[level-membership-for-neurology-category]
Chapter 72 Acquired Disorders Affecting the White Matter
Acute CNS Demyelination
A first demyelinating attack in childhood (referred to as an acquired demyelinating syndrome or ADS) may present either with neurological signs and symptoms attributable to a single CNS lesion (monofocal ADS), or with signs attributable to multiple CNS sites (polyfocal ADS). If polyfocal symptoms are accompanied by encephalopathy, the diagnosis of acute disseminated encephalomyelitis (ADEM) is conferred [Krupp et al., 2007].
The frequency of ADS presentations differs by patient age, and not all ADS presentations are equivalent in terms of subsequent MS risk. Data from a Canadian study of 219 children with ADS revealed the most common ADS presentation to be optic neuritis (23 percent), followed by ADEM (22 percent) and transverse myelitis (22 percent) [Banwell et al., 2009]. Children with ADEM were more likely to be under 10 years of age, whereas children with monofocal ADS (optic neuritis, transverse myelitis, and others) were more likely to be over 10 years of age.
Optic Neuritis
Optic neuritis (ON) should be considered in the differential diagnosis of any child presenting with acute or subacute visual loss (Figure 72-1). Typical clinical features of demyelinating ON include reduced visual acuity, a central visual field deficit (not always testable in young children), pain with eye movements, and red color desaturation [Lucchinetti et al., 1997]. Optic disc edema is sometimes seen, but this finding may be absent in retrobulbar ON. Concurrent or rapidly sequential transverse myelitis, magnetic resonance imaging (MRI) features of brainstem or diencephalic involvement in a pattern atypical for MS, and serum antibodies against aquaporin 4 support a diagnosis of NMO (discussed later in this chapter) [Wingerchuk et al., 2006].
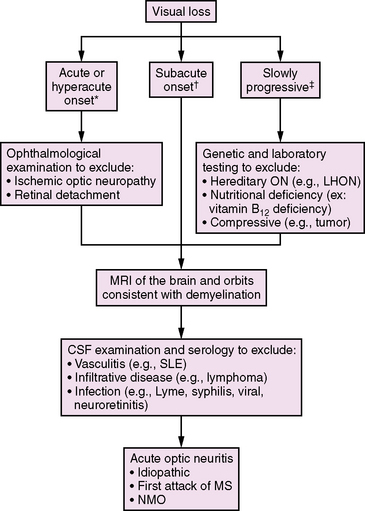
In a study of 36 children aged 2–17 years, ON was found to be more commonly unilateral (58 percent) than bilateral (42 percent) [Wilejto et al., 2006]. Neurological abnormalities apart from the optic nerve were common, affecting 13 children (36 percent). Neuroimaging studies of the optic nerve were abnormal in 55 percent, and 88 percent had abnormal visual-evoked potentials.
Optical coherence tomography (OCT) is a noninvasive technique that utilizes near-infrared light to measure retinal nerve fiber thickness, yielding a quantitative measure of axonal loss. This technique is currently being explored in both adults and children with ADS and MS as a way to provide an estimate of visual prognosis following ON [Frohman et al., 2006; Yeh et al., 2009a].
Following an episode of ON in childhood, approximately 80–85 percent of children achieve a full visual recovery [Hwang et al., 2002; Wilejto et al., 2006]. Children with ON have about a 36 percent risk of subsequent MS diagnosis overall, and MS may be more common in children presenting with bilateral optic nerve involvement [Lucchinetti et al., 1997; Wilejto et al., 2006]. MRI evidence of one or more T2-weighted lesions separate from the optic nerve is strongly predictive of MS risk, with up to 68 percent of such patients being diagnosed with MS within 2 years of their initial ON [Wilejto et al., 2006].
Transverse Myelitis
Demyelination of the spinal cord, accompanied by clinical signs of partial or complete cord impairment, is referred to as transverse myelitis (TM). The typical clinical features of TM include subacute onset of bilateral lower-extremity weakness, a spinal sensory level, and impaired bowel or bladder control. (Lesions above the sacral spine usually cause urinary retention and constipation.) L’Hermitte’s symptom (pain with forward neck flexion) frequently indicates a cervical spinal cord lesion. Paresis is often initially flaccid with associated hyporeflexia, and later hyperreflexia develops below the level of the lesion. These features are helpful in distinguishing TM from other diseases affecting the spinal cord (Figure 72-2).
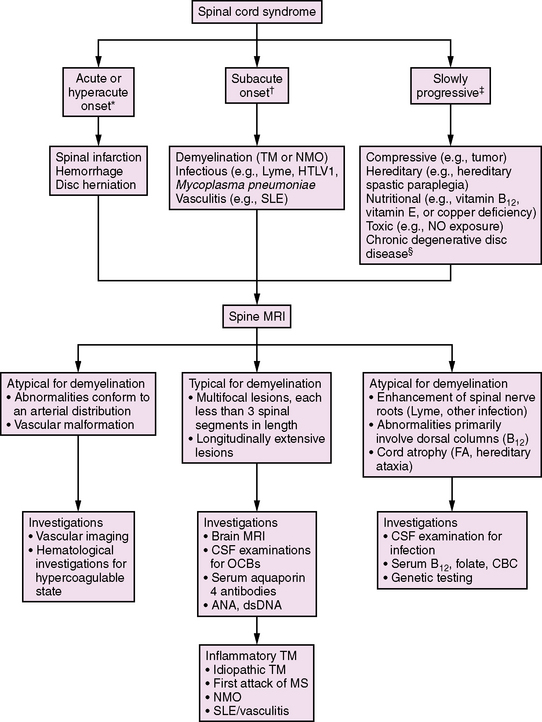
A review of 47 patients with TM showed that, at the time of maximal deficit, 89 percent of children either were unable to walk or required assistance with ventilation (or both) [Pidcock et al., 2007]. After 3 years of follow-up, 43 percent were unable to ambulate more than 30 feet, and 21 percent required a walker or ambulatory aid. Sixty-eight percent of children had residual bladder symptoms and 50 percent required bladder catheterization. Younger age at the onset of the TM event, complete paraplegia, and a shorter time (less than 24 hours) to maximal deficit have been associated with poorer functional outcomes [Dunne et al., 1986; Miyazawa et al., 2003; Pidcock et al., 2007]. The risk of MS following acute TM is approximately 2–8 percent [Dunne et al., 1986; Defresne et al., 2003; Miyazawa et al., 2003; Mikaeloff et al., 2004a; Pidcock et al., 2007].
Polyfocal Demyelination
Neurological symptoms suggesting multiple CNS areas of involvement may occur with or without concurrent encephalopathy (Figure 72-3). When encephalopathy (either behavioral change or alteration in level of consciousness) is present, the clinical syndrome is referred to as acute disseminated encephalomyelitis (ADEM) [Krupp et al., 2007]. ADEM tends to occur in younger patients, especially those under the age of 10 [Tenembaum et al., 2002]. Fever and a recent history of infection are often present [Rust, 2000; Tenembaum et al., 2002].
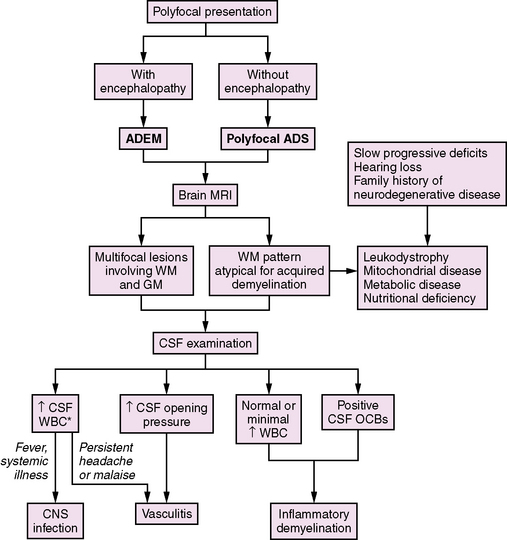
Recovery from ADEM can be incomplete, with 11–43 percent of children experiencing residual motor deficits [Dale et al., 2000; Tenembaum et al., 2002]. Cognitive deficits are often present, even in children who experience a full motor recovery [Hahn et al., 2003].
ADEM is most often a monophasic illness. However, approximately 10–29 percent of children with an initial presentation of ADEM will go on to have additional demyelinating attacks [Tenembaum et al., 2002; Mikaeloff et al., 2004]. The classification of further attacks of demyelination in children initially presenting with ADEM is reviewed later in this chapter.
Investigation of a Child with Acute Demyelination
Laboratory Investigations
Cerebrospinal fluid (CSF) analysis is important for excluding diseases other than demyelination, including infection and malignancy. CSF leukocyte counts in children with MS at first presentation are elevated (>4 cells/μL) in 66 percent of children and are typically below 30 cells/μL [Pohl et al., 2004]. Higher leukocyte counts are more consistent with acute infection, vasculitis, or NMO [Benseler and Schneider, 2004; Zaffaroni, 2004]. Elevated protein in the range of 100–720 mg/L may be seen, indicating disruption of the blood–CSF barrier [Pohl et al., 2004]. CSF oligoclonal bands (OCBs) are detected at the time of ADS in approximately 90 percent of children who are subsequently diagnosed with MS [Sindern et al., 1992; Ghezzi et al., 2002; Ozakbas et al., 2003; Pohl et al., 2004], which is slightly lower than the reported detection rate of 98 percent in adult MS patients [Reiber and Peter, 2001]. Isoelectric focusing methods have the highest yield for oligoclonal band detection [Freedman et al., 2005]. In contrast, CSF OCBs are detected in fewer than 10 percent of children with either ADEM [Tenembaum et al., 2002; Atzori et al., 2009] or NMO [Banwell et al., 2008; McKeon et al., 2008]. It should be noted that CSF immunoglobulin G synthesis can occur later in the disease course. Consequently, some MS patients who have initially negative CSF studies will later have detectable OCBs.
Evoked potential testing in the visual-evoked potential, brainstem auditory-evoked potential, and somatosensory-evoked potential pathways is useful for confirming the presence of demyelination and for detecting clinically silent disease. A study of 47 children with ON showed that visual-evoked potentials were abnormal in 88 percent [Wilejto et al., 2006]. In one study, almost 50 percent of 85 childhood patients with MS demonstrated clinically silent abnormalities in at least one of the evoked potential pathways, most commonly in the visual pathways [Pohl et al., 2006a].
Magnetic Resonance Imaging
MRI is a valuable tool to demonstrate the presence of inflammatory demyelination and to exclude other diagnoses [Miller et al., 1990] (Figure 72-4). Large lesions, ill-defined lesions, and lesions present in gray as well as white matter commonly occur in younger children, especially those with ADEM [Murthy et al., 2002; Garg, 2003; Mikaeloff et al., 2004b]. Several groups have highlighted the challenge in identifying MRI features sensitive enough to distinguish ADEM from MS at first attack [Schwarz et al., 2001; Mikaeloff et al., 2004b]. Recently proposed criteria suggest that the following features may be useful in making this distinction:
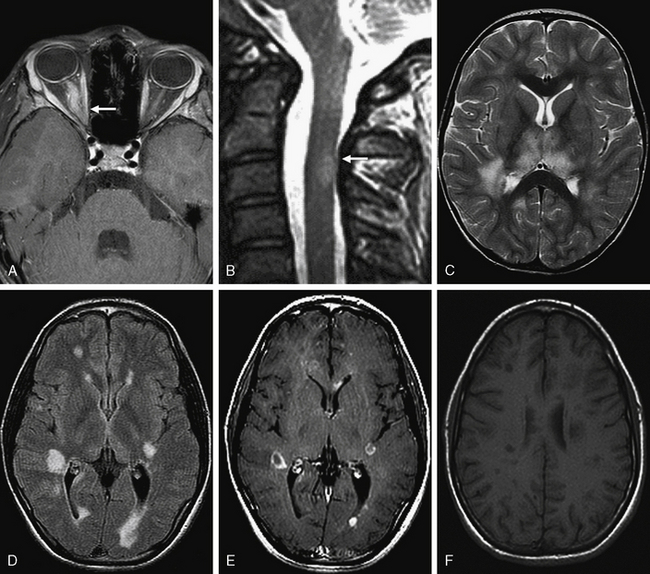
Serial MRI studies have been useful in showing dissemination in space within the CNS and time, and MRI characteristics have been incorporated into the diagnostic criteria for adult-onset and childhood-onset MS [McDonald et al., 2001; Barkhof et al., 2003; Polman et al., 2005; Krupp et al., 2007]. These criteria have between 52 and 76 percent sensitivity for distinguishing childhood MS from other diseases at first attack [Hahn et al., 2004; Mikaeloff et al., 2004b; Callen et al., 2009b]. Proposed revisions to these criteria for childhood MS have suggested that fewer lesions (≥5 T2 lesions, and either ≥2 periventricular lesions or ≥1 brainstem lesion) may be able to differentiate MS from other neurological disorders at first presentation with improved sensitivity and specificity [Callen et al., 2009b], but further validation is required.
Management of Acute Demyelination
Acute demyelination in children is generally treated with corticosteroids if symptoms are severe enough to interfere with daily functioning (Figure 72-5). While there are no published standardized trials for steroid therapy in children, typically 20–30 mg/kg/day of intravenous methylprednisolone (maximum 1 g) is given as a single daily intravenous dose for 3–5 days. If there is a significant improvement in symptoms, no further treatment is required. If there is an incomplete response to treatment but the symptoms are relatively mild, a tapering course of oral steroids (starting at 1 mg/kg/day and tapering over 21 days) is often considered. If children have significant residual symptoms after a course of intravenous steroids, a second 3–5-day course may be given at the same doses as outlined above. Alternatively, treatment with intravenous immunoglobulin (IVIg) at a dose of 2 g/kg over 2–5 days may be of benefit (class IV evidence) [Hahn et al., 1996; Nishikawa et al., 1999].
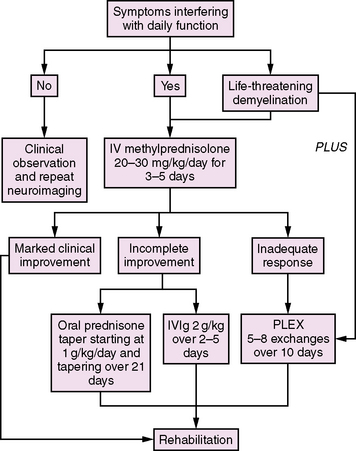
Fig. 72-5 Approach to the management of acute demyelination in childhood.
IV, intravenous; IVIG, intravenous immunoglobulin; PLEX, plasma exchange.
(Adapted from Thomas T, Banwell B. Multiple sclerosis in children. Semin Neurol 2008;28[1]:69–83.)
Rarely, acute demyelination in children can be life-threatening. Profound encephalopathy and respiratory depression may occur with brainstem and upper cervical spinal cord involvement. Plasma exchange may be useful in this situation, based on class I evidence in adult-onset MS [Weinshenker et al., 1999; Weinshenker, 2001]. A typical regimen of plasma exchange treatment would be 5–8 exchanges given over 10 days.
Acute Necrotizing Encephalopathy of Childhood
An acute necrotizing encephalopathy (ANE), with elevated serum aminotransaminases and bilateral symmetric thalamic lesions on neuroimaging, was first described in a Japanese child in 1979 (Mizuta et al., 1979), and since then there have been over 200 reports in the literature. The majority of children with ANE originate from the Far East, especially Japan, Taiwan, and Korea [Mizuguchi, 1997; Kim et al., 2004], with fewer cases reported in North America or Europe [Mastroyianni et al., 2006]. Recently, an affected kindred was described, representing the only case of male siblings with consanguineous parents [Marco et al., 2010] described in the United States. Familial (ANE1) and recurrent cases have recently been linked to mutations in RANBP2 (RAN-binding protein 2) [Neilson et al., 2009].
ANE typically affects children under the age of 5 [Mizuguchi, 1997; Mastroyianni et al., 2006]; in one series of 62, predominantly Japanese, patients, 46 percent of children were between 6 and 18 months of age [Mizuguchi et al., 1995]. In this same series, symptom onset occurred in winter months (December through February) in 51 percent of cases. Boys and girls appear to be equally affected [Mizuguchi et al., 1995; Mastroyianni et al., 2006].
A prodromal febrile illness is reported in up to 90 percent of children [Mizuguchi et al., 1995] and concurrent influenza A, influenza B, human herpesvirus (HHV)-6, Mycoplasma pneumoniae, herpes simplex, or measles infection has been documented in some patients [Mizuguchi, 1997; Mastroyianni et al., 2006]. Symptoms of an acute encephalopathy typically evolve 12–72 hours later and include decreased level of consciousness (28 percent), vomiting (20 percent), and seizures (40 percent) [Mizuguchi, 1997]. The disease is rapidly progressive and coma may ensue in less than 24 hours. Upper motor neuron signs, including hyperreflexia and extensor plantar responses, are seen acutely in two-thirds of patients [Mizuguchi, 1997; Mastroyianni et al., 2006].
Laboratory findings and neuroimaging features are helpful in distinguishing ANE from other conditions that may present in a similar manner, including ADEM (see earlier in this chapter). Laboratory findings in ANE include elevated serum aspartate aminotransferase, alanine aminotransferase, and lactate dehydrogenase in 50–95 percent of children [Mizuguchi, 1997; Mastroyianni et al., 2006]. Elevations in serum ammonia and bilirubin are uncommon. Metabolic acidosis may be present over 80 percent of children [Mizuguchi, 1997]. CSF examination demonstrates an opening pressure greater than 15 cm H2O with elevated CSF protein (>40 mg/dL) in the absence of pleocytosis in more than two-thirds of patients [Mizuguchi, 1997; Mastroyianni et al., 2006].
Neuroimaging characteristically reveals bilateral symmetric thalamic lesions, often accompanied by brainstem tegmentum, periventricular white-matter, putaminal, and cerebellar lesions. These lesions are initially hypodense on computed tomography (CT) and hypointense on T2-weighted MRI, later evolving into concentric lesions with a central area of hemorrhage that appears hyperdense on CT or hyperintense on T1-weighted MRI images [Mizuguchi et al., 1995; Mizuguchi, 1997; Mizuguchi et al., 2002]. Contrast enhancement around the area of hemorrhage becomes apparent by 2 weeks [Wong et al., 2006], and cavitation occurs in 50 percent of lesions [Kim et al., 2004; Wong et al., 2006]. Areas of diffusion restriction within acute lesions have been described [Albayram et al., 2004].
The outcome from ANE has historically been considered poor, with death in the acute phase occurring in up to one-third of patients [Mizuguchi, 1997]; however, more recently, patients with milder disease and fewer residual neurological sequelae have been reported [Kim et al., 2004; Okumura et al., 2006]. Patients may be left with residual neurological signs, including focal weakness, extraocular movement abnormalities, extrapyramidal movement disorders, or ataxia related to the sites of brain involvement visualized on MRI [Mizuguchi, 1997; Mastroyianni et al., 2006; Wong et al., 2006]. A normal CSF protein in the acute phase and lack of brainstem lesions on neuroimaging appear to be correlated with a more favorable long-term neurological outcome [Charney et al., 1979; Mizuguchi, 1997; Yoshikawa et al., 1999; Weitkamp et al., 2004; Wong et al., 2006].
While there are no consensus guidelines for the optimal medical management of children with ANE, a retrospective review of 34 affected Japanese children found that initiation of intravenous steroids (either 30 mg/kg of methylprednisolone given as a single daily dose for 3 days, or 0.6 mg/kg per day of dexamethasone administered in four divided doses for 2–4 days) within 24 hours of presentation in children without brainstem lesions was associated with a more favorable neurological outcome, as compared to children in whom steroid therapy was initiated after 24 hours or not at all [Okumura et al., 2009].
Relapsing Demyelinating Disorders
Multiple Sclerosis
Multiple sclerosis (MS), a chronic inflammatory and degenerative disorder of the CNS, is being increasingly diagnosed in children and adolescents. While historically considered a disease of young adulthood, it is now estimated that symptom onset prior to age 18 occurs in 3–10 percent of all MS patients [Duquette et al., 1987; Ghezzi et al., 1997; Boiko et al., 2002; Simone et al., 2002]. The increase in recognition of MS in the pediatric population relates in part to newly established clinical care and research programs in multiple countries [Mikaeloff et al., 2004a; Pohl et al., 2007; Banwell et al., 2009; Yeh et al., 2009b], and to the increased use of MRI, which has improved diagnostic certainty [Banwell et al., 2007b].
MS manifests with a relapsing-remitting disease course in over 95 percent of children [Banwell et al., 2007a; Banwell et al., 2007b]. Children experience more frequent relapses in the first few years of disease when compared to newly diagnosed adult MS patients [Ghezzi, 2004; Gorman et al., 2009], suggesting that the disease is highly inflammatory in the pediatric population. Secondary disease progression, diagnosed when accrual of disability occurs in the absence of discrete relapses, typically begins more than 15 years after presentation [Boiko et al., 2002; Simone et al., 2002].
As in adult patients, the diagnosis of MS in children requires demonstration of CNS demyelination separated both in space and in time [Poser et al., 1983; McDonald et al., 2001; Polman et al., 2005; Krupp et al., 2007) (Figure 72-6). MRI may be used to meet the dissemination in space or time requirement, as outlined in criteria published by McDonald et al. [McDonald et al., 2001; Polman et al., 2005]. Specifically, MRI evidence of dissemination in space requires the presence of at least three of the following:
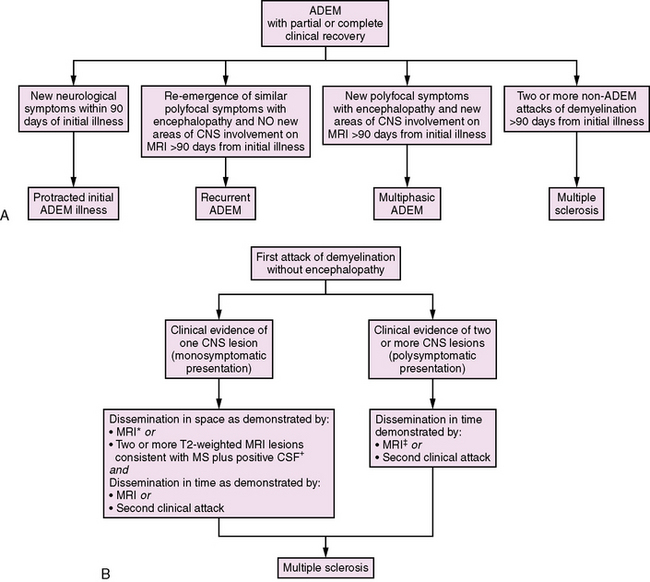
Fig. 72-6 Classification of further attacks of demyelination.
A, Approach to further attacks of demyelination in the child initially presenting with acute disseminated encephalomyelitis (ADEM) [Krupp et al., 2007]. B, Approach to further attacks of demyelination in the child presenting with a monofocal or polyfocal neurological syndrome without encephalopathy. *MRI criteria for dissemination in space require the presence of three of the following: 1. At least one gadolinium-enhancing lesion or nine T2 hyperintense lesions. 2. At least one infratentorial lesion. 3. At least one juxtacortical lesion. 4. At least three periventricular lesions. A spinal cord lesion can be considered the equivalent to an infratentorial brain lesion, and an enhancing spinal cord lesion is considered equivalent to an enhancing brain lesion. Individual spinal cord lesions can contribute together with individual brain lesions to reach the required number of T2 lesions [Polman et al., 2005]. †Isoelectric focusing evidence of cerebrospinal fluid oligoclonal IgG bands or increased IgG index, or both [Polman et al., 2005]. ‡MRI criteria for dissemination in time require either: 1. Detection of gadolinium enhancement at least 3 months after the onset of an initial clinical event, if not at the site corresponding to the initial event, or 2. Detection of a new T2 lesion if it appears at any time compared with a reference scan done at least 30 days after the onset of the initial clinical event [Polman et al., 2005].
Only two T2 lesions are required in the presence of abnormal CSF (isoelectric focusing evidence of CSF oligoclonal IgG bands or increased IgG index, or both) [Polman et al., 2005]. MRI can also be used to satisfy the criteria for dissemination in time in the absence of new clinical events in the presence of either:
MRI evaluation of lesion evolution in children following ADS is currently being explored.
Demographics and Epidemiology of MS in Children
Childhood-onset MS had been reported in many countries worldwide. While the exact incidence of pediatric MS remains to be determined, it is estimated that 3–10 percent of all MS patients experience their first clinical attack prior to the age of 18 years [Duquette et al., 1987; Boiko et al., 2002].
It has been well established that adult-onset MS is much more common in females than males. Sex differences are also apparent in pediatric MS, but appear to be related to age at first presentation. In children under the age of 10, the female to male ratio is approximately 1:1. After the age of 10, the female to male ratio increases to 3:1 [Ghezzi et al., 1997; Banwell et al., 2007a].
A family history of MS is reported in 5–10 percent of affected children [Simone et al., 2002; Mikaeloff et al., 2004a; Banwell et al., 2009). This is slightly lower than the 20–30 percent of adult MS patients who report MS in at least one first-degree relative [Robertson et al., 1996; Sadovnick et al., 1996]. Studies of familial MS risk in pediatric-onset MS patients are complicated by the fact that, due to their younger age, siblings, parents, and even grandparents may still be at risk of developing the disease.
Genetic and Environmental Risk Factors for MS in Children
Increased risk for MS in adults has been associated with the HLA DRB1*1501 locus (relative risk in heterozygotes = 3) [Ramagopalan and Ebers, 2008]. This has yet to be replicated in pediatric MS cohorts. Genome-wide association screens, performed in large adult MS cohorts, have recently identified single nucleotide polymorphisms in several non-HLA genes, including the IL-7 receptor, the IL-2 receptor alpha subunit, and KIF 1b (a kinesin motor protein) [Hafler et al., 2007; Aulchenko et al., 2008]. Polymorphisms in these genes confer a modest risk for adult-onset MS (odds ratios of 1.1–1.3). Genetic studies looking to identify risk genes for pediatric-onset disease will be of considerable interest in the future.
MS risk rises with increasing latitude [Ebers, 2008]. It has been hypothesized that reduced ultraviolet light exposure and consequent low vitamin D levels may account for this observation. Vitamin D levels are lower in adult MS patients as compared to healthy controls, especially in the summer [Soilu-Hanninen et al., 2005]. In children, low serum vitamin D concentrations at first presentation may predict a higher risk for subsequent MS diagnosis [Hanwell et al., 2008]. A recently described vitamin D response element within the promoter of the HLA DRB1*1501 gene provides an interesting link between an established genetic risk factor for MS and a putative environmental risk factor [Ramagopalan et al., 2009].
Viral exposures may also play a role in MS pathogenesis. Serological evidence of remote Epstein–Barr virus (EBV) has been observed in more than 85 percent of children with MS, as compared to 40–60 percent in healthy matched controls [Alotaibi et al., 2004; Pohl et al., 2006b; Banwell et al., 2007b). EBV may be involved in immune cell activation or in molecular mimicry between viral epitopes and myelin components (e.g., myelin basic protein) [Wucherpfennig and Strominger, 1995]. It is also possible that molecular mimicry or any other direct relationship between microbial infection and MS is not essential, and rather it is the influence of microbial infection on immune activation that contributes to MS risk.
Clinical Course of Multiple Sclerosis in Children
Over 95 percent of pediatric-onset MS patients experience an initial relapsing-remitting course [Boiko et al., 2002; Banwell et al., 2007a). Primary progressive MS, characterized by progressive clinical deterioration from onset in the absence of relapses, is extremely rare in children [Boiko et al., 2002]. Approximately 50 percent of children with MS will enter a secondary progressive stage of the disease after a disease duration of approximately 20 years [Boiko et al., 2002].
Immunomodulatory Therapy in Pediatric MS
Principles of Immunomodulatory therapy
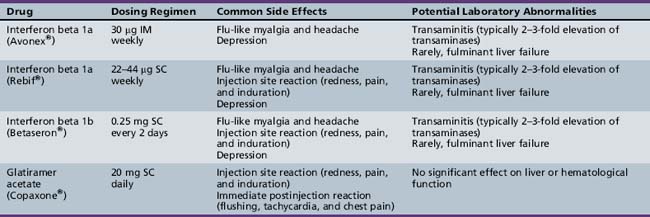
The main goal of the currently approved MS therapies is reduction in relapse rate, and modification of disease progression. To date, class I evidence (in adult-onset MS) is available for the following therapies [Goodin et al., 2002]: interferon beta 1a [PRISMS, 1998; Kappos et al., 2006; Koch-Henriksen et al., 2006], interferon beta 1b [Paty and Li, 1993; IFNB Multiple Schlerosis Study Group, 1995; Durelli et al., 2002; Koch-Henriksen et al., 2006], and glatiramer acetate [Wolinsky et al., 2002; Mikol et al., 2008]. All of these agents reduce relapse rates by approximately one-third in adult MS patients and are also associated with a reduction in the number of T2 MRI lesions. The dosing regimens and side effects of these medications are listed in Table 72-1. To date, there have been no comparable efficacy trials in children with MS. None the less, the available literature suggests that these medications are generally safe and well tolerated in the pediatric MS population [Waubant et al., 2001; Kornek et al., 2003; Ghezzi, 2005; Ghezzi et al., 2005; Pohl et al., 2005; Tenembaum and Segura, 2006; Banwell et al., 2006].
Second-line therapies
Immunosuppressive therapy is offered when frequent relapses occur despite compliance with interferon or glatiramer acetate. Cyclophosphamide [Makhani et al., 2009], azathioprine, methotrexate, and mitoxantrone have all been utilized in isolated cases or small case series of pediatric MS patients. There have been isolated case reports of natalizumab use in pediatric-onset MS [Huppke et al., 2008; Borriello et al., 2009], although it is currently not licensed for use in children. New immunomodulatory therapies (e.g., natalizumab, rituximab, alemtuzumab, and daclizumab) and oral therapies (e.g., cladribine and FTY720) have the potential for greater efficacy and tolerability than currently used immunomodulatory treatments. The side-effect profiles of these medications, including the risk of malignancy and infection, must be considered when choosing between these powerful immunosuppressive agents.
General Care Issues
The care of children with MS requires a multidisciplinary team that often includes a neurologist, nurse, occupational therapist, physiotherapist, psychologist, dietician, and social worker [Boyd and MacMillan, 2000]. The diagnosis of MS has important implications for social functioning, family functioning, and overall psychosocial well-being [MacAllister et al., 2007] and the care team plays an important role in providing education and support.
Recurrent and Multiphasic Acute Disseminated Encephalomyelitis
The classification of subsequent demyelinating events in children initially presenting with ADEM is summarized in Figure 72-6. The diagnosis of MS requires at least two subsequent non-ADEM events in a child with initial ADEM [Krupp et al., 2007]. Recurrence of ADEM with symptoms and signs similar to the first ADEM episode with no new areas of CNS involvement (either clinically or on MRI) more than 3 months from an initial ADEM episode is considered recurrent ADEM [Krupp et al., 2007]. By contrast, a second clinical episode meeting criteria for ADEM occurring more than 3 months from an initial ADEM event involving different areas of the CNS is defined as multiphasic ADEM [Krupp et al., 2007].
Neuromyelitis Optica
Neuromyelitis optica (NMO) is a severe, inflammatory demyelinating disorder of the central nervous system. While historically it has been debated whether or not NMO is a variant of MS, it is now generally considered a distinct entity with unique clinical, laboratory, and MRI features. The recent identification of a specific biomarker for NMO – NMO IgG – has resulted in improved diagnostic accuracy [Lennon et al., 2004].
Demographics and Epidemiology of Childhood NMO
The exact incidence of NMO in childhood is not known. In adults, NMO has a marked female predominance, with some studies reporting a female to male ratio as high as 9:1 [O’Riordan et al., 1996; Wingerchuk et al., 1999; de Seze et al., 2002; Ghezzi et al., 2004; Wingerchuk, 2009]. The median age of onset is 39 years, although there are published cases of childhood-onset disease [Banwell et al., 2008; Lotze et al., 2008; McKeon et al., 2008]. There is an over-representation of children reporting non-Caucasian ethnicity when compared to other demyelinating diseases such as MS [Banwell et al., 2008; Lotze et al., 2008; McKeon et al., 2008].
Clinical Features of NMO in Children
NMO is usually characterized by attacks of severe ON (visual acuity typically 20/200 or poorer) and TM, occurring either simultaneously or sequentially. When sequential, the attacks of ON and TM may be separated by months or even years [O’Riordan et al., 1996; Wingerchuk et al., 1999]. Although data are limited, it appears that approximately 53–100 percent of pediatric patients experience a relapsing course [Banwell et al., 2008; Lotze et al., 2008; McKeon et al., 2008].
Longitudinal studies of adult-onset NMO patients highlight the devastating nature of this disease; 50 percent of adults with NMO have no functional vision or require assistance with ambulation within 5 years of presentation [Wingerchuk et al., 1999]. In children with NMO, one study reported that 54 percent of patients had permanent visual impairment and 44 percent had limb weakness of sufficient severity so as to impact mobility after a median of 12 months of follow-up [McKeon et al., 2008].
Other autoimmune disorders, including systemic lupus erythematosus, type I diabetes, juvenile rheumatoid arthritis, and Graves’ disease, occur in 42–66 percent of children with NMO [Lotze et al., 2008; McKeon et al., 2008].
Laboratory Features
The most common CSF abnormality in NMO is a marked pleocytosis (typically greater than 50 leukocytes/μL), with either a neutrophilic or lymphocytic predominance [McKeon et al., 2008]. This is in contrast to the moderate lymphocytic pleocytosis typical of MS [Pohl et al., 2004]. CSF IgG oligoclonal bands are detected in less than 10 percent of children with NMO [Banwell et al., 2008; McKeon et al., 2008], as compared to 90 percent of children with MS [Pohl et al., 2004].
Multimodality-evoked potentials often show abnormalities in the visual-evoked potential pathway. In adults with NMO, markedly abnormal or absent visual-evoked potential responses indicate a worse visual prognosis [Watanabe et al., 2009].
Recently, a specific antibody, NMO IgG, has been identified in patients with NMO. NMO IgG is directed against aquaporin 4 water channels, which maintain CNS water homeostasis [Lennon et al., 2004]. This serum biomarker has a sensitivity of 73 percent and a specificity of 91 percent in differentiating NMO from other demyelinating disorders, such as MS, in adult NMO patients (Lennon et al., 2004) and is detected in 78 percent of children with relapsing NMO [Banwell et al., 2008]. NMO IgG has also been detected in children with isolated recurrent ON or TM [Banwell et al., 2008], diseases which are considered part of the NMO spectrum. The exact role of aquaporin 4 and associated antibodies in the pathogenesis of NMO and related disorders is still being explored.
MRI
In adult-onset NMO, brain MRI scans at first presentation are typically normal, apart from optic nerve enhancement in acute ON, or they show nonspecific brain MRI lesions atypical for MS [O’Riordan et al., 1996; Wingerchuk et al., 1999]. Brain MRI lesions appear to be more common in children with NMO as compared to adults, with 53–100 percent of children demonstrating brain abnormalities distinct from the optic nerves at presentation [Banwell et al., 2008; Lotze et al., 2008; McKeon et al., 2008]. Brain MRI lesions typically occur in the distribution of aquaporin 4 water channel expression, including periventricular, thalamic, hypothalamic, and brainstem periaqueductal regions (Figure 72-7). These lesions are symptomatic in approximately 33–45 percent of children, who may experience seizures, menstrual irregularities, hypersomnia, or the syndrome of inappropriate antidiuretic hormone secretion (SIADH) [Lotze et al., 2008; McKeon et al., 2008].
Spinal MRI in adult and pediatric patients with NMO, obtained during an acute attack of TM, typically reveals longitudinally extensive lesions that span three or more contiguous spinal segments (see Figure 72-7) [Wingerchuk et al., 1999; de Seze et al., 2002; Banwell et al., 2008; Lotze et al., 2008]. It is important to note, however, that up to 14 percent of children with MS experiencing an acute attack of TM will have lesions spanning more than three spinal segments [Banwell et al., 2008], and therefore longitudinal extension is not a specific feature for NMO in childhood.
Treatment of NMO in Children
Acute attacks of ON and TM are usually treated with corticosteroids, as outlined in the previous section on MS. If symptoms progress despite steroid therapy, IVIg or plasmapheresis may be considered [Lotze et al., 2008].
While there have been no large randomized clinical trials in either adult- or pediatric-onset NMO, long-term immunosuppression is generally recommended in order to prevent relapses [Wingerchuk and Weinshenker, 2005]. Azathioprine in combination with oral prednisone, IVIg, rituximab, mycophenylate mofetil, and mitoxantrone, has been shown in small case series of adult patients to reduce relapse frequency [Bakker and Metz, 2004; Cree et al., 2005; Mandler, 2006; Jacob et al., 2008].
References
 The complete list of references for this chapter is available online at www.expertconsult.com.
The complete list of references for this chapter is available online at www.expertconsult.com.
Albayram S., Bilgi Z., et al. Diffusion-weighted MR imaging findings of acute necrotizing encephalopathy. Am J Neuroradiol. 2004;25(5):792-797.
Alotaibi S., Kennedy J., et al. Epstein-Barr virus in pediatric multiple sclerosis. JAMA. 2004;291(15):1875-1879.
Atzori M., Battistella P.A., et al. Clinical and diagnostic aspects of multiple sclerosis and acute monophasic encephalomyelitis in pediatric patients: a single centre prospective study. Mult Scler. 2009;15(3):363-370.
Aulchenko Y.S., Hoppenbrouwers I.A., et al. Genetic variation in the KIF1B locus influences susceptibility to multiple sclerosis. Nat Genet. 2008;40(12):1402-1403.
Bakker J., Metz L. Devic’s neuromyelitis optica treated with intravenous gamma globulin (IVIG). Can J Neurol Sci. 2004;31(2):265-267.
Banwell B., Ghezzi A., et al. Multiple sclerosis in children: clinical diagnosis, therapeutic strategies, and future directions. Lancet Neurol. 2007;6(10):887-902.
Banwell B., Krupp L., et al. Clinical features and viral serologies in children with multiple sclerosis: a multinational observational study. Lancet Neurol. 2007;6(9):773-781.
Banwell B., Kennedy J., et al. Incidence of acquired demyelination of the CNS in Canadian children. Neurology. 2009;72(3):232-239.
Banwell B., Reder A.T., et al. Safety and tolerability of interferon beta-1b in pediatric multiple sclerosis. Neurology. 2006;66(4):472-476.
Banwell B., Tenembaum S., et al. Neuromyelitis optica-IgG in childhood inflammatory demyelinating CNS disorders. Neurology. 2008;70(5):344-352.
Barkhof F., Rocca M., et al. Validation of diagnostic magnetic resonance imaging criteria for multiple sclerosis and response to interferon beta1a. Ann Neurol. 2003;53(6):718-724.
Benseler S., Schneider R. Central nervous system vasculitis in children. Curr Opin Rheumatol. 2004;16(1):43-50.
Boiko A., Vorobeychik G., et al. Early onset multiple sclerosis: a longitudinal study. Neurology. 2002;59(7):1006-1010.
Borriello G., Prosperini L., et al. Natalizumab treatment in pediatric multiple sclerosis: a case report. Eur J Paediatr Neurol. 2009;13(1):67-71.
Boyd J.R., MacMillan L.J. Multiple sclerosis in childhood: understanding and caring for children with an “adult” disease. Axone. 2000;22(2):15-21.
Callen D.J., Shroff M.M., et al. Role of MRI in the differentiation of ADEM from MS in children. Neurology. 2009;72(11):968-973.
Callen D.J., Shroff M.M., et al. MRI in the diagnosis of pediatric multiple sclerosis. Neurology. 2009;72(11):961-967.
Charney E.B., Orecchio E.J., et al. Computerized tomography in infantile encephalitis. Am J Dis Child. 1979;133(8):803-805.
Cree B.A., Lamb S., et al. An open label study of the effects of rituximab in neuromyelitis optica. Neurology. 2005;64(7):1270-1272.
Dale R.C., de Sousa C., et al. Acute disseminated encephalomyelitis, multiphasic disseminated encephalomyelitis and multiple sclerosis in children. Brain. 2000;123(Pt 12):2407-2422.
de Seze J., Stojkovic T., et al. Devic’s neuromyelitis optica: clinical, laboratory, MRI and outcome profile. J Neurol Sci. 2002;197(1–2):57-61.
Defresne P., Hollenberg H., et al. Acute transverse myelitis in children: clinical course and prognostic factors. J Child Neurol. 2003;18(6):401-406.
Dunne K., Hopkins I.J., et al. Acute transverse myelopathy in childhood. Dev Med Child Neurol. 1986;28(2):198-204.
Duquette P., Murray T.J., et al. Multiple sclerosis in childhood: clinical profile in 125 patients. J Pediatr. 1987;111(3):359-363.
Durelli L., Verdun E., et al. Every-other-day interferon beta-1b versus once-weekly interferon beta-1a for multiple sclerosis: results of a 2-year prospective randomised multicentre study (INCOMIN). Lancet. 2002;359(9316):1453-1460.
Ebers G.C. Environmental factors and multiple sclerosis. Lancet Neurol. 2008;7(3):268-277.
Freedman M.S., Thompson E.J., et al. Recommended standard of cerebrospinal fluid analysis in the diagnosis of multiple sclerosis: a consensus statement. Arch Neurol. 2005;62(6):865-870.
Frohman E., Costello F., et al. Optical coherence tomography in multiple sclerosis. Lancet Neurol. 2006;5(10):853-863.
Garg R.K. Acute disseminated encephalomyelitis. Postgrad Med J. 2003;79(927):11-17.
Ghezzi A. Clinical characteristics of multiple sclerosis with early onset. Neurol Sci. 2004;25(Suppl 4):S336-S339.
Ghezzi A. Immunomodulatory treatment of early onset multiple sclerosis: results of an Italian Co-operative Study. Neurol Sci. 2005;26(Suppl 4):S183-S186.
Ghezzi A., Amato M.P., et al. Disease-modifying drugs in childhood-juvenile multiple sclerosis: results of an Italian co-operative study. Mult Scler. 2005;11(4):420-424.
Ghezzi A., Bergamaschi R., et al. Clinical characteristics, course and prognosis of relapsing Devic’s Neuromyelitis Optica. J Neurol. 2004;251(1):47-52.
Ghezzi A., Deplano V., et al. Multiple sclerosis in childhood: clinical features of 149 cases. Mult Scler. 1997;3(1):43-46.
Ghezzi A., Pozzilli C., et al. Prospective study of multiple sclerosis with early onset. Mult Scler. 2002;8(2):115-118.
Goodin D.S., Frohman E.M., et al. Disease modifying therapies in multiple sclerosis: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology and the MS Council for Clinical Practice Guidelines. Neurology. 2002;58(2):169-178.
Gorman M.P., Healy B.C., et al. Increased relapse rate in pediatric-onset compared with adult-onset multiple sclerosis. Arch Neurol. 2009;66(1):54-59.
Hafler D.A., Compston A., et al. Risk alleles for multiple sclerosis identified by a genomewide study. N Engl J Med. 2007;357(9):851-862.
Hahn C.D., Miles B.S., et al. Neurocognitive outcome after acute disseminated encephalomyelitis. Pediatr Neurol. 2003;29(2):117-123.
Hahn C.D., Shroff M.M., et al. MRI criteria for multiple sclerosis: Evaluation in a pediatric cohort. Neurology. 2004;62(5):806-808.
Hahn J.S., Siegler D.J., et al. Intravenous gammaglobulin therapy in recurrent acute disseminated encephalomyelitis. Neurology. 1996;46(4):1173-1174.
Hanwell H.E., Veith R., et al. Serum 25-hydroxyvitamin D status as a determinant of multiple sclerosis outcome following an initial demyelinating event in children (abstract). Multiple Sclerosis. 2008;14:S5-127.
Huppke P., Stark W., et al. Natalizumab use in pediatric multiple sclerosis. Arch Neurol. 2008;65(12):1655-1658.
Hwang J.M., Lee Y.J., et al. Optic neuritis in Asian children. J Pediatr Ophthalmol Strabismus. 2002;39(1):26-32.
IFNB Multiple Sclerosis Study Group, T. and T. University of British Columbia MS/MRI Analysis Group. Interferon beta-1b in the treatment of multiple sclerosis: final outcome of the randomized controlled trial. Neurology. 1995;45(7):1277-1285.
Jacob A., Weinshenker B.G., et al. Treatment of neuromyelitis optica with rituximab: retrospective analysis of 25 patients. Arch Neurol. 2008;65(11):1443-1448.
Kappos L., Traboulsee A., et al. Long-term subcutaneous interferon beta-1a therapy in patients with relapsing-remitting MS. Neurology. 2006;67(6):944-953.
Kim J.H., Kim I.O., et al. Acute necrotizing encephalopathy in Korean infants and children: imaging findings and diverse clinical outcome. Korean J Radiol. 2004;5(3):171-177.
Koch-Henriksen N., Sorensen P.S., et al. A randomized study of two interferon-beta treatments in relapsing-remitting multiple sclerosis. Neurology. 2006;66(7):1056-1060.
Kornek B., Bernert G., et al. Glatiramer acetate treatment in patients with childhood and juvenile onset multiple sclerosis. Neuropediatrics. 2003;34(3):120-126.
Krupp L.B., Banwell B., et al. Consensus definitions proposed for pediatric multiple sclerosis and related disorders. Neurology. 2007;68(16 Suppl 2):S7-S12.
Lennon V.A., Wingerchuk D.M., et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet. 2004;364(9451):2106-2112.
Lotze T.E., Northrop J.L., et al. Spectrum of pediatric neuromyelitis optica. Pediatrics. 2008;122(5):e1039-e1047.
Lucchinetti C.F., Kiers L., et al. Risk factors for developing multiple sclerosis after childhood optic neuritis. Neurology. 1997;49(5):1413-1418.
MacAllister W.S., Boyd J.R., et al. The psychosocial consequences of pediatric multiple sclerosis. Neurology. 2007;68(16 Suppl 2):S66-S69.
Makhani N., Gorman M.P., et al. Cyclophosphamide therapy in pediatric multiple sclerosis. Neurology. 2009.
Mandler R.N. Neuromyelitis optica – Devic’s syndrome, update. Autoimmun Rev. 2006;5(8):537-543.
Marco E.J., Anderson J.E., Neilson D.E., et al. Acute necrotizing encephalopathy in 3 brothers. Pediatrics. 2010;125(3):e693-e698. Epub 2010 Feb 8
Mastroyianni S.D., Gionnis D., et al. Acute necrotizing encephalopathy of childhood in non-Asian patients: report of three cases and literature review. J Child Neurol. 2006;21(10):872-879.
McDonald W.I., Compston A., et al. Recommended diagnostic criteria for multiple sclerosis: guidelines from the International Panel on the diagnosis of multiple sclerosis. Ann Neurol. 2001;50(1):121-127.
McKeon A., Lennon V.A., et al. CNS aquaporin-4 autoimmunity in children. Neurology. 2008;71(2):93-100.
Mikaeloff Y., Suissa S., et al. First episode of acute CNS inflammatory demyelination in childhood: prognostic factors for multiple sclerosis and disability. J Pediatr. 2004;144(2):246-252.
Mikaeloff Y., Adamsbaum C., et al. MRI prognostic factors for relapse after acute CNS inflammatory demyelination in childhood. Brain. 2004;127(Pt 9):1942-1947.
Mikol D.D., Barkhof F., et al. Comparison of subcutaneous interferon beta-1a with glatiramer acetate in patients with relapsing multiple sclerosis (the REbif vs Glatiramer Acetate in Relapsing MS Disease [REGARD] study): a multicentre, randomised, parallel, open-label trial. Lancet Neurol. 2008;7(10):903-914.
Miller D.H., Robb S.A., et al. Magnetic resonance imaging of inflammatory and demyelinating white-matter diseases of childhood. Dev Med Child Neurol. 1990;32(2):97-107.
Miyazawa R., Ikeuchi Y., et al. Determinants of prognosis of acute transverse myelitis in children. Pediatr Int. 2003;45(5):512-516.
Mizuguchi M. Acute necrotizing encephalopathy of childhood: a novel form of acute encephalopathy prevalent in Japan and Taiwan. Brain Dev. 1997;19(2):81-92.
Mizuguchi M., Abe J., et al. Acute necrotising encephalopathy of childhood: a new syndrome presenting with multifocal, symmetric brain lesions. J Neurol Neurosurg Psychiatry. 1995;58(5):555-561.
Mizuguchi M., Hayashi M., et al. Concentric structure of thalamic lesions in acute necrotizing encephalopathy. Neuroradiology. 2002;44(6):489-493.
Mizuta R., Izumi H., et al. A case of Reye’s syndrome with elevation in influenza A, CF antibody. Japanese Journal of Pediatrics. 1979(32):2144-2149.
Murthy S.N., Faden H.S., et al. Acute disseminated encephalomyelitis in children. Pediatrics. 2002;110(2 Pt 1):e21.
Neilson D.E., Adams M.D., Orr C.M., et al. Infection-triggered familial or recurrent cases of acute necrotizing encephalopathy caused by mutations in a component of the nuclear pore, RANBP2. Am J Hum Genet. 2009;84(1):44-51.
Nishikawa M., Ichiyama T., et al. Intravenous immunoglobulin therapy in acute disseminated encephalomyelitis. Pediatr Neurol. 1999;21(2):583-586.
O’Riordan J.I., Gallagher H.L., et al. Clinical, CSF, and MRI findings in Devic’s neuromyelitis optica. J Neurol Neurosurg Psychiatry. 1996;60(4):382-387.
Okumura A., Kidokoro H., et al. The mildest form of acute necrotizing encephalopathy associated with influenza A. Neuropediatrics. 2006;37(4):261-263.
Okumura A., Mizuguchi M., et al. Outcome of acute necrotizing encephalopathy in relation to treatment with corticosteroids and gammaglobulin. Brain Dev. 2009;31(3):221-227.
Ozakbas S., Idiman E., et al. Childhood and juvenile onset multiple sclerosis: clinical and paraclinical features. Brain Dev. 2003;25(4):233-236.
Paty D.W., Li D.K. Interferon beta-1b is effective in relapsing-remitting multiple sclerosis. II. MRI analysis results of a multicenter, randomized, double-blind, placebo-controlled trial. UBC MS/MRI Study Group and the IFNB Multiple Sclerosis Study Group. Neurology. 1993;43(4):662-667.
Pidcock F.S., Krishnan C., et al. Acute transverse myelitis in childhood: center-based analysis of 47 cases. Neurology. 2007;68(18):1474-1480.
Pohl D., Hennemuth I., et al. Paediatric multiple sclerosis and acute disseminated encephalomyelitis in Germany: results of a nationwide survey. Eur J Pediatr. 2007;166(5):405-412.
Pohl D., Rostasy K., et al. Pediatric multiple sclerosis: detection of clinically silent lesions by multimodal evoked potentials. J Pediatr. 2006;149(1):125-127.
Pohl D., Krone B., et al. High seroprevalence of Epstein-Barr virus in children with multiple sclerosis. Neurology. 2006;67(11):2063-2065.
Pohl D., Rostasy K., et al. Treatment of early onset multiple sclerosis with subcutaneous interferon beta-1a. Neurology. 2005;64(5):888-890.
Pohl D., Rostasy K., et al. CSF characteristics in early-onset multiple sclerosis. Neurology. 2004;63(10):1966-1967.
Polman C.H., Reingold S.C., et al. Diagnostic criteria for multiple sclerosis: 2005 revisions to the “McDonald Criteria”. Ann Neurol. 2005;58(6):840-846.
Poser C.M., Paty D.W., et al. New diagnostic criteria for multiple sclerosis: guidelines for research protocols. Ann Neurol. 1983;13(3):227-231.
PRISMS (Prevention of Relapses and Disability by Interferon beta-1a Subcutaneously in Multiple Sclerosis) Study Group, T. Randomised double-blind placebo-controlled study of interferon beta-1a in relapsing/remitting multiple sclerosis. Lancet. 1998;352(9139):1498-1504.
Ramagopalan S.V., Ebers G.C. Genes for multiple sclerosis. Lancet. 2008;371(9609):283-285.
Ramagopalan S.V., Maugeri N.J., et al. Expression of the multiple sclerosis-associated MHC class II Allele HLA-DRB1*1501 is regulated by vitamin D. PLoS Genet. 2009;5(2):e1000369.
Reiber H., Peter J.B. Cerebrospinal fluid analysis: disease-related data patterns and evaluation programs. J Neurol Sci. 2001;184(2):101-122.
Robertson N.P., Fraser M., et al. Age-adjusted recurrence risks for relatives of patients with multiple sclerosis. Brain. 1996;119(Pt 2):449-455.
Rust R.S. Multiple sclerosis, acute disseminated encephalomyelitis, and related conditions. Semin Pediatr Neurol. 2000;7(2):66-90.
Sadovnick A.D., Ebers G.C., et al. Evidence for genetic basis of multiple sclerosis. The Canadian Collaborative Study Group. Lancet. 1996;347(9017):1728-1730.
Schwarz S., Mohr A., et al. Acute disseminated encephalomyelitis: a follow-up study of 40 adult patients. Neurology. 2001;56(10):1313-1318.
Simone I.L., Carrara D., et al. Course and prognosis in early-onset MS: comparison with adult-onset forms. Neurology. 2002;59(12):1922-1928.
Sindern E., Haas J., et al. Early onset MS under the age of 16: clinical and paraclinical features. Acta Neurol Scand. 1992;86(3):280-284.
Soilu-Hanninen M., Airas L., et al. 25-Hydroxyvitamin D levels in serum at the onset of multiple sclerosis. Mult Scler. 2005;11(3):266-271.
Tenembaum S., Chamoles N., et al. Acute disseminated encephalomyelitis: a long-term follow-up study of 84 pediatric patients. Neurology. 2002;59(8):1224-1231.
Tenembaum S.N., Segura M.J. Interferon beta-1a treatment in childhood and juvenile-onset multiple sclerosis. Neurology. 2006;67(3):511-513.
Watanabe A., Matsushita T., et al. Multimodality-evoked potential study of anti-aquaporin-4 antibody-positive and -negative multiple sclerosis patients. J Neurol Sci. 2009;281(1-2):34-40.
Waubant E., Hietpas J., et al. Interferon beta-1a in children with multiple sclerosis is well tolerated. Neuropediatrics. 2001;32(4):211-213.
Weinshenker B.G. Plasma exchange for severe attacks of inflammatory demyelinating diseases of the central nervous system. J Clin Apher. 2001;16(1):39-42.
Weinshenker B.G., O’Brien P.C., et al. A randomized trial of plasma exchange in acute central nervous system inflammatory demyelinating disease. Ann Neurol. 1999;46(6):878-886.
Weitkamp J.H., Spring M.D., et al. Influenza A virus-associated acute necrotizing encephalopathy in the United States. Pediatr Infect Dis J. 2004;23(3):259-263.
Wilejto M., Shroff M., et al. The clinical features, MRI findings, and outcome of optic neuritis in children. Neurology. 2006;67(2):258-262.
Wingerchuk D.M. Neuromyelitis optica: effect of gender. J Neurol Sci. 2009;286(1-2):18-23.
Wingerchuk D.M., Hogancamp W.F., et al. The clinical course of neuromyelitis optica (Devic’s syndrome). Neurology. 1999;53(5):1107-1114.
Wingerchuk D.M., Lennon V.A., et al. Revised diagnostic criteria for neuromyelitis optica. Neurology. 2006;66(10):1485-1489.
Wingerchuk D.M., Weinshenker B.G. Neuromyelitis Optica. Curr Treat Options Neurol. 2005;7(3):173-182.
Wolinsky J.S., Comi G., et al. Copaxone’s effect on MRI-monitored disease in relapsing MS is reproducible and sustained. Neurology. 2002;59(8):1284-1286.
Wong A.M., Simon E.M., et al. Acute necrotizing encephalopathy of childhood: correlation of MR findings and clinical outcome. Am J Neuroradiol. 2006;27(9):1919-1923.
Wucherpfennig K.W., Strominger J.L. Molecular mimicry in T cell-mediated autoimmunity: viral peptides activate human T cell clones specific for myelin basic protein. Cell. 1995;80(5):695-705.
Yeh E.A., Weinstock-Guttman B., et al. Retinal nerve fiber thickness in inflammatory demyelinating diseases of childhood onset. Mult Scler. 2009;15(7):802-810.
Yeh E.A., Chitnis T., et al. Pediatric multiple sclerosis. Nat Rev Neurol. 2009;5(11):621-631.
Yoshikawa H., Watanabe T., et al. Clinical diversity in acute necrotizing encephalopathy. J Child Neurol. 1999;14(4):249-255.
Zaffaroni M. Cerebrospinal fluid findings in Devic’s neuromyelitis optica. Neurol Sci. 2004;25(Suppl 4):S368-S370.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Chapter 72 Acquired Disorders Affecting the White Matter
Acute CNS Demyelination
A first demyelinating attack in childhood (referred to as an acquired demyelinating syndrome or ADS) may present either with neurological signs and symptoms attributable to a single CNS lesion (monofocal ADS), or with signs attributable to multiple CNS sites (polyfocal ADS). If polyfocal symptoms are accompanied by encephalopathy, the diagnosis of acute disseminated encephalomyelitis (ADEM) is conferred [Krupp et al., 2007].
The frequency of ADS presentations differs by patient age, and not all ADS presentations are equivalent in terms of subsequent MS risk. Data from a Canadian study of 219 children with ADS revealed the most common ADS presentation to be optic neuritis (23 percent), followed by ADEM (22 percent) and transverse myelitis (22 percent) [Banwell et al., 2009]. Children with ADEM were more likely to be under 10 years of age, whereas children with monofocal ADS (optic neuritis, transverse myelitis, and others) were more likely to be over 10 years of age.
Optic Neuritis
Optic neuritis (ON) should be considered in the differential diagnosis of any child presenting with acute or subacute visual loss (Figure 72-1). Typical clinical features of demyelinating ON include reduced visual acuity, a central visual field deficit (not always testable in young children), pain with eye movements, and red color desaturation [Lucchinetti et al., 1997]. Optic disc edema is sometimes seen, but this finding may be absent in retrobulbar ON. Concurrent or rapidly sequential transverse myelitis, magnetic resonance imaging (MRI) features of brainstem or diencephalic involvement in a pattern atypical for MS, and serum antibodies against aquaporin 4 support a diagnosis of NMO (discussed later in this chapter) [Wingerchuk et al., 2006].
In a study of 36 children aged 2–17 years, ON was found to be more commonly unilateral (58 percent) than bilateral (42 percent) [Wilejto et al., 2006]. Neurological abnormalities apart from the optic nerve were common, affecting 13 children (36 percent). Neuroimaging studies of the optic nerve were abnormal in 55 percent, and 88 percent had abnormal visual-evoked potentials.
Optical coherence tomography (OCT) is a noninvasive technique that utilizes near-infrared light to measure retinal nerve fiber thickness, yielding a quantitative measure of axonal loss. This technique is currently being explored in both adults and children with ADS and MS as a way to provide an estimate of visual prognosis following ON [Frohman et al., 2006; Yeh et al., 2009a].
Following an episode of ON in childhood, approximately 80–85 percent of children achieve a full visual recovery [Hwang et al., 2002; Wilejto et al., 2006]. Children with ON have about a 36 percent risk of subsequent MS diagnosis overall, and MS may be more common in children presenting with bilateral optic nerve involvement [Lucchinetti et al., 1997; Wilejto et al., 2006]. MRI evidence of one or more T2-weighted lesions separate from the optic nerve is strongly predictive of MS risk, with up to 68 percent of such patients being diagnosed with MS within 2 years of their initial ON [Wilejto et al., 2006].
Transverse Myelitis
Demyelination of the spinal cord, accompanied by clinical signs of partial or complete cord impairment, is referred to as transverse myelitis (TM). The typical clinical features of TM include subacute onset of bilateral lower-extremity weakness, a spinal sensory level, and impaired bowel or bladder control. (Lesions above the sacral spine usually cause urinary retention and constipation.) L’Hermitte’s symptom (pain with forward neck flexion) frequently indicates a cervical spinal cord lesion. Paresis is often initially flaccid with associated hyporeflexia, and later hyperreflexia develops below the level of the lesion. These features are helpful in distinguishing TM from other diseases affecting the spinal cord (Figure 72-2).
A review of 47 patients with TM showed that, at the time of maximal deficit, 89 percent of children either were unable to walk or required assistance with ventilation (or both) [Pidcock et al., 2007]. After 3 years of follow-up, 43 percent were unable to ambulate more than 30 feet, and 21 percent required a walker or ambulatory aid. Sixty-eight percent of children had residual bladder symptoms and 50 percent required bladder catheterization. Younger age at the onset of the TM event, complete paraplegia, and a shorter time (less than 24 hours) to maximal deficit have been associated with poorer functional outcomes [Dunne et al., 1986; Miyazawa et al., 2003; Pidcock et al., 2007]. The risk of MS following acute TM is approximately 2–8 percent [Dunne et al., 1986; Defresne et al., 2003; Miyazawa et al., 2003; Mikaeloff et al., 2004a; Pidcock et al., 2007].
Polyfocal Demyelination
Neurological symptoms suggesting multiple CNS areas of involvement may occur with or without concurrent encephalopathy (Figure 72-3). When encephalopathy (either behavioral change or alteration in level of consciousness) is present, the clinical syndrome is referred to as acute disseminated encephalomyelitis (ADEM) [Krupp et al., 2007]. ADEM tends to occur in younger patients, especially those under the age of 10 [Tenembaum et al., 2002]. Fever and a recent history of infection are often present [Rust, 2000; Tenembaum et al., 2002].
Recovery from ADEM can be incomplete, with 11–43 percent of children experiencing residual motor deficits [Dale et al., 2000; Tenembaum et al., 2002]. Cognitive deficits are often present, even in children who experience a full motor recovery [Hahn et al., 2003].
ADEM is most often a monophasic illness. However, approximately 10–29 percent of children with an initial presentation of ADEM will go on to have additional demyelinating attacks [Tenembaum et al., 2002; Mikaeloff et al., 2004]. The classification of further attacks of demyelination in children initially presenting with ADEM is reviewed later in this chapter.
Investigation of a Child with Acute Demyelination
Laboratory Investigations
Cerebrospinal fluid (CSF) analysis is important for excluding diseases other than demyelination, including infection and malignancy. CSF leukocyte counts in children with MS at first presentation are elevated (>4 cells/μL) in 66 percent of children and are typically below 30 cells/μL [Pohl et al., 2004]. Higher leukocyte counts are more consistent with acute infection, vasculitis, or NMO [Benseler and Schneider, 2004; Zaffaroni, 2004]. Elevated protein in the range of 100–720 mg/L may be seen, indicating disruption of the blood–CSF barrier [Pohl et al., 2004]. CSF oligoclonal bands (OCBs) are detected at the time of ADS in approximately 90 percent of children who are subsequently diagnosed with MS [Sindern et al., 1992; Ghezzi et al., 2002; Ozakbas et al., 2003; Pohl et al., 2004], which is slightly lower than the reported detection rate of 98 percent in adult MS patients [Reiber and Peter, 2001]. Isoelectric focusing methods have the highest yield for oligoclonal band detection [Freedman et al., 2005]. In contrast, CSF OCBs are detected in fewer than 10 percent of children with either ADEM [Tenembaum et al., 2002; Atzori et al., 2009] or NMO [Banwell et al., 2008; McKeon et al., 2008]. It should be noted that CSF immunoglobulin G synthesis can occur later in the disease course. Consequently, some MS patients who have initially negative CSF studies will later have detectable OCBs.
Evoked potential testing in the visual-evoked potential, brainstem auditory-evoked potential, and somatosensory-evoked potential pathways is useful for confirming the presence of demyelination and for detecting clinically silent disease. A study of 47 children with ON showed that visual-evoked potentials were abnormal in 88 percent [Wilejto et al., 2006]. In one study, almost 50 percent of 85 childhood patients with MS demonstrated clinically silent abnormalities in at least one of the evoked potential pathways, most commonly in the visual pathways [Pohl et al., 2006a].
Magnetic Resonance Imaging
MRI is a valuable tool to demonstrate the presence of inflammatory demyelination and to exclude other diagnoses [Miller et al., 1990] (Figure 72-4). Large lesions, ill-defined lesions, and lesions present in gray as well as white matter commonly occur in younger children, especially those with ADEM [Murthy et al., 2002; Garg, 2003; Mikaeloff et al., 2004b]. Several groups have highlighted the challenge in identifying MRI features sensitive enough to distinguish ADEM from MS at first attack [Schwarz et al., 2001; Mikaeloff et al., 2004b]. Recently proposed criteria suggest that the following features may be useful in making this distinction:
Serial MRI studies have been useful in showing dissemination in space within the CNS and time, and MRI characteristics have been incorporated into the diagnostic criteria for adult-onset and childhood-onset MS [McDonald et al., 2001; Barkhof et al., 2003; Polman et al., 2005; Krupp et al., 2007]. These criteria have between 52 and 76 percent sensitivity for distinguishing childhood MS from other diseases at first attack [Hahn et al., 2004; Mikaeloff et al., 2004b; Callen et al., 2009b]. Proposed revisions to these criteria for childhood MS have suggested that fewer lesions (≥5 T2 lesions, and either ≥2 periventricular lesions or ≥1 brainstem lesion) may be able to differentiate MS from other neurological disorders at first presentation with improved sensitivity and specificity [Callen et al., 2009b], but further validation is required.
Management of Acute Demyelination
Acute demyelination in children is generally treated with corticosteroids if symptoms are severe enough to interfere with daily functioning (Figure 72-5). While there are no published standardized trials for steroid therapy in children, typically 20–30 mg/kg/day of intravenous methylprednisolone (maximum 1 g) is given as a single daily intravenous dose for 3–5 days. If there is a significant improvement in symptoms, no further treatment is required. If there is an incomplete response to treatment but the symptoms are relatively mild, a tapering course of oral steroids (starting at 1 mg/kg/day and tapering over 21 days) is often considered. If children have significant residual symptoms after a course of intravenous steroids, a second 3–5-day course may be given at the same doses as outlined above. Alternatively, treatment with intravenous immunoglobulin (IVIg) at a dose of 2 g/kg over 2–5 days may be of benefit (class IV evidence) [Hahn et al., 1996; Nishikawa et al., 1999].
Fig. 72-5 Approach to the management of acute demyelination in childhood.
IV, intravenous; IVIG, intravenous immunoglobulin; PLEX, plasma exchange.
(Adapted from Thomas T, Banwell B. Multiple sclerosis in children. Semin Neurol 2008;28[1]:69–83.)
Rarely, acute demyelination in children can be life-threatening. Profound encephalopathy and respiratory depression may occur with brainstem and upper cervical spinal cord involvement. Plasma exchange may be useful in this situation, based on class I evidence in adult-onset MS [Weinshenker et al., 1999; Weinshenker, 2001]. A typical regimen of plasma exchange treatment would be 5–8 exchanges given over 10 days.
Acute Necrotizing Encephalopathy of Childhood
An acute necrotizing encephalopathy (ANE), with elevated serum aminotransaminases and bilateral symmetric thalamic lesions on neuroimaging, was first described in a Japanese child in 1979 (Mizuta et al., 1979), and since then there have been over 200 reports in the literature. The majority of children with ANE originate from the Far East, especially Japan, Taiwan, and Korea [Mizuguchi, 1997; Kim et al., 2004], with fewer cases reported in North America or Europe [Mastroyianni et al., 2006]. Recently, an affected kindred was described, representing the only case of male siblings with consanguineous parents [Marco et al., 2010] described in the United States. Familial (ANE1) and recurrent cases have recently been linked to mutations in RANBP2 (RAN-binding protein 2) [Neilson et al., 2009].
ANE typically affects children under the age of 5 [Mizuguchi, 1997; Mastroyianni et al., 2006]; in one series of 62, predominantly Japanese, patients, 46 percent of children were between 6 and 18 months of age [Mizuguchi et al., 1995]. In this same series, symptom onset occurred in winter months (December through February) in 51 percent of cases. Boys and girls appear to be equally affected [Mizuguchi et al., 1995; Mastroyianni et al., 2006].
A prodromal febrile illness is reported in up to 90 percent of children [Mizuguchi et al., 1995] and concurrent influenza A, influenza B, human herpesvirus (HHV)-6, Mycoplasma pneumoniae, herpes simplex, or measles infection has been documented in some patients [Mizuguchi, 1997; Mastroyianni et al., 2006]. Symptoms of an acute encephalopathy typically evolve 12–72 hours later and include decreased level of consciousness (28 percent), vomiting (20 percent), and seizures (40 percent) [Mizuguchi, 1997]. The disease is rapidly progressive and coma may ensue in less than 24 hours. Upper motor neuron signs, including hyperreflexia and extensor plantar responses, are seen acutely in two-thirds of patients [Mizuguchi, 1997; Mastroyianni et al., 2006].
Laboratory findings and neuroimaging features are helpful in distinguishing ANE from other conditions that may present in a similar manner, including ADEM (see earlier in this chapter). Laboratory findings in ANE include elevated serum aspartate aminotransferase, alanine aminotransferase, and lactate dehydrogenase in 50–95 percent of children [Mizuguchi, 1997; Mastroyianni et al., 2006]. Elevations in serum ammonia and bilirubin are uncommon. Metabolic acidosis may be present over 80 percent of children [Mizuguchi, 1997]. CSF examination demonstrates an opening pressure greater than 15 cm H2O with elevated CSF protein (>40 mg/dL) in the absence of pleocytosis in more than two-thirds of patients [Mizuguchi, 1997; Mastroyianni et al., 2006].
Neuroimaging characteristically reveals bilateral symmetric thalamic lesions, often accompanied by brainstem tegmentum, periventricular white-matter, putaminal, and cerebellar lesions. These lesions are initially hypodense on computed tomography (CT) and hypointense on T2-weighted MRI, later evolving into concentric lesions with a central area of hemorrhage that appears hyperdense on CT or hyperintense on T1-weighted MRI images [Mizuguchi et al., 1995; Mizuguchi, 1997; Mizuguchi et al., 2002]. Contrast enhancement around the area of hemorrhage becomes apparent by 2 weeks [Wong et al., 2006], and cavitation occurs in 50 percent of lesions [Kim et al., 2004; Wong et al., 2006]. Areas of diffusion restriction within acute lesions have been described [Albayram et al., 2004].
The outcome from ANE has historically been considered poor, with death in the acute phase occurring in up to one-third of patients [Mizuguchi, 1997]; however, more recently, patients with milder disease and fewer residual neurological sequelae have been reported [Kim et al., 2004; Okumura et al., 2006]. Patients may be left with residual neurological signs, including focal weakness, extraocular movement abnormalities, extrapyramidal movement disorders, or ataxia related to the sites of brain involvement visualized on MRI [Mizuguchi, 1997; Mastroyianni et al., 2006; Wong et al., 2006]. A normal CSF protein in the acute phase and lack of brainstem lesions on neuroimaging appear to be correlated with a more favorable long-term neurological outcome [Charney et al., 1979; Mizuguchi, 1997; Yoshikawa et al., 1999; Weitkamp et al., 2004; Wong et al., 2006].
[/not-level-membership-for-neurology-category]
