Chapter 31 Acid-Base Disturbances
In this chapter, the principles of normal acid-base homeostasis are briefly reviewed and the common disturbances encountered in cardiac surgery patients are discussed. In a short summary such as this, many details are touched on only briefly or are oversimplified. In particular, the Stewart principle1 for analysis of acid-base disturbance is mentioned only in passing (Box 31-1).
BOX 31-1 The Stewart Physiochemical Approach to Acid-Base Analysis
The approach to acid-base analysis described by Stewart1 is based on the fundamental principles of physical chemistry and describes the mechanism of acid-base disturbances. These principles are as follows:
From these principles, one can derive the three independent variables influencing pH:
The main implication of the Stewart approach is that the concentration of hydrogen ions or bicarbonate cannot change unless one of the three independent variables changes. Similarly, changes caused by adding or removing hydrogen ions or bicarbonate in a compartment are accommodated by changes in the dissociation of water unless one or more of the independent variables changes. Acid-base disorders can therefore be classified in relation to the derangements of the independent variables. Metabolic acidoses are caused by a reduced SID or by an increased Atot, whereas metabolic alkaloses are caused by an increased SID or a reduced Atot. However, because no mechanisms appear to control Atot, changes in Atot are not considered to be acid-base disorders by some authors.12
ACID-BASE PHYSIOLOGY
Normal Regulation of pH
Buffers
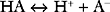
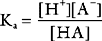
Buffering ability is maximal when the concentration of the weak acid and its conjugate base are equal; that is, when [HA] = [A−], which occurs when the hydrogen ion concentration of the solution is equal to the dissociation constant (Ka) of the buffer; put another way, when the pH of the solution is equal to the pKa of the buffer (where pKa = the −log10Ka). To function effectively, a buffer must have a pKa of within 1.0 unit of the pH of the solution being buffered.
Buffering Systems Within the Body.
The bicarbonate buffer system is as follows:
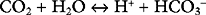
Despite not having the same form as Equation 33-1, the bicarbonate buffer system reacts to changes in pH in a similar manner: the addition of H+ shifts the equation to the left, generating CO2 and H2O; the removal of H+ shifts the equation to the right, generating HCO3− (and H+). The pKa of the bicarbonate buffer system is 6.1, which at first glance seems too far removed from the pH of plasma to be very effective. However, because the concentration of bicarbonate in extracellular fluid is high (24 mmol/l) and because the bicarbonate buffer system is open, with the ability to independently control carbon dioxide and bicarbonate concentrations, it is quantitatively the most important buffer of the extracellular fluid.
The histidine amino acid found on hemoglobin and plasma proteins (pKa 6.5) constitutes the majority of the nonbicarbonate buffering in the extracellular fluid. Although hemoglobin is an intracellular protein, the permeability of the red cell membrane is such that it can be considered an extracellular buffer.
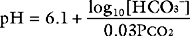
Role of the Lungs: Control of Paco2
The partial pressure of dissolved carbon dioxide in arterial blood (Paco2) is inversely proportional to alveolar ventilation and directly proportional to the rate of production of carbon dioxide (see Eq. 1-17). The carbon dioxide partial pressure in arterial blood is tightly controlled to a value close to 5.2 κPa (40 mmHg), corresponding to a concentration of dissolved gas of 1.2 mmol/l. A reduction in alveolar ventilation leads to an increase in Paco2, which drives the equilibrium of Equation 31-3 to the right, causing a fall in pH. This condition is termed respiratory acidosis. Conversely, an increase in alveolar ventilation drives the Paco2 down and the equilibrium of Equation 31-3 to the left, causing an increase in pH. This condition is termed respiratory alkalosis.
Role of the Kidneys: Control of Plasma HCO3− Concentration
On a normal diet, about 80 mmol/day of nonvolatile (non-carbon-dioxide or metabolic) acid is generated each day, mainly by the metabolism of proteins. This acid is buffered in the plasma by the bicarbonate buffer system and excreted as carbon dioxide gas, resulting in a net loss of bicarbonate. The cells of the distal convoluted tubule of the kidney regenerate this lost bicarbonate. In the process of regenerating bicarbonate, the distal tubular cells secrete an equimolar amount of hydrogen ions into the tubular fluid, resulting in acidification of the urine. With normal renal function, approximately 400 to 500 mmol of bicarbonate can be regenerated in this way (or, put another way, the kidneys can clear 400 to 500 mmol of H+ per day, providing that urinary buffering is ideal),2,3 resulting in a urinary pH as low as 4.5. In this way, the kidneys regulate the plasma bicarbonate concentration (normal range 24 to 28 mmol/l) in the face of a changing metabolic acid load. The larger the metabolic acid load, the lower the urinary pH. This renal mechanism takes some hours to days to be fully effective.
From Equation 31-3 it is seen that an excess of hydrogen ions leads to a fall in plasma bicarbonate concentration, a condition known as metabolic acidosis. Conversely, the loss of hydrogen ions results in an increase in plasma bicarbonate concentration, a condition known as a metabolic alkalosis.
Assessment of Acid-Base Status
Three parameters are used to assess acid-base status: pH, Paco2, and HCO3−. Both pH and Paco2 are measured directly by the blood-gas analyzer, whereas bicarbonate concentration is calculated (automatically) from the Henderson-Hasselbalch equation. Because bicarbonate concentration is influenced by changes in the partial pressure of carbon dioxide (due to shifts in the equilibrium shown in Equation 31-3), blood gas analyzers also report the standard bicarbonate or the base excess. The standard bicarbonate is what the actual bicarbonate concentration would be if the blood gas sample were equilibrated with carbon dioxide at 5.2 κPa in fully oxygenated blood at 37°C. The base excess is the amount (in mmol/l) of acid that would be required to titrate the blood sample to a pH of 7.4 at a Paco2 of 5.2 κPa at 37°C. A negative base excess indicates that there is excess acid in the blood; that is, a metabolic acidosis exists. Abnormalities of the standard bicarbonate and base excess provide a measure of the metabolic component of an acid-base disorder.
Primary, Compensated, and Mixed Acid-Base Disturbances
In addition to primary and compensated disturbances, two or more independent acid-base disturbances may occur simultaneously (e.g., respiratory acidosis due to respiratory failure and metabolic alkalosis due to potassium loss). These are termed mixed disturbances. To distinguish between compensated and mixed acid-base disturbances requires knowledge of the clinical context and of the expected changes in Paco2 and actual bicarbonate associated with compensation (Table 31-1). See stepwise evaluation of acid-base disturbances.
Table 31-1 Expected Responses to Primary Acid-Base Disorders
| Metabolic acidosis | Paco2 should equal 1.5 · bicarbonate concentration + 8. |
|---|---|
| Metabolic alkalosis | Paco2 should increase by 0.5 to 1.0 mmHg for each mmol/l gain in plasma bicarbonate above 25. |
| Acute respiratory acidosis | For every 10 mmHg increase in Paco2, actual bicarbonate should increase by 1 mmol/l. |
| Chronic respiratory acidosis | For every 10 mmHg increase in Paco2, actual bicarbonate should increase by 4 mmol/l, up to a maximum of 12 mmol/l. |
| Acute respiratory alkalosis | For every 10 mm Hg decrease in Paco2, actual bicarbonate should decrease by 2 mmol/l. |
| Chronic respiratory alkalosis | For every 10 mmHg decrease in Paco2, actual bicarbonate should decrease by 5 mmol/l. |
Note: Paco2 is indicated in mmHg. See text for details. The changes in actual bicarbonate that occur with acute respiratory acidosis and alkalosis are minor and result from shifts in the equilibrium demonstrated by Equation 31-3.
Consequences of Acidosis and Alkalosis
Acidemia
Respiratory acidosis causing acidemia is well tolerated down to a pH of about 7.1. Below a pH of 7.1, problems of impaired mentation, arrhythmias, and reduced myocardial contractility4 may develop. Reduced contractility may be explained in part by reduced cellular sensitivity to catecholamines.5 Patients with elevated pulmonary vascular resistance and right ventricular dysfunction may develop acute right ventricular failure because modest respiratory acidosis increases pulmonary vascular resistance.6 Acidosis is usually a consequence of severe respiratory, central nervous system, metabolic, or cardio-vascular abnormalities; it is these problems that demand urgent attention, rather than the acidosis per se.
ACID-BASE DISTURBANCES IN THE CARDIOTHORACIC INTENSIVE CARE UNIT
Respiratory Acidosis
Equation 1-17 shows that Paco2 depends on both alveolar ventilation and carbon dioxide production. Thus, respiratory acidosis may result from increased carbon dioxide production or inadequate alveolar ventilation. Increased carbon dioxide production arises as a result of increased metabolic rate caused by fever, sepsis, hyperthyroidism, and overfeeding—particularly when calories derive predominantly from carbohydrates. Excess carbon dioxide production rarely leads to respiratory acidosis alone because alveolar ventilation can increase, but it can exacerbate respiratory acidosis resulting from other causes.
Inadequate alveolar ventilation can arise from hypoventilation due to oversedation, residual neuromuscular blockade, or neurologic dysfunction. In a patient receiving mechanical ventilation, hypoventilation may occur because of inappropriate ventilator settings. One particular situation in which respiratory acidosis can develop unexpectedly is when lung compliance suddenly decreases (e.g., due to mucus plugging) in a patient receiving pressure-control ventilation (see Chapter 29).
Alveolar hypoventilation also arises when dead-space ventilation increases and minute ventilation fails to increase proportionately (see Eq. 1-16). This occurs when there is a ventilation-perfusion (V/Q) mismatch (see Chapter 1); lung units with a high V/Q ratio represent alveolar dead space. V/Q mismatch occurs with most acute and chronic lung diseases and imposes the requirement for increased minute ventilation to achieve a normal Paco2. However, many patients with acute respiratory compromise actually have a low Paco2 (i.e., respiratory alkalosis). This is because the minute ventilation increases out of proportion to the increase in alveolar dead space (see subsequent discussion under Respiratory Alkalosis). The presence of respiratory acidosis in the context of acute hypoxemic respiratory failure implies either reduced ventilatory drive (e.g., because of reduced level of consciousness) or significantly increased work of breathing (e.g., due to poor lung compliance), and it may indicate impending respiratory arrest. In contrast to the unstable nature of acute hypercarbic respiratory failure, chronic respiratory acidosis due to lung disease (e.g., chronic bronchitis) may remain stable for years.
Treatment of Respiratory Acidosis
When considering treatment for respiratory acidosis, the following issues must be considered:
Respiratory Alkalosis
Acute respiratory alkalosis results in a significant disturbance in pH. For instance, an increase in alveolar ventilation to twice baseline raises the pH by 0.23. Treatment is directed at the underlying condition. Respiratory alkalosis that develops during weaning from mechanical ventilation is discussed in Chapter 29.
Anion Gap
The anion gap is the difference between the measured cations and anions in plasma:
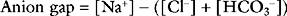
Normal anion gap acidosis results from either bicarbonate dilution (secondary to fluid resuscitation with 0.9% sodium chloride) or abnormal bicarbonate loss from the gut or the kidneys (renal tubular acidosis). With a normal anion gap metabolic acidosis, the reduction in bicarbonate is mirrored by an increase in chloride concentration. For this reason, a normal anion gap acidosis is sometimes called a hyperchloremic acidosis. Urinary pH should be low (usually less than 5) with a metabolic acidosis due to increased renal secretion of hydrogen ions. A urinary pH above 5.3 is suggestive of impaired urinary acidification, and usually indicates renal tubular acidosis. The causes of metabolic acidosis in the cardiothoracic ICU are summarized in Table 31-2.
| Increased anion gap | Ketoacidosis |
| Renal failure | |
| Lactic acidosis (see Table 31-3) | |
| Citrate | |
| Methanol, salicylate, other toxins | |
| Normal anion gap | Gastrointestinal bicarbonate loss |
| Renal tubular acidosis | |
| Excessive chloride administration (large-volume resuscitation with 0.9% sodium chloride) | |
| Postchronic hypocarbia |
Lactic Acidosis
In the presence of oxygen, glucose is metabolized into water and carbon dioxide within mitochondria. In the absence of oxygen, incomplete oxidation of glucose occurs, resulting in the production of lactate and hydrogen ions, which is known as lactic acidosis.7 (The incomplete oxidation of fatty acids results in the production of ketoacids.) Lactate production is a normal process that occurs in many tissues in the body, such as red blood cells, skin, brain, and muscle. In the liver, this lactate is metabolized to carbon dioxide and water or is used to produce glucose. Both processes consume hydrogen ions, and both generate bicarbonate (see Eq. 31-3). Thus, the lactate anion can be considered a precursor of bicarbonate. Lactate is normally present in low levels (<2 mmol/l) in plasma.
In the cardiothoracic ICU, lactic acidosis commonly occurs in patients with shock; a plasma lactate concentration of more than 5 mmol/l is a marker of increased likelihood of mortality.8
Lactic acidosis may also arise for other reasons, including:
Mesenteric infarction, hepatic ischemia, limb ischemia, and treatment with β2-agonists commonly coexist with shock. The causes of lactic acidosis in the cardiothoracic ICU are summarized in Table 31-3. In patients with renal failure receiving continuous hemofiltration, moderate hyperlactatemia (5 to 10 mmol/l) in the absence of acidemia is a normal finding when sodium lactate replacement fluid is used.
| Type A: Hypoxic | |
| Global ischemia | Shock |
| Severe anemia | |
| Severe hypoxemia | |
| Focal ischemia | Mesenteric infarction |
| Limb ischemia | |
| Type B: Nonhypoxic | |
| Delayed clearance of lactate | Hepatic or renal failure |
| Pyruvate dehydrogenase dysfunction | Sepsis, high catecholamine levels, thiamine deficiency |
| Uncoupling of oxidative phosphorylation | Protein breakdown, severe catabolism |
| Accelerated glycolysis | Sepsis, seizures, malignancies, parenteral nutrition |
| Lactate administration | Lactate-containing renal replacement fluid |
Treatment of lactic acidosis should be directed to the underlying cause. If a type A lactic acidosis is present, the cause of shock must be investigated and treated urgently. Therapy should be directed to increasing oxygen delivery and reducing oxygen demand. For unremitting lactic acidemia (pH <7.1) despite appropriate cardiovascular support, even in the absence of renal failure, renal replacement therapy with bicarbonate-buffered replacement solution may be indicated.9 Treatment with intravenous sodium bicarbonate is generally not indicated because it generates excess carbon dioxide, causes hyperosmolarity, worsens intracellular acidosis, may cause rebound metabolic alkalosis, and does not improve outcome.10,11 However, with severe lactic acidosis (pH <7.1), 50 to 100 mmol of IV sodium bicarbonate may temporarily arrest the fall of pH while other treatments are commenced. Other buffers, such as Tham (tris-hydroxymethyl aminomethane) or carbicarb, generate less carbon dioxide than sodium bicarbonate but have not been subjected to randomized clinical trials. In patients who are mechanically ventilated, increasing minute ventilation attenuates the fall in pH. However, this should not be undertaken at the cost of high airway pressures or large tidal volumes.
Normal Anion Gap Acidosis
The use of 0.9% sodium chloride as volume replacement can cause a hyperchloremic metabolic acidosis with a normal anion gap. Sodium chloride has a weak tendency to react with water-forming hydrochloric acid. Thus, the use of 0.9% sodium chloride to replace fluid losses, which effectively replaces bicarbonate with chloride, results in acidosis. Using the approach of Stewart (see Box 31-1), the strong ion difference is reduced, and the pH falls. If renal function is normal, this acidosis rapidly resolves as the kidneys regenerate bicarbonate and excrete excess chloride.
The cause of nonanion gap metabolic acidosis may be assessed by calculation of the urine anion gap:
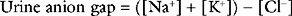
In contrast to lactic acidosis, treatment with sodium bicarbonate is indicated for normal anion gap metabolic acidosis.
Metabolic Alkalosis
Generation of Metabolic Alkalosis
In the cardiothoracic ICU, metabolic alkalosis most commonly arises for the following reasons:
The common causes of metabolic alkalosis are summarized in Table 31-4. Other causes, such as Conn or Cushing syndrome, occur rarely in a cardiac ICU.
| Alkali intake | Bicarbonate |
| Citrate (massive blood transfusion, anticoagulation for CVVH) | |
| Carbonates (antacids) | |
| Acetate (TPN, intermittent hemodialysis) | |
| Gastrointestinal acid loss | Vomiting |
| Nasogastric tube losses with gastroparesis | |
| Chloride-losing diarrhea | |
| Renal acid loss | Diuretics |
| Posthypercarbic alkalosis | |
| Hypovolemia | |
| Nonreabsorbable anions (e.g., penicillin) |
CVVH, Continuous venovenous hemofiltration; TPN, total parenteral nutrition.
Maintenance of Metabolic Alkalosis
The kidney has a great capacity to excrete excess bicarbonate, so maintenance of metabolic alkalosis requires excess renal bicarbonate retention. This occurs primarily in response to hypovolemia. Hypovolemia leads to activation of the renin-angiotensin-aldosterone system and a reduction in renal blood flow. In the distal nephron, aldosterone promotes sodium reabsorptions and excretion of either potassium or hydrogen ion (see Chapter 1). In hypokalemia, aldosterone augments the secretion of hydrogen ions (and therefore the production of plasma bicarbonate); in alkalosis, aldosterone promotes the loss of potassium. In the proximal nephron, hypovolemia results in enhanced sodium reabsorption via a sodium/hydrogen ion exchange mechanism. Thus, hypovolemia leads to abnormal hydrogen ion secretion (and therefore to bicarbonate reabsorption). To summarize: metabolic alkalosis is maintained by hypovolemia, particularly in the presence of hypokalemia. These mechanisms underlie an important physiologic principle: if necessary, circulating volume is defended in preference to acid-base and potassium homeostasis.
STEPWISE EVALUATION OF ACID-BASE DISTURBANCES
1 Stewart PA. Modern quantitative acid-base chemistry. Can J Physiol Pharmacol. 1983;61:1444-1461.
2 Halperin ML, Goldstein MB. Fluid, Electrolyte, and Acid-Base Physiology: A Problem-Based Approach. WB Saunders: Philadelphia, 1994.
3 Guyton AHJ. Textbook of Medical Physiology, ed 10, WB Saunders: Philadelphia, 2000.
4 Wildenthal K, Mierzwiak DS, Myers RW, et al. Effects of acute lactic acidosis on left ventricular performance. Am J Physiol. 1968;214:1352-1359.
5 Levraut J, Grimaud D, Offenstadt G, et al. Treatment of metabolic acidosis. Curr Opin Crit Care. 2003;9:260-265.
6 Fullerton DA, McIntyre RCJr, Kirson LE, et al. Impact of respiratory acid-base status in patients with pulmonary hypertension. Ann Thorac Surg. 1996;61:696-701.
7 Fall PJ, Szerlip HM. Lactic acidosis: from sour milk to septic shock. J Intens Care Med. 2005;20:255-271.
8 Luft D, Deichsel G, Schmulling RM, et al. Definition of clinically relevant lactic acidosis in patients with internal diseases. Am J Clin Pathol. 1983;80:484-489.
9 Honore PM, Jamez J, Wauthier M, et al. Prospective evaluation of short-term, high-volume isovolemic hemofiltration on the hemodynamic course and outcome in patients with intractable circulatory failure resulting from septic shock. Crit Care Med. 2000;28:3581-3587.
10 Mathieu D, Neviere R, Billard V, et al. Effects of bicarbonate therapy on hemodynamics and tissue oxygenation in patients with lactic acidosis: a prospective, controlled clinical study. Crit Care Med. 1991;19:1352-1356.
11 Cooper DJ, Walley KR, Wiggs BR, et al. Bicarbonate does not improve hemodynamics in critically ill patients who have lactic acidosis: a prospective, controlled clinical study. Ann Intern Med. 1990;112:492-498.
12 Siggaard-Andersen O, Fogh-Andersen N. Base excess or buffer base (strong ion difference) as measure of a non-respiratory acid-base disturbance. Acta Anaesthesiol Scand. 1995;107(suppl):123-128.