[level-membership-for-pathology-category]
CHAPTER 33 Acquired disorders affecting megakaryocytes and platelets
Structure and function of megakaryocytes and platelets
Human platelets
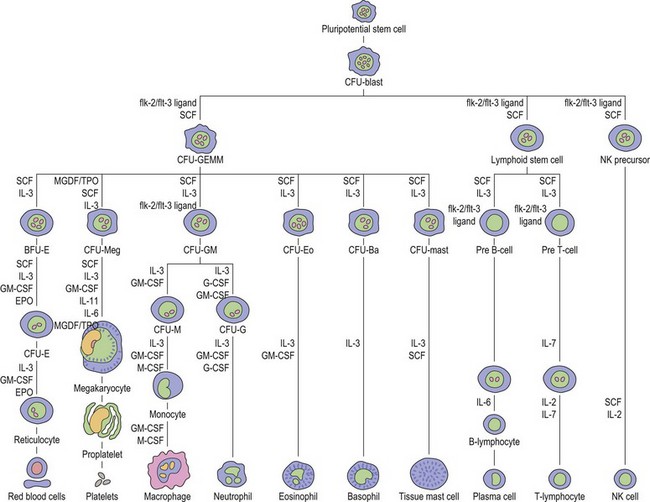
Thrombopoiesis, the generation of platelets from megakaryocytes in the bone marrow, is complex and incompletely understood. Megakaryocytes are large end-stage cells from which platelets bud. The earliest recognized committed progenitor is the burst-forming unit (BFU)-Meg.1 Fig. 33.1 shows megakaryocyte development from stem cell stage through to platelet production. BFU-Megs develop into colony-forming unit (CFU)-Megs in the presence of growth factors thrombopoietin (TPO), interleukin-3 (IL-3) and IL-11. Megakaryocyte nuclei are large polyploid structures with chromosome contents between diploid (2N) to 64N. Such polyploid status is achieved through a process termed nuclear endoduplication: that is, successive doubling of chromosome content in the absence of cell division. Platelets are produced from megakaryocytes that are 8N or greater.2 A single megakaryocyte can generate around 3000 platelets of which 20–30% are pooled in the spleen. In health the peripheral blood platelet count is 150–400 × 109/l but this fluctuates, for example, following heavy exercise, ‘stress’, and around the menstrual cycle. This transient rise in platelet count may be caused by mobilization of platelets pooled in the spleen. There are also racial differences in the ‘normal’ platelet count and some Mediterranean populations have platelet counts as low as 80 × 109/l in health. Platelets are produced at a rate of 35 000–44 000 per microliter per day3 and have a lifespan of 9–10 days.
Platelet structure and function
The integrin family of proteins
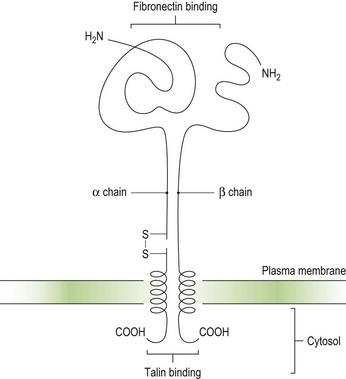
Integrins are key platelet membrane proteins and have been characterized on a large number of leukocytes and many other cells. For example, the fibronectin receptor on mammalian fibroblasts is one of the best characterized matrix receptor proteins4 (Fig. 33.2). This receptor, in common with all other integrins, is a heterodimer consisting of a non-covalently associated complex of two distinct high-molecular-weight polypeptides, α and β. The receptor functions as a transmembrane linker which mediates the interaction between the intracellular actin cytoskeleton and fibronectin in the extracellular matrix. Like all integrins, the fibronectin receptor recognizes so-called RGD (Arg-Gly-Asp) sequences in matrix components. In platelets, integrins recognize and bind a variety of proteins in order to form a hemostatic plug through a complex mechanism of platelet adhesion, shape change and activation of the clotting pathway.
Platelet integrins and related proteins
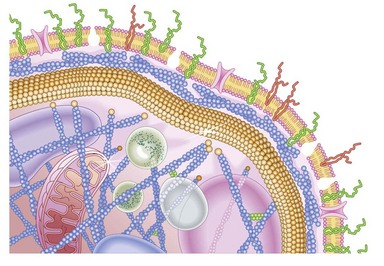
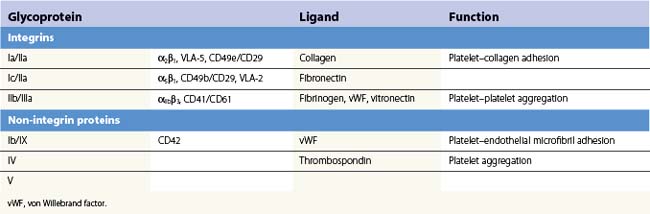
Platelets contain five integrin α subunits and two β subunits producing: αIIbβ3, ανβ3, α2β1, α5β1 and α6β1.5,6,7 These are shown in Fig. 33.3. Further detail is provided by Table 33.1. These proteins are essential for normal platelet function, and are often the target of immunological attack in disorders such as idiopathic thrombocytopenic purpura (ITP).
Platelet alloantigens
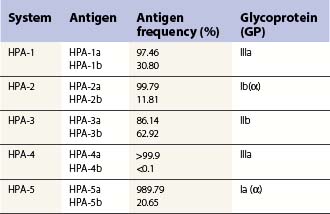
These can be platelet-specific or shared with other cells. Important shared antigens include HLA class I and ABH (blood group A and B) antigens. Platelet-specific antigens fall into five well-defined human platelet antigen (HPA) groups (Table 33.2; see also Chapter 37): HPA-1, HPA-2, HPA-3, HPA-4 and HPA-5, each of which has an α and β allele. Each platelet allotype represents a single amino acid substitution in the platelet glycoprotein molecule. Because some platelet glycoproteins carry epitopes that play a major role in platelet function, platelet alloantibodies may not only cause thrombocytopenia but also affect primary hemostasis.
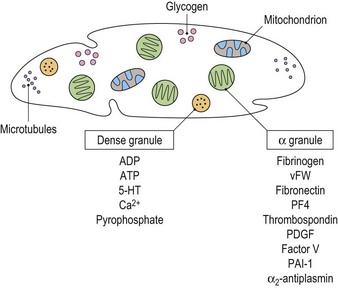
Cytoplasmic platelet constituents
Platelets contain two principal types of granule: dense bodies and α granules (Fig. 33.4). Dense bodies contain ADP, ATP, 5-HT, calcium and pyrophosphate. The α granules contain more than 300 releasable proteins including adhesion molecules, chemokines, cytokines, coagulation factors, fibrinolytic regulators, growth factors and pro- and anti-angiogenic factors such as PF4, β-thrombospondin, PDGF, vWF, fibrinogen, factor V and fibronectin. The contents of these granules are integral components of the platelet’s biological activities.
Biological function of platelets
The primary role of the platelet is the prevention of blood loss from damaged tissues and vessels, i.e. primary hemostasis. This is achieved through platelet activation, adhesion, shape change and aggregation. Platelets may also play a role in the maintenance of vascular integrity by the constitutive release of cytokines and growth factors from their granules that bind to endothelial cell surface receptors resulting in intracellular signaling that stabilizes the molecular complexes that form the junctions between adjacent endothelial cells.8
Platelet adhesion (Fig. 33.5A)
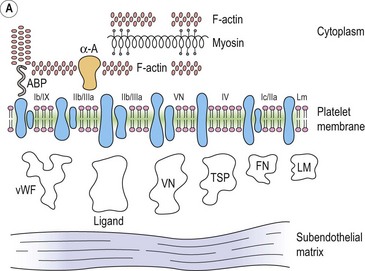
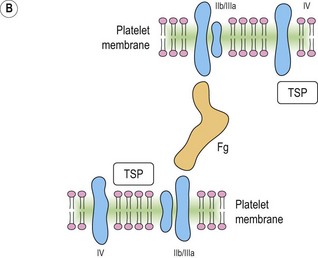
Platelet shape change
After adhesion to the subendothelium, platelets undergo a major shape change, from a discoid shape to one which is irregular, with projections (Fig. 33.6). This process is initially reversible but ultimately becomes irreversible.
Quantitative platelet abnormalities: thrombocytopenia
Thrombocytopenia, a reduction in platelet count, may be caused by:
Pseudothrombocytopenia
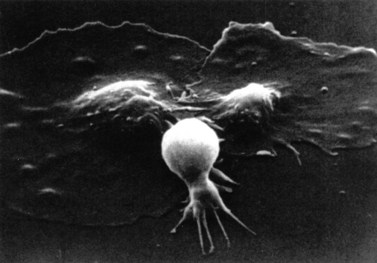
This describes patients in whom the peripheral blood platelet count is found to be spuriously low, and is caused by a variety of mechanisms. Platelet clumping caused by the anticoagulant ethylenediaminetetraacetic acid (EDTA) is the commonest cause, with an incidence of around 0.1%. EDTA-induced pseudothrombocytopenia is easily excluded by examination of a blood film which will confirm the presence of numerous large platelet clumps.9,10 Other causes of pseudothrombocytopenia include the presence of giant platelets, which are not counted as platelets by modern automated counters, and platelet satellitism (platelets attach themselves to monocytes or granulocytes) (Fig. 33.7).
Pooling of platelets in the spleen
Mechanism involved
In health, the spleen may pool up to one third of the total platelet mass, and in disease states this may rise to 90%.11 Although the peripheral blood platelet count may only be a fraction of the normal range, the patient generally has an overall normal platelet mass since production is entirely normal but the low peripheral counts simply reflect a larger than normal mass of platelets pooled in the spleen.
Thrombocytopenia due to failure of platelet production
Acquired amegakaryocytic thrombocytopenia
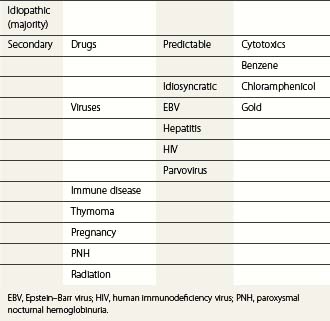
This describes severe thrombocytopenia caused by a selective reduction in megakaryocytes in an otherwise normal bone marrow, and is the result of damage to the megakaryocyte stem cell. The disorder is analogous to acquired pure red-cell aplasia (PRCA). Acquired amegakaryocytic thrombocytopenia may be caused by drugs, toxins and connective tissue disorders.12–14
Bone marrow failure syndromes
The bone marrow failure (BMF) disorders are those in which there is a failure of bone marrow precursor (stem) cells in contrast to those disorders such as myelodysplastic syndromes in which normal or increased numbers of abnormal cells are produced, or those in which the survival of the cells is reduced. The BMF syndromes are a diverse groups of disorders with a common endpoint in which there is loss of hematopoietic stem cells. BMF can be acquired or inherited (see Table 33.3).
Thrombocytopenia due to increased platelet destruction
Causes may be immunologic or non-immunologic.
Non-immunologic causes of thrombocytopenia
Disseminated intravascular coagulation (DIC)
DIC is characterized by excessive activation of the coagulation cascade (see also Chapter 35). In most cases DIC is an acute event, but chronic DIC is well described, although clinically less important. The main problem faced in patients with DIC is bleeding, which may be mild but is often severe with generalized oozing from venepuncture sites, central lines and other indwelling cannulae, gastrointestinal and genitourinary tracts. Microthrombi are found in 5–10% of cases, often affecting digits, with resulting peripheral gangrene.
Pathogenesis
DIC is triggered by the release or exposure of tissue thromboplastins which contain a high concentration of phospholipids following trauma, surgery, mismatched blood transfusion and a variety of other triggers (Table 33.4). In addition to systemic activation of coagulation (leading to fibrin clot formation, organ failure and consumption of platelets and coagulation factors that may cause bleeding), there is dysregulation of natural anticoagulant systems and fibrinolysis.
Table 33.4 Triggers for disseminated intravascular coagulation (DIC)
| Trauma | Including surgical |
| Dissemination of cancer cells | Malignancy, following administration of chemotherapy |
| Massive hemolysis | Post mismatched blood transfusion |
| Venoms | e.g. snake venoms |
| Endothelial injury | Gram-negative sepsis |
| Infections | |
| Burns | |
| Septicemia |
Thrombotic thrombocytopenic purpura and hemolytic uremic syndrome
Thrombotic microangiopathy refers to a state that is characterized pathologically by occlusive microvascular thrombosis and clinically by profound thrombocytopenia, microangiopathic hemolytic anemia and variable signs and symptoms of organ ischemia.16 The term primarily refers to two discrete but overlapping syndromes, thrombotic thrombocytopenic purpura (TTP) and hemolytic uremic syndrome (HUS).
TTP is a disseminated microangiopathy, first described by Moschowitz in 1924.17 It is uncommon, occurring primarily in adults and with an annual incidence in the US of 4–11 cases per million people. HUS has clinical and laboratory features that often overlap with those of TTP. However, HUS is more common in children, the renal abnormalities are more marked than in TTP (Table 33.5; see also Chapter 10) and the underlying pathogenetic mechanisms of the two conditions differ.
Table 33.5 Classification of thrombocytopenic purpura and hemolytic uremic syndrome.
| Idiopathic TTP/HUS | Classic adult TTP and childhood non-verotoxin-associated HUS-TTP | |
| Secondary TTP-HUS | Pregnancy-related | TTP, postpartum HUS |
| Verotoxin-induced | Escherichia coli and Shigella dysenteriae I | |
| Childhood HUS | ||
| Epidemic adult TTP-HUS | ||
| Malignant disease | Especially metastatic carcinomas | |
| Drug-induced | Chemotherapy agents, e.g. mitomycin C, cisplatin, and other drugs | |
| Immunosuppressive agents, e.g. cyclosporin, quinine, ticlopidine | ||
| Post-marrow/stem cell transplantation | Especially in conjunction with total body irradiation or high-dose (intensive) chemotherapy |
HUS, hemolytic uremic syndrome; TTP, thrombocytopenic purpura.
(Modified from George JN, El-Harake M 1995. Thrombocytopenia due to enhanced platelet destruction by non-immunologic mechanisms. In: Beutler E, Lichtman MA, Coller BS, Kipps TJ (eds), Williams Hematology, 5th edn. McGraw-Hill, New York, 1290–1315)
Clinical features of TTP
TTP is characterized by the pentad:
TTP typically has a sudden onset, with fever and neurologic symptoms including paralysis, coma, fits and psychiatric disturbance. There is usually purpura and the clinical picture is one of a fluctuating course. The causes of TTP can be subclassified into: 1) congenital; 2) idiopathic; and 3) non-idiopathic.16
Laboratory investigations
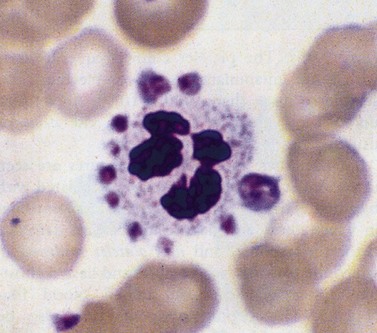
A full blood count and blood film will show anemia (hemoglobin ~ 8–9 g/dl) with polychromasia and other evidence of hemolysis. The film will usually show red-cell fragments and thrombocytopenia (Fig. 33.8). There is usually evidence of hemoglobinemia reflecting the presence of intravascular hemolysis. Lactate dehydrogenase will be elevated. Clotting tests are generally normal although occasionally there may be features of DIC. The direct Coombs test is negative. Renal failure is uncommon but elevated serum creatinine is seen in about a third of cases.
Pathogenesis
Failure to cleave large von Willebrand factor (vWF) multimers is thought to be crucial in the pathogenesis of TTP. Von Willebrand factor is synthesized in vascular endothelial cells and megakaryocytes and assembles into multimers linked by disulfide bonds which are stored in the Weibel–Palade bodies of endothelial cells and α-granules of platelets. A small proportion of these multimers is constitutively secreted by endothelial cells into the circulation, stabilizing factor VIII and mediating both platelet adhesion to sites of vascular injury and platelet aggregation in conditions of high shear. Endothelial cell stimulation causes unusually large vWF multimers to be secreted and these attach to the endothelial cell surface.16
Management
Plasma exchange with cryosupernatant or FFP is now regarded as the treatment of choice for TTP and should be instituted as soon as possible after diagnosis and continued on a daily basis with replacement of 1.0 to 1.5 times the predicted plasma volume of the patient. Plasma exchange should continue until there is normalization of the neurological state, the platelet count has been >150 × 109/l for at least 2 days, the LDH is normal and the hemoglobin rising.18 The disease is highly variable in its course and it is difficult to predict the clinical outcome at the outset. Patients require intensive care nursing, and may require ventilation. Hemodialysis may be required along with plasma exchange. A short course of high dose corticosteroids has also been recommended along with aspirin when the platelet count has recovered to 50 × 109/l.18 Platelet transfusions should be avoided except in cases of severe hemorrhage. Remission is achieved when the platelet count remains normal for 30 days after discontinuation of plasma exchange.19 Patients with severe deficiency of ADAMTS 13 appear to have an increased risk of relapse, most within a year. Rituximab has been used to try to reduce the risk of relapse.20,21
Hemolytic uremic syndrome
The disorder was first described in 1955 by Gasser,22 who reported on five patients (all infants) with acute renal failure, who died of renal cortical necrosis. HUS is classified into D+HUS and D-HUS.16 D+HUS accounts for >90% of cases and is caused by enteric infection with Shiga toxin-producing bacteria, most commonly Escherichia coli O157:H7 or occasionally Shigella dysenteriae I.22 It can occur at all ages but is seen typically in young children.22–24 HUS usually develops 4–6 days after the onset of diarrhea. There is a seasonal incidence, with the highest number of new cases reported in the summer months.25,26
D-HUS is uncommon and clinically heterogeneous. These individuals do not have an antecedent enteric infection with a Shiga toxin-producing organism and have a relatively poor prognosis with a mortality rate of about 25%.16
The histopathological features of HUS are different from those of TTP. The kidneys are preferentially involved and the thrombi are composed primarily of fibrin with few platelets and little vWF.16 Endothelial damage is pronounced in childhood D+HUS. Marked destruction of the renal cortex may occur and glomerular thrombosis is characteristic with an appearance suggesting capillary congestion rather than ischemia.16 D-HUS has been little studied but the renal lesions appear to differ with more pronounced mesangial involvement and less glomerular thrombosis than in D+HUS.16
Pathogenesis. Most D+HUS results from E. coli O157 infections caused by food-borne outbreaks from cattle. Unwashed fruit is another source of some outbreaks. Subunits of Shiga toxin produced by the bacteria bind to microvascular endothelial cells particularly in the kidney and on monocytes and platelets. The toxin is subsequently internalized and inhibits protein synthesis leading to cell death. Cytokine release is stimulated further increasing Shiga toxin binding. Resulting endothelial cell damage has prothrombotic consequences that lead to the histopathological changes described above.16
D-HUS in contrast is associated with complement dysregulation and mutations in one of the complement regulatory proteins (factor H, factor I, membrane cofactor protein (MCP) and factor B) have been described in about 50% of these patients.16 In some patients an autoantibody against factor H has been recognized and in about 5% of cases mutations that impair thrombomodulin function have been identified.27
Laboratory findings. There is evidence of acute renal failure and features of microangiopathic hemolysis.28 The blood count will generally show an elevated WBC, most usually neutrophilia; the prognosis is worst for patients with total neutrophil counts exceeding 20 × 109/l.25
Outlook. Unlike adult TTP, childhood D+HUS is rarely fatal and the mortality is around 3–5%. The outlook is less favorable in D-HUS. In cases with end-stage renal failure that have undergone renal transplantation, the outcome appears to correlate with the cause of the complement dysregulation. Recurrence of HUS has not been seen in transplanted kidneys in patients with MCP mutations whereas the condition did recur following renal transplantation in patients with factor H or factor I mutations, perhaps because these latter two factors are made in the liver. Combined liver and kidney transplants may be successful in some of these cases.16
Pre-eclampsia and HELLP syndrome
In addition to TTP and HUS there are a number of causes of thrombotic microangiopathy that can occur during pregnancy, in particular pre-eclampsia/eclampsia and the HELLP syndrome. Differentiation between these different conditions can be problematic. Pre-eclampsia occurs after 20 weeks of pregnancy and is associated with hypertension, edema, sodium retention, proteinuria and DIC. It may progress to eclampsia which is characterized by convulsions.29 Previously, pre-eclampsia/eclampsia and HELLP have been considered distinct disorders but it seems more likely that they represent a spectrum of a pathologic process.
HELLP syndrome
This disorder, characterized by hemolysis, elevated liver enzymes and low platelet count, occurs in 0.5–0.9% of all pregnancies,30,31 and is associated with a mortality rate of 1–4%.32 It occurs in up to 10% of cases of severe pre-eclampsia. Perinatal mortality is high and may reach 10–20%.32
Clinical features. HELLP is associated with generalized weakness, nausea and vomiting, right upper quadrant pain, headache and visual upset.33 In some cases HELLP may occur following delivery.36
Pathophysiology. There may be abnormalities of the placental vessels with resulting placental ischemia. This is associated with the systemic release of a thromboxanes, angiotensin, tumor necrosis factor-α (TNF-α) and other procoagulant proteins.31,33 DIC may result leading to thrombi which threaten major end-organs including placenta, renal, hepatic and central nervous systems. The thrombi lead to endothelial damage and there is microangiopathic hemolytic anemia (MAHA) and failure of the organs affected by the thrombotic process. Liver failure and occasionally liver rupture may result.34
Management. Prompt delivery, and control of the hypertension are required, in addition to correcting the factor deficiencies caused by the DIC. Corticosteroids have been used as well as plasma exchange and plasmapheresis.35 Maternal deaths, where these occur, are usually due to uncontrolled DIC. Cases in which thrombotic microangiopathy persists post-partum should be considered for plasma exchange.18
Thrombocytopenia caused by massive blood transfusion
Massive transfusion whereby a patient’s total blood volume is replaced over a short time period (24 h or less) may lead to thrombocytopenia, and although the degree of thrombocytopenia is largely related to the amount of blood transfused, the mechanism is not purely dilutional. Patients receiving 15 units of red cells within a 24-hour period develop mild thrombocytopenia, with platelet counts between 47 and 100 × 109/l, whereas patients transfused with 20 units of red cells over the same period develop more pronounced thrombocytopenia (25–61 × 109/l).36,37
Liver disease
Thrombocytopenia secondary to alcohol
Thrombocytopenia occurring in alcoholic patients may be caused by a variety of mechanisms including cirrhosis, splenomegaly and folic acid deficiency. However, thrombocytopenia may be found in the absence of any or all of these pathologies, and is probably due to the direct toxic effects of alcohol on the bone marrow itself, since ethanol is a poison and can suppress the production of platelets by the marrow.38,39
Thrombocytopenia in hepatocellular failure
In addition to the other coagulopathies induced by liver failure, thrombocytopenia is often present and generally reflects the hypersplenism that occurs in portal hypertension. In fulminant hepatic failure, there are abnormalities of both platelet structure and function. On laboratory testing there may be evidence of mild DIC, although this is not generally of major clinical importance.40 In addition, patients with chronic liver disease often have elevated levels of fibrin degradation products which interfere with platelet aggregation.
Thrombocytopenia caused by infection
There are numerous infections caused by a wide variety of pathogenic bacteria, fungi, viruses and protozoa that result in thrombocytopenia in humans. The mechanism underlying the thrombocytopenia is variable. In many cases there is suppression of marrow function, and this is particularly the case with viral infections and accounts for most cases of mild thrombocytopenia. Implicated viruses include mumps,41 varicella,42 EBV,43 rubella44 and many others.
Mechanism of thrombocytopenia
There is a variety of mechanisms involved in thrombocytopenia induced by infection. Following measles infection in children, there is a reduction in marrow megakaryocytes and by day 3 of the infection many of the megakaryocytes have vacuoles within the nucleus and cytoplasm.45
Thrombocytopenia induced by HIV infection
HIV infection probably induces thrombocytopenia through a variety of mechanisms, including immune-mediated thrombocytopenia with reduced platelet life span,46–48 and through direct infection of megakaryocytes themselves. This may occur at any stage in the course of HIV infection, and 40% of HIV positive individuals develop thrombocytopenia at some point in the illness.49–51 Unlike classical idiopathic thrombocytopenic purpura (ITP), males and females are affected equally and thrombocytopenia may be the presenting feature in 10% of HIV positive people.52
Immunologic causes of thrombocytopenia
Here we have restricted our discussion to the more important immune-mediated causes of thrombocytopenia, namely neonatal alloimmune thrombocytopenia (NAIT), post-transfusion purpura (PTP), ITP, drug-induced thrombocytopenia and heparin-induced thrombocytopenia (HIT) (Table 33.6). Since the targets involved in several of these disorders are human platelet alloantigens we outline briefly their salient features.
Table 33.6 Disorders associated with immune-mediated thrombocytopenia.
| Autoimmune | Idiopathic (ITP) Secondary immune Autoimmune disorders, e.g. SLE, rheumatoid, thyroid disease Lymphoproliferative disorders, e.g. CLL, NHL Cancer, e.g. solid tumors Miscellaneous, e.g. post-BMT, chemotherapy Viral infection e.g. HIV, measles, mumps, rubella, EBV, varicella Drug-induced |
| Alloimmune | Post-transfusion purpura Neonatal alloimmune thrombocytopenia |
BMT, bone-marrow transfer; CLL, chronic lymphocytic leukemia; EBV, Epstein–Barr virus; HIV, human immunodeficiency virus; ITP, idiopathic thrombocytopenic purpura, SLE, systemic lupus erythematosus; NHL, Non-Hodgkin lymphoma.
(From Chong BH 1998, Diagnosis treatment and pathophysiology of autoimmune thrombocytopenias. Critical Reviews in Oncology Hematology 20(3):271–296)
Molecular basis of HPA antigens
Platelets have a variety of cell surface antigens, some of which are shared by other cells, such as HLA class I, and blood groups A and B. Others are platelet-specific and are not found on any other type of cell. To date there are 19 alloantigen systems described on platelets, all of which map to membrane proteins (Table 33.2). Eleven of the 19 are carried on GPIIb/IIIa (αIIbβ3 integrin heterodimer), three are on GPIb/IX/V, two on GPIa/IIa (α2β1), and one on each of GPIV, GPV and CD109. The molecular basis for most of these is now elucidated (see Chapter 37).
Alloantibody-mediated thrombocytopenia
Neonatal alloimmune thrombocytopenia
Antibodies to HPA occur in 1 in 365 pregnancies and cause severe thrombocytopenia in 1 in 1100 neonates at term, accounting for around 20% of cases of thrombocytopenia in neonates. NAIT occurs when there is feto-maternal incompatibility for HPA. The condition was first described by van Loghem et al in 1959.54
Antigens involved in NAIT
In Caucasian females NAIT is most commonly due to anti-HPA-1a (98% of the population are HPA-1a+). Other implicated antigens include HPA-1b,55 101 HPA-2a,56 HPA-3a,57 HPA-3b,58 HPA-4a,59 HPA-4b,60 HPA-5b61 and HPA-5a.62
Diagnosis of NAIT
Maternal serum is tested against the father’s platelets in addition to normal control platelets. Occasionally, the maternal antibody titer is low at delivery and difficulty to detect using the above techniques. However, there are techniques available for the detection of low-titer maternal antibodies.63
Management
Future pregnancies
This depends on the father’s genotype for the particular HPA. For example, if he is homozygous then each pregnancy will be affected. In cases where the father is heterozygous, 50% of pregnancies will be affected and the fetus can be typed using DNA obtained from chorionic villus sampling.68
Post-transfusion purpura (PTP)
This rare disorder occurs 7–10 days following a red-cell transfusion in recipients who possess alloantibodies against platelet antigens of the donor. In addition to destroying the incoming platelets, the alloantibody also mediates destruction of the recipient’s own platelets (i.e. lacking the target antigen).69
Pathogenetic basis
Platelet alloantibodies may be present in the recipient against any one of the six major platelet antigens: anti-HPA-1a, anti-HPA-1b,70 anti-HPA3a,71 anti-bak-b,72 anti-HPA-4a or anti-HPA-5b.73 In most cases the antibody has specificity for HPA-1a (2% of the population is HPA-1a–).74
Clinical features
PTP affects multiparous women, although it has been reported in males,70 between the ages of 16 and 80 years. Patients have usually been exposed to platelet antigens through either pregnancy or transfusion or both. The platelet antigen most commonly involved is HPA-1a.
Laboratory features
Patients usually have a platelet count less than 10 × 109/l and the bone marrow will show normal or increased numbers of megakaryocytes. Diagnosis of PTP requires the demonstration of the presence of platelet-specific alloantibodies in the serum of the affected patient. Most patients with PTP are HPA-1a− and the presence of HPA-1a platelets in the transfused blood boosts the primary response.70,72,75,76 The antiplatelet antibodies produced are mainly of IgG1 and IgG3 class. Why patients destroy their own platelets which are HPA-1a− is unclear but is believed to involve IgG3. Possibly HPA-1a+ platelets release HPA-1a which combines with the newly-formed anti-HPA-1a. This complex is absorbed on to the surface of the patient’s HPA-1a− platelets.
Management
Corticosteroids may help the purpura but do not appear to be effective in increasing the platelet count. Intravenous immunoglobulin (IVIg) is the mainstay of treatment.69 Occasionally, plasma exchange may be required. If platelet are required these should be HPA-1a−. Fatal intracranial hemorrhage occurs in 10% but most patients recover within 1–6 weeks.69
Autoantibody-mediated thrombocytopenia
Immune thrombocytopenic purpura (ITP)
In ITP platelets are opsonized with antiplatelet autoantibodies and removed prematurely by the reticuloendothelial system (RES) leading to a reduced peripheral blood platelet count. In addition, in many patients platelet production is reduced. ITP is therefore a disorder of platelet destruction and relative failure of production.77 The etiology of ITP is obscure and the clinical course is variable and unpredictable. ITP has an incidence of 5.8–6.6 new cases per million population per year in the US,78 with a similar incidence in the UK. Childhood ITP is generally seasonal and typically follows a trivial viral infection or vaccination, and in most cases is transient, requiring no treatment with spontaneous recovery in 80% of cases. In the adult (generally chronic) form there is usually no obvious antecedent illness and most patients have chronic thrombocytopenia; spontaneous recovery is uncommon.79 In most cases of adult ITP the platelet glycoprotein (GP) antigen targets are GPIIb/IIIa and GPIb/IX.80
The terminology used in ITP has been updated following a consensus meeting of international experts. The term ‘acute ITP’ is no longer in use, and chronic ITP, which used to define ITP persisting longer than 6 months, now refers to ITP lasting beyond 12 months81 (Fig. 33.9).

Self-limiting ITP is the most common form of ITP found in children, with an annual incidence of between 3 and 8 per 100 000 per year.82,83
Pathophysiology
It is believed that this type of ITP is most likely due to an inappropriate immune response to an environmental trigger; the nature of this trigger is not yet identified.84,85 The disorder may represent an abnormality of antigen-presenting cells, with an increase in the numbers of CD4+ and CD8+ cells. The platelets are rapidly destroyed by the immune complexes that bind to the Fc receptors on the platelets, or due to autoantibodies that bind to the antigenic site on the platelets. Platelets that are coated with antibody or immune complexes are rapidly cleared by the reticuloendothelial system.
Clinical features
The children affected by ITP are, in general, well. The disorder is commonest in children between the ages of 2 and 5 years and, unlike adult chronic ITP, there is no sex predominance. A viral illness predates the development of ITP in most cases of childhood ITP. Physical signs of thrombocytopenia usually take the form of bruising or petechial hemorrhage. Splenomegaly, hepatomegaly and lymphadenopathy are not features of newly diagnosed ITP. A recent UK study reported that in some 76% of children with ITP the disorder was mild with bruising and occasional epistaxis.82 Around 4% had no bleeding symptoms and only 3% had severe bleeding from the gastrointestinal tract, nose or vagina requiring hospitalization. In around 15% of children the disease persisted longer than 6 months and fell into the ‘chronic ITP’ group. The chronic form was found to be commoner in older children and in females.
Laboratory investigation
The blood count will show an isolated thrombocytopenia but should otherwise be normal. The platelet count is often less than 10 × 109/l.82 Anemia may be present in cases in which there has been significant bleeding.
Thrombopoietin (TPO) levels
These are not measured routinely, but in cases where TPO has been assayed in ITP the levels tend not to be elevated.86–88
Management
Most children with ITP need no therapy. There is no set threshold for medical intervention and what constitutes a ‘safe’ platelet count is not known.89 From studies of the natural history of patients with newly diagnosed ITP, we know that patients with this disorder have far fewer bleeding problems than those patients with comparable platelet counts caused by other diseases, such as acute leukemia or aplastic anemia. This, in part, reflects the fact that platelet function in ITP is extremely good, with a large proportion of reticulated (young) platelets in the peripheral blood.90 If therapy is required to elevate the platelet count then the options comprise oral corticosteroids, intravenous immunoglobulin and splenectomy.
Chronic immune thrombocytopenic purpura (ITP)
Clinical features of ITP
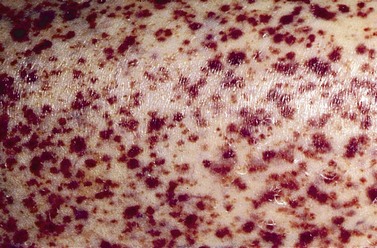
This is the most common form of ITP in adults. Patients may be asymptomatic or may have purpura, bruising or mucosal bleeding including gum bleeding, retinal hemorrhage, epistaxis, melena or menorrhagia (Fig. 33.10). The degree of bleeding is largely dependent on the platelet count, and patients with platelet counts below 10 × 109/l are at greatest risk of bleeding. Splenomegaly is not a feature of ITP and if present, tends to suggest a diagnosis other than ITP.
Pathophysiology of ITP
ITP is an autoimmune disease characterized by increased platelet destruction due to the presence of antiplatelet antibodies. This results in increased platelet clearance by the RES. Several investigators have demonstrated specific autoantibodies against platelet membrane antigens, thus confirming the autoimmune nature of the disorder.91,92 The cause is unknown but it appears likely that in a genetically predisposed individual, a trigger such as infection leads to loss of self-tolerance93 (Fig. 33.11).
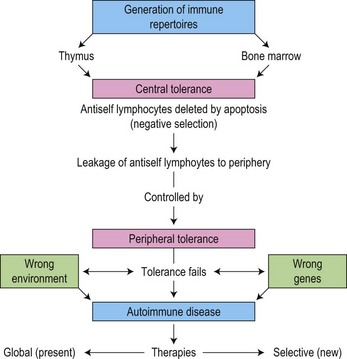
(From Mackay IR. Tolerance and autoimmunity. British Medical Journal. 2000;321:93–96, with permission.93)
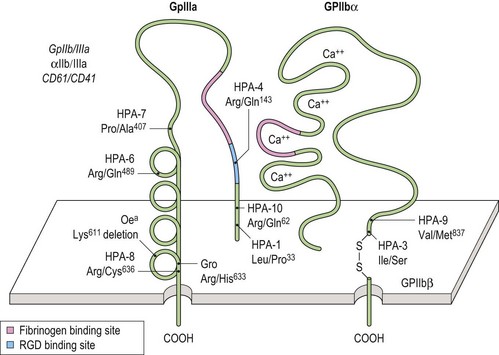
Glycoprotein (GP)-specific autoantibodies may be important in the pathogenesis of chronic ITP;94 from available data GPIIb/IIIa appear to play a major role in the development of chronic ITP in 30–40% of cases.62,95 Fig. 33.12 illustrates the structure of GPIIb/IIIa schematically. Previous investigators have looked for autoantigenic epitopes on the GPIIb/IIIa molecule using competitive binding between human autoantibodies and mouse monoclonal antibodies (MoAbs).96,97
Implicated epitopes
Kekomaki et al. have shown that the 33kDa chymotryptic core fragment of IIIa is a frequent target in chronic ITP.98 Fujisawa and colleagues have used synthetic peptides corresponding to IIIa sequences and have shown that in five of 13 sera from patients with chronic ITP binding was to residues 721–744 or 742–762, corresponding to the carboxy terminal of IIIa.97
GP-specific human MoAbs have been developed as important tools in the search for GP autoepitopes in chronic ITP.94,99 Some investigators have localized certain autoantigenic epitopes to regions of IIb or IIIa but blocking experiments using murine MoAbs have produced contradictory data in terms of homogeneity of the IIb/IIIa antigenic repertoire.96 Only a few cryptic epitopes on IIb/IIIa have been recognized using GP-specific human MoAbs.
Antibody class
The autoantibodies involved in ITP are generally IgG, but IgA and IgM autoantibodies have also been reported.62
Diagnosis
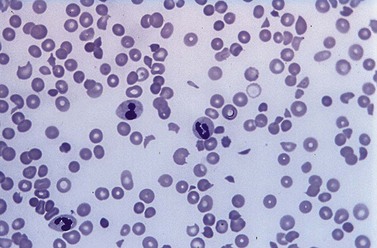
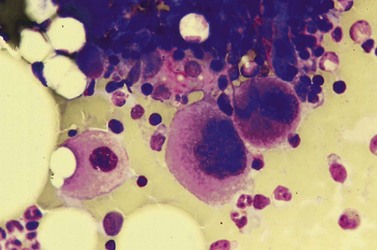
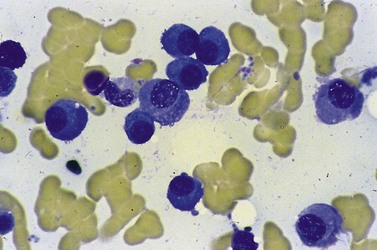
Despite advances in serologic and other techniques the diagnosis of ITP remains largely clinical, and one of exclusion. Secondary causes include systemic lupus erythematosus (SLE), lymphoproliferative disease, HIV infection and others. Standard investigative tests include full blood count which will confirm the presence of isolated thrombocytopenia, blood film to ensure there are no red cell fragments or other diseases such as leukemia or parasitic infections, and an autoimmune profile, to exclude a secondary cause for the thrombocytopenia. A bone marrow aspirate is often carried out in adults, but not usually in children, and will usually show normal or increased numbers of megakaryocytes in an otherwise normal marrow (Fig. 33.13).
Standard first-line therapy
Therapy is seldom necessary for patients whose platelet counts exceed 20–30 × 109/l and in whom there are few spontaneous bleeding episodes47 unless they are undergoing any procedure likely to induce blood loss.100 Standard treatments, including oral prednisolone,32 IVIg,79,101,102 and splenectomy, will elevate the platelet count sufficiently in the majority of adults. However, some 20–25% of adults with ITP are refractory to first-line therapy.
Chronic ITP failing to respond to therapy
This defines those patients who fail to respond to first-line treatment or require unacceptably high doses of corticosteroids to maintain a safe platelet count. A number of agents have been used as second-line therapy for ITP including high-dose steroids, high-dose IVIg, intravenous anti-D, vinca alkaloids, danazol, azathioprine, combination chemotherapy and dapsone. An excellent summary is provided by McMillan.78
Experimental therapies
For those who fail to respond to standard first- and second-line therapy and who require treatment the options are limited and include: interferon-α,79 cyclosporin A, CAMPATH 1H, and protein A columns.103
Thrombopoietin receptor agonists (TPO-mimetics)
Traditionally anti-platelet autoantibodies accelerating platelet clearance from the peripheral circulation have been recognized as the primary pathophysiologic mechanism in chronic immune thrombocytopenia (ITP). Recently, increasing evidence supports the coexistence of insufficient megakaryopoiesis. Inadequate low thrombopoietin (TPO) levels are associated with insufficient proliferation and differentiation of megakaryocytes, decreased proplatelet formation and subsequent platelet release.104 The successful isolation and cloning of thrombopoietin (TPO) in the mid-1990s and identification of its key role in platelet production was a major breakthrough, rapidly followed by the development of the recombinant thrombopoietins, recombinant human TPO and a pegylated truncated product, PEG-rHuMGDF. Both agents increased platelet counts but development was halted because of the development of antibodies that cross-reacted with native TPO, resulting in prolonged treatment-refractory thrombocytopenia. Second generation thrombopoietin receptor agonists were developed with no sequence homology to native TPO and these have now been extensively used with no significant side-effects.105 Two agents are currently available and have been used in early clinical studies and are licensed in some countries at the time of writing. Romiplostim (AMG 531, Nplate®; Amgen, Thousand Oaks, CA, USA) is a novel recombinant thrombopoiesis-stimulating Fc-peptide fusion protein (‘peptibody’) and eltrombopag, a non-peptide, synthetic TPO-receptor agonist (GSK, London, UK; marketed as Promacta in the US and Revolade in the UK). These two novel activators of thrombopoietin receptors have been used in several phase III studies and both agents demonstrate increase of platelet counts in about 80% of chronic ITP patients within 2–3 weeks. These agents substantially broaden the therapeutic options for patients with chronic ITP although long-term results are still pending.106,107
Secondary immune thrombocytopenia
Immune-mediated thrombocytopenia, similar to ITP, may occur in patients with other underlying autoimmune diseases such as systemic lupus erythematosus (SLE). Table 33.7 summarizes the main causes of immune-mediated thrombocytopenia caused by autoantibodies.
| Idiopathic | ITP (newly diagnosed, peristent and chronic) |
| Secondary to other autoimmune or inflammatory disorders | SLE Other autoimmune disorders Lymphoproliferative disease, e.g. CLL Post-BMT |
| Associated with viral infections | e.g. HIV |
| Drug-induced | e.g. quinine, quinidine, heparin, gold salts |
BMT, bone-marrow transfer; CLL, chronic lymphocytic leukemia; HIV, human immunodeficiency virus; ITP, idiopathic thrombocytopenic purpura; SLE, systemic lupus erythematosus.
Drug-induced thrombocytopenia
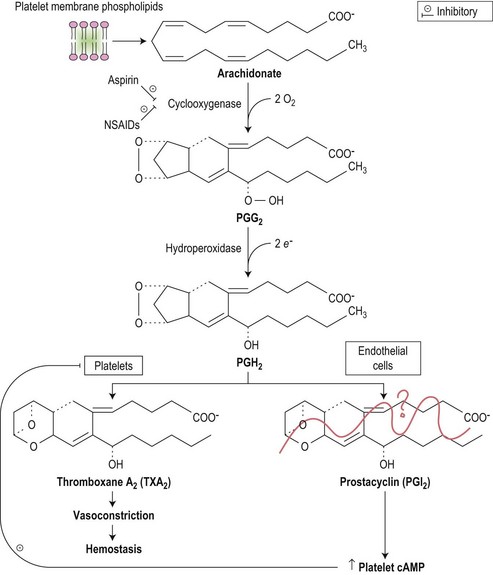
Drugs may induce thrombocytopenia through a variety of mechanisms, both immune and non-immune. Only drug-induced immune thrombocytopenia is discussed here; drug-induced qualitative abnormalities are discussed later in the chapter. In drug-induced immune-mediated thrombocytopenia, drug-dependent antibodies most commonly recognize epitopes of glycoproteins Ib-IX and IIb-IIIa.108 Fig. 33.14 shows the pathways involved in prostaglandin metabolism highlighting the sites of action of aspirin and non-steroidal anti-inflammatory drugs (NSAIDs).
Clinical features
Thrombocytopenia is an uncommon drug-related side-effect. The predominant features are of bleeding or bruising. The number of drugs suspected of causing thrombocytopenia is quite large but strong evidence is only available for a few of these including: quinine,109,110 quinidine,111 sulfonamides,112 trimethoprim, abciximab, and gold salts.113 The incidence of thrombocytopenia in the cases of abciximab is relatively common at 0.5–1% after first exposure rising to 10–14% after second exposure.114 The incidence is also high with gold at about 1%.114
Investigations
Serologic tests may confirm the presence of drug-dependent antibodies. However, there are many patients in whom immune-mediated platelet destruction is believed to be occurring who have no detectable drug-dependent antibodies,73 and the reverse is also true, whereby patients have drug-dependent antibodies in their plasma but with no accompanying thrombocytopenia.115 Overall, therefore, the diagnosis of drug-induced thrombocytopenia through the action of drug-dependent antibodies remains largely clinical.
Heparin-induced thrombocytopenia (HIT)
Heparin-induced thrombocytopenia (HIT) is a common and serious complication of heparin therapy. It has an immunologic basis that is not yet fully understood and, in contrast to other drug-induced causes of thrombocytopenia, is associated with a thrombotic rather than a bleeding tendency. This condition was previously called type II HIT to distinguish it from the mild and clinically benign type I HIT which is sometimes observed early in the course of heparin administration, is non-immune mediated and self-limiting, and is not associated with a bleeding or clotting tendency (Fig. 33.15).
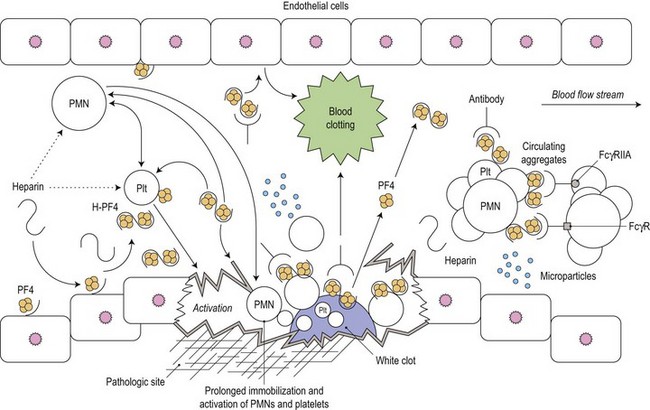
(From Amiral J, Meyer D. Heparin-induced thrombocytopenia: diagnostic tests and biological mechanisms. Baillière’s Clinical Haematology. 1998;11:447–460, with permission.144)
Pathogenesis
PF4 is a chemokine produced by megakaryocytes and released from platelet α-granules upon platelet activation. Released PF4 binds cell-surface glycosaminoglycans and heparin. Formation of the PF4-heparin complex causes a conformational change in the protein leading to exposure of cryptic epitopes to which the antibodies in HIT are usually generated. The anti-PF4/heparin antibodies, which are of IgG isotype, bind to PF4 through their F(ab) domains. Serial PF4 molecules align on the platelet surface and several IgG molecules bind, leading to the formation of large immune complexes that cross-link the platelet FcγIIa receptors, and resulting in platelet aggregation and an associated platelet procoagulant response.116 HIT antibodies also bind to endothelial cells and monocytes resulting in additional procoagulant effects through increased expression of tissue factor and formation of platelet-monocyte aggregates leading to thrombin generation. Therefore in HIT the clinical manifestations are the result of both FcγIIa-dependent platelet activation and activation of the coagulation system through tissue factor expression and platelet activation.
Clinical features
The risk of HIT is greater with 5 or more days of exposure to unfractionated heparin (1–3%) than to low-molecular-weight heparin (0–0.8%).117 The risk is also greater in certain patient groups such as surgical patients than in others, e.g. obstetrics. In HIT the platelet count usually falls by >50% and this typically occurs 5–14 days after the start of therapeutic or prophylactic heparin administration (the first day of heparin administration is day 0). The platelet count generally reaches a nadir in the range 20–100 × 109/l and it is very unusual for it to drop below 10–15 × 109/l in contrast to some other drug-induced thrombocytopenias. HIT can also be of rapid onset, developing within 24 hours in patients who have had heparin exposure within the previous 3 months. Occasionally delayed-onset HIT can occur beginning several days to weeks after heparin exposure and caused by antibodies that activate platelets independently of heparin. Although hemorrhage is uncommon, because of the enhanced thrombin generation as described above, there is a high rate of thrombotic events affecting major vessels. In patients treated with heparin for ischemic vascular disease, arterial thrombosis is common, and in those receiving heparin for thrombotic diseases such as deep vein thrombosis (DVT), the associated thrombus is usually venous. Even in the presence of heparin there may be considerable extension of the DVT, which may prove fatal. The mortality rate is high at 30%. Other manifestations of HIT occasionally occur, for example skin lesions (erythema with or without central necrosis) at sites of heparin injections or acute systemic reactions after intravenous heparin administration. DIC may also be seen in some patients. Thus the development of any of these features, new or recurrent thrombosis, or the development of thrombocytopenia during or following heparin administration should always prompt consideration of HIT.
Diagnosis
The differential diagnosis of HIT is wide and includes sepsis, multi-organ failure, malignancy, the antiphospholipid syndrome and other drugs that cause thrombocytopenia. HIT is a clinicopathological syndrome and the diagnosis is based on a combination of clinical and laboratory features. The laboratory tests are of two types, measurement of antibody binding to PF4/heparin complexes or detection of heparin-dependent activation by patient serum.116 The former is performed by enzyme-immunoassay (EIA) and is highly sensitive but with a specificity that ranges from 50% to 90%. The strength of the assay correlates with the probability of HIT – the diagnosis is likely in those that are strongly positive (>1.0 optical density (OD)) but unlikely in those that are weakly positive (0.4–1.0 OD).116 In the functional assays, aggregation of donor platelets by patient serum or plasma plus heparin is assessed. The optimal assays of heparin-dependent platelet activation use washed platelets. The sensitivity of the functional assays for HIT is generally lower than the EIAs but the specificity is higher and can reach 95–99% with assays that use washed platelets. The specificity of both types of assay is further enhanced by the finding that a positive result at low heparin concentration is inhibited at high heparin concentration due to disruption of PF4/heparin complexes.
Combining clinical assessment and appropriate laboratory testing is essential in the diagnosis of HIT.116 The first stage is estimation of the pretest probability, for example, using the so-called 4Ts scoring system which takes account of: 1) degree of thrombocytopenia; 2) timing of thrombocytopenia; 3) presence of thrombosis or other clinical manifestations of HIT; 4) other potential causes of thrombocytopenia. In patients with a low pretest probability no laboratory testing is required and heparin can be continued. If the EIA is negative HIT is very unlikely and heparin can be continued. If the EIA is weakly positive at low heparin concentration and reactivity is not inhibited at high concentration, HIT is unlikely and heparin can be continued. A strongly positive IgG EIA indicates an increased risk of platelet activating antibodies and a platelet activation assay should be performed. If this is positive, HIT is extremely likely. Finally the clinical situation should be reassessed to support or exclude the diagnosis.
Other drugs causing immune-mediated platelet destruction
There are many drugs implicated in immune-mediated thrombocytopenia. The evidence is strongest for quinine, quinidine, heparin and gold salts,113 as already discussed. Other implicated agents, though with fewer reports to date, include: α-methyldopa,118 diclofenac,119 rifampin,120 carbamazepine121 and sulfonamides. Readers are referred to George et al. for an excellent review of the topic.122
Acquired functional abnormalities of platelets
Bleeding problems may arise through either inadequate numbers of platelets or functional abnormalities of the platelets themselves. This section discusses disorders in which there are abnormalities of platelet function along with their pathogenetic basis (Table 33.8).
Table 33.8 Drugs and disorders interfering with platelet function
| Systemic disorders | Uremia Cardiac by-pass surgery |
| Hematologic disorders | Myeloproliferative diseases Leukemia Myelodysplasia Paraproteinemias, including multiple myeloma |
| Drugs | Aspirin Antibiotics Anticoagulants Others |
Uremia in renal failure
Bleeding may be a feature of either acute or chronic renal failure,123,124 with spontaneous bleeding into the skin, mucous membranes including the gastrointestinal or genitourinary tracts, central nervous system and other sites (see also Chapter 35).
Pathogenesis
Platelet function, in the presence of uremia, is abnormal,123,125–127 and a variety of laboratory studies have shown that all aspects of platelet activity are affected, including platelet adhesion, aggregation and procoagulant activity.128–130
The normal process of platelet adhesion has been shown to involve contact of platelets to endothelial structures. This is dependent on the binding of vWF to GP lb/IX.124 In the presence of uremia there may be a qualitative or quantitative abnormality of vWF or GPlb/IX itself.
Management
The most important factor to consider is whether the patient is actively bleeding, rather than abnormalities detected using bleeding time, and other coagulation assays. Hemodialysis itself will help correct the bleeding induced by the uremia.127 Desmopressin (DDAVP) which promotes the release of vWF from vascular endothelial cells may reduce the bleeding time in some uremic patients.126
Myeloproliferative disorders
The myeloproliferative diseases (MPDs) are neoplastic hematologic stem cell disorders and include essential thrombocythemia (ET) (Fig. 33.16), polycythemia rubra vera (PRV), idiopathic myelofibrosis (IMF) and chronic myeloid leukemia (CML). These disorders are associated with both bleeding and thrombosis.
Pathophysiology
In PRV there is a rise in whole blood viscosity through elevation of the hematocrit which may contribute to thrombosis.133,134 Abnormalities in platelet function have been reported in MPDs, and the bleeding time is prolonged in a minority of patients. However, bleeding may occur even if the template bleeding time is normal.133
Platelet abnormalities in the myeloproliferative disorders
Platelets may be larger than normal, although in some cases they are smaller. Their survival may be reduced, especially in essential thrombocythemia. Platelet aggregation is abnormal with the standard aggregants including ADP and collagen, though this is not a feature of thrombocytosis when the underlying cause is reactive, thereby excluding thrombocytosis per se as a cause of the abnormal aggregation.134 It may, in fact, be a secondary consequence of the conversion of arachidonic acid to prostaglandin endoperoxides or lipooxygenase products,135 or a decrease in platelet responsiveness to thromboxane A2.136
Management
For PRV, a reduction in hematocrit, aiming for a level less than 0.45 (45%) is an approach adopted by most hematologists.137 Ongoing clinical trials may help determine the optimal level of hematocrit. The bone marrow may be suppressed effectively using hydroxyurea or busulfan with an overall reduction in platelet count in patients with essential thrombocythemia.138,139 However, even though the peripheral platelet count is lowered, this does not necessarily correct the associated platelet abnormalities. The management of the patient who is actively bleeding is more complex.
Platelet abnormalities in leukemia and myelodysplastic syndromes
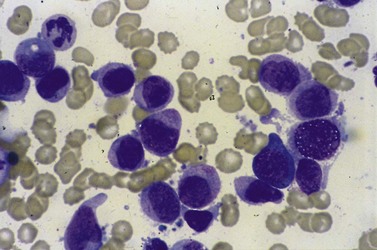
Bleeding is common in both types of disease, and is often due to thrombocytopenia. However, functional abnormalities of platelets are also recognized. In acute myeloid leukemia (AML) for example, platelets may be larger than usual, with abnormalities of shape and the appearance of their granules (Fig. 33.17).
Management
Patients with functional platelet abnormalities generally respond to platelet transfusion. Correction of the underlying malignancy with definitive therapy will also improve the acquired bleeding diathesis. Similar abnormalities are also seen in MDS and treatment generally consists of regular platelet transfusions140,141 (Fig. 33.18).
Drugs that interfere with platelet function
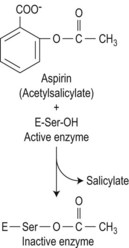
Drugs may cause bleeding through the induction of profound thrombocytopenia. In addition, there are numerous agents that may induce bleeding through the interference with the normal function of platelets. Thus, although the platelet count may be entirely normal, the platelets are rendered functionally defective. Aspirin and non-steroidal anti-inflammatory agents (NSAIDs) are the most common cause of acquired platelet dysfunction (Table 33.9). Their effects are mediated through irreversibly inhibiting cyclooxygenase activity in the platelet resulting in impairment of the granule release reaction and defective aggregation. Aspirin, in particular, acetylates the serine residue at position 530 of prostaglandin synthase, the enzyme responsible for converting arachidonate to prostaglandin cyclic endoperoxides, and thereby inhibits the synthesis of prostacyclin and thromboxane A2 (Fig. 33.19). These effects are seen in both the platelet and endothelium, but the effect of aspirin on platelet function is detectable for several days after the drug is stopped since there is a lag phase before new platelets lacking the drug enter the circulation. Endothelial cells, on the other hand, are able to generate prostaglandin synthase much more rapidly.
| NSAIDs | Aspirin Diclofenac Mefenamic acid Others |
Cyclooxygenase inhibitors |
| Antibiotics | Penicillins Cephalosporins Nitrofurantoin |
In high doses, particularly in ill patients, many antibiotics may interfere with platelet aggregation |
| Anticoagulants | Heparin Epsilon aminocaproic acid |
|
| Drugs that increase platelet cAMP | Dipyridamole Iloprost |
Dipyridamole is a phosphodiesterase inhibitor |
| Cardiovascular system drugs | Diltiazem Isosorbide dinitrate Nifedipine Propranolol |
|
| Psychotropics | Tricyclic antidepressants such as imipramine and amitriptyline Phenothiazines, e.g. chlorpromazine, promethazine |
|
| Anesthetics | Local and general anesthetics (e.g. halothane) | |
| Anticancer drugs | Chemotherapeutic agents such as mithramycin, BCNU and daunorubicin | |
| Anticoagulants | Heparin and coumadin | |
| Miscellaneous | Dextrans Ticlopidine Lipid-lowering drugs, e.g. clofibrate Quinidine Ethanol |
NSAIDs, non-steroid anti-inflammatory drugs.
(Modified from Rao AK, Carvalho ACA 1994. In: Colman RW et al (eds), In Hemostasis and Thrombosis: Principles and Practice, 3rd edn. JB Lippincott, Philadelphia.)
1 Long MW, Gragowski LL, Heffner CH, Boxer LA. Phorbol diesters stimulate the development of an early murine progenitor cell. The burst-forming unit-megakaryocyte. J Clin Invest. 1985;76:431-438.
2 Jackson CW. Megakaryocyte endomitosis: a review. Int J Cell Cloning. 1990;8:224-226.
3 Harker LA, Finch CA. Thrombokinetics in man. J Clin Invest. 1969;48:963-974.
4 Hynes RO. Integrins: a family of cell surface receptors. Cell. 1987;48:549-554.
5 Kasirer-Friede A, Kahn ML, Shattil SJ. Platelet integrins and immunoreceptors. Immunol Rev. 2007;218:247-264.
6 Kieffer N, Phillips DR. Platelet membrane glycoproteins: functions in cellular interactions. Annu Rev Cell Biol. 1990;6:329-357.
7 Kunicki TJ, Newman PJ. The molecular immunology of human platelet proteins. Blood. 1992;80:1386-1404.
8 Nachman RL, Rafii S. Platelets, petechiae, and preservation of the vascular wall. N Engl J Med. 2008;359:1261-1270.
9 Payne BA, Pierre RV. Pseudothrombocytopenia: a laboratory artifact with potentially serious consequences. Mayo Clin Proc. 1984;59:123-125.
10 Savage RA. Pseudoleukocytosis due to EDTA-induced platelet clumping. Am J Clin Pathol. 1984;81:317-322.
11 Aster RH. Pooling of platelets in the spleen: role in the pathogenesis of ‘hypersplenic’ thrombocytopenia. J Clin Invest. 1966;45:645-657.
12 Gewirtz AM, Hoffman R. Transitory hypomegakaryocytic thrombocytopenia: aetiological association with ethanol abuse and implications regarding regulation of human megakaryocytopoiesis. Br J Haematol. 1986;62:333-344.
13 Manoharan A, Williams NT, Sparrow R. Acquired amegakaryocytic thrombocytopenia: report of a case and review of literature. Q J Med. 1989;70:243-252.
14 Boggs DR. Amegakaryocytic thrombocytopenia. Am J Hematol. 1985;20:413-416.
15 Levi M, ten Cate H, van der Poll T, van Deventer SJ. Pathogenesis of disseminated intravascular coagulation in sepsis. JAMA. 1993;270:975-979.
16 Zheng XL, Sadler JE. Pathogenesis of thrombotic microangiopathies. Annu Rev Pathol. 2008;3:249-277.
17 Moschowitz E. Hyaline thrombosis of the terminal arterioles and capillaries: a hitherto undescribed disease. Proceedings of the New York Pathological Society. 1924;24:21-24.
18 Allford SL, Hunt BJ, Rose P, Machin SJ. Guidelines on the diagnosis and management of the thrombotic microangiopathic haemolytic anaemias. Br J Haematol. 2003;120:556-573.
19 George JN. Clinical practice. Thrombotic thrombocytopenic purpura. NEJM. 2006;354(18):1927-1935.
20 Schleinitz N, Ebbo M, Mazodier K, et al. Rituximab as preventive therapy of a clinical relapse in TTP with ADAMTS13 inhibitor. Am J Hematol. 2007;82:417-418.
21 Scott SM, Szczepiorkowski ZM. Rituximab for TTP. Am J Hematol. 2005;80:87-88.
22 Gasser C, Gautier E, Steck A, et al. [Hemolytic-uremic syndrome: bilateral necrosis of the renal cortex in acute acquired hemolytic anemia.]. Schweiz Med Wochenschr. 1955;85:905-909.
23 Ashkenazi S. Role of bacterial cytotoxins in hemolytic uremic syndrome and thrombotic thrombocytopenic purpura. Annu Rev Med. 1993;44:11-18.
24 Neill MA, Agosti J, Rosen H. Hemorrhagic colitis with Escherichia coli 0157:H7 preceding adult hemolytic uremic syndrome. Arch Intern Med. 1985;145:2215-2217.
25 Martin DL, MacDonald KL, White KE, et al. The epidemiology and clinical aspects of the hemolytic uremic syndrome in Minnesota. N Engl J Med. 1990;323:1161-1167.
26 Milford DV, Taylor CM, Guttridge B, et al. Haemolytic uraemic syndromes in the British Isles 1985–8: association with verocytotoxin producing Escherichia coli. Part 1: Clinical and epidemiological aspects. Arch Dis Child. 1990;65:716-721.
27 Delvaeye M, Noris M, De Vriese A, et al. Thrombomodulin mutations in atypical hemolytic-uremic syndrome. N Engl J Med. 2009;361:345-357.
28 Rowe PC, Orrbine E, Wells GA, McLaine PN. Epidemiology of hemolytic-uremic syndrome in Canadian children from 1986 to 1988. The Canadian Pediatric Kidney Disease Reference Centre. J Pediatr. 1991;119:218-224.
29 Brenner BM. Vascular injury to the kidney. In: Fauci AS, Braunwald E, Isselbacher KJ, editors. Principles of Internal Medicine. New York: McGraw–Hill, 1998.
30 Ishibashi M, Ito N, Fujita M, et al. Endothelin-1 as an aggravating factor of disseminated intravascular coagulation associated with malignant neoplasms. Cancer. 1994;73:191-195.
31 Jones SL. HELLP! A cry for laboratory assistance: a comprehensive review of the HELLP syndrome highlighting the role of the laboratory. Hematopathol Mol Hematol. 1998;11:147-171.
32 Portis R, Jacobs MA, Skerman JH, Skerman EB. HELLP syndrome (hemolysis, elevated liver enzymes, and low platelets) pathophysiology and anesthetic considerations. AANA J. 1997;65:37-47.
33 Stone JH. HELLP syndrome: hemolysis, elevated liver enzymes, and low platelets. JAMA. 1998;280:559-562.
34 Sheikh RA, Yasmeen S, Pauly MP, Riegler JL. Spontaneous intrahepatic hemorrhage and hepatic rupture in the HELLP syndrome: four cases and a review. Journal of Clinical Gastroenterology. 1999;28:323-328.
35 Hamada S, Takishita Y, Tamura T, et al. Plasma exchange in a patient with postpartum HELLP syndrome. J Obstet Gynaecol Res. 1996;22:371-374.
36 Counts RB, Haisch C, Simon TL, et al. Hemostasis in massively transfused trauma patients. Ann Surg. 1979;190:91-99.
37 Leslie SD, Toy PT. Laboratory hemostatic abnormalities in massively transfused patients given red blood cells and crystalloid. Am J Clin Pathol. 1991;96:770-773.
38 Lindenbaum J, Hargrove RL. Thrombocytopenia in alcoholics. Ann Intern Med. 1968;68:526-532.
39 Post RM, Desforges JF. Thrombocytopenia and alcoholism. Ann Intern Med. 1968;68:1230-1236.
40 Kelly DA, Summerfield JA. Hemostasis in liver disease. Semin Liver Dis. 1987;7:182-191.
41 Ninomiya N, Maeda T, Matsuda I. Thrombocytopenic purpura occurring during the early phase of a mumps infection. Helv Paediatr Acta. 1977;32:87-89.
42 Espinoza C, Kuhn C. Viral infection of megakaryocytes in varicella with purpura. Am J Clin Pathol. 1974;61:203-208.
43 Angle RM, Alt HL. Thrombocytopenic purpura complicating infectious mononucleosis. Blood 1050;5:499.
44 Bayer WL, Sherman FE, Michaels RH, et al. Purpura in congenital and acquired rubella. N Engl J Med. 1965;273:1362-1366.
45 Oski FA, Naiman JL. Effect of live measles vaccine on the platelet count. N Engl J Med. 1966;275:352-356.
46 Ballem PJ, Belzberg A, Devine DV, et al. Kinetic studies of the mechanism of thrombocytopenia in patients with human immunodeficiency virus infection. N Engl J Med. 1992;327:1779-1784.
47 Yang R, Zhong CH. Pathogenesis and management of chronic idiopathic thrombocytopenic purpura: an update. International Journal of Hematology. 2000;71:1824.
48 Walsh CM, Nardi MA, Karpatkin S. On the mechanism of thrombocytopenic purpura in sexually active homosexual men. N Engl J Med. 1984;311:635-639.
49 Abrams DI, Chinn EK, Lewis BJ, et al. Hematologic manifestations in homosexual men with Kaposi’s sarcoma. Am J Clin Pathol. 1984;81:13-18.
50 Frontiera M, Myers AM. Peripheral blood and bone marrow abnormalities in the acquired immunodeficiency syndrome. West J Med. 1987;147:157-160.
51 Treacy M, Lai L, Costello C, Clark A. Peripheral blood and bone marrow abnormalities in patients with HIV related disease. Br J Haematol. 1987;65:289-294.
52 Rossi G, Gorla R, Stellini R, et al. Prevalence, clinical, and laboratory features of thrombocytopenia among HIV-infected individuals. AIDS Res Hum Retroviruses. 1990;6:261-269.
53 Stabler SP, Allen RH, Savage DG, Lindenbaum J. Clinical spectrum and diagnosis of cobalamin deficiency. Blood. 1990;76:871-881.
54 van Loghem JJ, Dorfmeyer H, van de Hart M, Schreuder F. Serological and genetical studies on the platelet antigen (Zw). Vox Sanguinis. 1959;4:161-169.
55 Mueller-Eckhardt C, Becker T, Weisheit M, et al. Neonatal alloimmune thrombocytopenia due to fetomaternal Zwb incompatibility. Vox Sang. 1986;50:94-96.
56 Bizzaro N, Dianese G. Neonatal alloimmune amegakaryocytosis. Case report. Vox Sang. 1988;54:112-114.
57 von dem Borne AE, von Riesz E, Verheugt FW, et al. Baka, a new platelet-specific antigen involved in neonatal allo-immune thrombocytopenia. Vox Sang. 1980;39:113-120.
58 McGrath K, Minchinton R, Cunningham I, Ayberk H. Platelet anti-Bakb antibody associated with neonatal alloimmune thrombocytopenia. Vox Sang. 1989;57:182-184.
59 Friedman JM, Aster RH. Neonatal alloimmune thrombocytopenic purpura and congenital porencephaly in two siblings associated with a ‘new’ maternal antiplatelet antibody. Blood. 1985;65:1412-1415.
60 Shibata Y, Matsuda I, Miyaji T, Ichikawa Y. Yuka, a new platelet antigen involved in two cases of neonatal alloimmune thrombocytopenia. Vox Sang. 1986;50:177-180.
61 Kaplan C, Morel-Kopp MC, Kroll H, et al. HPA-5b (Br(a)) neonatal alloimmune thrombocytopenia: clinical and immunological analysis of 39 cases. Br J Haematol. 1991;78:425-429.
62 Kiefel V, Shechter Y, Atias D, et al. Neonatal alloimmune thrombocytopenia due to anti-Brb (HPA-5a). Report of three cases in two families. Vox Sang. 1991;60:244-245.
63 Mueller-Eckhardt C, Kayser W, Forster C, et al. Improved assay for detection of platelet-specific PlA1 antibodies in neonatal alloimmune thrombocytopenia. Vox Sang. 1982;43:76-81.
64 Katz J, Hodder FS, Aster RS, et al. Neonatal isoimmune thrombocytopenia. The natural course and management and the detection of maternal antibody. Clin Pediatr (Phila). 1984;23:159-162.
65 Adner MM, Fisch GR, Starobin SG, Aster RH. Use of ‘compatible’ platelet transfusions in treatment of congenital isoimmune thrombocytopenic purpura. N Engl J Med. 1969;280:244-247.
66 Suarez CR, Anderson C. High-dose intravenous gammaglobulin (IVG) in neonatal immune thrombocytopenia. Am J Hematol. 1987;26:247-253.
67 Pearson HA, Shulman NR, Marder VJ, Cone TE. Isoimmune neonatal thrombocytopenic purpura: clinical and therapeutic considerations. Blood. 1964;23:154.
68 McFarland JG, Aster RH, Bussel JB, et al. Prenatal diagnosis of neonatal alloimmune thrombocytopenia using allele-specific oligonucleotide probes. Blood. 1991;78:2276-2282.
69 Mueller-Eckhardt C. Post-transfusion purpura. Br J Haematol. 1986;64:419-424.
70 Taaning E, Morling N, Ovesen H, Svejgaard A. Post transfusion purpura and anti-Zwb (-P1A2). Tissue Antigens. 1985;26:143-146.
71 Keimowitz RM, Collins J, Davis K, Aster RH. Post-transfusion purpura associated with alloimmunization against the platelet-specific antigen, Baka. Am J Hematol. 1986;21:79-88.
72 Kickler TS, Ness PM, Herman JH, Bell WR. Studies on the pathophysiology of posttransfusion purpura. Blood. 1986;68:347-350.
73 Christie DJ, Pulkrabek S, Putnam JL, et al. Posttransfusion purpura due to an alloantibody reactive with glycoprotein Ia/IIa (anti-HPA-5b). Blood. 1991;77:2785-2789.
74 Seidenfeld AM, Owen J, Glynn MF. Post-transfusion purpura cured by steroid therapy in a man. Can Med Assoc J. 1978;118:1285-1286.
75 Simon T, Collins J, Kunicki T. Post-transfusion purpura with antiplatelet antibody specific for the platelet antigen Pena. Blood. 1986;68:117a.
76 Waters AH. Post-transfusion purpura. Blood Rev. 1989;3:83-87.
77 Nugent D, McMillan R, Nichol JL, Slichter SJ. Pathogenesis of chronic immune thrombocytopenia: increased platelet destruction and/or decreased platelet production. Br J Haematol. 2009;146:585-596.
78 McMillan R. Therapy for adults with refractory chronic immune thrombocytopenic purpura. Ann Intern Med. 1997;126:307-314.
79 George JN, Woolf SH, Raskob GE, et al. Idiopathic thrombocytopenic purpura: a practice guideline developed by explicit methods for the American Society of Hematology. Blood. 1996;88:3-40.
80 Warner MN, Moore JC, Warkentin TE, et al. A prospective study of protein-specific assays used to investigate idiopathic thrombocytopenic purpura. Br J Haematol. 1999;104:442-447.
81 Rodeghiero F, Stasi R, Gernsheimer T, et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood. 2009;113:2386-2393.
82 Bolton-Maggs PH, Moon I. Assessment of UK practice for management of acute childhood idiopathic thrombocytopenic purpura against published guidelines. Lancet. 1997;350:620-623.
83 Buchanan GR. The nontreatment of childhood idiopathic thrombocytopenic purpura. Eur J Pediatr. 1987;146:107-112.
84 Imbach P. Immune thrombocytopenia in children: the immune character of destructive thrombocytopenia and the treatment of bleeding. Semin Thromb Hemost. 1995;21:305-312.
85 Imbach PA, Kuhne T, Hollander G. Immunologic aspects in the pathogenesis and treatment of immune thrombocytopenic purpura in children. [published erratum appears in Curr Opin Pediatr 1997 Jun;9(3):298]. Curr Opin Pediatr. 1997;9:35-40.
86 Baatout S. Thrombopoietin. A review. Haemostasis. 1997;27:1-8.
87 Kaushansky K. The molecular mechanisms that control thrombopoiesis. J Clin Invest. 2005;115:3339-3347.
88 Kaushansky K. Thrombopoietin and the hematopoietic stem cell. Ann N Y Acad Sci. 2005;1044:139-141.
89 Lilleyman JS. Intracranial haemorrhage in idiopathic thrombocytopenic purpura. Paediatric Haematology Forum of the British Society for Haematology. Arch Dis Child. 1994;71:251-253.
90 Kienast J, Schmitz G. Flow cytometric analysis of thiazole orange uptake by platelets: a diagnostic aid in the evaluation of thrombocytopenic disorders. Blood. 1990;75:116-121.
91 Woods VLJ, Kurata Y, Montgomery RR, et al. Autoantibodies against platelet glycoprotein Ib in patients with chronic immune thrombocytopenic purpura. Blood. 1984;64:156-160.
92 Woods VLJ, Oh EH, Mason D, McMillan R. Autoantibodies against the platelet glycoprotein IIb/IIIa complex in patients with chronic ITP. Blood. 1984;63:368-375.
93 Mackay IR. Science, medicine, and the future: tolerance and autoimmunity. BMJ. 2000;321:93-96.
94 Hou M, Stockelberg D, Kutti J, Wadenvik H. Glycoprotein IIb/IIIa autoantigenic repertoire in chronic idiopathic thrombocytopenic purpura. Br J Haematol. 1995;91:971-975.
95 McMillan R, Tani P, Millard F, et al. Platelet-associated and plasma anti-glycoprotein autoantibodies in chronic ITP. Blood. 1987;70:1040-1045.
96 Varon D, Karpatkin S. A monoclonal anti-platelet antibody with decreased reactivity for autoimmune thrombocytopenic platelets. Proc Natl Acad Sci USA. 1983;80:6992-6995.
97 Fujisawa K, Tani P, O’Toole TE, et al. Different specificities of platelet-associated and plasma autoantibodies to platelet GPIIb-IIIa in patients with chronic immune thrombocytopenic purpura. Blood. 1992;79:1441-1446.
98 Kekomaki R, Dawson B, McFarland J, Kunicki TJ. Localization of human platelet autoantigens to the cysteine-rich region of glycoprotein IIIa. J Clin Invest. 1991;88:847-854.
99 Nugent DJ, Kunicki TJ, Berglund C, Bernstein ID. A human monoclonal autoantibody recognizes a neoantigen on glycoprotein IIIa expressed on stored and activated platelets. Blood. 1987;70:16-22.
100 Young NS, Barrett AJ. The treatment of severe acquired aplastic anemia. Blood. 1995;85:3367-3377.
101 Provan A. Management of adult idiopathic thrombocytopenic purpura. Prescribers’ Journal. 1992;32:193-200.
102 Provan D, Newland A. Idiopathic thrombocytopenic purpura in adults. J Pediatr Hematol Oncol. 2003;25(Suppl. 1)):S34-S38.
103 Cahill MR, Macey MG, Cavenagh JD, Newland AC. Protein A immunoadsorption in chronic refractory ITP reverses increased platelet activation but fails to achieve sustained clinical benefit. Br J Haematol. 1998;100:358-364.
104 Provan D, Stasi R, Newland AC, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood. 2010;115:168-186.
105 Newland A. Thrombopoietin receptor agonists in the treatment of thrombocytopenia. Curr Opin Hematol. 2009;16:357-364.
106 Bussel JB, Provan D, Shamsi T, et al. Effect of eltrombopag on platelet counts and bleeding during treatment of chronic idiopathic thrombocytopenic purpura: a randomised, double-blind, placebo-controlled trial. Lancet. 2009;373:641-648.
107 Molineux G, Newland A. Development of romiplostim for the treatment of patients with chronic immune thrombocytopenia: from bench to bedside. Br J Haematol.. 2010;150(1):9-20.
108 Garratty G. Drug-induced immune cytopenia. Transfus Med Rev. 1993;7:213-214.
109 Connellan JM, Deacon S, Thurlow PJ. Changes in platelet function and reactivity induced by quinine in relation to quinine (drug) induced immune thrombocytopenia. Thromb Res. 1991;61:501-514.
110 Kunicki TJ, Christie DJ, Aster RH. The human platelet receptor(s) for quinine/quinidine-dependent antibodies. Blood Cells. 1983;9:293-301.
111 Salom IL. Purpura due to inhaled quinidine. JAMA. 1991;266:1220.
112 Curtis BR, McFarland JG, Wu GG, et al. Antibodies in sulfonamide-induced immune thrombocytopenia recognize calcium-dependent epitopes on the glycoprotein IIb/IIIa complex. Blood. 1994;84:176-183.
113 Coblyn JS, Weinblatt M, Holdsworth D, Glass D. Gold-induced thrombocytopenia. A clinical and immunogenetic study of twenty-three patients. Ann Intern Med. 1981;95:178-181.
114 ASTER AND BORIGIE, NEJM, 2007
115 Warkentin TE, Chong BH, Greinacher A. Heparin-induced thrombocytopenia: towards consensus. Thromb Haemost. 1998;79:1-7.
116 Greinacher A. Heparin-induced thrombocytopenia. J Thromb Haemost. 2009;7(Suppl. 1):9-12.
117 Shantsila E, Lip GY, Chong BH. Heparin-induced thrombocytopenia. A contemporary clinical approach to diagnosis and management. Chest. 2009;135:1651-1664.
118 Manohitharajah SM, Jenkins WJ, Roberts PD, Clarke RC. Methyldopa and associated thrombocytopenia. Br Med J. 1971;1:494.
119 Epstein M, Vickars L, Stein H. Diclofenac induced immune thrombocytopenia. J Rheumatol. 1990;17:1403-1404.
120 Burnette PK, Ameer B, Hoang V, Phifer W. Rifampin-associated thrombocytopenia secondary to poor compliance. DICP. 1989;23:382-384.
121 Casasin T, Allende A, Macia M, Guell R. Two episodes of carbamazepine-induced severe thrombocytopenia in the same child. Ann Pharmacother. 1992;26:715-716.
122 George JN, El-Harake M. Thrombocytopenia due to enhanced platelet destruction by non-immunologic mechanisms. In: Beutler E, Lichtman MA, Coller BS, Kipps TJ, editors. Williams Hematology. New York: McGraw-Hill, 1995.
123 Rabiner SF. Uremic bleeding. Prog Hemost Thromb. 1972;1:233-250.
124 Remuzzi G. Bleeding disorders in uremia: pathophysiology and treatment. Adv Nephrol Necker Hosp. 1989;18:171-186.
125 Castaldi PA, Rozenberg MC, Stewart JH. The bleeding disorder of uraemia. A qualitative platelet defect. Lancet. 1966;2:66-69.
126 Livio M, Benigni A, Remuzzi G. Coagulation abnormalities in uremia. Semin Nephrol. 1985;5:82-90.
127 Remuzzi G, Livio M, Marchiaro G, et al. Bleeding in renal failure: altered platelet function in chronic uraemia only partially corrected by haemodialysis. Nephron. 1978;22:347-353.
128 Castillo R, Lozano T, Escolar G, et al. Defective platelet adhesion on vessel subendothelium in uremic patients. Blood. 1986;68:337-342.
129 Zwaginga JJ, Ijsseldijk MJ, Beeser-Visser N, et al. High von Willebrand factor concentration compensates a relative adhesion defect in uremic blood. Blood. 1990;75:1498-1508.
130 Zwaginga JJ, Ijsseldijk MJ, de Groot PG, et al. Defects in platelet adhesion and aggregate formation in uremic bleeding disorder can be attributed to factors in plasma. Arterioscler Thromb. 1991;11:733-744.
131 Weiss HJ, Turitto VT, Baumgartner HR. Effect of shear rate on platelet interaction with subendothelium in citrated and native blood. I. Shear rate – dependent decrease of adhesion in von Willebrand’s disease and the Bernard–Soulier syndrome. J Lab Clin Med. 1978;92:750-764.
132 Murphy S. Polycythemia vera. Dis Mon. 1992;38:153-212.
133 Schafer AI. Essential thrombocythemia. Prog Hemost Thromb. 1991;10:69-96.
134 Ginsburg AD. Platelet function in patients with high platelet counts. Ann Intern Med. 1975;82:506-511.
135 Schafer AI. Deficiency of platelet lipoxygenase activity in myeloproliferative disorders. N Engl J Med. 1982;306:381-386.
136 Okuma M, Takayama H, Uchino H. Subnormal platelet response to thromboxane A2 in a patient with chronic myeloid leukaemia. Br J Haematol. 1982;51:469-477.
137 Kaplan ME, Mack K, Goldberg JD, et al. Long-term management of polycythemia vera with hydroxyurea: a progress report. Semin Hematol. 1986;23:167-171.
138 Kessler CM, Klein HG, Havlik RJ. Uncontrolled thrombocytosis in chronic myeloproliferative disorders. Br J Haematol. 1982;50:157-167.
139 Murphy S, Iland H, Rosenthal D, Laszlo J. Essential thrombocythemia: an interim report from the Polycythemia Vera Study Group. Semin Hematol. 1986;23:177-182.
140 Cowan DH, Haut MJ. Platelet function in acute leukemia. J Lab Clin Med. 1972;79:893-905.
141 Meschengieser S, Blanco A, Maugeri N, et al. Platelet function and intraplatelet von Willebrand factor antigen and fibrinogen in myelodysplastic syndromes. Thromb Res. 1987;46:601-606.
142 Mackie IJ. The biology of haemostasis and thrombosis. In: Ledingham JGG, Warrell DA, editors. Concise Oxford Textbook of Medicine. Oxford: Oxford University Press, 2000.
143 Roth GJ. Developing relationships: arterial platelet adhesion, glycoprotein Ib, and leucine-rich glycoproteins. Blood. 1991;77:5-19.
144 Amiral J, Meyer D. Heparin-induced thrombocytopenia: diagnostic tests and biological mechanisms. Baillière’s Clinical Haematology. 1998;11:447-460.
[/level-membership-for-pathology-category][not-level-membership-for-pathology-category]
CHAPTER 33 Acquired disorders affecting megakaryocytes and platelets
Structure and function of megakaryocytes and platelets
Human platelets
Thrombopoiesis, the generation of platelets from megakaryocytes in the bone marrow, is complex and incompletely understood. Megakaryocytes are large end-stage cells from which platelets bud. The earliest recognized committed progenitor is the burst-forming unit (BFU)-Meg.1 Fig. 33.1 shows megakaryocyte development from stem cell stage through to platelet production. BFU-Megs develop into colony-forming unit (CFU)-Megs in the presence of growth factors thrombopoietin (TPO), interleukin-3 (IL-3) and IL-11. Megakaryocyte nuclei are large polyploid structures with chromosome contents between diploid (2N) to 64N. Such polyploid status is achieved through a process termed nuclear endoduplication: that is, successive doubling of chromosome content in the absence of cell division. Platelets are produced from megakaryocytes that are 8N or greater.2 A single megakaryocyte can generate around 3000 platelets of which 20–30% are pooled in the spleen. In health the peripheral blood platelet count is 150–400 × 109/l but this fluctuates, for example, following heavy exercise, ‘stress’, and around the menstrual cycle. This transient rise in platelet count may be caused by mobilization of platelets pooled in the spleen. There are also racial differences in the ‘normal’ platelet count and some Mediterranean populations have platelet counts as low as 80 × 109/l in health. Platelets are produced at a rate of 35 000–44 000 per microliter per day3 and have a lifespan of 9–10 days.
Platelet structure and function
The integrin family of proteins
Integrins are key platelet membrane proteins and have been characterized on a large number of leukocytes and many other cells. For example, the fibronectin receptor on mammalian fibroblasts is one of the best characterized matrix receptor proteins4 (Fig. 33.2). This receptor, in common with all other integrins, is a heterodimer consisting of a non-covalently associated complex of two distinct high-molecular-weight polypeptides, α and β. The receptor functions as a transmembrane linker which mediates the interaction between the intracellular actin cytoskeleton and fibronectin in the extracellular matrix. Like all integrins, the fibronectin receptor recognizes so-called RGD (Arg-Gly-Asp) sequences in matrix components. In platelets, integrins recognize and bind a variety of proteins in order to form a hemostatic plug through a complex mechanism of platelet adhesion, shape change and activation of the clotting pathway.
Platelet integrins and related proteins
Platelets contain five integrin α subunits and two β subunits producing: αIIbβ3, ανβ3, α2β1, α5β1 and α6β1.5,6,7 These are shown in Fig. 33.3. Further detail is provided by Table 33.1. These proteins are essential for normal platelet function, and are often the target of immunological attack in disorders such as idiopathic thrombocytopenic purpura (ITP).
Platelet alloantigens
These can be platelet-specific or shared with other cells. Important shared antigens include HLA class I and ABH (blood group A and B) antigens. Platelet-specific antigens fall into five well-defined human platelet antigen (HPA) groups (Table 33.2; see also Chapter 37): HPA-1, HPA-2, HPA-3, HPA-4 and HPA-5, each of which has an α and β allele. Each platelet allotype represents a single amino acid substitution in the platelet glycoprotein molecule. Because some platelet glycoproteins carry epitopes that play a major role in platelet function, platelet alloantibodies may not only cause thrombocytopenia but also affect primary hemostasis.
Cytoplasmic platelet constituents
Platelets contain two principal types of granule: dense bodies and α granules (Fig. 33.4). Dense bodies contain ADP, ATP, 5-HT, calcium and pyrophosphate. The α granules contain more than 300 releasable proteins including adhesion molecules, chemokines, cytokines, coagulation factors, fibrinolytic regulators, growth factors and pro- and anti-angiogenic factors such as PF4, β-thrombospondin, PDGF, vWF, fibrinogen, factor V and fibronectin. The contents of these granules are integral components of the platelet’s biological activities.
Biological function of platelets
The primary role of the platelet is the prevention of blood loss from damaged tissues and vessels, i.e. primary hemostasis. This is achieved through platelet activation, adhesion, shape change and aggregation. Platelets may also play a role in the maintenance of vascular integrity by the constitutive release of cytokines and growth factors from their granules that bind to endothelial cell surface receptors resulting in intracellular signaling that stabilizes the molecular complexes that form the junctions between adjacent endothelial cells.8
Platelet adhesion (Fig. 33.5A)
Platelet shape change
After adhesion to the subendothelium, platelets undergo a major shape change, from a discoid shape to one which is irregular, with projections (Fig. 33.6). This process is initially reversible but ultimately becomes irreversible.
Quantitative platelet abnormalities: thrombocytopenia
Thrombocytopenia, a reduction in platelet count, may be caused by:
Pseudothrombocytopenia
This describes patients in whom the peripheral blood platelet count is found to be spuriously low, and is caused by a variety of mechanisms. Platelet clumping caused by the anticoagulant ethylenediaminetetraacetic acid (EDTA) is the commonest cause, with an incidence of around 0.1%. EDTA-induced pseudothrombocytopenia is easily excluded by examination of a blood film which will confirm the presence of numerous large platelet clumps.9,10 Other causes of pseudothrombocytopenia include the presence of giant platelets, which are not counted as platelets by modern automated counters, and platelet satellitism (platelets attach themselves to monocytes or granulocytes) (Fig. 33.7).
Pooling of platelets in the spleen
Mechanism involved
In health, the spleen may pool up to one third of the total platelet mass, and in disease states this may rise to 90%.11 Although the peripheral blood platelet count may only be a fraction of the normal range, the patient generally has an overall normal platelet mass since production is entirely normal but the low peripheral counts simply reflect a larger than normal mass of platelets pooled in the spleen.
Thrombocytopenia due to failure of platelet production
Acquired amegakaryocytic thrombocytopenia
This describes severe thrombocytopenia caused by a selective reduction in megakaryocytes in an otherwise normal bone marrow, and is the result of damage to the megakaryocyte stem cell. The disorder is analogous to acquired pure red-cell aplasia (PRCA). Acquired amegakaryocytic thrombocytopenia may be caused by drugs, toxins and connective tissue disorders.12–14
Bone marrow failure syndromes
The bone marrow failure (BMF) disorders are those in which there is a failure of bone marrow precursor (stem) cells in contrast to those disorders such as myelodysplastic syndromes in which normal or increased numbers of abnormal cells are produced, or those in which the survival of the cells is reduced. The BMF syndromes are a diverse groups of disorders with a common endpoint in which there is loss of hematopoietic stem cells. BMF can be acquired or inherited (see Table 33.3).
Thrombocytopenia due to increased platelet destruction
Causes may be immunologic or non-immunologic.
Non-immunologic causes of thrombocytopenia
Disseminated intravascular coagulation (DIC)
DIC is characterized by excessive activation of the coagulation cascade (see also Chapter 35). In most cases DIC is an acute event, but chronic DIC is well described, although clinically less important. The main problem faced in patients with DIC is bleeding, which may be mild but is often severe with generalized oozing from venepuncture sites, central lines and other indwelling cannulae, gastrointestinal and genitourinary tracts. Microthrombi are found in 5–10% of cases, often affecting digits, with resulting peripheral gangrene.
Pathogenesis
DIC is triggered by the release or exposure of tissue thromboplastins which contain a high concentration of phospholipids following trauma, surgery, mismatched blood transfusion and a variety of other triggers (Table 33.4). In addition to systemic activation of coagulation (leading to fibrin clot formation, organ failure and consumption of platelets and coagulation factors that may cause bleeding), there is dysregulation of natural anticoagulant systems and fibrinolysis.
Table 33.4 Triggers for disseminated intravascular coagulation (DIC)
| Trauma | Including surgical |
| Dissemination of cancer cells | Malignancy, following administration of chemotherapy |
| Massive hemolysis | Post mismatched blood transfusion |
| Venoms | e.g. snake venoms |
| Endothelial injury | Gram-negative sepsis |
| Infections | |
| Burns | |
| Septicemia |
Thrombotic thrombocytopenic purpura and hemolytic uremic syndrome
Thrombotic microangiopathy refers to a state that is characterized pathologically by occlusive microvascular thrombosis and clinically by profound thrombocytopenia, microangiopathic hemolytic anemia and variable signs and symptoms of organ ischemia.16 The term primarily refers to two discrete but overlapping syndromes, thrombotic thrombocytopenic purpura (TTP) and hemolytic uremic syndrome (HUS).
TTP is a disseminated microangiopathy, first described by Moschowitz in 1924.17 It is uncommon, occurring primarily in adults and with an annual incidence in the US of 4–11 cases per million people. HUS has clinical and laboratory features that often overlap with those of TTP. However, HUS is more common in children, the renal abnormalities are more marked than in TTP (Table 33.5; see also Chapter 10) and the underlying pathogenetic mechanisms of the two conditions differ.
Table 33.5 Classification of thrombocytopenic purpura and hemolytic uremic syndrome.
| Idiopathic TTP/HUS | Classic adult TTP and childhood non-verotoxin-associated HUS-TTP | |
| Secondary TTP-HUS | Pregnancy-related | TTP, postpartum HUS |
| Verotoxin-induced | Escherichia coli and Shigella dysenteriae I | |
| Childhood HUS | ||
| Epidemic adult TTP-HUS | ||
| Malignant disease | Especially metastatic carcinomas | |
| Drug-induced | Chemotherapy agents, e.g. mitomycin C, cisplatin, and other drugs | |
| Immunosuppressive agents, e.g. cyclosporin, quinine, ticlopidine | ||
| Post-marrow/stem cell transplantation | Especially in conjunction with total body irradiation or high-dose (intensive) chemotherapy |
HUS, hemolytic uremic syndrome; TTP, thrombocytopenic purpura.
(Modified from George JN, El-Harake M 1995. Thrombocytopenia due to enhanced platelet destruction by non-immunologic mechanisms. In: Beutler E, Lichtman MA, Coller BS, Kipps TJ (eds), Williams Hematology, 5th edn. McGraw-Hill, New York, 1290–1315)
Clinical features of TTP
TTP is characterized by the pentad:
TTP typically has a sudden onset, with fever and neurologic symptoms including paralysis, coma, fits and psychiatric disturbance. There is usually purpura and the clinical picture is one of a fluctuating course. The causes of TTP can be subclassified into: 1) congenital; 2) idiopathic; and 3) non-idiopathic.16
Laboratory investigations
A full blood count and blood film will show anemia (hemoglobin ~ 8–9 g/dl) with polychromasia and other evidence of hemolysis. The film will usually show red-cell fragments and thrombocytopenia (Fig. 33.8). There is usually evidence of hemoglobinemia reflecting the presence of intravascular hemolysis. Lactate dehydrogenase will be elevated. Clotting tests are generally normal although occasionally there may be features of DIC. The direct Coombs test is negative. Renal failure is uncommon but elevated serum creatinine is seen in about a third of cases.
Pathogenesis
Failure to cleave large von Willebrand factor (vWF) multimers is thought to be crucial in the pathogenesis of TTP. Von Willebrand factor is synthesized in vascular endothelial cells and megakaryocytes and assembles into multimers linked by disulfide bonds which are stored in the Weibel–Palade bodies of endothelial cells and α-granules of platelets. A small proportion of these multimers is constitutively secreted by endothelial cells into the circulation, stabilizing factor VIII and mediating both platelet adhesion to sites of vascular injury and platelet aggregation in conditions of high shear. Endothelial cell stimulation causes unusually large vWF multimers to be secreted and these attach to the endothelial cell surface.16
Management
Plasma exchange with cryosupernatant or FFP is now regarded as the treatment of choice for TTP and should be instituted as soon as possible after diagnosis and continued on a daily basis with replacement of 1.0 to 1.5 times the predicted plasma volume of the patient. Plasma exchange should continue until there is normalization of the neurological state, the platelet count has been >150 × 109/l for at least 2 days, the LDH is normal and the hemoglobin rising.18 The disease is highly variable in its course and it is difficult to predict the clinical outcome at the outset. Patients require intensive care nursing, and may require ventilation. Hemodialysis may be required along with plasma exchange. A short course of high dose corticosteroids has also been recommended along with aspirin when the platelet count has recovered to 50 × 109/l.18 Platelet transfusions should be avoided except in cases of severe hemorrhage. Remission is achieved when the platelet count remains normal for 30 days after discontinuation of plasma exchange.19 Patients with severe deficiency of ADAMTS 13 appear to have an increased risk of relapse, most within a year. Rituximab has been used to try to reduce the risk of relapse.20,21
Hemolytic uremic syndrome
The disorder was first described in 1955 by Gasser,22 who reported on five patients (all infants) with acute renal failure, who died of renal cortical necrosis. HUS is classified into D+HUS and D-HUS.16 D+HUS accounts for >90% of cases and is caused by enteric infection with Shiga toxin-producing bacteria, most commonly Escherichia coli O157:H7 or occasionally Shigella dysenteriae I.22 It can occur at all ages but is seen typically in young children.22–24 HUS usually develops 4–6 days after the onset of diarrhea. There is a seasonal incidence, with the highest number of new cases reported in the summer months.25,26
D-HUS is uncommon and clinically heterogeneous. These individuals do not have an antecedent enteric infection with a Shiga toxin-producing organism and have a relatively poor prognosis with a mortality rate of about 25%.16
The histopathological features of HUS are different from those of TTP. The kidneys are preferentially involved and the thrombi are composed primarily of fibrin with few platelets and little vWF.16 Endothelial damage is pronounced in childhood D+HUS. Marked destruction of the renal cortex may occur and glomerular thrombosis is characteristic with an appearance suggesting capillary congestion rather than ischemia.16 D-HUS has been little studied but the renal lesions appear to differ with more pronounced mesangial involvement and less glomerular thrombosis than in D+HUS.16
Pathogenesis. Most D+HUS results from E. coli O157 infections caused by food-borne outbreaks from cattle. Unwashed fruit is another source of some outbreaks. Subunits of Shiga toxin produced by the bacteria bind to microvascular endothelial cells particularly in the kidney and on monocytes and platelets. The toxin is subsequently internalized and inhibits protein synthesis leading to cell death. Cytokine release is stimulated further increasing Shiga toxin binding. Resulting endothelial cell damage has prothrombotic consequences that lead to the histopathological changes described above.16
D-HUS in contrast is associated with complement dysregulation and mutations in one of the complement regulatory proteins (factor H, factor I, membrane cofactor protein (MCP) and factor B) have been described in about 50% of these patients.16 In some patients an autoantibody against factor H has been recognized and in about 5% of cases mutations that impair thrombomodulin function have been identified.27
Laboratory findings. There is evidence of acute renal failure and features of microangiopathic hemolysis.28 The blood count will generally show an elevated WBC, most usually neutrophilia; the prognosis is worst for patients with total neutrophil counts exceeding 20 × 109/l.25
Outlook. Unlike adult TTP, childhood D+HUS is rarely fatal and the mortality is around 3–5%. The outlook is less favorable in D-HUS. In cases with end-stage renal failure that have undergone renal transplantation, the outcome appears to correlate with the cause of the complement dysregulation. Recurrence of HUS has not been seen in transplanted kidneys in patients with MCP mutations whereas the condition did recur following renal transplantation in patients with factor H or factor I mutations, perhaps because these latter two factors are made in the liver. Combined liver and kidney transplants may be successful in some of these cases.16
Pre-eclampsia and HELLP syndrome
In addition to TTP and HUS there are a number of causes of thrombotic microangiopathy that can occur during pregnancy, in particular pre-eclampsia/eclampsia and the HELLP syndrome. Differentiation between these different conditions can be problematic. Pre-eclampsia occurs after 20 weeks of pregnancy and is associated with hypertension, edema, sodium retention, proteinuria and DIC. It may progress to eclampsia which is characterized by convulsions.29 Previously, pre-eclampsia/eclampsia and HELLP have been considered distinct disorders but it seems more likely that they represent a spectrum of a pathologic process.
HELLP syndrome
This disorder, characterized by hemolysis, elevated liver enzymes and low platelet count, occurs in 0.5–0.9% of all pregnancies,30,31 and is associated with a mortality rate of 1–4%.32 It occurs in up to 10% of cases of severe pre-eclampsia. Perinatal mortality is high and may reach 10–20%.32
Clinical features. HELLP is associated with generalized weakness, nausea and vomiting, right upper quadrant pain, headache and visual upset.33 In some cases HELLP may occur following delivery.36
[/not-level-membership-for-pathology-category]
