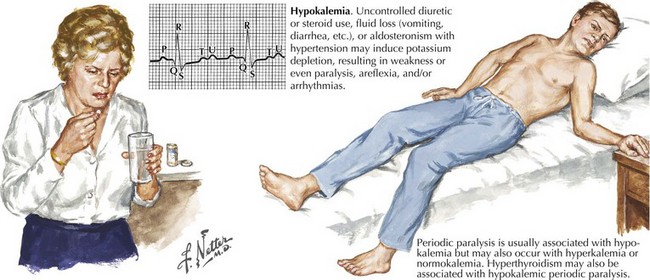
Hypokalemia is a rare metabolic cause of an acute myopathy (Fig. 76-8). The presentation may mimic Guillain–Barré syndrome. ICU observation is recommended because of potential serious cardiac arrhythmias that the severe hypokalemia may induce. The differential diagnosis includes various potassium-losing diuretics and corticosteroids and other medications (e.g., laxatives, lithium, or amphotericin). Chronic alcoholism, rarely hyperaldosteronism or a villous adenoma of the colon, and very excessive intakes of licorice, are other important causes of hypokalemia-induced weakness.
Endocrine Myopathies
Clinical Vignette
Cushing syndrome due to hyperadrenocorticism is either primary, iatrogenic, or rarely secondary to excessive pituitary secretion of ACTH (Fig. 76-9). This is one of the more common causes of an endocrine myopathy. Patients with Cushing syndrome, irrespective of etiology, experience proximal muscle weakness with atrophy usually starting in the hip girdles. Distal, bulbar, and ocular muscles are usually unaffected. Women seem to be more susceptible than men. Alternate-day corticosteroid dosing schedules and enriched protein diets may reduce susceptibility to iatrogenic induced Cushing syndrome. The serum CK level is usually normal. EMG is normal in iatrogenic steroid myopathy but is occasionally “myopathic” in patients with true Cushing syndrome. Muscle biopsy demonstrates a nonspecific type II muscle fiber atrophy. The pathogenesis of the myopathy is poorly understood but may be related to increased protein catabolism.
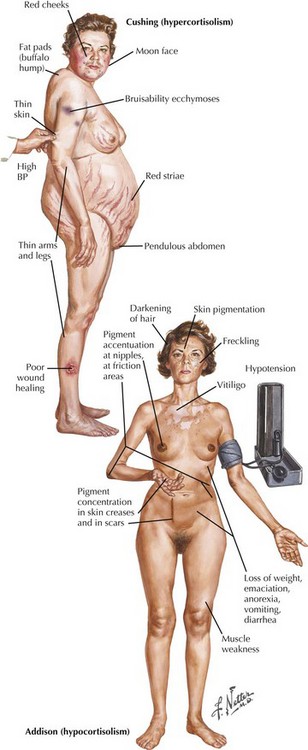
Primary adrenocortical insufficiency or Addison disease may be associated with a myopathy. Addison disease is characterized by weight loss, bronzing of the skin, hypotension, and hyperkalemia (see Fig. 76-9). Muscle weakness may be an early symptom of this disease and may be due to the associated hyperkalemia.
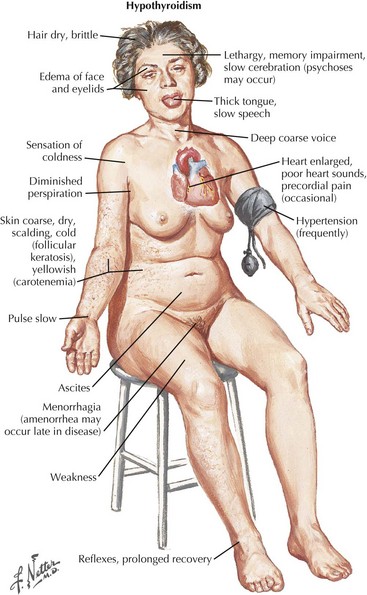
Thyroid dysfunction is another important consideration in the differential diagnosis of adult-onset myopathies. Hypothyroidism-associated myopathy is characterized by proximal weakness, fatigue, slowed movements and reflexes, stiffness, myalgia, and muscle cramps (Fig. 76-10). An elevated CK, sometimes up to 10 times normal, is a common finding in hypothyroid patients.
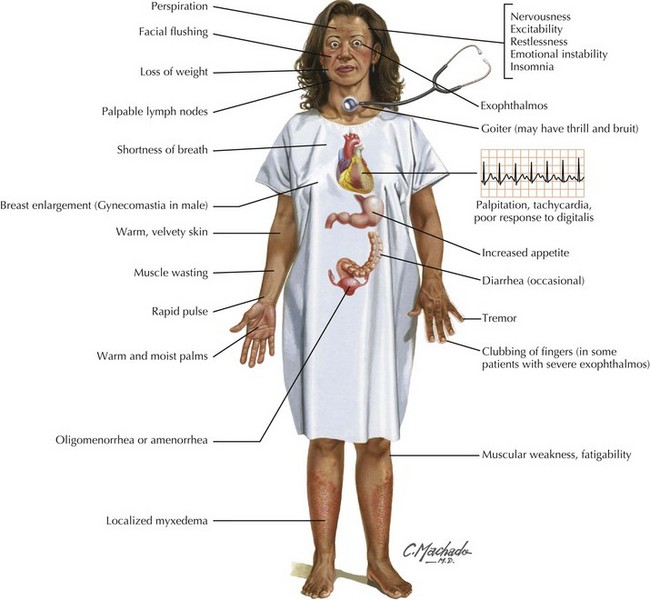
Hyperthyroidism-induced myopathy may also present with weakness, and the incidence of weakness in patients with thyrotoxicosis is high (up to 82%). Patients with thyrotoxicosis tend to have proximal muscle weakness and fatigue as prominent complaints (Fig. 76-11). Serum enzyme levels including CPK and AST tend to be normal.
Ahmed W, Khan N, Glueck CJ, et al. Low serum 25 (OH) vitamin D levels (<32 ng/mL) are associated with reversible myositis-myalgia in statin-treated patients. Transl Res. 2009 Jan;153(1):11-16.
Amato AA, Barohn RJ. Inclusion body myositis: old and new concepts. J Neurol Neurosurg Psychiatry. 2009;80:1186-1193.
Amato AA, Russell JA. Neuromuscular Disorders. New York: McGraw-Hill; 2008.
Davies NP, Hanna MG. The skeletal muscle channelopathies: distinct entities and overlapping syndromes. Curr Opin Neurol. 2003;16:559-568.
DiMauro S, Lamperti C. Muscle glycogenoses. Muscle Nerve. 2001;24:984-999.
Engel AG. Metabolic and endocrine myopathies. In Walton NJ, editor: Disorders of voluntary muscle, 5th ed, Edinburgh: Churchill-Livingstone, 1988.
Engel AG, Banker BQ, editors. Myology. New York: McGraw-Hill, 1986.
Fardet L, Dupuy A, Gain M, et al. Factors associated with underlying malignancy in a retrospective cohort of 121 patients with dermatomyositis. Medicine (Baltimore). 2009 Mar;88(2):91-97.
Felice KJ, Schneebaum AB, Jones HR. McArdle’s disease with late onset symptoms. J Neurol Neurosurg Psychiatry. 1992;55:407-408.
Ferrante MA, Wilbourn AJ. Myopathies. In: Levin KH, Luders HO, editors. Comprehensive Clinical Neurophysiology. Philadelphia, Pa: WB Saunders; 2000:268-281.
Griggs RC, Engel WK, Resnick JS. Acetazolamide treatment of periodic paralysis. Ann Int Med. 1970;73:39-48.
Griggs RC, Mendell JR, Miller RG. Evaluation and Treatment of Myopathy. Philadelphia, Pa: FA Davis Co; 1995.
Haller RG, Knochel JP. Metabolic myopathies. In: Johnson RT, Griffin JW, editors. Current Therapy in Neurologic Disease. St. Louis, Mo: Mosby-Year Book; 1993:397-402.
Jones HR, Darras B, De Vivo DC. Neuromuscular disorders of infancy, childhood, and adolescence: a clinician’s approach. Elsevier Health Sciences. 2002.
Katirji B, Al-Jaberi MM. Creatine kinase revisited. J Clin Neuromusc Dis. 2001;2:158-163.
McManis PG, Lambert EH, Daube JR. The exercise test in periodic paralysis. Muscle Nerve. 1986;8:704-710.
Miller A. Muscle disease. American Academy of Neurology. Continuum. Philadelphia, Pa: Lippincott Williams & Wilkins; 2006:12. No 3
Moxley RTIII. Channelopathies affecting skeletal muscle in childhood: myotonic disorders including myotonic dystrophy and periodic paralysis. In: Jones HR, De Vivo DC, Darras BT, editors. Neuromuscular Disorders of Infancy, Childhood, and Adolescence. Philadelphia, Pa: Butterworth-Heinemann; 2003:1017-1035.
Muthukrishnan J, Jha S, Modi KD, et al. Symptomatic primary hyperparathyroidism: a retrospective analysis of fifty one cases from a single centre. J Assoc Physicians India. 2008 Jul;56:503-507.
Al-Said YA, Al-Rached HS, Al-Qahtani HA, et al. Severe proximal myopathy with remarkable recovery after vitamin D treatment. Can J Neurol Sci. 2009 May;36(3):336-339.
Simmons A, Peterlin BL, Boyer PJ, et al. Muscle biopsy in the evaluation of patients with modestly elevated creatine kinase levels. Muscle Nerve. 2003;27:242-244.
Tonin P, Lewis P, Servidei S, et al. Metabolic causes of myoglobinuria. Ann Neurol. 1990;27:181-185.
Warren JD, Blumberg PC, Thompson PD. Rhabdomyolysis. Muscle Nerve. 2002;25:332-347.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
76 Acquired Myopathies
Clinical Vignette
The common nongenetically determined myopathies are classified into those having a primary inflammatory process, an underlying endocrinopathy, a toxic pathophysiology, or an underlying associated systemic disorder. Much less commonly, a few infectious agents, such as trichinosis, may lead to a primary myopathy. Myopathies typically present with symmetric symptoms and signs of muscle weakness affecting the proximal limbs and paraspinal musculature (Fig. 76-1). Asymmetric, distal, generalized, or regional patterns of weakness also occur in certain distinct myopathies such as inclusion body myositis (IBM). Less commonly, ventilatory muscles or cardiac muscles are primarily affected. Myopathies occasionally present with periodic weakness, exercise-induced muscle pain, or stiffness.
Diagnostic Approach
Laboratory Evaluation
The serum CK is characteristically increased in many myopathies; this may vary from a 2- to 50-fold increase, although in most myopathies CKs are usually in the 500–5000 IU/mL range (Fig. 76-2). When this enzyme is abnormally elevated, its serum levels do not closely parallel disease severity or activity. Serum aldolase levels are also frequently elevated in myopathies; its increase generally parallels the increase in CK, although many clinical neuromuscular specialists do not routinely order an aldolase level. However on occasion it may be elevated with a normal CK as illustrated in the Cushing syndrome vignette reported later in this chapter.
Patients with persistently increased CK levels sometimes associated with muscle pain but without clinically demonstrable weakness, family history, or exposure to potentially myotoxic substances are classified as having hyperCKemia. Despite thorough clinical and laboratory examination, it is often difficult to assign a specific pathophysiologic mechanism to this finding. HyperCKemia is often an elusive clinical challenge. However, it is important to emphasize that although no diagnosis per se is defined, the finding of hyperCKemia deserves serious consideration. Such individuals are at an increased risk of developing malignant hyperthermia (MH) if they require surgery under general anesthesia. Certain induction agents, particularly the halogenated ones, namely halothane, are particularly prone to inducing this life-threatening complication in patients with hyperCKemia. Therefore, we suggest that our hyperCKemia patients wear a MedAlert bracelet to always call the attention of anesthesiologists to this finding and thus potentially prevent an episode of MH (Fig. 76-3).
The serum myositis-specific and myositis-associated antibodies are other testing parameters that are useful in the evaluation of some patients with a myopathy. However, as these are present in fewer than half of all patients with polymyositis and dermatomyositis, routine serologic testing for these antibodies is of limited use. The presence of anti-Jo-1 (antibody to histidyl t-RNA synthetase) antibodies suggests potential end organ comorbidity, for example, interstitial lung disease (Fig. 76-4). Signal recognition particle antibodies are most often associated with necrotizing myopathies and may suggest a poor treatment response.
Electromyography
EMG evaluation of patients with suspected myopathies is important (Fig. 76-5, and see Fig. 76-2). Results of routine nerve conduction studies are normal in myopathies, with the exception of diminished compound muscle action potential amplitudes in more severe disorders. The primary EMG abnormalities in the myopathies are classically found at the time of the needle examination. Classic findings of a myopathy include the presence of abnormally low amplitude, short duration, and polyphasic motor unit potentials (MUPs). It is typical for these patients to have both an early recruitment and increased numbers of MUPs early on in the muscle activation for a given effort. Destruction of myofibrils or muscle membrane results in abnormal insertional activity, particularly fibrillation potentials and complex repetitive discharges. Inflammatory myopathies, several dystrophies, and various myotonic muscle disorders may be distinguished by the presence of myotonic potentials on needle EMG.
Muscle Biopsy
Muscle biopsy is the definitive diagnostic tool for many myopathies (Fig. 76-6). The selection of the biopsy site is important; muscles that are unaffected, that are severely affected (are at end stage), or have been recently subjected to EMG evaluation should be avoided. Muscles commonly biopsied include the vastus lateralis, deltoid, and biceps brachii. The gastrocnemius muscle is often avoided due to the possibility of incidentally discovered neurogenic atrophy. The upper lumbosacral muscles, thoracic paraspinal muscles, such as the multifidus, and much less commonly the cervical paraspinal muscles provide an alternative site for biopsy. On reflection one recognizes that these muscles are indeed the most proximal ones and thus more prone to show early changes of an active myopathic process.
The muscle biopsy specimen per se is divided into separate aliquots for formalin fixation, paraffin embedding, and immediate freezing. The formalin-fixed piece is stained with hematoxylin and eosin (H and E) because this permits a rapid means for initial evaluation. This is especially useful for identifying inflammatory myopathies where such a diagnosis offers the potential for successful therapeutic intervention. Frozen specimens are best for other stains, including nicotinamide adenosine dinucleotide dehydrogenase (NADH), modified Gomori trichrome, adenosine triphosphatase, and lipid and glycogen stains (Fig. 76-7 and see Fig. 76-5).
