Acid-Base, Electrolyte, and Metabolic Abnormalities
Acid-Base Homeostasis
Normal Acid-Base Physiology
Normal biochemical and physiologic function requires that the extracellular pH be maintained within a very narrow range. Although the “normal” range of pH in clinical laboratories is 7.35 to 7.45 pH units, the actual pH in vivo varies considerably less.1 This tight control is maintained by a complex homeostatic mechanism involving buffers and the elimination of volatile acid by respiration.
The principal extracellular buffer system is the carbonic acid/bicarbonate pair. The equilibrium relationships of the components of this system are illustrated as follows:1
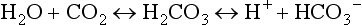
From these relationships, the Henderson-Hasselbalch equation is derived:
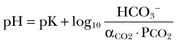
In this equation, αCO2 is the solubility coefficient of CO2 (0.03), and pK is the equilibrium constant for this buffer pair (6.1). Rearrangement yields the Henderson equation:
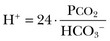
Acid-base homeostasis depends on compensation for a primary disturbance. Compensation for a respiratory disturbance is metabolic, and compensation for a metabolic disturbance is respiratory. Furthermore, it is clear from the previous equations that in order to mitigate the change in proton concentration or pH, the direction of the compensation must be the same as the direction of the primary disturbance. Thus, consumption of bicarbonate will be accompanied by hyperventilation and a consequent reduction in PCO2. A simple acid-base disturbance is considered to consist of the primary disturbance and its normal compensation. A complex acid-base disturbance consists of more than one primary disturbance. In order to detect complex acid-base disturbances, one must be familiar with both the direction and magnitude of normal compensation (shown in Table 57.1).2 More than one metabolic disturbance may coexist (e.g., metabolic acidosis and metabolic alkalosis), but only one respiratory disturbance is possible at a time.

In the present section, we will discuss disorders that affect the metabolic component of acid-base homeostasis: metabolic acidosis and metabolic alkalosis. Respiratory disturbances affecting acid-base balance will be discussed elsewhere (Chapters 37 and 40).
Metabolic Acidosis
Definition and Classification
An adult eating a normal diet generates 16,000 to 20,000 mmol of acid a day.3 Almost all of that acid is in the form of carbonic acid, resulting from CO2 and water generation in the metabolism of carbohydrates and fats. Individuals with normal ventilatory capacity eliminate this prodigious acid load through the lungs, thus the term volatile acid. The remainder of the daily acid load, about 1 mmol/kg body weight per day, derives from metabolism of phosphate- and sulfate-rich protein (yielding phosphoric and sulfuric acid). These nonvolatile or fixed acids are buffered, primarily by extracellular bicarbonate under normal circumstances. The kidneys are responsible for regenerating the consumed bicarbonate by secreting hydrogen ions (protons) in the distal nephron. These secreted protons must be buffered in the tubule lumen in order to allow elimination of the daily fixed acid load within the physiologic constraint of the minimum urinary pH. The urinary buffers are composed of the filtered sodium salts of the phosphoric acid and ammonia, which is synthesized in the proximal tubule and acidified in the collecting duct to form ammonium (NH4+). Under conditions of acid loading, the normal kidney reabsorbs all the filtered bicarbonate in the proximal tubule. Urinary net acid excretion therefore comprises phosphoric acid (so-called titratable acidity, because it is quantified by titrating the urine with alkali to pH 7.40) and ammonium, less any excreted bicarbonate.4
Many factors modify the kidney’s capacity to regulate acid-base balance. For example, renal ammoniagenesis is stimulated by acidemia, and inhibited by alkalemia, and thus participates in a homeostatic feedback loop.1 Hyperkalemia inhibits and hypokalemia stimulates renal ammoniagenesis. Hypokalemia further stimulates acid secretion by activating the Na+-H+ exchanger in the proximal tubule and the H+/K+-ATPase in the collecting duct. Finally, aldosterone stimulates both proton and K+ secretion in the collecting duct. For these reasons, hypokalemia tends to perpetuate a metabolic alkalosis, and hyperkalemia a metabolic acidosis.1
Metabolic acidosis can be caused by excessive production of fixed acid, decreased renal secretion of fixed acid, or loss of bicarbonate, either through the kidney or through the intestine.4 The net effect of any of these processes is a reduction in the blood bicarbonate concentration. The plasma anion gap helps to distinguish among the various causes of metabolic acidosis. Of course, because of charge neutrality, the sum of the concentration of all cations in the plasma is equal to the sum of all the anions. By convention, however, the anion gap is defined as the difference between the plasma sodium concentration and the sum of the bicarbonate and chloride concentrations. It represents the concentration of anions that are normally unmeasured by a basic metabolic chemistry panel.5 The anion gap normally is about 8 mmol/L, but it varies widely according to the methods employed by the clinical chemistry laboratory.6 The anion gap is composed mainly of albumin, along with phosphates, sulfates, and organic anions.
There are two important pitfalls in the interpretation of the anion gap. First, because the anion gap is proportional to the plasma albumin concentration, hypoalbuminemia (common in critically ill patients) will lower the “baseline” anion gap (by approximately 2.5 mmol/L for each g/dL decline in the albumin concentration).7 Thus, profound hypoalbuminemia may falsely lower the anion gap, and thus mask a high anion gap acidosis. Second, alkalemia increases the anion gap by causing lactate generation and by titrating plasma buffers, most notably albumin.8 (Thus, in respiratory alkalosis, the bicarbonate concentration will be low in compensation, and the anion gap may be elevated, giving a false impression of a high anion gap metabolic acidosis by inspection of the electrolytes alone.)
If bicarbonate is lost (e.g., through diarrhea), or hydrochloric acid is gained (e.g., renal tubular acidosis or administration of unbuffered amino acid solutions9), the bicarbonate concentration falls with a commensurate increase in the plasma chloride concentration; thus the anion gap is unchanged. If, on the other hand, bicarbonate is lost in buffering an organic acid such as lactic acid or a ketoacid, the decrement in the bicarbonate concentration is more or less matched by an increase in the anion gap. These processes are illustrated in Figure 57.1.
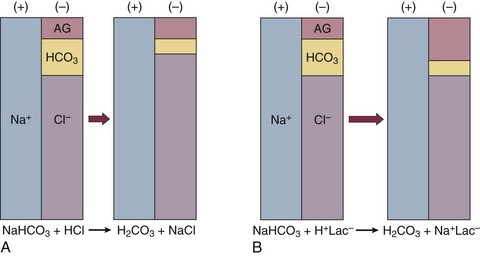
Box 57.1 lists the causes of hyperchloremic metabolic acidosis. Two diagnoses are of particular interest in the critical care arena. First is the posthypocapnic metabolic acidosis, in which bicarbonate falls in compensation for a chronic respiratory alkalosis. When “normal” ventilation is restored, the pH falls until bicarbonate can be retained, giving the appearance of a hyperchloremic metabolic acidosis. This emphasizes the importance of observation over time in the analysis of acid-base status. The second entity of interest is a so-called dilutional hyperchloremic acidosis. This is seen in patients who are rapidly resuscitated with large volumes of isotonic saline solution. The acidosis traditionally has been attributed to dilution of blood bicarbonate. Analysis based on physical-chemistry principles may better explain the phenomenon (see later).10
The differential diagnosis of high anion gap metabolic acidosis is limited (Box 57.2). The most common cause in critically ill patients is a lactic acidosis. The causes of lactic acidosis are numerous. As shown in Box 57.3, they are divided into type A (imbalance between tissue oxygen demand and supply) and type B (impaired oxygen utilization).5 Diabetic ketoacidosis (DKA) (Chapter 58) and intoxications (Chapter 68) are discussed elsewhere. Two causes of high anion gap acidosis recently added to the differential diagnosis, and of particular relevance to intensivists, are pyroglutamic acidosis and intoxication with propylene glycol.
Pyroglutamic acid is a metabolic intermediate in the γ-glutamyl cycle, one product of which is glutathione. Pyroglutamic acidosis may be congenital (caused by one of several enzyme deficiencies) or acquired.8 The acquired syndrome may be caused by acetaminophen (which depletes glutathione, leading to uninhibited pyroglutamic acid synthesis), β-lactam antibiotics, or glycine deficiency. The acidosis may be profound and the anion gap greater than 30 mmol/L.11 Definitive diagnosis is made by urinary screen for organic acids. In practice, however, circumstantial evidence suggests the acquired syndrome and the diagnosis is supported by a favorable response to appropriate intervention.
Propylene glycol is a solvent for medications, many of which are commonly infused intravenously in critically ill patients, such as lorazepam, nitroglycerin, etomidate, and phenytoin. Propylene glycol is metabolized by alcohol dehydrogenase to lactic acid. High anion gap acidosis has been associated with high- and even low-dose infusions, particularly of lorazepam.7,12 Thus, development of a high anion gap acidosis in a critically ill patient should prompt a search for a source of propylene glycol, because withdrawal of the agent will promptly alleviate the acidosis.
Consequences of Acidemia
It has been generally accepted that severe acidemia (pH < 7.20) is associated with a variety of deleterious effects. Of particular concern are the cardiovascular effects, including pressure-resistant arterial vasodilation, venoconstriction, diminished myocardial contractility, and impaired hepatic and renal perfusion.13 (Some controversy exists as to which of these effects are directly caused by acidemia.4) A predisposition to malignant arrhythmias has been reported in vitro and in animal models. Finally, numerous metabolic derangements have been attributed to the effect of acidemia on key enzymes in metabolic pathways, resulting in sympathetic hyperactivity with diminished catecholamine responsiveness; insulin resistance and suppressed glycolysis; and reduced hepatic lactic acid uptake and metabolism.14
Diagnosis of Acid-Base Disorders
Acid-base disorders are revealed most commonly through the basic metabolic chemistry panel, when the plasma bicarbonate concentration is noted to be outside the normal range. If the bicarbonate is low, and if the anion gap is clearly elevated on that sample, a diagnosis of high anion gap metabolic acidosis can be made with some confidence, keeping in mind the pitfalls in the interpretation of the anion gap mentioned earlier.7
Once the primary disturbance has been identified, the astute clinician, recognizing the possibility of a mixed disturbance, is obligated to ask, “Is that all there is?” This question can be answered only by an understanding of the rules of normal compensation for simple acid-base disorders (see Table 57.1).2 Knowing at least the expected direction of compensation will allow the clinician to diagnose the most obvious mixed disturbances. For example, if the pH is low, the bicarbonate is low, and the PCO2 is above 40 mm Hg, there is clearly a mixed metabolic and respiratory acidosis. Similarly, if the pH is high, the bicarbonate is high, and the PCO2 is below 40 mm Hg, the diagnosis is a mixed respiratory and metabolic alkalosis. More subtle mixed disorders can be diagnosed only by understanding not only the expected direction, but the expected magnitude of compensation. This will allow one to conclude, for example, whether the hyperventilation in a patient with metabolic acidosis is appropriate (expected compensation), inadequate (a separate respiratory acidosis), or excessive (a separate respiratory alkalosis).
The preceding method permits the diagnosis of simple and dual acid-base disorders. Triple acid-base disorders can be diagnosed only by comparing the change in the anion gap with the change in the plasma bicarbonate concentration. Most simply conceived, the fall in the bicarbonate should equal the rise in the anion gap (see Fig. 57.1). If the rise in the anion gap exceeds the fall in the bicarbonate, a metabolic alkalosis is said to be present in addition to the high anion gap acidosis. Conversely, if the fall in the bicarbonate exceeds the rise in the anion gap, mixed hyperchloremic and high anion gap acidoses are said to coexist. Although this analysis is useful in the case of large discrepancies, in more subtle cases it is confounded by theoretical and practical considerations.5,15
The classical approach to acid-base disorders described earlier has been challenged recently by proponents of a physical-chemistry approach described originally by Stewart.16 According to this method, the pH of the blood depends on the ionization of water by the difference in the concentration of so-called strong ions (the strong ion difference, or SID). The SID offers a quantitative approach to measuring the degree of acidosis in hyperchloremic metabolic acidosis. Although there is evidence that this approach may offer prognostic capabilities in patients with severe sepsis and septic shock with hyperchloremic metabolic acidosis, the complexity of the equations for calculating the SID may make this method cumbersome in clinical settings.17 The main utility of this construct in the critical care setting seems to be its explanation of a hyperchloremic metabolic acidosis in patients who receive large volumes of isotonic saline.
Treatment of Metabolic Acidosis
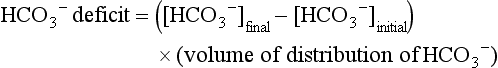
The difficulty in accurately estimating this value arises from two factors: First, the apparent volume of distribution of bicarbonate varies more than twofold from 50% of body weight to 100% of body weight and is inversely proportional to the initial bicarbonate concentration.18 Second, there are often many simultaneous processes in a critically ill patient that tend to ameliorate or exacerbate the metabolic acidosis, such as vomiting, shock, and liver failure. In order to avoid overshoot alkalemia, it is prudent to estimate the volume of distribution to be 50% of the body weight14 and to target an increase in the bicarbonate concentration of no more than 8 mmol/L over 12 to 24 hours.
Sodium bicarbonate generally is considered to be the alkalinizing agent of choice for severe acidemia. Alternative alkalinizing agents such as citrate, acetate, and lactate, which under normal circumstances are oxidized in the liver to bicarbonate, should not be used to treat acidemia in patients with suspected or confirmed hepatic impairment or circulatory compromise. The sodium bicarbonate should be administered as a continuous infusion, the concentration of which should be guided by the patient’s serum sodium concentration. Bolus injection of undiluted ampules of sodium bicarbonate (1000 mmol/L) should be used with great restraint and only in patients with the most severe acidemia because of the risk of hyperosmolality. Large volumes of any bicarbonate solution can lead to volume overload, a reduction in the ionized calcium concentration (see “Hypocalcemia”), and increased generation of CO2. This last effect will tend to cause a respiratory acidosis in patients with ventilatory insufficiency. Plasma electrolytes and blood gases must be monitored frequently to guide adjustments in the composition of the solution and its rate of infusion.
Tris(hydroxymethyl)aminomethane, or THAM, is an amino alcohol that buffers without generating CO2. It has the advantage, therefore, of avoiding a superimposed respiratory acidosis. It has been used successfully in animals and humans with various metabolic acidoses.13,19 It is eliminated by the kidney, and thus should be used with caution in the setting of renal insufficiency. Risks include hyperkalemia, hypoglycemia, and hepatic necrosis (in neonates).7
The treatment of choice for lactic acidosis is reversal of the underlying cause of the acidosis (see Box 57.3). Pending resolution of the underlying disorder, however, the intensivist is often confronted with an unstable patient who is profoundly acidemic. Treatment at this stage is controversial.5,13 The debate has focused on the potentially deleterious effects of bicarbonate administration in lactic acidosis.20 In addition to the effects mentioned earlier, bicarbonate in animal models of lactic acidosis has been associated with increased lactate generation, reduction in intracellular pH, increased venous PCO2, and reduction in cardiac output. (This last effect correlates well with the reduction in ionized calcium concentration.13) Studies in humans likewise show no improvement in cardiac output, morbidity rate, or mortality rate with bicarbonate.13 Continuous venovenous hemodialysis (e.g., CVVHD) may be a promising tool for treating lactic acidosis, because it provides large amounts of bicarbonate without the risks of volume overload or hypocalcemia. There are several reported cases of successful treatment of metformin-associated lactic acidosis using continuous hemodialysis.21,22 Because of its superior short-term clearance compared with continuous renal replacement therapy, however, conventional hemodialysis remains the preferred treatment for metformin intoxication.23 Treatment of DKA and the acidoses associated with other intoxications are discussed in Chapters 58 and 68, respectively.
Metabolic Alkalosis
Definition and Classification
Metabolic alkalosis is a process leading to accumulation of extracellular bicarbonate that, if unopposed, will result in an increase in the plasma pH (alkalemia). It can be caused either by a gain of bicarbonate or a loss of fixed acid from the ECF. The causes of metabolic alkalosis have been described.24 In its pure form, it is accompanied by hypoventilation (CO2 retention).2
From a pathophysiologic perspective, metabolic alkalosis is divided into those factors that generate the alkalosis and those factors that maintain or perpetuate it.24,25 Metabolic alkalosis is generated by addition of bicarbonate to the blood. This can occur either by loss of acid from the body or by addition of exogenous alkali. Loss of acid may be from the stomach (e.g., vomiting or nasogastric suction) or kidney. Renal acid loss is enhanced by a high rate of sodium delivery to the distal nephron, high circulating mineralocorticoid levels, potassium depletion, and high rates of ammoniagenesis.
Because of the kidney’s prodigious ability to excrete bicarbonate, however, addition of bicarbonate to the blood is not sufficient to cause a sustained metabolic alkalosis. Some mechanism(s) to maintain the alkalosis must prevail. The most common mechanism contributing to the maintenance of metabolic alkalosis is volume depletion, either absolute or relative (e.g., congestive heart failure), which (1) reduces glomerular filtration, (2) enhances tubular bicarbonate reabsorption, and (3) causes secondary hyperaldosteronism, further enhancing urinary acidification. Another common perpetuating factor is potassium depletion, which stimulates proton secretion at several sites along the nephron.24–26
Most cases of clinically significant metabolic alkalosis are maintained by loss of chloride or potassium. Although total body sodium (and hence, volume) derangements are not directly responsible for the generation and maintenance of the metabolic alkalosis, potassium and chloride depletion are commonly seen in settings of volume depletion or excess. Therefore, from a clinical standpoint, it is useful to approach the patient with metabolic alkalosis centering on the history and physical examination, with special attention to the ECF volume status, followed by sequential analysis of blood chemistries.26 Causes of metabolic alkalosis are shown in Box 57.4. One entity unique to critically ill patients is posthypercapnic metabolic alkalosis. This syndrome is caused by abrupt treatment (usually with tracheal intubation and mechanical ventilation) of a chronic respiratory acidosis. The renal bicarbonate retention that compensated for the chronic respiratory acidosis persists (because of volume depletion) after restoration of a normal PCO2, resulting in the high pH and high plasma bicarbonate characteristic of metabolic alkalosis. The key to the diagnosis is the history and sequential analysis of blood chemistries.24
Clinical Consequences
Alkalemia in critically ill patients is associated with increased mortality rate.27 Patients with combined metabolic and respiratory alkalosis have a higher mortality rate than those with respiratory alkalosis alone, and mortality rate in alkalemia is roughly proportional to the pH.27 Although no causal relationship between alkalemia and mortality rate has been established, the pathophysiology of alkalemia is far from benign.25
First, metabolic alkalosis suppresses ventilation, causing CO2 retention and relative hypoxemia.28 Second, alkalemia acutely increases hemoglobin’s oxygen affinity (Bohr effect). Third, respiratory alkalosis causes vasoconstriction, particularly in the cerebral circulation.25 All these processes tend to decrease tissue oxygen delivery.29 (Note that chronic alkalemia inhibits 2,3-diphosphoglycerate synthesis, allowing normalization of the oxyhemoglobin desaturation curve, mitigating tissue hypoxia to some extent.) These alterations in tissue oxygen delivery could be responsible at least in part for some of the clinical manifestations of metabolic alkalosis.
Because alkalemia causes a decrease in ionized calcium concentration (see discussion under “Calcium Homeostasis”), many of the neuromuscular manifestations of metabolic alkalosis overlap with those of hypocalcemia, including paresthesias, tetany, and a predisposition to seizures.1 The acutely diminished tissue oxygen delivery to the brain may contribute to initial confusion and obtundation seen with metabolic alkalosis.
Metabolic alkalosis often is accompanied by hypokalemia and hypomagnesemia. Thus, there is an association between alkalosis and arrythmias,24 but an independent effect of the alkalosis on cardiac arrythmogenesis has not been established.
Increases in blood lactate concentration may occur in patients with metabolic alkalosis due to upregulation of phosphofructokinase and thus glycolysis, and because of tissue hypoxia (see earlier).8 With severe metabolic alkalosis (arterial pH above 7.55), the tissue hypoxia may be so marked that compensatory hypoventilation will be overridden by hypoxic drive, resulting in a normal to low arterial PCO2 and elevated blood lactate levels (so-called “lactic alkalosis”).30
Treatment
Treatment of metabolic alkalosis entails correcting the factor(s) responsible for its maintenance and, if possible, correcting the factor that generated the alkalosis. Once the underlying diagnosis is clear (see Box 57.4), therapy is usually straightforward. If the metabolic alkalosis is maintained by chloride depletion and ECF volume contraction, the intravascular volume should be restored to normal, usually with intravenous isotonic saline.24,25 Potassium should be given, as KCl, to replace any deficits (see “Disorders of Potassium Homeostasis”), because potassium depletion perpetuates the metabolic alkalosis. If nasogastric suction cannot be stopped, acid loss can be reduced by the use of H2 blockers and proton pump inhibitors.
Treating patients with metabolic alkalosis in the setting of volume overload and diminished effective circulating volume (e.g., congestive heart failure, hepatic cirrhosis) is more challenging, because saline infusion is contraindicated. Unless hyperkalemia is present, chloride should be replenished with KCl supplementation. In rare cases of concurent hyperkalemia, acetazolamide (a carbonic anydrase inhibitor) may be of benefit as it produces a bicarbonate diuresis. Acetazolamide should be avoided in patients with hypokalemia, because the alkaline diuresis will cause renal potassium wasting.24 Another potential complication of acetazolamide administration, particularly in patients with impending ventilatory failure, is worsening of hypercapnia owing to inhibition of red blood cell carbonic anhydrase and impaired CO2 transport.25 Hydrochloric acid infusion, as a 0.1 to 0.25 N solution, has been used with success in patients with severe metabolic alkalosis (pH > 7.55 and systemic instability such as encephalopathy or cardiac arrhythmia24) refractory to conventional measures.25,31 Correction of the metabolic disturbances has been reported with infusion of 0.25 N HCl at 100 mL/hour over about 12 hours.31 Extreme care must be taken to ensure that the infusion catheter is properly positioned within the vena cava, because the solution is highly caustic. Plasma chemistries must be monitored frequently in order to avoid overcorrection. If renal function is severely impaired or medical therapy is not possible, hemodialysis against a low-bicarbonate bath may be used.24
Potassium Homeostasis
Normal Potassium Physiology
Disorders of potassium (K) homeostasis are common in hospitalized patients and may be associated with severe adverse clinical outcomes, including death.32,33 Prevention and proper treatment of hyper- and hypokalemia depend on an understanding of the underlying physiology.
The total body potassium content of a 70-kg adult is about 3500 mmol, of which only 2% (about 70 mmol) is extracellular.34 This uneven distribution reflects the large potassium concentration gradient between the intracellular (Ki ≈140 mmol/L) and the extracellular (Ke ≈ 4.5 mmol/L) space, a gradient that is maintained by the intrinsic ion permeabilities of cell membranes and by Na+/K+-ATPase, the sodium-potassium pump.35 The Ke : Ki ratio largely determines the resting membrane potential of cells and thus is crucial for proper function of excitable tissues (muscle and nerve).35 Small absolute changes in Ke will perturb the ratio significantly. Therefore, disturbances of Ke (measured as changes in plasma potassium concentration, or PK) may have serious, even fatal, consequences mainly in the form of excitable tissue dysfunction.
Regulation of Internal Potassium Balance
Internal potassium balance serves to protect against changes in Ke; potassium tends to move out of cells during potassium depletion and into cells following potassium intake. This process tends to prevent drastic alterations of Ke : Ki.36,37 The factors that influence internal potassium balance include hormones, acid-base status, plasma tonicity, exercise, and cell integrity (Box 57.5).
The direction and magnitude of an acid-base-related change in PK depend on the nature and the duration of the disturbance. The most consistent and pronounced relationship between changes in pH and PK occurs in acute mineral (hyperchloremic) acidosis, where there is a strong inverse relationship between these two variables.38–40 Interestingly, hypokalemia is seen with prolonged mineral acidosis in patients with normal renal function and reflects increased renal potassium excretion.39 Unlike mineral acidoses, however, even severe acute organic (high anion gap) acidoses are not usually associated with hyperkalemia.41–44 Indeed, organic acidoses, such as lactic acidosis, actually tend to cause cellular potassium uptake.41 Nonetheless, factors coincident with the acidosis may alter PK. For example, mesenteric ischemia may result in both lactic acidosis (from anaerobic metabolism) and hyperkalemia. Even the hyperkalemia so commonly seen in patients with DKA does not result from the acidemia; rather, it appears to be a consequence of the characteristic insulin deficiency and hyperglycemia (see discussion of hypertonicity in the next paragraph).44 Respiratory disturbances typically alter PK less than metabolic disturbances. Alkaloses, respiratory or metabolic, have less effect on PK than their corresponding acidoses.38 Bicarbonate administration, which was once thought to reduce the PK by stimulating cellular potassium uptake,45 is now known to have very little if any immediate effect on internal potassium balance,46,47 except perhaps in patients with preexisting severe metabolic acidosis.41 It is clear, however, that longstanding alkalemia causes urinary potassium losses that may over time result in profound potassium depletion.48
Hypertonicity, as seen with hypertonic fluid administration49 or diabetic hyperglycemic states,50 leads to hyperkalemia, probably as a result of potassium efflux from cells by way of solvent drag. Fatal hyperkalemia has been attributed to this phenomenon in diabetic patients with end-stage renal disease (ESRD).51
Exercise causes a transient shift of potassium out of cells. Clinically significant hyperkalemia may result from exercise52,53 (and clinically misleading local venous hyperkalemia results from fist clenching during phlebotomy54).
Regulation of External Potassium Balance
In contrast to the prodigious capacity of the kidney to excrete potassium,55 renal potassium conservation is imperfect and explains why significant potassium depletion and hypokalemia may result from dietary potassium deficiency alone.56
Normally, 90% to 95% of dietary potassium is eliminated through the kidney, and only about 5% to 10% through the intestine. It is the kidney that is almost entirely responsible for matching potassium output to potassium intake in order to maintain total body potassium constant.57 The majority of potassium excreted by the kidney derives from potassium secretion in the distal nephron (connecting tubule and collecting duct).57 Virtually all regulation of potassium excretion takes place at this site in the nephron, under the influence of two principal factors: the rate of flow and sodium delivery through that part of the nephron, and the effect of aldosterone.57 Potassium secretion is directly proportional to flow rate and sodium delivery through the distal nephron, explaining in part why diuretic use often is accompanied by hypokalemia.
Metabolic acidosis with acidemia results in inhibition of renal potassium secretion.41 In contrast, metabolic alkalosis and bicarbonate delivery to the distal nephron stimulate kaliuresis by increasing the electrochemical “driving force” for potassium secretion.57 Other anions that are poorly reabsorbed in the distal nephron (e.g., synthetic penicillins) have a similar effect to stimulate potassium secretion.58
It is well established that aldosterone participates in a homeostatic feedback loop with PK such that increases in PK stimulate adrenal aldosterone production, which in turn reduces PK primarily by stimulating renal potassium excretion.57 Hypokalemia is a prominent feature of primary aldosteronism (Conn syndrome) because the high circulating aldosterone levels are accompanied by volume expansion and thus a high rate of sodium delivery to the distal nephron. When circulating aldosterone levels are high due to volume depletion (secondary aldosteronism), the increase in distal potassium secretion is offset by a decrease in distal nephron flow, thus mitigating renal potassium loss. Indeed, it is only when patients with secondary hyperaldosteronism (e.g., in congestive heart failure or hepatic cirrhosis) are treated with diuretic drugs that distal nephron flow is increased and hypokalemia may ensue.
Magnesium deficiency is associated with renal potassium wasting and may result in severe potassium depletion.59 Because magnesium, like calcium, acts to stabilize excitable membranes, the deleterious effects of hypokalemia on the myocardium are magnified by concurrent hypomagnesemia (see “Clinical Manifestations” later).60
The effect of dexamethasone (a pure glucocorticoid) to enhance renal potassium excretion appears to result entirely from hemodynamic changes that cause an increase in glomerular filtration rate and distal flow rate. All other glucocorticoids tend to further stimulate potassium secretion in proportion to their mineralocorticoid activity.57
Disorders of Potassium Homeostasis
Acute Hyperkalemia (Box 57.6)
Excessive Potassium Intake
Given an acute potassium load, a normal individual will excrete about 50% in the urine and transport about 90% of the remainder into cells over 4 to 6 hours.61 It is possible to overwhelm this adaptive mechanism such that if too much potassium is taken in too quickly, significant hyperkalemia will result. Such events are almost always iatrogenic (i.e., overly aggressive potassium replacement therapy).62 One’s ability to tolerate a potassium load declines with disordered internal balance (see later) and impaired renal potassium excretory capacity.63 In such circumstances, an otherwise tolerable increase in potassium intake may cause clinically significant hyperkalemia: Doses of oral potassium supplements as small as 30 to 45 mmol have resulted in severe hyperkalemia in patients with impaired external or internal potassium homeostasis.64
KCl, used as a supplement, is the drug most commonly implicated in acute hyperkalemia.63,65 Banked blood represents a trivial potassium load under most circumstances, because a unit of fresh banked blood, either whole or packed cells, contains only about 7 mmol of potassium.66 (The potassium concentration in banked blood does increase substantially as the blood ages, however.67) Thus, severe hyperkalemia would result only from massive transfusion of compatible blood.67,68 Infants69 or patients with renal insufficiency may develop hyperkalemia from an otherwise tolerable transfusion.
Patients undergoing open heart surgery are exposed to cardioplegic solutions containing KCl typically at about 16 mmol/L,70 which may lead to clinically significant hyperkalemia in the postoperative period, especially in patients with diabetes mellitus with or without renal failure.71
Abnormal Potassium Distribution
Among the most impressive syndromes associated with acute hyperkalemia are those involving rapid cell lysis. The tumor lysis syndrome results from treatment of chemosensitive bulky tumors with release of intracellular contents, including potassium, into the ECF.72 Extreme hyperkalemia even causing sudden death73 has featured prominently in some series of patients. Most of such patients were in renal failure from acute uric acid nephropathy, thus impairing their ability to excrete the potassium load.73 Rhabdomyolysis, either traumatic or nontraumatic, may result in sudden massive influx of potassium to the extracellular space.74 Hyperkalemia is present in about 40% of patients upon presentation with rhabdomyolysis75 and is more common among patients whose course is complicated by oliguric acute renal failure.76 Rhabdomyolysis is commonly associated with the use of alcohol75 and cocaine.77 Extreme hyperkalemia in this latter context has been reported.78 Statin drugs are frequently associated with rhabdomyolysis,79 rarely causing extreme hyperkalemia.80 Other circumstances that may result in redistributive hyperkalemia include severe extensive burns, hemolytic transfusion reactions, and mesenteric ischemia or infarction.
Pharmacologic Agents
Two drugs may rarely cause acute hyperkalemia by redistribution: digitalis glycosides and succinylcholine. Massive digitalis overdose has been associated with extreme hyperkalemia.81,82 Succinylcholine depolarizes the motor end plate and in normal individuals causes a trivial amount of potassium leak from muscle, resulting in an increase in PK by about 0.5 mmol/L.83 In patients with neuromuscular disorders, muscle damage, or prolonged immobilization, however, muscle depolarization may be more widespread, causing severe hyperkalemia.84 Prolonged use of nondepolarizing neuromuscular blockers in critically ill patients may predispose to succinylcholine-induced hyperkalemia.85
Hyperkalemic Periodic Paralysis
This rare syndrome of episodic hyperkalemia and paralysis is caused by a mutation of the skeletal muscle sodium channel, inherited in an autosomal dominant pattern.86 Attacks may be precipitated by exercise, fasting, exposure to cold, and potassium administration, and prevented by frequent carbohydrate snacks. Attacks are usually brief and treatment consists of carbohydrate ingestion. Severe attacks may require intravenous glucose infusions.87
Acute Renal Failure
Hyperkalemia accompanies acute renal failure in 30% to 50% of cases. It is seen most commonly in oliguric renal failure. Contributing factors include tissue destruction (e.g., tumor lysis syndrome, rhabdomyolysis) and increased catabolism.88
Pseudohyperkalemia
Pseudohyperkalemia refers to a measured potassium level that is higher than that circulating in the patient’s blood. It has a number of possible causes. First, it may be caused by efflux of potassium out of blood cells in the test tube after phlebotomy. This may be seen in a serum specimen in cases of thrombocytosis89 or leukocytosis,90 when the clot causes cell lysis in vitro. These days, many clinical laboratories measure electrolytes in plasma (unclotted) specimens. Even under these conditions, extreme leukocytosis may cause pseudohyperkalemia if the specimen is chilled for a long time before the plasma is separated, leading to passive potassium leak from cells.91 Hemolysis during specimen collection will falsely raise PK or plasma potassium concentration by liberating intraerythrocyte potassium. Second, if the patient’s arm is exercised by fist clenching with a tourniquet in place before the specimen is drawn, the sampled blood potassium concentration will rise significantly as a result of local muscle release of intracellular potassium.54
Acute Hypokalemia
Hypokalemia that develops over hours is virtually always the result of redistribution of potassium from the extracellular to the intracellular space. The causes of acute hypokalemia are summarized in Box 57.7. Selected causes are discussed as follows.
Treatment of Diabetic Ketoacidosis
It is well recognized that patients presenting in DKA are always severely depleted in total body potassium as a result of glucose-driven osmotic diuresis, poor nutrition, and vomiting during the development of DKA.44 Paradoxically, most patients in DKA have a normal PK upon admission.92 Insulin deficiency and hyperglycemia appear to account for the preservation of a normal PK despite severe total body potassium depletion.44 Once therapy for DKA is instituted, however, PK typically plummets as potassium is rapidly taken up by cells. Potassium replacement at rates up to 120 mmol per hour have been reported, with total potassium supplementation of 600 to 800 mmol within the first 24 hours of treatment.93 Hypokalemia in this setting may lead to respiratory arrest.94
Refeeding
A situation analogous to DKA arises during aggressive refeeding after prolonged starvation or with aggressive “hyperalimentation” of chronically ill patients. The glucose-stimulated hyperinsulinemia and tissue anabolism shift potassium into cells, rapidly depleting extracellular potassium.95 Death in the setting of refeeding has been reported and may be partly due to rapid cellular uptake of other ions (e.g., phosphorus, magnesium).96
Pharmacologic Agents
Specific β2-adrenergic receptor agonists (e.g., albuterol) may cause electrophysiologically significant hypokalemia, especially when given to patients who are potassium depleted from the use of diuretic drugs.97 Epinephrine, given intravenously in a dose about 5% of that recommended for cardiac resuscitation, causes a fall in PK by about 1 mmol/L.98 Such a dose achieves plasma levels of epinephrine comparable to those seen after acute myocardial infarction and may explain the transient hypokalemia following resuscitation from cardiac arrest even without the use of exogenous epinephrine (postresuscitation hypokalemia).99,100 A rare cause of severe hypokalemia is poisoning with soluble barium salts such as chloride, carbonate, hydroxide, and sulfide. Soluble barium salts are used in pesticides and some depilatories, which may be ingested accidentally or intentionally.101 Thiopentone, a barbiturate used to induce coma for refractory intracranial hypertension, is associated with redistributive hypokalemia in the majority of treated patients within 12 hours of initiating therapy.102
Hypokalemic Periodic Paralysis
Three forms of this rare syndrome have been described: familial, sporadic, and thyrotoxic.103,104 All have in common attacks of muscle weakness accompanied by acute hypokalemia caused by cellular potassium uptake. Death may occur due to ventilatory failure or cardiac dysrhythmias. The familial variety resulting from a skeletal muscle calcium channelopathy86 is inherited in an autosomal dominant pattern, with onset of clinical manifestations typically in the second decade of life. Attacks may occur after carbohydrate or salt ingestion or exercise. Administration of potassium orally or intavenously will abort an acute attack but is ineffective in preventing attacks.103 The sporadic variety of hypokalemic periodic paralysis is identical to the familial form except for the absence of a hereditary pattern. Thyrotoxic periodic paralysis was first described in Asians but is now recognized to be nearly ubiquitous.104 The usual onset of symptoms is in the third decade. Severe hypophosphatemia may accompany the hypokalemia.105 Treatment of the disorder is the same as treatment of hyperthyroidism.
Pseudohypokalemia
Severe leukocytosis may cause spuriously low plasma potassium concentrations if blood cells are left in contact with the plasma for a long time at room temperature or higher. This phenomenon results from ongoing cell metabolism in vitro with glucose and potassium uptake.59 Unexpected hypokalemia and hypoglycemia in the setting of leukocytosis should alert the clinician to this phenomenon.
Chronic Hyperkalemia
Renal Failure
Patients with chronic kidney disease tend to maintain a normal PK until renal function declines to about 10% of normal.106 Aldosterone and insulin both appear to play a role in the extrarenal potassium adaptation in chronic kidney disease.107 This explains why patients with chronic kidney disease who are mineralocorticoid or insulin deficient have a particular predisposition to hyperkalemia.108
Mineralocorticoid Deficiency
Mineralocorticoid deficiency may result from global adrenal insufficiency (Addison’s disease) or from selective defects in the renin-angiotensin-aldosterone axis (see Chapter 59). Hyperkalemia in the setting of unexplained hypotension should immediately raise one’s suspicion for adrenal insufficiency. A common setting for isolated mineralocorticoid deficiency is the syndrome of hyporeninemic hypoaldosteronism.108 This syndrome is most often seen in elderly patients with diabetes mellitus and moderate renal insufficiency. Hyperkalemia is a universal finding. An associated hyperchloremic metabolic acidosis (type IV renal tubular acidosis) is characteristic.108 In addition to diabetes mellitus, two other systemic diseases are associated with this syndrome: the acquired immunodeficiency syndrome109,110 and systemic lupus erythematosus.111
Aldosterone deficiency may be induced by a variety of pharmacologic agents acting at different sites in the renin-angiotensin-aldosterone axis. β-Adrenergic receptor blockers and, to a greater extent, cyclooxygenase inhibitors (COX-1 and COX-2) predispose patients to hyperkalemia by suppressing renin release.112 As a general rule, COX inhibitors should be avoided in patients with renal insufficiency or who are otherwise prone to hyperkalemia either because of diabetes or the use of other implicated drugs. Converting enzyme inhibitors and angiotensin receptor blockers decrease aldosterone biosynthesis. These drugs are reported to be implicated in 10% to 38% of hyperkalemia in hospitalized patients.113 Volume depletion and hypotension increase the risk of hyperkalemia with all these agents.
High- and low-dose heparin therapy decreases circulating aldosterone levels by selectively inhibiting aldosterone biosynthesis.114 Hyperkalemia is seen with the use of low-molecular-weight heparins as well, particularly in patients with diabetes mellitus.115
Renal Potassium Secretory Defect
An isolated defect in renal potassium secretion (often with a renal tubular acidosis) is associated with sickle cell disease or trait,116 systemic lupus erythematosus,111 and after renal transplantation.117 In this last circumstance, the hyperkalemia is exacerbated by the use of cyclosporine and tacrolimus for immunosuppresion.118,119 A syndrome of hyperkalemic (type IV) distal renal tubular acidosis is seen in patients with urinary tract obstruction.120
The so-called potassium-sparing diuretics (spironolactone, eplerenone, amiloride, and triamterene) impair renal potassium excretion by blocking sodium reabsorption in the distal nephron. Two antibiotics, pentamidine121 and trimethoprim,122,123 cause hyperkalemia, occasionally severe, by blocking sodium reabsorption in the distal nephron.
Chronic Hypokalemia
Inadequate Potassium Intake
Because renal potassium conservation is not perfect, severe dietary potassium restriction will cause hypokalemia in 3 to 7 days in normal humans.56 In one series of hypokalemic hospitalized patients, inadequate potassium supplementation during intravenous therapy contributed to the development of severe hypokalemia in 45% of cases and was the sole cause in 6%.124 Other disorders associated with nutritional hypokalemia include anorexia nervosa, alcoholism, and malignancy.
Excessive Potassium Losses
Hypokalemia may develop as a result of both upper and lower GI fluid losses, but the pathogenesis is quite different in the two situations. With diarrhea, the potassium is lost from the gut.125 Gastric fluid losses (e.g., vomiting or gastric suction) are associated with hypokalemia. Paradoxically, however, most of the potassium losses are renal, not gastric. Gastric fluid potassium concentration is only 5 to 10 mmol/L. Thus, only massive gastric fluid losses would, alone, significantly deplete total body potassium stores. The gastric fluid losses, however, stimulate renal potassium secretion in several ways. First, by generating a metabolic alkalosis and increasing bicarbonate delivery to the distal nephron, potassium secretion is stimulated. The metabolic alkalosis also leads to cellular proton loss and potassium uptake that, in renal epithelial cells, enhances potassium secretion. Finally, the volume contraction that usually accompanies GI fluid losses causes secondary aldosteronism, which further augments urinary potassium losses. Thus, in this situation, urinary potassium concentration is typically high while urinary chloride concentration is low due to volume contraction. (Urinary sodium losses may be high because of natriuresis obligated by the bicarbonaturia.)
All diuretics work by inhibiting sodium and chloride reabsorption by the nephron. Those drugs that act proximal to the potassium secretory site in the nephron promote a kaliuresis by increasing delivery of fluid distally and causing secondary aldosteronism. Thus, hypokalemia frequently accompanies the use of the two most common classes of diuretics: thiazides and loop diuretics.126 Carbonic anhydrase inhibitors exert an additional kaliuretic effect by shunting bicarbonate-rich, chloride-poor fluid to the distal nephron.127 Combining two potassium-wasting diuretics for added diuretic effect (e.g., furosemide plus metolazone) can result in severe hypokalemia. In such cases, PK has been found to fall below 3.5 mmol/L in over 80% of patients and below 3.0 mmol/L in more than half.126
Various antibiotic agents may cause renal potassium wasting and thereby hypokalemia; 90% of patients receiving amphotericin B require potassium supplementation.128 Penicillin antibiotics, particularly polyanionic derivatives such as carbenicillin and ticarcillin, have been associated with hypokalemia.129
Mineralocorticoids predispose to hypokalemia by stimulating renal potassium excretion. Mineralocorticoid excess may be primary130 (Conn syndrome) or secondary to diminished real or “effective” circulating volume. All glucocorticoid drugs except dexamethasone possess some mineralocorticoid activity. Therefore, prolonged administration of these agents can cause severe hypokalemia. In edematous patients with secondary aldosteronism (e.g., congestive heart failure, hepatic cirrhosis) hypokalemia commonly ensues only when diuretic therapy enhances distal nephron flow rate.
Magnesium deficiency is associated with renal potassium wasting and may result in severe potassium depletion (see later). Because magnesium, like calcium, acts to stabilize excitable membranes, the deleterious effects of hypokalemia on the myocardium are magnified by concurrent hypomagnesemia.131 The intensive care setting is fraught with potential causes of hypomagnesemia (see discussion under “Magnesium Homeostasis”).
Hypercalcemia causes a salt and water diuresis and is therefore commonly associated with renal hypokalemia (see “Calcium Homeostasis”).132 In one series, one third of hypercalcemic patients were hypokalemic with no other predisposing factors; the prevalence was 52% in patients with hypercalcemia of malignancy. PK was inversely proportional to the plasma calcium concentration.133
Several inborn tubular transport abnormalities are associated with chronic hypokalemia (and metabolic alkalosis). Bartter and Gitelman syndromes are associated with volume contraction and normal blood pressure, and Liddle syndrome with hypertension.134
Clinical Manifestations of Potassium Imbalance
Clinical Manifestations of Hyperkalemia
Cardiac Effects
Hyperkalemia depolarizes the cell membrane, slows ventricular conduction, and decreases the duration of the action potential. These changes produce the classic electrocardiogram (ECG) manifestations of hyperkalemia including (in order of their usual appearance) peaked T waves, prolongation of the PR interval, widening of the QRS complex, loss of the P wave, “sine wave” configuration or ventricular fibrillation, and asystole.135,136 These ECG changes may be modified by a multitude of factors such as ECF, pH, calcium concentration, sodium concentration, and the rate of rise of PK.135
ECG changes may not accompany changes in PK. If present, these ECG changes certainly suggest hyperkalemia. However, in the absence of the classic ECG changes, the clinician should not be lulled into a false sense of security when evaluating a hyperkalemic patient. Normal ECGs occur despite extreme hyperkalemia,137 and the first cardiac manifestation of hyperkalemia may be ventricular fibrillation.138 Consequently, PK greater than 6.5 mmol/L, even with a normal ECG, should be treated as an emergency (see “Treatment of Potassium Imbalance”).
Clinical Manifestations of Hypokalemia
Cardiac Effects
Hypokalemia hyperpolarizes the cell membrane and prolongs the cardiac action potential.142 These changes are associated with the following ECG manifestations: ST-segment depression, a decrease in T-wave amplitude, and an increase in U-wave amplitude.135,136 However, because all of these changes are nonspecific, the ECG is an even less reliable index of hypokalemia than it is of hyperkalemia.
Hypokalemia may be associated with an increased incidence of arrhythmias and conduction defects. It is well established that potassium depletion increases the cardiac toxicity of digitalis glycosides.143 However, controversy exists as to whether hypokalemia per se induces ventricular arrhythmias in patients not taking digitalis. There is an increase in benign ventricular ectopy in hypokalemic patients without acute myocardial ischemia.144 The clinical importance of this observation is unclear. In individuals hospitalized with acute myocardial infarction, however, a correlation between hypokalemia and ventricular tachycardia and fibrillation was observed.145 Because potassium repletion did not reduce the occurrence of these arrhythmias, it is unlikely that hypokalemia was the sole arrhythmogenic factor.145 A recent study of patients presenting with acute myocardial infarction showed a U-shaped relationship between mean in-hospital PK and mortality rate, such that PK less than 3.5 mmol/L or greater than 4.5 mmol/L were each associated with higher in-hospital mortality rates. The lowest mortality rate was seen in patients with PK 3.5 to 4.0 mmol/L, implying that potassium supplementation to achieve higher concentrations was not justifiable.146
Neuromuscular Effects
Modest hypokalemia generally presents as weakness, myalgias, muscle fatigue, and “restless” legs. With more severe hypokalemia (less than 2 mmol/L), paralysis may supervene. This usually involves the extremities but may progress to include the trunk and muscles of ventilation. As with hyperkalemia, cranial nerves typically are spared and sensory function usually remains intact.59 It is important to note that these manifestations may be masked by concomitant hypocalcemia and may only appear when calcium is replenished. Conversely, in patients with hypokalemia and hypocalcemia, tetany may develop only after potassium replacement.59 Smooth muscle dysfunction (ileus, gastroparesis) is more commonly seen with hypokalemia than with hyperkalemia.
In addition to the effects of potassium depletion on the electrical properties of the neuromuscular system, profound hypokalemia may result in muscle injury and frank rhabdomyolysis, even in bed-bound patients.147
Evaluation of Disorders of Potassium Homeostasis
Evaluation of Acute Hyperkalemia
When PK rises abruptly or if PK is high (greater than 6.5 mmol/L) on initial presentation of the patient, the first step is to obtain an ECG to look for electrophysiologic evidence of hyperkalemia. In the presence of such signs, treatment for hyperkalemia should begin urgently (see “Treatment of Potassium Imbalance”). At the same time, an unclotted blood sample should be obtained, using meticulous phlebotomy technique, for another set of electrolytes, glucose, blood urea nitrogen (BUN) and creatinine, and complete blood count (CBC). Urine should be tested for heme pigments to exclude acute rhabdomyolysis or hemolysis. The patient’s list of medications and diet should be reviewed promptly, looking for exogenous sources of potassium and drugs that may impair potassium tolerance (see Box 57.6).
Evaluation of Chronic Hyperkalemia
Figure 57.2 outlines an approach to the patient with hyperkalemia lasting for days. Failure to stimulate cortisol release with a cosyntropin stimulation test (see Chapter 59) supports a diagnosis of Addison’s disease. Absent that diagnosis, the patient is likely to have either selective aldosterone deficiency or tubular unresponsiveness to aldosterone. Assessment of the renin-angiotensin-aldosterone axis is most simply done by measuring plasma renin activity (PRA) and aldosterone levels in the basal and diuretic/posture-stimulated state.108 Tubular unresponsiveness to aldosterone is assessed by measuring the potassium secretory effect of 9a-fludrocortisone (9a-F, Florinef). One index of the driving force for potassium secretion that may be useful in this regard is the transtubular potassium gradient (TTKG), calculated as follows:
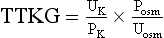
where Posm and Uosm are plasma and urine osmolalities, respectively.150 A spot urine potassium : creatinine ratio may be used instead of the TTKG.
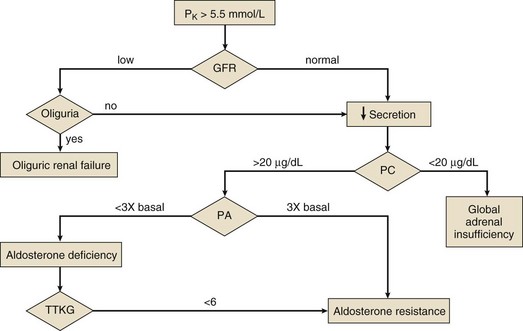
Figure 57.2 Diagnostic evaluation of chronic hyperkalemia. GFR, glomerular filtration rate; PA, stimulated plasma aldosterone (see text); PC, stimulated plasma cortisol (see Chapter 59); TTKG, transtubular potassium gradient (see text).
Evaluation of Acute Hypokalemia
Hypokalemia accompanied by serious cardiac or neuromuscular manifestations is an emergency. Likewise, urgent therapy is indicated for profound hypokalemia (PK less than 2.0 mmol/L) even in the absence of clinical complications. In addition, moderate hypokalemia (PK less than 3.0 mmol/L) in patients taking digitalis,143 and perhaps with acute myocardial ischemia,145 should be treated urgently because of the risk of ventricular arrhythmias. In all these situations, it is imperative that the blood specimen be obtained and handled properly, especially in patients with leukocytosis, because rapid administration of potassium to a patient with pseudohypokalemia may cause severe hyperkalemia.
Evaluation of Chronic Hypokalemia
Once acute hypokalemia and transient potassium redistribution have been excluded, one should next determine whether the kidney is responding appropriately to the potassium deficit or whether it is contributing to the problem. This is best done by measuring the 24-hour urinary excretion of potassium during potassium repletion (Fig. 57.3). Potassium excretion less than 20 mmol per day suggests appropriate renal potassium conservation and points to extrarenal (lower GI or skin) potassium losses, recovery from diuretic-induced hypokalemia, or chronically potassium-deficient diet. Excretion of greater than 20 mmol per day is evidence of inadequate renal potassium conservation indicating a renal cause of the hypokalemia. Renal potassium losses associated with normal systemic blood pressure are most commonly seen with the use of thiazide or loop diuretics and are accompanied by a metabolic alkalosis. Other causes of hypokalemia with metabolic alkalosis in a normotensive patient include gastric fluid loss and Bartter and Gitelman syndromes. These are separable most often by history, but if not, the urinary chloride measurement will be helpful, being low with gastric fluid losses. Renal hypokalemia may accompany a renal tubular acidosis, in which case the plasma bicarbonate will be low.
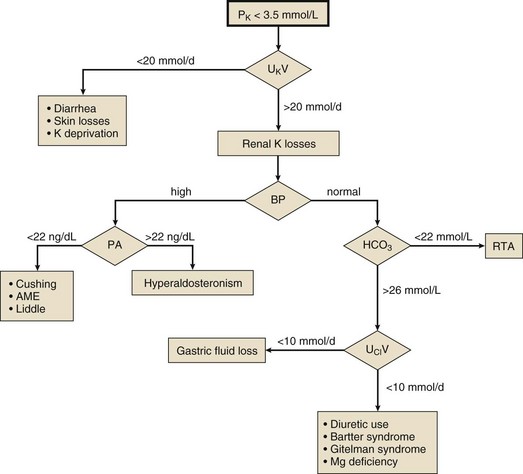
Mineralocorticoid excess may be the cause if the renal potassium loss is associated with systemic hypertension, and the renin-aldosterone axis should be studied with basal and saline-suppressed blood hormone measurements. High PRA and aldosterone levels suggest renal artery stenosis, malignant hypertension, or rarely a renin-secreting tumor. Low PRA and high aldosterone levels indicate primary aldosteronism. When both PRA and aldosterone levels are low, one should suspect the syndrome of apparent mineralocorticoid excess, Cushing syndrome, or rarely Liddle syndrome. Note that Cushing syndrome due to ectopic adrenocorticotropic hormone (ACTH) secretion often is not accompanied by typical cushingoid features.151
Treatment of Potassium Imbalance
Treatment of Acute Hyperkalemia
In considering when hyperkalemia constitutes an emergency, two points should be kept in mind. First, the electrophysiologic effects of hyperkalemia are directly proportional to both the absolute PK and its rate of rise.135 Second, although the ECG manifestations of hyperkalemia are generally progressive and proportional to the PK, ventricular fibrillation may be the first ECG disturbance of hyperkalemia;138 conversely, a normal ECG may be seen with extreme hyperkalemia.137 Thus, it is apparent that neither the ECG nor the PK alone is an adequate index of the urgency of hyperkalemia, and that the clinical context must be considered when assessing a hyperkalemic patient. Because most patients manifest hyperkalemic ECG changes at PK greater than 6.7 mmol/L,136 hyperkalemia should be treated emergently for (1) PK greater than 6.5 mmol/L and (2) ECG manifestations of hyperkalemia regardless of the PK.152
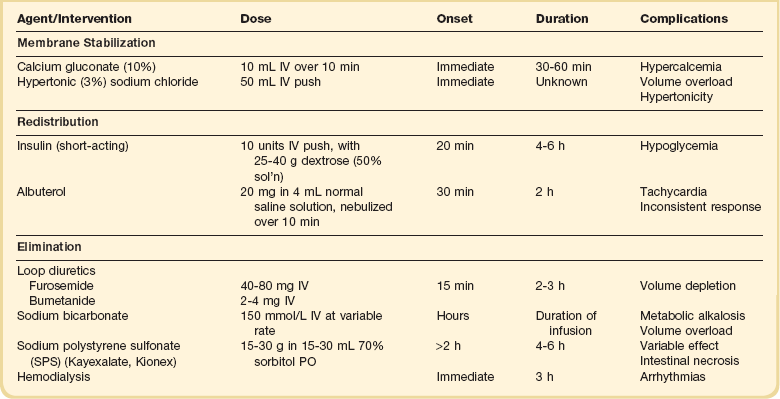
Therapy of acute or severe hyperkalemia is directed at preventing or ameliorating its untoward electrophysiologic effects on the myocardium. The goals of therapy, in chronologic order, are as follows (Table 57.2):
1. Antagonize the effect of potassium on excitable cell membranes.
Membrane Antagonism
Calcium.
Calcium directly antagonizes the myocardial effects of hyperkalemia without lowering PK.153 During treatment with calcium, the ECG should be monitored continuously. The dose may be repeated in 5 minutes if there is no improvement in the ECG, or if the ECG deteriorates after an initial improvement.152 There are several case reports of sudden death in patients given intravenous calcium while also receiving digitalis glycosides.154 Although these observations do not provide clear guidance, it may be wise to administer intravenous calcium with caution to patients known or strongly suspected of having toxic levels of digitalis glycosides.
Hypertonic Saline.
Intravenous hypertonic sodium chloride has been shown to reverse the ECG changes of hyperkalemia in patients with concurrent hyponatremia.155 Whether hypertonic saline is effective in the treatment of eunatremic patients has not been established. Moreover, the extracellular volume load imposed by hypertonic saline argues against its use.
Redistribution of Potassium into Cells
Insulin.
Insulin reliably lowers PK in a dose-dependent manner. An intravenous dose of 10 units of regular insulin given as a bolus along with an intravenous bolus of dextrose (25-40 g as a 50% solution) to adult patients lowers the PK by about 1 mmol/L.156,157 After the initial bolus, a dextrose infusion should be started, because a single bolus of 25 g of dextrose has been shown to be inadequate to prevent hypoglycemia at 60 minutes.156 There seems to be no advantage of a continuous insulin infusion over a bolus injection.40 Insulin should be used without dextrose in hyperglycemic patients; indeed, the cause of the hyperkalemia in those patients may be the hyperglycemia itself.50
Albuterol.
PK has been shown to decline by 0.6 mmol/L after inhalation of 10 mg of albuterol, and by about 1.0 mmol/L after 20 mg in patients with ESRD.158 The effect of insulin is additive with that of albuterol, with the combination reported to result in a decline in PK by about 1.2 mmol/L at 60 minutes.156 Even among patients not taking beta blockers, as many as 40% appear to be resistant to the hypokalemic effect of albuterol.156,158 For that reason, albuterol should never be used alone for the treatment of urgent hyperkalemia.
Bicarbonate.
The putative benefits of a bolus injection of sodium bicarbonate in the emergency treatment of hyperkalemia pervaded the literature until the past decade. Ironically, this dogma was based on studies using a prolonged (4-6 hours) infusion of bicarbonate.45 It has now been clearly demonstrated that short-term bicarbonate infusion does not reduce PK in patients with dialysis-dependent kidney failure, implying that it does not cause potassium shift into cells.46,47,159
Elimination of Potassium from the Body
Enhanced Renal Elimination.
Hyperkalemia occurs most often in patients with renal insufficiency. However, renal potassium excretion may be enhanced even in patients with moderate renal failure by increasing distal nephron flow. This may be accomplished with saline or sodium bicarbonate infusions and may be enhanced further by the use of loop diuretics. Diuretic-induced volume contraction must be avoided because this will lead to decreased distal nephron flow and reduced potassium excretion.152
Exchange Resin.
Sodium polystyrene sulfonate (SPS, Kayexalate, Kionex) is a cation exchange resin that exhanges sodium for secreted potassium in the colon. Each gram of resin binds approximately 0.65 mmol of potassium in vivo, although the effect is highly variable and unpredictable.40 The resin causes constipation and hence almost always is given with a cathartic. It is more effective when given orally than by retention enema.40
There are two concerns with the use of SPS for the treatment of urgent hyperkalemia. The first is its slow effect. When given orally, the onset of action is at least 2 hours and the maximum effect may not be seen for 6 hours or more. One recent study in hemodialysis patients failed to show any effect on PK after an oral dose of SPS.160 The second concern with SPS is its possible toxicity. There are numerous case reports of patients who developed intestinal necrosis after exposure to SPS in sorbitol as an enema161–163 and orally.164 A retrospective study estimated the incidence of colonic necrosis to be 1.8% among postoperative patients.164 For these reasons, some authorities consider the use of SPS to be unjustifiable.165
Dialysis.
Hemodialysis is the dialytic method of choice for removal of potassium from the body. PK falls by over 1 mmol/L in the first 60 minutes of hemodialysis and a total of 2 mmol/L by 180 minutes, after which it reaches a plateau.40 Rebound always occurs after dialysis, with 35% of the reduction abolished after an hour and nearly 70% after 6 hours.40 There is controversy as to whether dialysis for severe hyperkalemia precipitates serious ventricular arrhythmias. Because of the possibility, patients dialyzed for severe hyperkalemia should have continuous ECG monitoring.40 The rate of potassium removal with peritoneal dialysis is much slower than with hemodialysis.40
Treatment of Chronic Hyperkalemia
Further therapy of the persistently hyperkalemic patient should be guided by the diagnostic evaluation outlined in Figure 57.2. In cases of mineralocorticoid unresponsiveness or when mineralocorticoid treatment is complicated by fluid overload, a thiazide or loop diuretic can be added to the regimen. This will restore normal volume status and enhance renal tubular potassium secretion in many mineralocorticoid-resistant patients. It is crucial to avoid diuretic-induced volume depletion, however, because this will exacerbate the renal potassium secretory defect.
Patients who fail to respond to the previously mentioned measures with an increase in TTKG or urine potassium : creatinine ratio and a decrease in PK may be given sodium bicarbonate, which will stimulate renal potassium secretion. This is especially appropriate for patients whose chronic hyperkalemia is accompanied by a renal tubular acidosis (type IV RTA). The usual dose is 1 to 2 mmol bicarbonate per kg body weight per day in three or four divided doses.152
Treatment of Acute Hypokalemia
A low PK almost always indicates a large total body potassium deficit. In fact, PK decreases by approximately 0.3 mmol/L for each decrement of 100 mmol total body potassium.166 But if potassium is replenished too quickly, the homeostatic mechanisms that defend PK will be overwhelmed and PK will rise abruptly. The rate of rise of PK with potassium administration can be greatly altered by factors that affect internal potassium balance. For example, during treatment of DKA with insulin, cellular uptake of potassium may be massive, obligating enormous replacement doses of potassium. Conversely, insulin deficiency markedly impairs tolerance to a potassium load.61
See “Evaluation of Acute Hypokalemia” for definitions of urgent hypokalemia. Limited information exists on which to base a rational prescription of KCl in an emergency.61,167,168
Based on the available literature, we can estimate that nondiabetic patients with normal renal function should respond well to a 1- to 2-hour infusion of KCl at 0.6 mmol/kg/hour given intravenously in saline. In patients with renal failure of any degree, the infusion rate should be halved (0.3 mmol/kg/hour). Patients with diabetes mellitus not being treated for DKA or hyperglycemia should receive no more than 0.2 mmol/kg/hour, or about 0.1 mmol/kg/hour in the setting of renal failure. For severe hypokalemia, the ECG should be monitored continuously and the infusion stopped immediately if signs of hyperkalemia develop. The maximum increase in PK is seen at the end of the infusion, and about 50% of the increase is lost over the next 2 to 3 hours when a new steady state is achieved. Thus, PK should be measured at the end of the infusion. If the patient is still dangerously hypokalemic at this point, additional potassium may be given. If at the end of the infusion PK is in an acceptable range, the measurement should be repeated 2 to 3 hours later when disposal of potassium load is complete in order to determine the need for further treatment.152
Hypokalemia that is not life threatening is best treated with oral potassium replacement. It is important to recognize that GI absorption of an oral dose of KCl elixir is essentially complete. Dangerous hyperkalemia can occur in entirely normal individuals following KCl ingestion.169 The maximum increase in PK is seen 1.5 to 2 hours after an oral potassium load. Thus, a sensible oral dose of KCl in moderate hypokalemia should probably not exceed the hourly intravenous doses proposed earlier. There is no reason to give a simultaneous oral and intravenous potassium dose; serious hyperkalemia may ensue.
Water Homeostasis
Hyponatremia and hypernatremia reflect disorders of water homeostasis. They are common disorders in critically ill patients and are associated with increased morbidity and mortality rates.170,171
Physiology of Water Homeostasis
Tonicity or effective osmolality describes the capacity of particles in solution to effect water movement across a semipermeable membrane such as the cell membrane. The normal response to water ingestion (of sufficient magnitude to lower the plasma osmolality even slightly) is the excretion of maximally dilute urine (urine osmolality <100 mOsm/kg). The underlying physiologic sequence is as follows: The plasma hypotonicity is sensed by the cells making up the hypothalamic osmostat. These hypothalamic nuclei then proportionately reduce their synthesis of AVP, also known as ADH, leading to diminished AVP release into the circulation by the posterior pituitary. The lower circulating AVP concentration, in turn, results in the insertion of proportionately fewer water channels into the collecting duct of the kidney. This, in turn, creates a more water-impermeable conduit, preventing water reabsorption and allowing excretion of the dilute urine elaborated by the more proximal segments of the nephron.172
Conversely, plasma hypertonicity leads to higher circulating AVP concentration and proportionately higher water permeability of the collecting duct, and the excretion of a concentrated urine.172
Figure 57.4 shows the relationship between plasma osmolality, plasma AVP concentration, and urine osmolality. The normal “set point” is a plasma osmolality of about 285 mOsm/kg. Notice that the minimum urine osmolality is about 50 mOsm/kg, and the maximum is about 1200 mOsm/kg.173
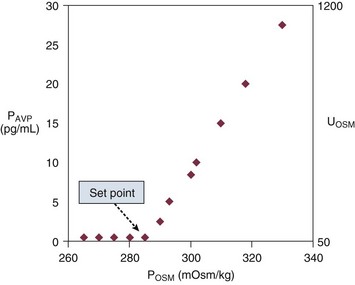
When plasma osmolality rises beyond 290 to 295 mOsm/kg, the thirst center of the hypothalamus is stimulated. At that point, neurologically intact individuals with access to water will drink until the plasma osmolality returns to normal.173
Nonosmotic Vasopressin Release
It is important to recognize that plasma osmolality is not the only determinant of AVP synthesis and release. Low arterial blood pressure and low effective arterial volume powerfully stimulate AVP release.173 This baroreceptor-mediated AVP release is teleologic, because water retention is an important component in the defense against hypovolemia. So primal is this circulatory defense that the baroreceptor stimulation predominates over any osmolal effect on AVP release.173 Thus, a volume-contracted or hypotensive individual will have high circulating AVP levels even if his plasma osmolality is low. In addition, circulating AVP levels rise with pain, stress, nausea, hypoxia, hypercapnia, and a variety of medications, most notably epinephrine and high doses of narcotic analgesics.173
Hyponatremia
Epidemology and Clinical Manifestations
Hyponatremia (plasma sodium concentration < 135 mmol/L) is one of the most common electrolyte disorders, found in approximately 3% of hospitalized patients and as many as 30% of patients in ICUs.170
The clinical manifestations of hyponatremia are largely attributed to intracellular volume expansion (cellular edema), which occurs only when hyponatremia is associated with hypotonicity. Intracellular volume expansion is of greatest consequence in the brain, where it is translated into increased intracranial pressure because of the rigid calvarium.174
Pathophysiology
The pathophysiology of hypotonic hyponatremia has important implications for its management. Most cells—especially brain cells—have adaptive mechanisms for mitigating tonicity-related volume changes.174 Cell volume peaks 1 to 2 hours after the onset of acute hypotonicity. Thereafter, solute and water are lost from cells, and cell volume returns toward normal. After several days of sustained hypotonicity, cell volume is restored nearly to normal.174
The morbidity and mortality risks associated with hypotonic hyponatremia are influenced by several factors, including the magnitude and rate of development of the hyponatremia, the patient’s age and gender, and the nature and severity of any underlying diseases.174 The very young and very old, women, and alcoholics appear to be at particular risk.175 Cell-volume adaptation to hypotonicity may be deficient in premenopausal women, who suffer more frequent and more severe neurologic consequences than men with equivalent degrees of hypotonicity.176
Neurologic symptoms usually do not occur until the plasma sodium concentration falls below 125 mmol/L, at which point the patient may complain of anorexia, nausea, and malaise. Between 120 and 110 mmol/L, headache, lethargy, confusion, agitation, and obtundation may be seen. More severe symptoms (seizures, coma) may occur with levels below 110 mmol/L.177 Focal neurologic findings are unusual but do occur, and transtentorial cerebral herniation has been described in severe cases, especially in young women following surgery.176 In that setting, hypoxemia is common and often is associated with noncardiogenic pulmonary edema.178 Hypoxia appears to exacerbate the cerebral damage in hyponatremia.179
Although symptoms generally resolve with correction of the hypotonicity, permanent neurologic deficits may occur, particularly in acute severe hypotonicity, when the brain’s volume-regulatory defenses may be overwhelmed.176 Profound hypotonicity that develops in less than 24 hours may be associated with residual neurologic deficits and has a 50% mortality rate in some populations.176 In contrast, when hypotonicity develops more gradually, symptoms are both less common and less severe. Indeed, patients with chronic hyponatremia, even in the range of 115 to 120 mmol/L, may be completely asymptomatic.174
Differential Diagnosis
Hyponatremia may coexist with a normal, high, or low plasma osmolality. Thus, the diagnostic algorithm for hyponatremia (see Fig. 57.2) begins with an assessment of the plasma osmolality (Posm). This may be estimated by the following formula:
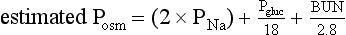
where Pgluc is the plasma glucose concentration and BUN is blood urea nitrogen concentration, both in mg/dL. If there is a suspicion that an unmeasured, osmotically effective solute may be implicated (e.g., mannitol or glycerol), the Posm should be measured directly.
Isotonic hyponatremia (also known as factitious hyponatremia or pseudohyponatremia) is a laboratory artifact seen with analytic techniques that measure the mass of sodium per unit volume of plasma sampled.180 It is seen in the presence of marked hypertriglyceridemia or paraproteinemia, when the measurement method involves a predilution step. Direct potentiometry (which uses an ion-selective electrode in undiluted plasma) avoids this problem.180
Hypotonic hyponatremia is almost always caused by an inability of the kidney to excrete sufficient electrolyte-free water to match water intake. This may occur either because the normal diluting capacity of the kidney is overwhelmed by excessive water intake or because the diluting capacity of the kidney is impaired. These alternatives usually can be distinguished by measuring the urine osmolality. A urine osmolality less than 100 mOsm/kg in a patient with hypotonic hyponatremia points to excessive water intake as the cause (Fig. 57.5). It is a prodigious feat for an individual eating a normal diet to overwhelm the normal diluting capacity of the kidney. Estimates are that one can ingest (and excrete) about 20 L of water a day without affecting the plasma osmolality appreciably.173 Thus, patients who develop hyponatremia from so-called psychogenic or primary polydipsia—usually patients with obsessive-compulsive disorder or psychosis—typically have concurrent urinary diluting defects, either in association with the underlying mental illness or perhaps as a side effect of psychotropic or anticonvulsant medications.181
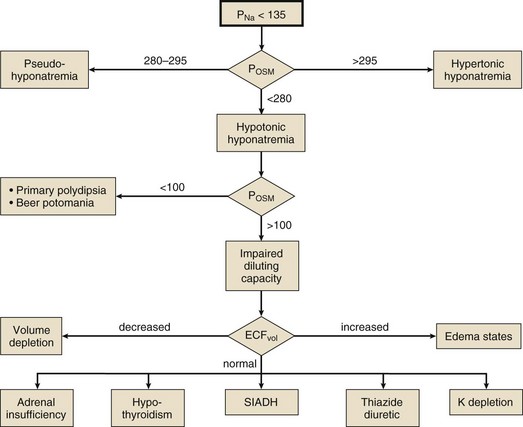
Not all patients with hypotonic hyponatremia and a dilute urine have primary polydipsia. The patient may be ingesting a diet so deficient in protein and salt that he excretes very little solute in the urine. In that situation (called beer potomania for obvious reasons,182 although the syndrome has been seen in other patients with very low daily solute intake183) the low daily solute excretion limits the total amount of water that can be eliminated even with a maximally dilute urine (i.e., maximum urine volume = solute excretion ÷ minimum Uosm). This might reduce the maximum water excretion to only 3 to 4 L/day, a quantity easily exceeded by an enthusiastic beer drinker.
Euvolemic Hyponatremia
Patients with pure water excess appear clinically euvolemic because excess water distributes throughout the total body water space; only one third of total body water is extracellular (and only one twelfth is intravascular). The only evidence of the slight intravascular volume expansion is low BUN and plasma uric acid concentration.184 The paradigm of euvolemic hyponatremia with a concentrated urine is the syndrome of inappropriate antidiuretic hormone secretion (SIADH). It is characterized by elevated circulating AVP (ADH) levels that are inappropriate to vasopressin’s two physiologic stimuli (i.e., osmotic or hemodynamic).185 Hypotonic hyponatremia in patients with SIADH develops to the extent that water ingestion exceeds water eliminated by insensible, GI, and renal routes. Because the normal response to extracellular hypotonicity is the elaboration of maximally dilute urine (urine osmolality < 100 mOsm/kg), the urine need only be inappropriately concentrated (i.e., >100 mOsm/kg) to be compatible with a diagnosis of SIADH.
Because hypothyroidism186 and glucocorticoid insufficiency187 may impair urinary dilution, patients in whom a diagnosis of SIADH is entertained should undergo appropriate tests of thyroid and adrenocortical function. (see Chapter 59.)
Once a diagnosis of SIADH is made, its cause must be established, because the cause may have important implications in its own right and may be easily remediable. Box 57.8 lists important causes of SIADH. They fall into five major categories: intracranial abnormalities, intrathoracic abnormalities, tumors, drugs, and idiopathic. An important variant of SIADH is the reset osmostat syndrome,188 in which vasopressin levels are regulated normally by tonicity but around a lower “set point” than normal. This syndrome is seen most often in patients who are severely debilitated (e.g., malnutrition, metastatic cancer, advanced tuberculosis) and may account for up to one third of cases of SIADH. The diagnosis of reset osmostat syndrome has important therapeutic implications, as will be discussed later.
Hypovolemic Hyponatremia
The cause of the volume contraction usually is obvious (e.g., hemorrhage, vomiting, diarrhea, diuretics). When it is not, the urine sodium concentration can be helpful in distinguishing between renal and extrarenal solute losses. Renal losses (e.g., as a result of diuretic medications) are usually reflected by sodium wasting, and extrarenal losses are usually accompanied by sodium conservation (urine sodium concentration < 10 mM). Exceptions occur in the recovery phase after diuretic therapy and in metabolic alkalosis due to vomiting. In the latter situation, the urine chloride concentration tends to be very low and is the best indicator of extracellular volume depletion.189,190
Cerebral salt wasting may be responsible for hypovolemic hyponatremia in patients with intracranial pathology (e.g., tumors, hemorrhage). The pathogenesis of the urinary salt wasting is incompletely understood. The mechanism of hyponatremia in this setting is similar to that of other hypovolemic states. As a hyponatremic syndrome in patients with central nervous system disease, cerebral salt wasting is often difficult to distinguish from SIADH because urinary sodium excretion tends to be high. Particularly confusing in patients with cerebral salt wasting is the finding of hypouricemia, which is thought to reflect impaired solute reabsorption in the proximal tubule.191 The key features that distinguish cerebral salt wasting from SIADH are volume depletion and urinary sodium excretion inappropriate to the patient’s volume status.191
The hyponatremia associated with diuretic treatment is multifactorial in origin. Insofar as diuretics produce overt volume depletion, they can cause hyponatremia by the mechanisms discussed previously. Thiazides have been associated with the development of acute severe, symptomatic hyponatremia, particularly in small, elderly women, in the absence of overt signs of volume depletion.192 The cause of this often precipitous syndrome remains uncertain.193
Hypervolemic Hyponatremia
Hypervolemic hyponatremia generally is seen in patients who cannot excrete sodium normally because they have either severe renal failure or one of the pathologic edema-forming states (e.g., congestive heart failure, hepatic cirrhosis, nephrotic syndrome). Patients with advanced chronic kidney disease are predisposed to hyponatremia.194 Acute oliguric renal failure or end-stage (dialysis-dependent) renal failure will be accompanied by hyponatremia to the extent that water intake exceeds insensible and GI water elimination (see Chapter 55).
Hyponatremia is seen in over 20% of patients presenting with decompensated congestive heart failure and over 30% of patients admitted to hospital with complications of hepatic cirrhosis.195 The hormonal milieu of such patients is typical of intravascular volume depletion, even though the absolute intravascular volume typically is increased. Thus, these disorders are said to be characterized by reduced effective circulating volume.196 Because of the perceived intravascular volume depletion, renal diluting ability is compromised for reasons similar to those in hypovolemic hyponatremia.
Hyponatremia in Endurance-Sports Athletes
The incidence of hyponatremia, with serum sodium less than 135 mmol/L, is approximately 15% in marathon and triathalon athletes; 0.6% of runners develop severe hyponatremia with serum sodium levels less than 120 mEq/L.195 The three major risk factors associated with hyponatremia in this setting includes body mass index (BMI) less than 20, racing time more than 4 hours, and weight gain related to excessive fluid intake during the race. Fluid overload is the most important factor in the development of hyponatremia for runners.195
Management and Complications
Hyponatremia per se requires treatment only when it is associated with hypotonicity. Hypertonic hyponatremia responds to the treatment of the underlying disorder, most commonly a hyperosmolar hyperglycemic state (see Chapter 58).
The therapy of hypotonic hyponatremia must be tailored to (1) the patient’s signs and symptoms, and (2) the duration of the disorder.175 Severe hyponatremia (plasma sodium concentration < 115 mmol/L) can be life threatening, especially if it develops rapidly.177,197 The therapy of symptomatic hyponatremia, irrespective of cause, is directed at raising ECF tonicity to shift water out of the intracellular space, thereby ameliorating cerebral edema. The rate of correction, however, must be carefully regulated. Overly rapid correction, particularly in patients with chronic hyponatremia, in whom cell volume adaptations may be complete, can produce osmotic demyelination syndrome.198,199 Osmotic demyelination syndrome is associated with a variety of sometimes irreversible neurologic deficits (e.g., dysarthria, dysphagia, behavioral disturbances, ataxia, quadriplegia, coma), which typically develop 3 to 10 days after treatment.200 Additional risk factors for osmotic demyelination include hypokalemia, malnutrition, alcoholism, advanced age, female gender, and the postoperative state, particularly after orthotopic liver transplantation.201–203
For patients with chronic hyponatremia (>48 hours duration) or hyponatremia of unknown duration, the plasma sodium concentration should be raised by a maximum of 0.5 mmol/L/hour, 8 to 10 mmol/L in the first 24 hours199 and 20 mmol/L over the first 48 hours. Care should be taken to avoid overcorrecting the plasma sodium concentration.187,200 In grave situations (plasma sodium concentration < 105 mmol/L or in the presence of seizure or coma), initial therapy can be more aggressive (targeting a change in the plasma sodium concentration of 1 to 2 mmol/L/hour for the first few hours), but the recommended daily target should not be exceeded.187,200
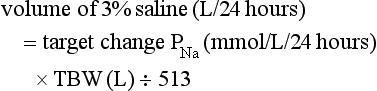
For example, in a 70-kg man with a plasma sodium concentration of 105 mmol/L and total body water (TBW) of 42 L (60% of body weight), the amount of sodium needed to raise the plasma sodium concentration by 10 mmol/L is 10 × 42, or 420 mmol. Therefore, 420 ÷ 513 or 0.82 L of 3% saline would be required in the first 24 hours, or 34 mL/hour. It is important to recognize that the calculation provides only a very rough guideline. The plasma sodium concentration must be monitored frequently during treatment to adjust the rate of correction. If the rate of correction begins to exceed the target rate, the hypertonic saline infusion should be stopped; rarely, it may be necessary to administer water (enterally or intravenously) or even desmopressin204 in order to prevent overly rapid correction or overcorrection. Rapid extracellular volume expansion with hypertonic saline may precipitate pulmonary edema, particularly in patients with underlying heart disease. Thus, patients receiving 3% saline should be assessed frequently for evidence of volume overload. One may administer a loop diuretic if necessary, recognizing that this will enhance electrolyte-free water clearance and accelerate the correction. Rarely, administration of isotonic (normal) saline to patients with SIADH paradoxically may lower the plasma sodium concentration if the urine osmolality remains high—a process that has been called desalination.205
If the cause of SIADH cannot be corrected and if water restriction is poorly tolerated or ineffective, a specific vasopressin (V2) receptor antagonist (VRA) can be used.206 Conivaptan (parenteral) and tolvaptan (oral) were the first VRAs to be approved by the U.S. Food and Drug Administration for clinical use. These agents have changed the management of patients with SIADH.207 Tolvaptan reliably increases PNa concentration, but hyponatremia recurs within 1 week after the medication is discontinued.208 For patients admitted with hyponatremia related to decompensated congestive heart failure, tolvaptan along with diuretic therapy improves most signs and symptoms of heart failure within 1 week,209 although no effect on 24-month mortality rate or heart failure morbidity has been demonstrated.210 Before the introduction of VRAs, demeclocycline (a tetracycline antibiotic that increases electrolyte-free water excretion by inhibiting vasopressin-mediated water reabsorption in the collecting duct) was commonly used to treat patients with SIADH. Demeclocycline is contraindicated in patients with renal disease, hepatic cirrhosis, or congestive heart failure because drug-related renal insufficiency has been described in these situations.206 Neither VRAs nor demeclocycline are indicated for the treatment of acute, severe hyponatremia.
The treatment of diuretic-induced hyponatremia is straightforward: Withdrawing the offending drug, liberalizing salt intake, and replenishing body potassium stores usually correct the disorder. Severe symptomatic hyponatremia in this setting should be treated with hypertonic saline as detailed earlier. Patients must be watched carefully after correction of the hyponatremia because relapse may occur for up to a week.193
Resolution of the hyponatremia associated with any of the pathologic edematous disorders ultimately depends on effective treatment of the underlying disease. Regardless of the specific therapy of the underlying disorder, the mainstay of therapy for the hyponatremic edematous patient remains salt and water restriction. Diuretics are often a double-edged sword in the hyponatremic edematous patient: They may be needed to treat pulmonary vascular congestion, peripheral edema, and ascites but if used to excess can produce further decrements in effective arterial blood volume and exacerbate water retention. Strategies directed at increasing effective arterial blood volume (e.g., afterload reduction with angiotensin-converting enzyme inhibitors211,212) have had some success in increasing electrolyte-free water excretion and ameliorating hyponatremia in patients with congestive heart failure. VRAs are likely to facilitate treatment of hypervolemic hyponatremia (see earlier).207
Hypernatremia
Epidemiology and Clinical Manifestations
Hypernatremia is common in critically ill patients, being present on admission in about 9% of patients and developing during the course of the ICU stay in another 6%.213 It is associated with a significantly higher mortality rate than is seen in patients without hypernatremia.213
Sustained hypernatremia develops in patients whose water output exceeds their input. Water ingestion can defend against the development of hypernatremia even when water losses are prodigious. For that reason, hypernatremia upon presentation to the hospital occurs most commonly in patients who are incapacitated: those who have impaired thirst sensation, who cannot access water, or who cannot express their need for water (e.g., infants and patients with neurologic impairments). Similar predispositions prevail among critically ill patients. Thus, the development of hypernatremia in hospitalized patients is considered to be iatrogenic, reflecting an incomplete understanding of the factors that lead to hypernatremia.213 The increased mortality rate seen in patients with hypernatremia probably is due to their underlying vulnerabilities rather than an effect of the hypernatremia itself.214
The clinical manifestations of hypernatremia are proportional to the magnitude and rate of rise of the plasma sodium concentration and are attributable to intracellular volume contraction. To counteract cellular volume contraction, cells begin to adapt within minutes by allowing the influx of electrolytes, thus mitigating cell shrinkage. When hypernatremia lasts more than a few hours, brain cells generate new organic osmolytes. This leads to further water movement back into brain cells, restoring cell volume nearly to normal after about 3 days.215 Thus, chronic progressive hypernatremia is associated with fewer and milder symptoms than acute severe hypernatremia. Most often, patients with longstanding hypernatremia present with weakness, lethargy, and confusion. Seizure and coma may supervene. Acute severe hypernatremia in infants and small children is associated with intracranial bleeding,216 presumably caused by brain shrinkage and traction on the penetrating vessels. There is some controversy, however, as to whether the hypernatremia in that situation is the cause or the effect of the intracranial hemorrhage.217
Differential Diagnosis
The plasma sodium concentration reflects the ratio of body sodium content to total body water. Thus, hypernatremia (plasma sodium concentration > 145 mmol/L) can result from loss of pure water alone, loss of hyponatric* fluid, or a gain of sodium or hypernatric fluid. It is important to distinguish among these paths to hypernatremia because they have diagnostic and therapeutic implications (Fig. 57.6).
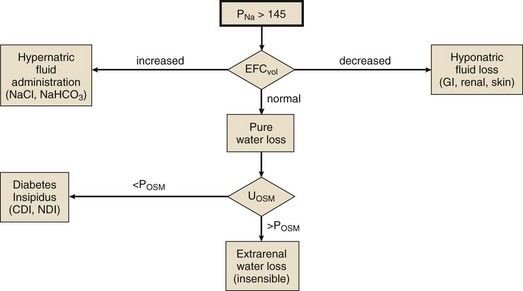
Figure 57.6 Diagnostic evaluation of hypernatremia. See Figure 57.3 legend for abbreviations. CDI, central diabetes insipidus; GI, gastrointestinal; NDI, nephrogenic diabetes insipidus.
Euvolemic Hypernatremia
Hypernatremic patients who appear euvolemic most likely have pure water loss as an explanation for their hypernatremia. This is because the water is lost from all body compartments proportionately; only one twelfth of the water loss is intravascular. For example, a 60-kg woman with a 3 L pure water loss would experience an intravascular loss of only 250 mL (clinically imperceptible) but would develop a plasma sodium concentration of 155 mmol/L.* Pure water can be lost either through the skin and respiratory tract (so-called insensible losses) or in urine.
Insensible losses amount to about 10 mL/kg/day under normal environmental conditions in an afebrile individual with a normal respiratory rate. A hot environment, fever, or rapid respiratory rate may double that rate.173 Note that a patient on a mechanical ventilator using humidified gas will lose no water through the respiratory tract.
The loss of large amounts of dilute, electrolyte-free water in the urine is typical of diabetes insipidus (DI). DI may be central (CDI) or nephrogenic (NDI) depending on whether the defect is in vasopressin release from the posterior pituitary or in renal response to circulating vasopressin, respectively. The causes of DI are shown in Box 57.9. Most cases of CDI, especially those following trauma or intracranial surgery, are self-limited, lasting 3 to 5 days. Of special interest to intensivists is a classic triphasic syndrome that may be seen following severe head trauma:
1. Initially, there is abrupt cessation of vasopressin release from the posterior pituitary, accompanied by polyuria.
2. About a week later, an antidiuretic phase ensues, characterized by urinary concentration and water retention with a tendency toward hyponatremia, lasting 5 to 6 days. This appears to result from the release of stored vasopressin from the degenerating hypothalamic neurons.
3. Persistent CDI recurs when the vasopressin stores are depleted.218
Hypovolemic Hypernatremia
A common cause of hypovolemic hypernatremia is the loss of GI fluids.219 Most GI fluids have an electrolyte concentration below that of plasma: The concentration of sodium plus potassium in stool is roughly constant at 110 to 120 mmol/L over a wide range of stool volume.125 Gastric fluid has an even lower electrolyte concentration: about 40 to 50 mmol/L total cation concentration.220 Diuresis, either osmotic (glucose-, mannitol-, or urea-induced) or medication-induced, causes the loss of urine with an electrolyte concentration less than that of plasma, leading to volume contraction and hypernatremia. The loss of sweat, which contains some sodium, can cause hypovolemic hypernatremia in individuals who exercise vigorously in a hot environment. If the cause of the fluid loss is not apparent from the history or the physical examination, a urinary chloride concentration less than 10 mmol/L in the face of hypovolemic hypernatremia suggests that the electrolyte loss is extrarenal (cutaneous or GI).
Hypervolemic Hypernatremia
Hypervolemic hypernatremia is relatively uncommon. It results most often from the administration of hypertonic sodium salts to patients without free access to water.219 Patients show signs of extracellular volume expansion (e.g., hypertension, edema, congestive heart failure, and pulmonary edema). In infants, this syndrome has been caused by erroneous preparation of dietary formula using salt instead of sugar; in adult outpatients, it may be caused by ingestion of a concentrated salt solution, usually for its emetic effect.221 The risk of death is substantial and seems to be proportional to the plasma sodium concentration.221
In hospitalized adults, hypervolemic hypernatremia it is most often iatrogenic, caused by intravenous administration of undiluted sodium bicarbonate (formulated at 1 mEq/mL or 1000 mmol/L) or sodium chloride (3% [513 mmol/L] or 23.5% [4019 mmol/L]). Not all hypervolemic hypernatremia results from the administration of hypertonic fluids. It may be seen in a volume-expanded patient who then loses hypotonic fluid.222
Treatment
If the water losses are urinary (see Fig. 57.6) and due to CDI, ADH should be administered. Several formulations are available (Table 57.3). In the acute (postsurgical or posttraumatic) setting, L-arginine vasopressin may be used either subcutaneously or intravenously, although the latter route may be associated with hypertension and coronary spasm and should therefore be used with extreme caution.223 The advantage of vasopressin in this setting is its short half-life, which allows the physician to repeatedly assess the need for continued hormone replacement, especially when the disorder may be self-limited. Desmopressin (DDAVP) is a synthetic analog of vasopressin that has no vasoconstrictor properties, thus avoiding the risks of hypertension and myocardial ischemia. The treatment of the urinary water losses associated with NDI are best treated with thiazide diuretics with or without COX inhibitors. Because most of these agents are orally administered, treatment of NDI in the critically ill patient often consists of urinary water replacement until he or she is able to take medications by mouth.
Table 57.3
Pharmacologic Treatment of Central Diabetes Insipidus
Adapted from Singer I, Oster JR, Fishman LM: The management of diabetes insipidus in adults. Arch Intern Med 1997;157(12):1293-1301.
The current body water deficit can be estimated by the following formula:
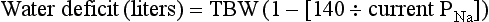
where TBW is total body water in liters (estimated as about 0.5 × lean body weight [kg] in women and 0.6 × lean body weight in men). For example, a 60-kg woman presenting with a plasma sodium concentration of 160 mmol/L is estimated to have a total body water deficit of 30 (1 – 0.875), or 3.75 L. This formula provides only a rough estimate of the water deficit.
The rate of water replacement should be proportional to the rapidity with which the hypernatremia developed.215 Thus, if the hypernatremia had developed over only a few hours (such as in postsurgical or posttraumatic DI), it can be corrected just as quickly. On the other hand, hypernatremia of more than a day’s duration, or of unknown duration, must be correctly slowly in order to avoid cerebral edema. In general, one should aim to correct half the water deficit in the first 24 hours and the remainder over the next 24 to 48 hours.
Water is best administered enterally, as tap water. If that route is unavailable, 5% dextrose in water (D5W) may be used, with the understanding that the capacity to metabolize glucose is limited to about 15 g/hour in a critically ill adult.224 Thus, even in nondiabetic patients, the administration of more than 300 mL/hour of D5W is likely to result in hyperglycemia, which may be relatively resistant to insulin administration. Hyperglycemia will exacerbate urinary water losses by causing an osmotic diuresis. Half-normal (0.45%) saline may be a good alternative, as long as one recognizes that only half the administered volume is electrolyte-free water and that the sodium load may cause unwanted volume expansion.